

Abstract
Campylobacter fetus is a causative agent of intestinal illness and, occasionally, severe systemic infections and meningitis. C. fetus currently comprises three subspecies: C. fetus subspecies fetus (Cff), C. fetus subspecies venerealis (Cfv), and C. fetus subspecies testudinum (Cft). Cff and Cfv are primarily associated with mammals whereas Cft is associated with reptiles.
To offer an alternative to laborious sequence-based techniques such as multilocus sequence typing (MLST) and polymerase chain reaction (PCR)-ribotyping for this species, the purpose of the study was to develop a typing scheme based on proteotyping.
In total, 41 representative C. fetus strains were analyzed by intact cell mass spectrometry and compared to MLST results. Biomarkers detected in the mass spectrum of C. fetus subsp. fetus reference strain LMG 6442 (NCTC 10842) as well as corresponding isoforms were associated with the respective amino acid sequences and added to the C. fetus proteotyping scheme.
In combination, the 9 identified biomarkers allow the differentiation of Cft subspecies strains from Cff and Cfv subspecies strains. Biomarkers to distinguish between Cff and Cfv were not found. The results of the study show the potential of proteotyping to differentiate different subspecies, but also the limitations of the method.
Keywords: MALDI-TOF MS, Campylobacter fetus, below species differentiation, ICMS, proteotyping, MLST
Introduction
Campylobacter spp. can cause gastrointestinal and extra-intestinal infections [1]. Although the majority of cases (≥90%) of intestinal campylobacteriosis are caused by Campylobacter jejuni and Campylobacter coli, a small number of these cases are also caused by Campylobacter fetus [2–5]. Among these, C. fetus is the most common cause of Campylobacter bacteremia. The frequency of detection in blood cultures varies between 19% and 53% [6–8] of all campylobacterioses. The reported case fatality rate of invasive C. fetus infections is at 14% [9]. Due to the high incidence rate of campylobacteriosis worldwide, this shows that C. fetus infections occur frequently and have the potential to become a significant public health issue. However, relatively little is known about the infection sources and the people at risk, so far. Most reported C. fetus infections were observed in AIDS patients and other immunocompromised individuals [1, 10].
C. fetus is a Gram-negative, microaerophilic bacterium, growing between 25 °C and 37 °C. Clinical symptoms of human C. fetus infection vary from acute diarrhea to systemic illness [11, 12], and the presentation of these symptoms depends on localization of the disseminated pathogen. Septicemia with fever, but without apparent localized infection, for example, is reported in 24% to 41% of cases [7, 9]. Other manifestations can be the result of neurological infections (i.e., meningoencephalitis, meningitis, or brain abscesses), arthritis, lung abscesses, osteomyelitis, and perinatal infections (i.e., abortion, infection in utero, or placentitis) [12]. Furthermore, C. fetus infections may also cause vascular pathology (i.e., endocarditis, pericarditis, vasculitis, and mycotic aneurysms) [13].
Currently, 3 subspecies of C. fetus are known. These are C. fetus subspecies fetus (Cff), C. fetus subspecies venerealis (Cfv), and C. fetus subspecies testudinum (Cft). For Cfv, also the biovar intermedius (Cfvi) has been identified in previous studies [14, 15]. Subspecies Cff and Cfv are primarily associated with mammals [13, 14], whereas the third subspecies Cft is associated with reptiles [15, 16]. Cff and Cfv are genetically very closely related [17, 18] but differ in host adaption. Cff can cause sporadic infections in humans and abortion in sheep and cattle and can be isolated from different sites in different hosts [19]. Occurrence of Cfv is restricted to the genital tract of cattle and is furthermore responsible for bovine genital campylobacteriosis (BGC). This syndrome is characterized by fertility problems in cattle [20]. Previous studies have demonstrated a substantial genetic divergence between strains of reptile and mammal origins [21, 22], and molecular and phenotypic characterization of human cases and 3 reptiles identified a new subspecies and proposed the name C. fetus subsp. testudinum subsp. nov. [15, 23].
In recent years, intact cell matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry (ICMS) became a standard method for microbial species identification in clinical diagnostic laboratories [24, 25]. MALDI-TOF mass spectrometry (MS) also offers the opportunity to classify unknown bacterial isolates by identifying similarities in mass spectra of unknown bacteria and biomarkers in existing databases, a procedure referred to as phyloproteomics [26]. Typing methods, which are based on mass spectrometric analysis, are generally known as proteotyping [27] and have previously been used for characterization of microbial communities, tissues, individual proteins, viruses, and bacteria for several years now [28–31]. Among clinically relevant bacteria Salmonella serotypes, Clostridioides difficile polymerase chain reaction (PCR) ribotypes and methicillin-resistant Staphylococcus aureus lineages have been shown to be detectable by proteotyping, to name just a few [32–34].
Previous studies of our working group demonstrated the potential of bacterial subtyping on Campylobacter species in the clinical context, as it was possible to differentiate clinically relevant from clinically less relevant subgroups (Figure 1) [35–39]. At the heart of our approach is a list of allelic isoforms that resulted from non-synonymous mutations and posttranslational modifications in biomarker gene sequences, which are detectable as mass shifts in MALDI-TOF spectra. In this way, a combination of amino-acid sequences specific for each of the isolates to be typed can be derived, in a similar manner as for multilocus sequence typing (MLST). By using proteotyping, only the changes in mass associated with a certain set of allelic isoforms of the same protein are taken into account for the derivation of phylogeny, whereas the visibility or absence of particular masses, as well as their intensity, is not considered. This improves the measurement accuracy, wherefore ICMS is a very promising subtyping approach and a realistic alternative to currently used sequence-based techniques [37].
Figure 1.
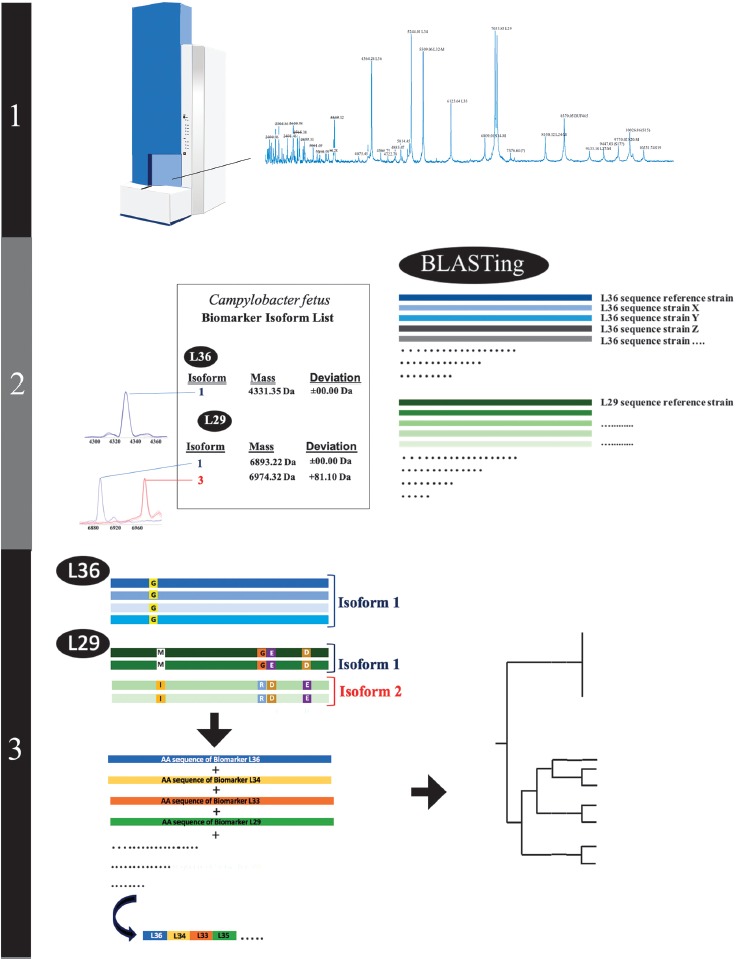
Illustration of the different proteotyping steps. 1) Recording of the ICMS mass spectra of the C. fetus test cohort and reference strain Cff LMG 6442 (NCTC 10842). 2) Establishment of a C. fetus-specific allelic isoform list by blasting the genome sequences obtained from the NCBI database against the genome of the C. fetus reference strain. Subsequently, allelic isoforms in the test cohort are identified by comparing with the newly established allelic isoform list. 3) For each strain in the test cohort, a specific set of biomarker isoforms is obtained. Subsequently the amino acid sequences of the biomarkers are fused into a single sequence that results in specific proteotyping-based sequence type for each of the strains and allows the calculation of an proteotyping-derived taxonomic dendrogram
The goal of this study was to complete the set of typing schemes for clinically relevant Campylobacter species by developing a C. fetus-specific proteotyping scheme. A set of 41 C. fetus isolates covering all currently known subspecies of C. fetus was used. All isolates were characterized by proteotyping and MLST, followed by the deduction of the phylogenetic relations.
Materials and Methods
C. fetus Isolates
The test cohort was compiled in way that all subspecies of the bacterial species were represented. In total, 41 C. fetus isolates were included in our study: 20 Cff, 11 Cfv, 7 Cft, and 3 Cfvi isolates (Table 1). The isolates were of different biological origins, namely, preputial washing of cattle (4 Cff, 7 Cfv), vaginal mucus of cattle (2 Cfv), fetuses of cattle (2 Cfv), cattle (not further specified, 3 Cfv), bovine sperm (1 Cff), bull genitals (1 Cff), calf fetus (2 Cff), intestinal content of a calf (1 Cff), intestinal content of a pig (1 Cff), fetus brain of a sheep (1Cff), reptile cloak swab (3 Cft), human blood culture (7 Cff, 4 Cft), and 2 Cff strains of unknown origin. Animal isolates were provided by the Friedrich-Loeffler-Institut Bundesforschungsinstitut für Tiergesundheit, Jena, Germany. The following strains were received from the Belgian coordinated collections of microorganisms (BCCM; http://bccm.belspo.be/about-us/bccm-lmg): LMG6443 (Cfv), LMG6442 (Cff), LMG6570 (Cfv), LMG27499 (Cft), LMG06569 (Cff), LMG06571 (Cff), and LMG06727 (Cff). Human blood culture isolates were provided by the routine diagnostic laboratory of the University Medical Center, Göttingen, Germany (Table 1).
Table 1.
List of C. fetus isolates used in the study
| Isolate | Origin | Region | Date | Other strain designations | MLST-ST |
|---|---|---|---|---|---|
| Cfv0018 | Preputial washing | Lower-Saxony | 28.04.2009 | 4 | |
| Cfv145/05 | Preputial washing | S-Bavaria | 02.08.2005 | 4 | |
| Cfv0114 | Vaginal sample cattle | Lower-Saxony | 19.12.2006 | 4 | |
| Cfv151/05 | Preputial washing | S-Bavaria | 10.08.2005 | 4 | |
| Cfv93/05 | Preputial washing | Thuringia | 10.05.2005 | 6 | |
| Cff94/05 | Preputial washing | Thuringia | 12.05.2005 | 6 | |
| Cfvi 96/05 | Preputial washing | Thuringia | 12.05.2005 | 4 | |
| Cff225/04 | Fetus calf | Thuringia | 16.12.2004 | 3 | |
| Cff512/99 | Calf intestinal content | Thuringia | 24.09.1999 | 5 | |
| Cfv63/05 | Preputial washing | N-Bavaria | 6.03.2005 | 4 | |
| Cfv11/05 | Preputial washing | N-Bavaria | 21.01.2005 | 4 | |
| CfvBS122/05 | Fetus, cattle | Baden-W. | 14.06.2005 | 4 | |
| Cfv07BS0007 | Preputial washing | Baden-W. | 26.09.2007 | 4 | |
| Cfv134/65 | Fetus cattle | S-Bavaria | 12.07.2005 | 4 | |
| Cff201/05 | - | Thuringia | 23.11.2005 | 2 | |
| Cff91/05 | Preputial washing | Thuringia | 12.05.2005 | 6 | |
| Cff155/60s | Preputial washing | Baden-W. | 07.09.2006 | 6 | |
| Cff222/04 | Bovine sperm | Saxony | 16.12.2004 | 2 | |
| Cff45361 | Human blood culture | Germany | 3 | ||
| Cff169361 | Human blood culture | Germany | 3 | ||
| Cff148/5361 | Human blood culture | Germany | 3 | ||
| CfvLMG6443 | Cow, vaginal mucus | United Kingdom | 1962 | ATCC 19438; CCUG 538; CIP 68.29; JCM 2528; NCDO 1876; NCTC 10354; Park X/161/5 |
4 |
| CffLMG6442 | Sheep fetus brain | Sweden, Göteborg | 1972 | ATCC 27374; CCTM La3023; CCUG 6823A; CECT 564; CIP 53.96; JCM 2527; LMG 8849; NCTC 10842; NIAH 1049; Vinzent strain Mouton 1 |
3 |
| CfvLMG6570 | Cattle | Belgium | 1985 | CCUG 7477; CIP 53.105; Florent 483; NIDO 483 |
4 |
| Cff71721 | Human blood culture | Germany, Duderstadt | 2016 | 3 | |
| Cff82014 | Human blood culture | Germany, Herzberg am Harz | 2015 | 68 | |
| CftLMG27499 | Human blood culture | USA New York | 2003 | ATCC BAA-2539; Blaser 03–427 | 15 |
| CffLMG06569 | Calf fetus | Belgium | 1985 | CCUG 17693; CIP 68.8; Florent 7572; NIDO 7572 |
11 |
| CffLMG06571 | Bull genitals | Belgium | 1985 | CCUG 17694; De Keyser 2125/4; NIDO 2125/4 |
3 |
| CffLMG06727 | Belgium | 1985 | CCUG 17695A; LMG 6628 t1 | 2 |
Bacterial Culture Conditions
C. fetus isolates used in the experiments were kept as cryobank stocks (Mast Diagnostica, Reinfeld, Germany) at –80 °C. For the subsequent MALDITOF MS analysis, the isolates were incubated under microaerophilic conditions (5% O2, 10% CO2, and 85% N2) in Mueller–Hinton agar supplemented with horse blood at 37 °C for 2–3 days.
Preparation of Matrix Solution
As part of the measurement preparation α-cyano-4-hydroxy-cinnamic acid (HCCA) purified matrix substance (Bruker Daltonics, Bremen, Germany) was dissolved in standard solvent (acetonitrile 50%, trifluoroacetic acid 2.5% in ddH2O) to 10 mg HCCA/mL. Purified recombinant human insulin (Sigma-Aldrich, Taufkirchen, Germany) was added to the HCCA solution as an internal calibrant to a final concentration of 10 pg/μL. The exact mass of the internal calibrant was experimentally determined (m/z = 5806.1) with reference to the Bruker Test Standard (BTS). The calibrant did not overlap with any of the biomarker masses of interest and allowed a very precise internal mass calibration of the spectra.
MALDI-TOF Mass Spectrometry
To prepare samples for the measurements, 2 different variants were used: smear preparation and formic acid/acetonitrile extraction. Smear preparation by experience yields clearer peaks in the m/z range <10,000 Da, whereas the extraction variant allows more precise analysis in the field >10,000 Da [39].
The samples for the measurements were prepared as described before [37, 39]. In the measurement process, 600 spectra (mass range 2 to 20 kDa) were obtained in 100-shot steps on an Autoflex III system and summed up. If the MALDI Biotyper (Database release 2016) identification score values were ≥2.00, they were considered correct.
Identification of Biomarkers in ICMS Spectra
The obtained mass spectra were analyzed by standard algorithms of FlexAnalysis (Bruker Daltonics, Bremen, Germany). Initially, spectra were internally calibrated to the spiked human insulin peak. Subsequently, the baseline was subtracted, and the spectra were smoothened (standard MBT method).
For determination of the theoretical average weight of the amino acid sequences corresponding to the respective open reading frames of ribosomal proteins, the amino acid sequences were uploaded one by one to the ExPASy Bioinformatics Resource Portal (https://web.expasy.org/compute_pi/), where a molecular weight calculator tool is provided.
Proteins used for previous proteotyping schemes sometimes underwent posttranslational modifications [40, 41]; therefore, further molecular weights were calculated for each biomarker, taking into account potential proteolytic removal of the N-terminal methionine (–131.04 Da), acetylation, phosphorylation, formylation, and methylation (Table 2).
Table 2.
Theoretical biomarker masses predicted by the genome sequence of C. fetus reference strain LMG 6442 (NCTC 10842) under consideration of possible posttranslational modifications
| Biomarker | [-Met M + H+] | [-Met mM + H+] | [-Met + PO4 M + H+] | [M + H+] | [fM + H+] |
|---|---|---|---|---|---|
| L36 | 4197 | 4211 | 4277 | 4332 | 4360 |
| L34 | 5083 | 5097 | 5163 | 5218 | 5246 |
| L32-M | 5527 | 5541 | 5607 | 5662 | 5690 |
| L33-M | 6202 | 6216 | 6282 | 6337 | 6365 |
| S14-M | 6725 | 6739 | 6805 | 6860 | 6888 |
| L29 | 6759 | 6773 | 6839 | 6894 | 6922 |
| L24-M | 8023 | 8037 | 8103 | 8158 | 8186 |
| S20-M | 9738 | 9752 | 9818 | 9873 | 9901 |
| S19-M | 10,274 | 10,288 | 10,354 | 10,409 | 10,437 |
[-Met mM + H+] = methylated mass - demethioninated form.
[-Met +PO4 M + H+] = phosphorylated mass - demethioninated form.
[M + H+] = unmodified mass.
[fM + H+] = formylated mass.
Biomarker masses observed in the reference genome of reference strain LMG 6442 (NCTC 10842) (Figure 2) were matched to the calculated masses. In contrast, biomarker masses are observed in the spectrum of clinical isolates, which could not be assigned to the calculated masses from the C. fetus reference genome, and the spectra were considered as novel isoforms of the particular biomarker.
Figure 2.
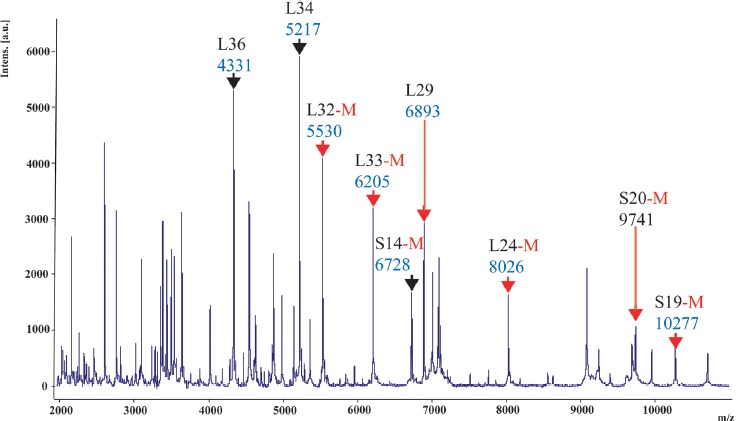
ICMS spectrum of C. fetus subsp. fetus reference strain LMG 6442 (NCTC 10842). Singularly charged biomarkers that were part of the C. fetus proteotyping scheme labeled with a black arrow or, in the case of an N-terminal methionine cleavage (posttranslational modification) with a red arrow. Multiple charged ions are not marked in this illustration
For each isolate of the C. fetus test cohort, all biomarker genes were amplified by PCR using primers listed in Table 3, and the amplicon was sequenced (Microsynth Seqlab, Göttingen, Germany). To confirm the respective allelic isoforms, the gene sequences obtained from the amplicons were translated in silico, and the amino acid sequences were subsequently aligned.
Table 3.
Primers used for sequencing of C. fetus genes coding for ribosomal proteins included in the proteotyping scheme
| Biomarker | Gene | Forward primer [5′→3′] | Reverse primer [5′→3′] | Amplicon length [bp] |
|---|---|---|---|---|
| L36 | RpmJ | CGGGTGATCGCGTTAAAGTT | TACGAATCGCAGCAGCTTCA | 522 |
| L34 | RpmH | AGTTATGCCGCAAACACCTAT | TTTTCAAGCCCTGCTTTTGCT | 699 |
| L32-M | RpmF | ACCACTATTGTGATAGATGCGGT | ACATCAGTAGCACTTTCTCCCA | 596 |
| L33-M | RpmG | CCCAGTTGCACTTGAAGAAGG | ACGATCGCTACAACAGCAAAT | 539 |
| S14-M | RpsN | AGGACTTCCGTGGTCTTCCA | ACGCTTCTACCACGTTCGTC | 624 |
| L29 | RpmC | CGCCAGATAGAATCAGCTCGT | GCGGAAGCTTTTTCTAGCAC | 701 |
| L24-M | RpIX | TTTGACGAAAATGCAGCCGT | ACTGGGAAGCCTTCACGAAC | 621 |
| S20-M | RpsT | TTCTCCGGCTCTGCCTCTAA | GCGAGTTCGCCTAGTTCTGG | 736 |
| S19 | RpsS | GGGCAAACGTAACTATCGGC | GAACAGGACCGGCATCTACT | 752 |
Multilocus Sequence Typing (MLST)
For MLST, a procedure modified from the original typing schemes was used [18, 23]. In brief, the annealing temperature of the PCR was decreased from 48 °C to 47 °C, and the glyA2 oligonucleotide primer for the amplification of the glyA locus was replaced with the primer glyS4 [18]. After concatenating of the MLST gene sequences for each strain, the software MEGA X was also used to construct an MLST-based UPGMA dendrogram [42].
Phylogenetic and Phyloproteomic Analyses
An amino acid sequence list of all allelic isoforms of the 9 identified biomarkers was compiled (Table 4). GenBank accession numbers for the biomarker sequences observed in this study are listed in Table 5.
Table 4.
C. fetus-specific allelic isoform list
| Locus | Full name/product (ORF Locus tag in LMG 6442) |
Calc. average mass [Da] |
Frequency in database | ||
|---|---|---|---|---|---|
| RpmJ/L36 | |||||
| Sequence | MKVRPSVKKMCDKCKIVKRKGIVHVICENPKHKQRQG (37aa) | ||||
| 1a | reference isoform LMG 6442 (NCTC 10842) | 4331.35 | ±0.00 | 100.000% (67/67) | |
| RpmH/L34 | |||||
| Sequence | MKRTYQPHKTPKKRTHGFRGRMKTKNGRKVINARRAKGRKRLAA (44aa) | ||||
| 1a | reference isoform LMG 6442 (NCTC 10842) | 5217.26 | ±0.00 | 47.761% (32/67) | |
| 2a | MKRTYQPHKTPKKRTHGFRERMKTKNGRKVINARRAKGRKRLAA(44aa) | 5289.32 | +72.06 | 32.836% (22/67) | |
| 3 | MKRTYQPHKTPKKRTHGFRERMRTKNGRKVLNARRAKGRKRLAA(44aa) | 5317.33 | +100.07 | 19.403% (13/67) | |
| RpmF/L32-M] | |||||
| Sequence | (M)AVPKRRVSHTRAAKRRTHYKVTLPMPVKDKDGSWKMPHRINKTTGEYa (48aa) | ||||
| 1a | reference isoform LMG 6442 (NCTC 10842) | 5530.47 | ±0.00 | 77.941% (53/68) | |
| 2 | (M)AVPKRRVSHTRAAKRRTHYKVTLPMPVKDKDGSWKMPHRMNKTTGEY (48aa) | 5548.50 | +18.03 | 17.647% (12/68) | |
| 3 | (M)AVPKRRVSHTRAAKRRTHYKVTLPMPVKDKDGSWKMPHRINKITGEY (48aa) | 5542.52 | +12.05 | 1.471% (1/68) | |
| 4 | (M)AVPKRRVSHTRAAKCRTHYKVTLPMPVKDKDGSWKMPHRINKTTGEY (48aa) | 5477.42 | –53.05 | 1.471% (1/68) | |
| 5 | (M)AVPKRLVSHTRAAKRRTHYKVTLPMPVKNKDGSWKMPHRINKTTGEY (48aa) | 5486.45 | –44.02 | 1.471% (1/68) | |
| 6a | (M)AVPKRRVSHTRAAKRRTHYKITLPMPVKDKDGSWKMPHRINKTTGEY (48aa) | 5544.49 | +14.02 | New sequence | |
| RpmG/L33-M | |||||
| Sequence | (M)ASANRVKIGLKCAECNDINYTTTKNSKTTTEKLELKYCPRLKKHTVHKEVKLK (55aa) | ||||
| 1a | reference isoform LMG 6442 (NCTC 10842) | 6205.31 | ±0.00 | 46.970% (31/66) | |
| 2 | (M)ASANRIKIGLKCVECGDINYTTTKNSKKTTEKLELKKYCPRLKKHTEHKEVKLK (55aa) | 6247.39 | +42.08 | 18.182% (12/66) | |
| 3a | (M)ASANRVKIGLKCAECNDINYTTTKNSKKTTEKLELKKYCPRLKKHTVHKEVKLK (55aa) | 6232.38 | +27.07 | 33.333% (22/66) | |
| 4 | (M)ASVNRIKIGLKCVECGDINYTTTKNSKKTTEKLELKKYCPRLKKHTEHKEVKLK (55aa) | 6275.44 | +70.13 | 1.515% (1/66) | |
| 5a | (M)ASANRVKIGLKCAECNDINYTTTKNSKTTTEKSELKKYCPRLKKHTVHKEVKLK (55aa) | 6179.23 | –26.08 | New sequence | |
| RpsN/S14-M | |||||
| Sequence | (M)AKKSMIAKAARKPKFSARGYTRCQICGRPHSVYKFGICRVCLRKMANEGLIPGLKKASW (61aa) | ||||
| 1a | reference isoform LMG 6442 (NCTC 10842) | 6728.11 | ±0.00 | 80.303% (53/66) | |
| 2 | (M)AKKSMIAKAARKPKFSVRGYTRCQICGRPHSVYKDFGICRVCLRKMANEGLIPGLKKASW (61aa) | 6756.16 | +28.05 | 3.030% (2/66) | |
| 3 | (M)AKKSMIAKAARAPKFSSRGYTRCQICGRPHSVYKDFGICRVCLRKMANEGLIPGLKKASW (61aa) | 6687.01 | +41.10 | 16.667% (11/66) | |
| RpmC/L29 | |||||
| Sequence | MKYIDISAKSMSELNALLKEKKVLLFTLRQKLKTMQLTNPNEIGETKKDIARINTAISAAK (61aa) | ||||
| 1a | reference isoform LMG 6442 (NCTC 10842) | 6893.22 | ±0.00 | 47.761% (32/67) | |
| 2 | MKYTEISAKSVSELTALLKEKKVLLFTLRQKLKTMQLTNPNEIRDTKKEIARINTAI SAAK (61aa) | 6949.27 | +56.05 | 19.403% (13/67) | |
| 3a | MKYIDISAKSISELNALLKEKKVLLFTLRQKLKTMQLTNPNEIRDTKKEIARINTAISAAK(61aa) | 6974.32 | +81.10 | 32.836% (22/67) | |
| RplX/L24-M | |||||
| Sequence | (M)AVKYKIKKGDEVKVIAGDDKGKVAKVIAVLPKKGQVIVEGVKVAKKAVKPTEKNPNGGFISKEMPIDISNVAKVEG (77aa) | ||||
| 1a | reference isoform LMG 6442 (NCTC 10842) | 8026.59 | ±0.00 | 47.751% (32/67) | |
| 2 | (M)AIKYKIKKGDEVKVIAGDDKGKVAKVLAVLPKKGQVIVEGVKVAKKAVKPTDKNPNGGFVSKEMPIDISNVAKVEG (77aa) | 8012.56 | –14.03 | 17.910% (12/67) | |
| 3a | (M)AVKYKIKKGDEVKVIAGDDKGKVAKVIAVLPKKGQVIVEGVKVAKKAVKPTDKNPNGGFISKEMPIDISNVAKVEG (77aa) | 8012.56 | –14.03 | 32.836% (22/67) | |
| 4 | (M)AIKYKIKKGDEVKVIAGDDKGKVAKVLAVLPKKGQVIVEGIKVAKKAVKPTDKNPNGGFVSKEMPIDISNVSKVEG (77aa) | 8042.59 | +16.00 | 1.493% (1/67) | |
| RpsT/S20-M | |||||
| Sequence | (M)ANHKSAEKRARQTIKRTERNRFYRTRLKNLTKAVRVAVASGDKDAALVALKDANKNFHSFVSKGFLKKETASRKVSRLAKLVSTLAA (88aa) | ||||
| 1a | reference isoform LMG 6442 (NCTC 10842) | 9741.33 | ±0.00 | 48.529% (33/68) | |
| 2 | (M)ANHKSAEKRARQTIKRTERNRFYRTRLKNLTKAVRVAVANGDKDAALLALKDVNKNFHSFVSKGFLKKETASRKVSRLAKLVSTLAA (88aa) | 9810.43 | +69.10 | 16.176% (11/68) | |
| 3 | (M)ANHKSAEKRARQTIKRTERNRFYRTRLKNLTKAVRVAVANGDKDAALLALKDVNKNFHSFVSKGFLKKKTASRKVSRLAKLVSTLAA (88aa) | 9809.49 | +68.14 | 1.471% (1/68) | |
| 4a | (M)ANHKSAEKRARQTIKRTERNRFYRTRLKNLTKAVRVAVTSGDKDAALLALKDVNKNFHSFVSKGFLKKETASRKVSRLAKLVSTLAA (88aa) | 9813.43 | +72.10 | 32.353% (22/68) | |
| 5 | (M)ANHKSAEKRARQTIKRTERNRFYRTRLKNLTKAVRVAVANGDKDAALLALKDVNKNFHSFVSKGFLKKETASRKVGRLAKLVSTLAA (88aa) | 9780.41 | +39.08 | 1.471% (1/68) | |
| RpsS/S19-M | |||||
| Sequence | (M)ARSLKKGPFVDDHVMKKVLAAKAANDNKPIKTWSRSMIIPEMIGLFNVHNGKGFIPVYVTENHIGYKLGEFAPTRTFKGHKGSQKKIGK (93aa) | ||||
| 1a | reference isoform LMG 6442 (NCTC 10842) | 10277.10 | ±0.00 | 47.761% (32/67) | |
| 2 | (M)ARSLKKGPFVDDHVMKKVLAAKAANDNKPIKTWSRRSTIIPEMIGLTFNVHNGKSFIPVYVTENHIGYKLGEFAPTRTFKGHKGSVQKKIGK (93aa) | 10277.04 | –0.06 | 14.925% (10/67) | |
| 3a | (M)ARSLKKGPFVDDHVMKKVLAAKAANDNKPIKTWSRRSMIIPEMIGLTFNVHNGKSFIPVYVTENHIGYKLGEFAPTRTFKGHKGSVQKKIGK (93aa) | 10307.12 | +30.02 | 32.836% (22/67) | |
| 4 | (M)ARSLKKGPFVDDHVMEKVLAAKATNDNKPIKTWSRRSTIIPEMIGLTFNVHNGKSFIPVYVTENHIGYKLGEFAPTRTFKGHKGSVQKKIGK (93aa) | 10308.00 | +30.90 | 4.478% (3/67) | |
aObserved in test population AA numbering including start-methionine, if mass spectrometry indicates its absence it is written in brackets (M).
Table 5.
Accession numbers of C. fetus-specific proteotyping biomarker isoforms
| Biomarker | Isoform | Gene Bank Accession | Locus Tag | Protein ID |
|---|---|---|---|---|
| L36 | 1 | MK463617 | ||
| L34 | 1 | CP000487.1:557520–557,654 | CFF8240_0551 | ABK82017.1 |
| L34 | 2 | CP027287.1:608973–609,107 | C6B32_03095 | AVK80859.1 |
| L32-M | 1 | CP000487.1:210702–210,848 | CFF8240_0235 | ABK81894.1 |
| L32-M | 6 | MK463615 | ||
| L33-M | 1 | CP000487.1:1313847–1,313,949 | CFF8240_1324 | ABK82614.1 |
| L33-M | 3 | CP027287.1:c1398913–1,398,746 | C6B32_06940 | AVK81560.1 |
| L33-M | 5 | MK463616 | ||
| S14-M | 1 | CP000487.1:39526–39,711 | CFF8240_0047 | ABK82398.1 |
| L29 | 1 | CP000487.1:37925–38,110 | CFF8240_0042 | ABK82084.1 |
| L29 | 3 | CP027287.1:36898–37,083 | C6B32_00200 | AVK80319.1 |
| L24-M | 1 | CP000487.1:38746–38,979 | CFF8240_0045 | ABK83333.1 |
| L24-M | 3 | CP027287.1:37719–37,952 | C6B32_00215 | AVK80322.1 |
| S20-M | 1 | CP000487.1:1678191–1,678,457 | CFF8240_1718 | ABK82453.1 |
| S20-M | 4 | CP027287.1:1762618–1,762,884 | C6B32_08820 | AVK81906.1 |
| S19-M | 1 | CP000487.1:36187–36,468 | CFF8240_0038 | ABK81869.1 |
| S19-M | 3 | CP027287.1:35160–35,441 | C6B32_00180 | AVK80315.1 |
To analyze the biomarkers' protein sequences translated from the National Center for Biotechnology Information (NCBI) nucleotide database (Geneious V10.1.3) they were concatenated for each strain and an unweighted pair group method with arithmetic mean (UPGMA) dendrogram (MEGA X) was constructed [42].
Ethical Approval
Ethical approval for the study was obtained from Ethics Commission of the University Medical Center Göttingen, Germany. No humans, animals, or personalized data were used for this study.
Results and Discussion
In 2015, our working group set up a new proteotyping workflow for the proteotyping of microorganisms (Figure 1) [37]. Now, the established procedure was used to develop a C. fetus-specific proteotyping scheme. According to the standard workflow, masses emerging in the mass spectrum of the genome sequenced Cff reference strain LMG 6442 (NCTC 10842) were analyzed, and MS biomarker ions were related with gene products consistent with the observed mass. By evaluating the 67 C. fetus nucleotide sequences available in the NCBI database, a collection of allelic isoforms for all biomarkers observed in the reference spectrum was set up (Table 4). In accordance with the established proteotyping procedure, mass spectra of all strains included in the test cohort were recorded. Subsequently, spectra were edited (baseline subtraction and smoothing) and overlaid with the spectrum of Cff reference strain LMG 6442 (NCTC 10842). Recorded biomarker masses were matched with the calculated average protein masses, and mass shifts in relation to the masses of the references strain were analyzed. After concatenation of amino acid sequences of the biomarkers included in the C. fetus typing scheme, a UPGMA tree based on these strain-specific proteotyping-based types was calculated.
Identification of Biomarker Ions
In total, the analysis based on the genome of Cff reference strain LMG 6442 (NCTC 10842) yielded nine, single charged biomarker masses between m/z = 4300 and 10,300, which were presumptively correlated with a specific gene product. To provide reliable statements on reproducibility of our measurements, the standard deviation was calculated on the basis of 6 measurements. The highest standard deviation (0.959) was observed for isoform 1 of biomarker S20-M, whereas the lowest standard deviation (0.271) was observed for isoform 5 of biomarker L33-M (Table 6). The following biomarkers were identified: L36 (4331.35 Da), L34 (4217.26 Da), L32-M (5530.47 Da), L33-M (6205.31 Da), S14-M (6728.11 Da), L29 (6893.22 Da), L24-M (8026.59 Da), S20-M (9741.33 Da), and S19-M (10,277.10 Da). De-methionation was observed for biomarkers L32-M, L33-M, S14-M, L24-M, S19-M, and S20-M (Table 2, Figures 2 & 3). In the case of MLST, the established markers are distributed over the whole genome of the reference strain. As the biomarkers identified in this study show a comparable distribution, they were suitable for the deduction of phylogenetic relations.
Table 6.
Measured and calculated biomarker masses
| Biomarker | Isoform | Measured mass (Da) | Standard deviation | Δ Measured mass/average mass | Monoisotopic mass (Da) | Average mass (Da) |
|---|---|---|---|---|---|---|
| L36 | Isoform 1 | 4331 | 0.765 | 0.35 | 4328.40 | 4331.35 |
| L34 | Isoform 1 | 5217 | 0.425 | 0.26 | 5214.02 | 5217.26 |
| L34 | Isoform 2 | 5290 | 0.593 | 0.68 | 5286.04 | 5289.32 |
| L32-M | Isoform 1 | 5530 | 0.478 | 0.47 | 5526.99 | 5530.47 |
| L32-M | Isoform 6 | 5544 | 0.475 | 0.49 | 5541.01 | 5544.49 |
| L33-M | Isoform 1 | 6205 | 0.867 | 0.31 | 6201.37 | 6205.31 |
| L33-M | Isoform 3 | 6232 | 0.381 | 0.38 | 6228.42 | 6232.38 |
| L33-M | Isoform 5 | 6179 | 0.271 | 0.23 | 6175.32 | 6179.23 |
| S14-M | Isoform 1 | 6728 | 0.445 | 0.11 | 6854.63 | 6728.11 |
| L29 | Isoform 1 | 6893 | 0.321 | 0.22 | 6888.84 | 6893.22 |
| L29 | Isoform 3 | 6975 | 0.877 | 0.68 | 6969.96 | 6974.32 |
| L24-M | Isoform 1 | 8026 | 0.928 | 0.59 | 8021.62 | 8026.59 |
| L24-M | Isoform 3 | 8012 | 0.620 | 0.56 | 8007.61 | 8012.56 |
| S20-M | Isoform 1 | 9741 | 0.959 | 0.33 | 9735.50 | 9741.33 |
| S20-M | Isoform 4 | 9813 | 0.361 | 0.43 | 9807.56 | 9813.43 |
| S19-M | Isoform 1 | 10,277 | 0.499 | 0.10 | 10270.58 | 10277.10 |
| S19-M | Isoform 3 | 10,308 | 0.635 | 0.88 | 10300.59 | 10307.12 |
Figure 3.
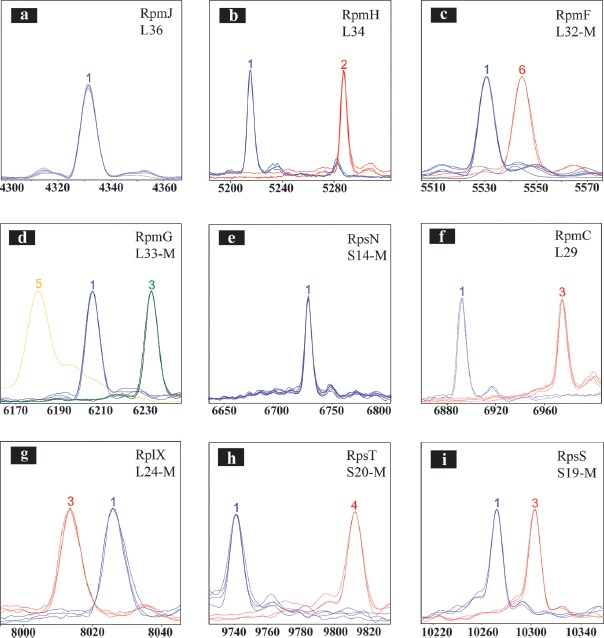
Overview of C. fetus proteotyping biomarkers (a–i). To illustrate the observed mass differences of the allelic isoforms, spectra of different proteotyping-based sequence types were overlaid using the FlexAnalysis evaluation tool. X-Axis: mass [Da]–charge ratio, scale 200 Da. Y-Axis: intensity [10x arbitrary units]. For the graphical illustration, the peak intensity of high but respectively lower peaks was adjusted. Color code of the spectra: Spectra of strains with the isoform of C. fetus subsp. fetus reference strain LMG 6442 (NCTC 10842) are blue, whereas the differing isoforms are colored red, green, and yellow. If the N-terminal methionine of a ribosomal protein was cleaved, the respective illustration is provided with an “-M”
Comparing C. fetus proteotyping biomarkers to biomarkers identified within the context of C. jejuni subsp. jejuni, C. jejuni subsp. doylei, and C. coli proteotyping [37–39], several differences can be noted: In the case of C. jejuni subsp. jejuni, 19 biomarkers were identified and associated with the respective peak in the ICMS spectrum, whereas less than half (9) were found for C. fetus. Furthermore, biomarker L33 lacked N-terminal methionine in the case of C. fetus (L33-M) but it was present in C. jejuni subsp. jejuni, C. jejuni subsp. doylei, and C. coli. These observations confirm the results published by Fagerquist et al., in which posttranslational modification patterns are microbial species-specific. Within the isolate collection, biomarker mass shifts were observed in 7 out of 9 biomarkers [43].
Establishment of an Allelic Isoform Database
Following the identification of biomarker ions, an amino acid sequence isoform list for each of the biomarkers identified in the previous step was compiled. In this context, we analyzed the 67 C. fetus genome sequences that can be found on NCBI. The number of identified isoforms for the respective biomarker varied. The highest number was 6, whereas 1 biomarker showed just a single isoform. Differences were also observed regarding frequency of occurrence; whereas some isoforms occurred in >99% of the cases, other isoforms were only found once. Regarding single occurrence of isoforms, a sequencing error is possible. Except for biomarker L36, all identified biomarkers showed at least 3 different isoforms, demonstrating their suitability in the C. fetus subtyping context.
The amino acid sequences of all biomarker isoform are listed in Table 4. Variations of the amino acid sequences obtained by alignment of the sequences are indicated in red; additionally, the computed average protein mass for each isoform is listed. It should be noted that due to some draft genomes in GenBank, the number of available sequences may vary, as there were no contigs with the sequences coding for each biomarker in all genomes.
MLST and Proteotyping of the Isolate Collection
To proof functionality of the C. fetus proteotyping scheme the test cohort (41 C. fetus strains) was typed by MLST, as well as proteotyping. The composition of the test cohort was such that all known subspecies of the species were covered. The isolate collection comprised the following 14 MLST sequence types: ST2 (3 isolates), ST3 (7 isolates), ST4 (14 isolates), ST5 (2 isolates), ST6 (4 isolates), ST11 (1 isolate), ST15 (2 isolates), ST16 (1 isolate), ST20 (2 isolates), ST27 (1 isolate), ST30 (1 isolate), ST31 (1 isolate), ST66 (1 isolate), and ST68 (1 isolate, Table 1).
The concatenated amino acid sequences of the different biomarkers yielded four proteotyping-derived types (Figure 4, right dendrogram). Proteotyping-derived type A comprised most of the Cff and Cfv isolates (31/41). More precisely, it comprised 3 MLST-ST2 isolates, 7 MLST-ST3 isolates, 14 MLST-ST4 isolates, 4 MLST-ST6 isolates, 1 MLST-ST11 isolate, and 1 MLST-ST68 isolate.
Figure 4.
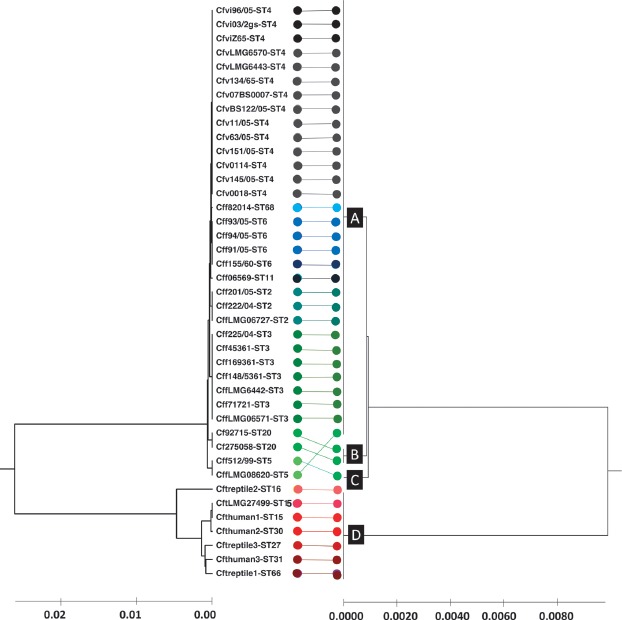
Comparison of MLST- and proteotyping-derived phylogenies. On the left: Evolutionary tree calculated based on MLST by means of the maximum composite likelihood method (UPGMA). In total, 14 different MLST sequence types were identified which are illustrated in different colors. On the right: Evolutionary tree based on proteotyping and calculated using UPGMA. Four different proteotyping-derived types were identified. Type A contains most of the C. fetus subsp. fetus and C. fetus subsp. venerealis strains. Type B and C contain 2 MLST ST 5 and one MLST ST 20 strain. The most interesting proteotyping-derived type is type D, which contains all C. fetus subsp. testudinum strains and thereby allows the differentiation of the subspecies from other C. fetus subspecies. The different proteotyping-based sequence types are marked at the branches of the evolutionary tree (A, B, C, and D).
Proteotyping-derived type B consisted of two Cff MLST-ST 20 isolates, while proteotyping-derived type C consisted of one MLST-ST5 isolate (Cff).
The most interesting findings were that proteotyping-derived type D consisted only of Cft isolates. Regarding MLST sequence types, it comprised particularly 1 isolate of ST16, 2 isolates of ST15, 1 isolate of ST27, 1 isolate of ST30, 1 isolate of sequence type 31, and 1 isolate of ST61.
Identification of Allelic Isoforms
The test cohort was measured in exactly the same manner as it was done for the reference strain LMG 6442 (NCTC 10842). The evaluation of the measurements of mass spectra of the strains was done based on the comparison with the spectrum of this reference strain. Observed mass shifts were compared to the sequence list of amino acid isoforms, whereby a particular allelic isoform could be identified.
If two different isoforms with the same mutation at different positions were observed, which though did not differ regarding mass difference to the reference isoform, the variants were further examined by DNA sequencing. In the test cohort, 3 allelic isoforms for biomarker L33-M (RpmG) and 2 for biomarkers L34 (RpmH), L32-M (RpmF), L29 (RpmC), L24-M (RplX), S20-M (RpsT), and S19-M (RpsS) were detected. For biomarkers L36 (RpmJ) and S14-M (RpsN), only one allelic isoform was identified (Table 2, Figure 3).
Construction of an UPGMA-Dendrogram
To deduce the phylogenetic relationships of the species, amino acid sequences of the 9 identified proteotyping biomarkers were fused into a single sequence. The concatenated sequence was then further processed with the MEGA X software to calculate a phyloproteomic tree (UPGMA). The 9 identified biomarkers allowed a clear differentiation of a group of Cff and Cfv strains from a group of utterly Cft strains. In order to assess the quality of the proteotyping results, another UPGMA tree was calculated based on MLST data (Figure 4). Comparative analysis of the trees revealed some differences between the two resulting phylogenies. While the test cohort was differentiated into 14 MLST sequence types, the proteotyping-based analysis led to a division into only 4 different groups. The most interesting finding was that proteotyping-based type D comprised all of the Cft isolates, showing that our approach here is comparable to the quality of the current gold standard MLST.
Unfortunately, the MLST-ST4 corresponding to the subspecies Cfv could not be differentiated by means of proteotyping. Here, proteotyping proves to be inferior to MLST in its discriminatory resolution.
A previous study by Fitzgerald et al. showed that it is possible to distinguish Cft from other C. fetus subspecies. Based on multiple unidentified biomarker peaks, a dendrogram was calculated using Pearson correlation [15]. A factor, which reduces the informative value of these results, was the lack of knowledge about the proteins responsible for each of the discriminating peaks.
In contrast to this study, we were able to identify at least 9 defined ribosomal proteins as biomarkers. As Cft strains exhibited different biomarker isoforms compared to the other two C. fetus subspecies, they could be clearly differentiated. PCR and subsequent Sanger sequencing of the respective biomarkers further confirmed these differences.
Regarding the limitations of proteotyping, the number of sequence data available is decisive for the quality of the typing scheme. In the case of C. fetus, much less sequences (67) were available as compared to C. jejuni subsp. jejuni (more than 3000) [37]. Another factor affecting the quality of the typing scheme is the number of biomarkers it comprises. Further studies should therefore focus on the identification of additional reliable biomarkers that can be included in the existing scheme.
The prerequisite for the application of the technique is the visibility of all biomarkers of the typing scheme. If this is not the case, it is advisable to use sequence-based techniques.
Conclusion
As the results obtained so far demonstrate, proteotyping is a promising tool for microbial typing at the species, subspecies, and even below subspecies levels. A smart bioinformatics solution and the development of an easy-to-handle user interface would allow the application of the technique in daily diagnostic routine, as the corresponding equipment for proteotyping is available in modern clinical laboratories anyway. The rapidly growing sequence databases due to next generation sequencing (NGS) are opening up a wide range of opportunities for the development of further proteotyping schemes that possibly allow a rapid detection in the case of a disease outbreak.
Footnotes
Funding Sources
This work was funded by the Federal State of Lower Saxony, Niedersächsisches Vorab (VWZN2889/3215/3266). The Open Access Support Program of the Deutsche Forschungsgemeinschaft and the publication fund of the Georg-August-Universität Göttingen funded publication of this paper.
Authors' Contributions
M.F.E. and M.K. contributed equally to this work. M.F.E., O.B., and A.E.Z. wrote the manuscript and established the biomarker isoform database in silico. M.K. performed MALDI measurements and confirmatory PCRs. H.H., L.vd.G.B. and A.E.Z. collected bacterial isolates and performed data interpretation, bioinformatics, and correction of the manuscript. O.B., U.G. and A.E.Z. designed the experiments and evaluated the data.
Conflicts of Interest
There are no conflicts of interest.
References
- 1.Forbes BA, Sahm DF, Weissfeld AS. Diagnostic microbiology. Bailey & Scott’s Diagnostic Microbiology; 2002. pp. 11–14. [Google Scholar]
- 2.Gillespie IA, O'Brien SJ, Frost JA, Adak GK, Horby P, Swan AV, et al. A Case-Case Comparison of Campylobacter coli and Campylobacter jejuni Infection: A Tool for Generating Hypotheses. Emerg Infect Dis. 2002;8:937–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nielsen H, Hansen KK, Gradel KO, Kristensen B, Ejlertsen T, Østergaard C, et al. Bacteraemia as a result of Campylobacter species: a population-based study of epidemiology and clinical risk factors. Clin Microbiol Infect. 2010;16:57–61. [DOI] [PubMed] [Google Scholar]
- 4.Havelaar AH, Kirk MD, Torgerson PR, Gibb HJ, Hald T, Lake RJ, et al. Group on behalf of WHOFDBER: World Health Organization Global Estimates and Regional Comparisons of the Burden of Foodborne Disease in 2010. PLOS Med. 2015;12:e1001923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.World Health Organization. WHO estimates of the global burden of foodborne diseases: foodborne disease burden epidemiology reference group 2007–2015. 2015. [Google Scholar]
- 6.Guerrant RL, Lahita RG, Winn WC, Roberts RB. Campylobacteriosis in man: Pathogenic mechanisms and review of 91 bloodstream infections. Am J Med. 1978;65:584–92. [DOI] [PubMed] [Google Scholar]
- 7.Pacanowski J, Lalande V, Lacombe K, Boudraa C, Lesprit P, Legrand P, et al. Campylobacter Bacteremia: Clinical Features and Factors Associated with Fatal Outcome. Clin Infect Dis. 2008;47:790–96. [DOI] [PubMed] [Google Scholar]
- 8.Fernández-Cruz A, Muñoz P, Mohedano R, Valerio M, Marin M, Alcalá L, et al. Campylobacter Bacteremia: Clinical Characteristics, Incidence, and Outcome Over 23 Years. Medicine (Baltimore). 2010;89:319. [DOI] [PubMed] [Google Scholar]
- 9.Gazaigne L, Legrand P, Renaud B, Bourra B, Taillandier E, BrunBuisson C, et al. Campylobacter fetus bloodstream infection: risk factors and clinical features. Eur J Clin Microbiol Infect Dis Off Publ Eur Soc Clin Microbiol. 2008;27:185–89. [DOI] [PubMed] [Google Scholar]
- 10.Elshafie SS, Asim M, Ashour A, Elhiday AH, Mohsen T, Doiphode S. Campylobacter peritonitis complicating continuous ambulatory peritoneal dialysis: report of three cases and review of the literature. Perit Dial Int. 2010;30:99–104. [DOI] [PubMed] [Google Scholar]
- 11.Klein BS, Vergeront JM, Blaser MJ, Edmonds P, Brenner DJ, Janssen D, et al. Campylobacter infection associated with raw milk. An outbreak of gastroenteritis due to Campylobacter jejuni and thermotolerant Campylobacter fetus subsp. fetus. JAMA. 1986;255:361–64. [DOI] [PubMed] [Google Scholar]
- 12.Man SM. The clinical importance of emerging Campylobacter species. Nat Rev Gastroenterol Hepatol. 2011;8:669–85. [DOI] [PubMed] [Google Scholar]
- 13.Wagenaar JA, van Bergen MAP, Blaser MJ, Tauxe RV, Newell DG, van Putten JPM. Campylobacter fetus infections in humans: exposure and disease. Clin Infect Dis Off Publ Infect Dis Soc Am. 2014;58:1579–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Véron M, Chatelain R. Taxonomic Study of the Genus Campylobacter Sebald and Véron and Designation of the Neotype Strain for the Type Species, Campylobacter fetus (Smith and Taylor) Sebald and Véron. Int J Syst Evol Microbiol. 1973;23:122–34. [Google Scholar]
- 15.Fitzgerald C, Z chao Tu, Patrick M, Stiles T, Lawson AJ, Santovenia M, et al. Campylobacter fetus subsp. testudinum subsp. nov., isolated from humans and reptiles. Int J Syst Evol Microbiol. 2014;64:2944–48. [DOI] [PubMed] [Google Scholar]
- 16.Gilbert MJ, Kik M, Timmerman AJ, Severs TT, Kusters JG, Duim B, et al. Occurrence, Diversity, and Host Association of Intestinal Campylobacter, Arcobacter, and Helicobacter in Reptiles. PLOS ONE. 2014;9:e101599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.On SLW, Harrington CS. Evaluation of numerical analysis of PFGE-DNA profiles for differentiating Campylobacter fetus subspecies by comparison with phenotypic, PCR and 16S rDNA sequencing methods. J Appl Microbiol. 2001;90:285–93. [DOI] [PubMed] [Google Scholar]
- 18.van Bergen M a. P, Linnane S, van Putten JPM, Wagenaar JA. Global detection and identification of Campylobacter fetus subsp. venerealis. Rev Sci Tech Int Off Epizoot. 2005;24:1017–26. [PubMed] [Google Scholar]
- 19.Thompson SA, Blaser MJ. Pathogenesis of Campylobacter fetus infections. Campylobacter. Washington, DC: American Society Microbiolgy Press; 2000. pp. 321–47. [Google Scholar]
- 20.Dekeyser J. Bovine genital campylobacteriosis. Campylobacter infection in man and animals. Boca Raton: CRC Press; 1984. pp. 181–91. [Google Scholar]
- 21.Van der Graaf-van Bloois L, Miller WG, Yee E, Duim B, Wagenaar JA. Whole genome sequencing of Campylobacter fetus subspecies. American Society for Microbiology General Meeting, San Francisco, CA: 2012. pp. 16–19. [Google Scholar]
- 22.Tu Z-C, Zeitlin G, Gagner J-P, Keo T, Hanna BA, Blaser MJ. Campylobacter fetus of Reptile Origin as a Human Pathogen. J Clin Microbiol. 2004;42:4405–07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dingle KE, Blaser MJ, Tu Z-C, Pruckler J, Fitzgerald C, van Bergen MAP, et al. Genetic Relationships among Reptilian and Mammalian Campylobacter fetus Strains Determined by Multilocus Sequence Typing. J Clin Microbiol. 2010;48:977–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seng P, Rolain JM, Fournier PE, La Scola B, Drancourt M, Raoult D. MALDI-TOF-mass spectrometry applications in clinical microbiology. Future Microbiol. 2010;5:1733–54. [DOI] [PubMed] [Google Scholar]
- 25.Zingue D, Flaudrops C, Drancourt M. Direct matrix-assisted laser desorption ionisation time-of-flight mass spectrometry identification of mycobacteria from colonies. Eur J Clin Microbiol Infect Dis Off Publ Eur Soc Clin Microbiol. 2016;35:1983–87. [DOI] [PubMed] [Google Scholar]
- 26.Conway GC, Smole SC, Sarracino DA, Arbeit RD, Leopold PE. Phyloproteomics: species identification of Enterobacteriaceae using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. J Mol Microbiol Biotechnol. 2001;3:103–12. [PubMed] [Google Scholar]
- 27.Karlsson R, Gonzales-Siles L, Boulund F, Svensson-Stadler L, Skovbjerg S, Karlsson A, et al. Proteotyping: Proteomic characterization, classification and identification of microorganisms–A prospectus. Syst Appl Microbiol. 2015;38:246–57. [DOI] [PubMed] [Google Scholar]
- 28.Hugo A, Baxter DJ, Cannon WR, Kalyanaraman A, Kulkarni G, Callister SJ. Proteotyping of microbial communities by optimization of tandem mass spectrometry data interpretation. Pac Symp Biocomput Pac Symp Biocomput. 2012. 225–34. [PubMed] [Google Scholar]
- 29.Shillingford JM, Miyoshi K, Robinson GW, Bierie B, Cao Y, Karin M, et al. Proteotyping of mammary tissue from transgenic and gene knockout mice with immunohistochemical markers: a tool to define developmental lesions. J Histochem Cytochem OffJ Histochem Soc. 2003;51:555–65. [DOI] [PubMed] [Google Scholar]
- 30.Rodriguez C, Quero C, Dominguez A, Trigo M, Posada de la Paz M, Gelpi E, et al. Proteotyping of human haptoglobin by MALDI-TOF profiling: Phenotype distribution in a population of toxic oil syndrome patients. Proteomics. 2006;6:S272–81. [DOI] [PubMed] [Google Scholar]
- 31.Schwahn AB, Wong JWH, Downard KM. Rapid differentiation of seasonal and pandemic H1N1 influenza through proteotyping of viral neuraminidase with mass spectrometry. Anal Chem. 2010;82:4584–90. [DOI] [PubMed] [Google Scholar]
- 32.Ojima-Kato T, Yamamoto N, Takahashi H, Tamura H. Matrix-assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry (MALDI-TOF MS) Can Precisely Discriminate the Lineages of Listeria monocytogenes and Species of Listeria. PloS One. 2016;11:e0159730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reil M, Erhard M, Kuijper EJ, Kist M, Zaiss H, Witte W, et al. Recognition of Clostridium difficile PCR-ribotypes 001, 027 and 126/078 using an extended MALDI-TOF MS system. Eur J Clin Microbiol Infect Dis. 2011;30:1431–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wolters M, Rohde H, Maier T, Belmar-Campos C, Franke G, Scherpe S, et al. MALDI-TOF MS fingerprinting allows for discrimination of major methicillin-resistant Staphylococcus aureus lineages. Int J Med Microbiol. 2011;301:64–8. [DOI] [PubMed] [Google Scholar]
- 35.Kuhns M, Zautner AE, Rabsch W, Zimmermann O, Weig M, Bader O, et al. Rapid discrimination of Salmonella enterica serovar Typhi from other serovars by MALDI-TOF mass spectrometry. PLoS One. 2012;7:e40004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zautner AE, Masanta WO, Tareen AM, Weig M, Lugert R, Gross U, et al. Discrimination of multilocus sequence typing-based Campylobacter jejuni subgroups by MALDI-TOF mass spectrometry. BMC Microbiol. 2013;13:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zautner AE, Masanta WO, Weig M, Groß U, Bader O. Mass Spectrometry-based PhyloProteomics (MSPP): A novel microbial typing Method. Sci Rep. 2015;5:13431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zautner AE, Lugert R, Masanta WO, Weig M, Groß U, et al. Subtyping of Campylobacter jejuni ssp. doylei Isolates Using Mass Spectrometry-based PhyloProteomics (MSPP). JoVE J Vis Exp. 2016. e54165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Emele MF, Možina SS, Lugert R, Bohne W, Masanta WO, Riedel T, et al. Proteotyping as alternate typing method to differentiate Campylobacter coli clades. Sci Rep. 2019;9:4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gonzales T, Robert-Baudouy J. Bacterial aminopeptidases: properties and functions. FEMS Microbiol Rev. 1996;18:319–44. [DOI] [PubMed] [Google Scholar]
- 41.Varland S, Osberg C, Arnesen T. N-terminal modifications of cellular proteins: The enzymes involved, their substrate specificities and biological effects. Proteomics. 2015;15:2385–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol. 2018;35:1547–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fagerquist CK, Bates AH, Heath S, King BC, Garbus BR, Harden LA, et al. Sub-speciating Campylobacter jejuni by proteomic analysis of its protein biomarkers and their post-translational modifications. J Proteome Res. 2006;5:2527–38. [DOI] [PubMed] [Google Scholar]