

Abstract
Spatiotemporal control over biochemical signaling processes involving G protein‐coupled receptors (GPCRs) is highly desired for dissecting their complex intracellular signaling. We developed sixteen photoswitchable ligands for the human histamine H3 receptor (hH3R). Upon illumination, key compound 65 decreases its affinity for the hH3R by 8.5‐fold and its potency in hH3R‐mediated Gi protein activation by over 20‐fold, with the trans and cis isomer both acting as full agonist. In real‐time two‐electrode voltage clamp experiments in Xenopus oocytes, 65 shows rapid light‐induced modulation of hH3R activity. Ligand 65 shows good binding selectivity amongst the histamine receptor subfamily and has good photolytic stability. In all, 65 (VUF15000) is the first photoswitchable GPCR agonist confirmed to be modulated through its affinity and potency upon photoswitching while maintaining its intrinsic activity, rendering it a new chemical biology tool for spatiotemporal control of GPCR activation.
Keywords: agonism, dynamic modulation, H3R, photopharmacology, VUF15000
In recent years, photopharmacology has been gaining momentum as a strategy to optically control biochemical processes.1 The use of light as an external trigger to change ligand shape and consequently its pharmacological properties allows the probing of biological systems with great spatiotemporal resolution.2 The azobenzene moiety is often used in photoswitchable ligands1a due to its limited size, high photostability, and tunability of the absorption wavelength λ max.3 Its thermodynamically stable trans isomer typically involves a flat elongated structure, whereas its photoinduced cis configuration has a bent geometry with a considerably shorter end‐to‐end distance.4 Whereas photopharmacology is well established in the field of enzyme and ion channel modulation, it is an upcoming technology for G protein‐coupled receptors (GPCRs).1a GPCRs constitute one of the largest families of transmembrane proteins, their dysfunction is associated with a plethora of diseases and consequently GPCRs are one of the most successful classes of drug targets.5 Recently, various GPCRs have been successfully targeted using photopharmacology approaches, including μ‐opioid,6 CXCR3,7 CB1,8 H3R,9 mGlu5,10 and GLP1.11 Yet, almost all these examples include at least one but more frequently two antagonistic/partial agonist isomeric forms. In contrast, freely diffusible affinity and potency photoswitches in which both isomers act as full agonists are scarce,1a even though such compounds would be very useful for photopharmacology approaches and complementary to agonist‐to‐antagonist switches.
The histamine H3R receptor is an intensively studied GPCR that is known to play an important role in sleep disorders and cognition‐related diseases, such as Alzheimer's and Parkinson's disease. The first H3R antagonist pitolisant (Wakix®) has been approved by the European Medicines Agency for the treatment of narcolepsy.12 Recently, we published a toolbox of photoswitchable antagonists9 that competitively inhibit histamine‐induced H3R activity. In the current work, we aimed to develop high‐potency H3R photoswitchable agonists that can simplify spatiotemporal studies of the signaling network of the H3R. We disclose unique photoswitchable H3R agonists that can be optically converted to isomers differing in their affinity and potency.
The scaffold design was inspired by the hH3R full agonist VUF5980 previously published by our lab13 (Figure 1). To date, virtually every published hH3R full agonist contains a 4‐substituted imidazole moiety combined with a basic or neutral side chain, as is the case for VUF5980. We left the imidazole portion of the molecule unchanged, and considered the diphenylacetylene moiety to be an attractive candidate for an “azologization” strategy.3 Introducing the azobenzene at this position allows for great flexibility in the diversification of the scaffold. Based on the steep structure‐activity relationship observed with VUF5980,13 it was postulated that small changes would have significant impact on the affinity and potency for hH3R. Therefore, primarily the azobenzene was decorated with small substituents (i.e., methyl and fluorine groups) on both phenyl rings.
Figure 1.
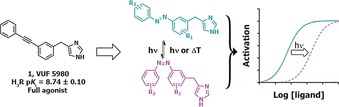
General design and concept of photoswitchable H3R full agonists.
To synthesize the ligands, imidazole‐4‐carbaldehyde 2 was protected using N,N‐dimethylsulfamoylchloride (DMS‐Cl, Scheme 1) to afford 3, which was reduced to 4. Alcohol 4 was converted to chloride 5 using in situ mesylation. A diverse set of anilines 6–15 was oxidized to the corresponding nitrosobenzenes 16–25 using Oxone™. After work‐up, they were directly used in a Mills reaction with 3‐amino‐phenylboronic acid pinacol esters 26–32 to yield azobenzene–pinacol esters 33–48. Cross coupling with chloride 5 afforded 49–64 in generally good yields. Acidic deprotection yielded final compounds 65–80, which were used for biological evaluation.
Scheme 1.
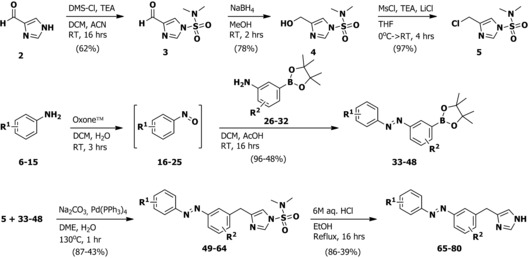
General synthetic scheme for photoswitchable H3R agonists. See the Supporting Information for detailed experimental procedures.
Compounds 65–80 all have λ max values for the π–π* transition of the trans isomer between 313 and 330 nm (Table 1). The observed limited variation is due to the absence of strong electron‐donating or ‐withdrawing substituents. Similarly, λ max values for the n–π* transition of the cis isomer differed marginally, ranging between 417 and 430 nm. Upon continuous illumination at 360±20 nm, the values for the photostationary states (PSS) of 65–80 ranged from 92.3 to 97.5 % cis, except for 69, which has 82.6 % cis. Compounds 65–80 all showed slow thermal relaxation at room temperature (20 °C, Table 1). The observed thermal relaxation half‐lives were impractically long for direct quantification, therefore extrapolations of high temperature thermal relaxation were used to quantify half‐lives at 20 °C (Table 1 and Supporting Information, Figure S1).14 Compound 69 showed the fastest thermal relaxation in 50 mm Tris‐HCl pH 7.4 buffer, with a half‐life of 26.6 days, while 72 showed the slowest relaxation, with a half‐life of 147 days.
Table 1.
Structure‐affinity relationship and photochemical properties of photoswitchable azobenzene‐derived H3R agonists.
| Compound number |
R1 | R2 | pK
i
trans ± SEM |
pK
i at PSS cis ± SEM |
pK
i shift ± SEM |
λ
max
trans
[b]
[nm] |
λ
max
cis
[b]
[nm] |
t
1/2
[c]
[days] |
PSS[d]
± SEM |
|
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
|
8.74±0.10[a] | – | – | – | – | – | |||
| 65 | H | H | 8.42±0.04 | 7.49±0.05 | −0.93±0.06 | 320 | 427 | 106 | 96.1±1.9 | |
| 66 | 2‐F | H | 8.28±0.08 | 7.09±0.03 | −1.19±0.04 | 323 | 425 | 128 | 95.7±0.27 | |
| 67 | 3‐F | H | 8.35±0.09 | 7.42±0.05 | −0.93±0.04 | 320 | 425 | 101 | 94.1±1.3 | |
| 68 | 4‐F | H | 7.69±0.08 | 6.51±0.08 | −1.18±0.09 | 322 | 426 | 95.9 | 95.9±1.6 | |
| 69 | 2,6‐F | H | 8.00±0.02 | 7.26±0.09 | −0.74±0.10 | 313 | 417 | 26.6 | 82.6±1.9 | |
| 70 | 2‐Cl | H | 7.86±0.03 | 6.85±0.04 | −1.02±0.03 | 324 | 420 | 96.1 | 95.3±0.22 | |
| 71 | 4‐Cl | H | 6.76±0.07 | 5.98±0.07 | −0.78±0.10 | 326 | 428 | 29.7 | 97.5±0.48 | |
| 72 | H | 2‐Me | 5.57±0.09 | 5.45±0.03 | −0.12±0.10 | 323 | 428 | 147 | 92.3±4.9 | |
| 73 | H | 4‐Me | 6.90±0.06 | 5.77±0.13 | −1.13±0.08 | 327 | 430 | 42.9 | 96.3±1.1 | |
| 74 | H | 5‐Me | 5.75±0.03 | 5.13±0.17 | −0.62±0.19 | 322 | 427 | 122 | 94.5±1.4 | |
| 75 | H | 6‐Me | 7.15±0.03 | 5.94±0.06 | −1.21±0.04 | 324 | 426 | 125 | 95.8±0.92 | |
| 76 | 2‐Me | H | 7.72±0.03 | 6.40±0.04 | −1.32±0.05 | 326 | 426 | 35.6 | 96.5±1.6 | |
| 77 | 3‐Me | H | 7.39±0.08 | 6.46±0.06 | −0.94±0.09 | 323 | 428 | 77.0 | 95.4±0.39 | |
| 78 | 4‐Me | H | 5.72±0.14 | 5.71±0.06 | −0.01±0.16 | 330 | 429 | 34.1 | 94.0±4.4 | |
| 79 | H | 4‐F | 7.81±0.07 | 6.54±0.06 | −1.27±0.02 | 324 | 425 | 91.7 | 96.6±0.51 | |
| 80 | H | 6‐F | 8.39±0.06 | 7.36±0.03 | −1.03±0.03 | 322 | 427 | 84.6 | 94.6±1.3 | |
[a] Adapted from Wijtmans et al.13 [b] Determined at 25 μm in 50 mm Tris‐HCl pH 7.4 buffer + 1 % [D6]DMSO. [c] Thermal relaxation half‐life times, as determined by the method of Ahmed et al.14 in 50 mm Tris‐HCl pH 7.4 buffer + 1 % [D6]DMSO, extrapolating to 20 °C. Arrhenius plots are available in Figure S1 in the Supporting Information. [d] Photostationary state area percentages after illumination at 360±20 nm at 1 mm in [D6]DMSO and as determined by LC‐MS analysis at 254 nm. All pharmacology experiments were performed at least in triplicate.
Based on its favorable pharmacological profile (see below) and synthetic tractability, compound 65 was subjected to in‐depth photochemical characterization using 1H NMR and LC‐MS analysis during illumination at 360±20 nm. The well‐resolved signal of the benzylic CH2 group provided a clear handle for quantification in 1H NMR analysis (Figure 2 A). An overestimation of isomerization percentage is observed in LC‐MS analysis at 254 nm compared to 1H NMR analysis (Figure 2 B and Supporting Information, Table S1), which can be explained by the differences in extinction coefficients for the trans and cis isomer at 254 nm (Figure 2 C and Supporting Information, Figures S2 and S3). Compound 65 showed excellent resistance to photobleaching during more than 1000 isomerization cycles (Figure 2 D). The dynamic isomerization was studied using UV/Vis spectroscopy under alternating illumination (Figure 2 E). At 25 μm of 65 in 50 mm Tris‐HCl pH 7.4 buffer + 1 % [D6]DMSO, a half‐life of 4.2±0.16 s for 360±20 nm and 5.7±0.19 s for 434±9 nm was observed.
Figure 2.
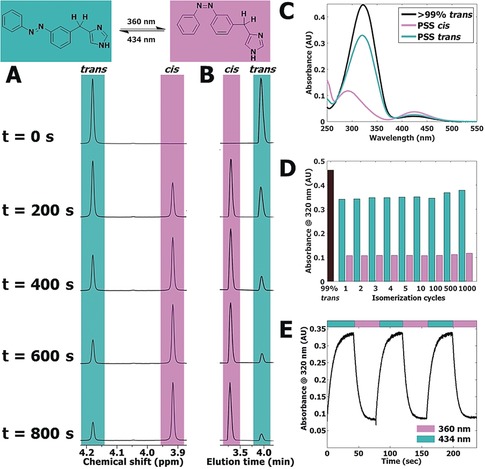
A) Representative part of 1H NMR spectra of 10 mm 65 in [D6]DMSO illuminated at 360±20 nm displayed at various time points (seconds). The presented peak belongs to the hydrogen atoms explicitly drawn in the structure shown above the spectrum. Full spectra are available in Figure S4 in the Supporting Information. B) Representative part of LC‐MS chromatograms belonging to the illuminated NMR sample in Figure 2 A. Full chromatograms are available in Figure S5 in the Supporting Information. C) UV/Vis spectra of 25 μm of 65 in 50 mm Tris‐HCl pH 7.4 buffer + 1 % [D6]DMSO. PSS cis represents a sample which has been illuminated for 300 s using 360±20 nm light. PSS trans represents subsequent illumination for 300 s using 434±9 nm light. D) Repeated isomerization of 25 μm of 65 in 50 mm Tris‐HCl pH 7.4 buffer + 1 % [D6]DMSO analyzed at 320 nm. PSS cis was obtained by illuminating 65 for 40 s at 360±20 nm. PSS trans was obtained by illuminating 65 for 40 s at 434±9 nm. E) Absorbance at 320 nm of 25 μm of 65 in 50 mm Tris‐HCl pH 7.4 buffer + 1 % [D6]DMSO. UV/Vis spectra were obtained at 1 s intervals under alternating illumination at 360±20 nm and 434±9 nm perpendicular to the light source of the UV/Vis spectrometer.
The long thermal relaxation half‐lives allowed for detailed pharmacological evaluation using hH3R competition binding as well as functional experiments. For this, the compound solutions were either illuminated at 360±20 nm to reach a PSS cis or kept in the dark to ensure more than 99 % trans isomer. The affinity of both isomers for the hH3R was assessed in competition binding with [3H]‐Nα‐methylhistamine (NAMH). All compounds displayed hH3R binding affinity, which decreased upon illumination, reaching up to a 21‐fold affinity difference in the case of 76. In terms of absolute affinity, 65 displayed the highest affinities for the hH3R with a pK i value of 8.42±0.04 for its trans isomer and a pK i value of 7.49±0.05 for its cis isomer, resulting in an 8.5‐fold shift upon illumination (Figure 3 A and Table 1). Fluorine‐substituted analogues 67 and 80 performed similarly to 65 in competition binding, displaying only a marginally lower affinity (Table 1). Notably, para‐methyl substitution on the R1 position (78) decreased the binding affinity and abrogated the photoisomerization‐induced affinity shift compared to 65 (Table 1). Reduction of the size of the para‐substituents to either chlorine (71) or fluorine (68) moieties gradually rescued hH3R affinity and reestablished the shift in affinity to 6‐ and 15‐fold, respectively. Methylation at either the ortho (76) or meta (77) position of R1 still resulted in decent binding affinities and high (21‐fold) to good (8.5‐fold) affinity shifts upon illumination. Addition of substituents at the R2 position resulted in a clear affinity cliff, with fluorine substitutions (79 and 80) still being allowed, but the addition of a methyl substituent (72–75) highly decreases the binding affinity of the cis isomer. Moreover, for the trans isomers, 4‐Me (73) and 6‐Me (75) substitution was still tolerated yet showed a log‐unit decrease in hH3R affinity compared to 65, while 2‐Me (72) and 5‐Me (74) groups highly reduced hH3R affinity and consequently reduced or even abolished (72) the affinity shift (Table 1).
Figure 3.
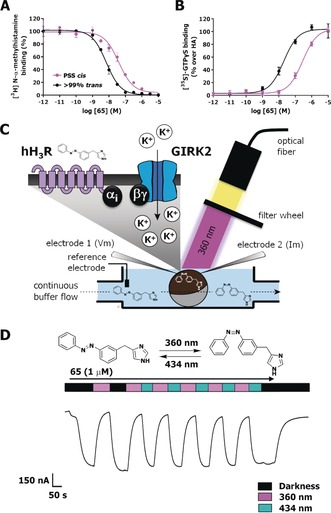
Representative curves of 65 (A) in competition binding with [3H]‐NAMH or (B) in Gi protein activation, as measured by [35S]‐GTPγS accumulation on HEK293T cell homogenates transiently expressing hH3R. Black lines refer to a sample containing more than 99 % trans 65, while magenta lines refer to a sample of 65 illuminated to PSS cis with 360±20 nm prior to the assay. C) Schematic drawing of the TEVC setup used for dynamic hH3R and GIRK current activation in Xenopus laevis oocytes expressing hH3R and GIRK. D) Representative part of a GIRK‐mediated current trace during continuous perfusion with 1 μm 65, while illuminating the oocyte with alternating 360±20 and 434±9 nm wavelength, as measured by TEVC. Error bars shown are mean±SD.
Based on the observed affinities and photo‐induced affinity shifts, the efficacy in stimulating hH3R‐mediated Gi protein activation was evaluated for ligands 65 and 76 in a [35S]‐GTPγS binding assay. The highest‐affinity ligand 65 (pK i trans=8.42±0.04) also displayed the highest potency (pEC50 trans=7.60±0.13) to induce Gi activation, which upon photoisomerization decreased 20‐fold (pEC50 at PSS cis: 6.30±0.13), with both isomers being full agonists and having intrinsic activities of α=1.0±0.03, compared to histamine (Figure 3 B). Since the observed shift in hH3R affinity was 8.5‐fold, the larger (20‐fold) shift in functional potency indicates that for 65 the efficacy (propensity to activate a GPCR15) is also affected upon trans–cis isomerization. Interestingly, a large photo‐induced decrease in potency of 23‐fold was also obtained for 76 (pEC50 trans: 6.78±0.11, PSS cis: 5.41±0.11, α=1.00±0.0). This shift in potency of 76 is completely explained by the observed change of its affinity (see above).
Compound 65 (VUF15 000) was selected as tool compound for further analysis, as it has good synthetic tractability and its superior potency is a clear advantage for pharmacological studies. As the imidazole‐based pharmacophore/scaffold used in the design of these photoswitchable ligands is prone to interact with other histamine receptor subtypes,13 65 was tested for its subtype selectivity. Binding of 65 was more than 300‐fold selective for hH3R over hH1R and hH2R (Supporting Information, Table S2), while a 30‐fold selectivity was observed over its closest homologue hH4R (Supporting Information, Table S2). Interestingly, 65 displayed high nm (trans) to low μm (PSS cis) binding affinities for both mouse and rat H3R, with a 4‐fold and 8‐fold shift in binding affinity upon photoisomerization, respectively (Supporting Information, Table S2).
Real‐time photomodulation of hH3R activity by 65 was measured using two‐electrode voltage clamp (TEVC) experiments on Xenopus laevis oocytes expressing both hH3R and G protein‐coupled inwardly rectifying potassium (GIRK)‐channels (Figure 3 C). In this expression system, histamine application resulted in hH3R‐mediated GIRK activation, which was insensitive to optical modulation.9 As expected based on our data with the [35S]‐GTPγS binding assay, trans‐65 elicited an agonistic response in this system, which could be reduced by switching to the less active cis isomer upon illumination at 360±20 nm. Retrieval of the agonistic response could be provoked by either actively switching the cis isomer back into its trans isomer by illuminating at 434±9 nm or by stopping illumination, due to continuous perfusion of the trans isomer (Figure 3 D). Dynamic photoswitching of 65 could be performed repeatedly, illustrating that the use of two specific wavelengths allows optical control of the hH3R activation mediated by 65. Furthermore, photoswitchable agonist 65 showed rapid hH3R activation and deactivation kinetics, aiding in its use in in vivo experimentation.
In summary, we have synthesized and characterized 16 photoswitchable hH3R agonists that change their affinity and potency upon illumination, indicating a successful azologization strategy. All possess long thermal relaxation half‐lives at room temperature making them useful for a variety of pharmacological studies. Compound 65 (VUF15 000) was selected as key compound on the basis of synthetic tractability and highest absolute hH3R affinity. Moreover, upon illumination, 65 displays a high potency and a 20‐fold potency shift, while maintaining full intrinsic activity in Gi protein activation, making it especially attractive as a tool compound. With a 20‐fold shift in potency, 65 is one of the best photoswitchable GPCR agonists reported so far. Electrophysiology experiments showed the dynamic optical modulation of hH3R activation induced by 65 in real time, setting the stage for further unraveling of the downstream signaling of hH3R with great spatiotemporal precision. Recently, photopharmacology approaches with freely diffusible GPCR ligands have, for the first time, been used successfully in vivo to modulate tadpole and zebrafish behavior10, 16 and to elucidate the role of the metabotropic glutamate receptor 4 in the nervous system using a mouse model of chronic pain.17 In view of the widespread distribution of the H3R in the central and peripheral nervous system, photopharmacology approaches with tools such as 65 offer new means (complementary to optogenetic approaches18) to investigate the spatial and temporal details of H3R modulation of important processes, for example, arousal, cognition and neuropathic pain.12a–12e
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge The Netherlands Organisation for Scientific Research (NWO) for financial support (TOPPUNT, “7 ways to 7TMR modulation (7‐to‐7)”, 718.014.002). All authors participate in the European Cooperation in Science and Technology Action CM1207 [GPCR‐Ligand Interactions, Structures, and Transmembrane Signaling: A European Research Network (GLISTEN)]. We thank Hans Custers for HRMS analyses, Andrea van de Stolpe for setting up the photochemistry equipment and Fons Lefeber (Leiden University) for NMR assistance. Kristoffer Sahlholm (Karolinska institute) is kindly acknowledged for providing the pcDNA3.1‐Kir3.1 and ‐Kir3.4 plasmids.
N. J. Hauwert, T. A. M. Mocking, D. Da Costa Pereira, K. Lion, Y. Huppelschoten, H. F. Vischer, I. J. P. De Esch, M. Wijtmans, R. Leurs, Angew. Chem. Int. Ed. 2019, 58, 4531.
Contributor Information
Dr. Maikel Wijtmans, Email: M.Wijtmans@vu.nl.
Prof. Dr. Rob Leurs, Email: R.Leurs@vu.nl.
References
- 1.
- 1a. Hüll K., Morstein J., Trauner D., Chem. Rev. 2018, 118, 10710–10747; [DOI] [PubMed] [Google Scholar]
- 1b. Hoorens M. W. H., Szymanski W., Trends Biochem. Sci. 2018, 43, 567–575. [DOI] [PubMed] [Google Scholar]
- 2. Szymanski W., Beierle J. M., Kistemaker H. A., Velema W. A., Feringa B. L., Chem. Rev. 2013, 113, 6114–6178. [DOI] [PubMed] [Google Scholar]
- 3. Broichhagen J., Frank J. A., Trauner D., Acc. Chem. Res. 2015, 48, 1947–1960. [DOI] [PubMed] [Google Scholar]
- 4. Beharry A. A., Woolley G. A., Chem. Soc. Rev. 2011, 40, 4422–4437. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Hauser A. S., Chavali S., Masuho I., Jahn L. J., Martemyanov K. A., Gloriam D. E., Babu M. M., Cell 2018, 172, 41–54 e19; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Sriram K., Insel P. A., Mol. Pharmacol. 2018, 93, 251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schönberger M., Trauner D., Angew. Chem. Int. Ed. 2014, 53, 3264–3267; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3329–3332. [Google Scholar]
- 7. Gómez-Santacana X., de Munnik S. M., Vijayachandran P., Da Costa Pereira D., Bebelman J. P. M., de Esch I. J. P., Vischer H. F., Wijtmans M., Leurs R., Angew. Chem. Int. Ed. 2018, 57, 11608–11612; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 11782–11786. [Google Scholar]
- 8. Westphal M., Schafroth M. A., Sarott R., Imhof M., Bold C., Leippe P., Dhopeshwarkar A., Grandner J., Katritch V., Mackie K., Trauner D., Carreira E. M., Frank J. A., J. Am. Chem. Soc. 2017, 139, 18206–18212. [DOI] [PubMed] [Google Scholar]
- 9. Hauwert N. J., Mocking T. A. M., Da Costa Pereira D., Kooistra A. J., Wijnen L. M., Vreeker G. C. M., Verweij E. W. E., De Boer A. H., Smit M. J., De Graaf C., Vischer H. F., de Esch I. J. P., Wijtmans M., Leurs R., J. Am. Chem. Soc. 2018, 140, 4232–4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pittolo S., Gomez-Santacana X., Eckelt K., Rovira X., Dalton J., Goudet C., Pin J. P., Llobet A., Giraldo J., Llebaria A., Gorostiza P., Nat. Chem. Biol. 2014, 10, 813–815. [DOI] [PubMed] [Google Scholar]
- 11. Broichhagen J., Podewin T., Meyer-Berg H., von Ohlen Y., Johnston N. R., Jones B. J., Bloom S. R., Rutter G. A., Hoffmann-Roder A., Hodson D. J., Trauner D., Angew. Chem. Int. Ed. 2015, 54, 15565–15569; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15786–15790. [Google Scholar]
- 12.
- 12a. Leurs R., Bakker R. A., Timmerman H., de Esch I. J. P., Nat. Rev. Drug Discovery 2005, 4, 107–120; [DOI] [PubMed] [Google Scholar]
- 12b. Panula P., Chazot P. L., Cowart M., Gutzmer R., Leurs R., Liu W. L., Stark H., Thurmond R. L., Haas H. L., Pharmacol. Rev. 2015, 67, 601–655; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12c. Khanfar M. A., Affini A., Lutsenko K., Nikolic K., Butini S., Stark H., Front. Neurosci. 2016, 10, 201; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12d. Panula P., Nuutinen S., Nat. Rev. Neurosci. 2013, 14, 472–487; [DOI] [PubMed] [Google Scholar]
- 12e. Tiligada E., Kyriakidis K., Chazot P. L., Passani M. B., CNS Neurosci. Ther. 2011, 17, 620–628; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12f. Kollb-Sielecka M., Demolis P., Emmerich J., Markey G., Salmonson T., Haas M., Sleep Med. 2017, 33, 125–129. [DOI] [PubMed] [Google Scholar]
- 13. Wijtmans M., Celanire S., Snip E., Gillard M. R., Gelens E., Collart P. P., Venhuis B. J., Christophe B., Hulscher S., van der Goot H., Lebon F., Timmerman H., Bakker R. A., Lallemand B. I., Leurs R., Talaga P. E., de Esch I. J. P., J. Med. Chem. 2008, 51, 2944–2953. [DOI] [PubMed] [Google Scholar]
- 14. Ahmed Z., Siiskonen A., Virkki M., Priimagi A., Chem. Commun. 2017, 53, 12520–12523. [DOI] [PubMed] [Google Scholar]
- 15. Kenakin T. P., Pharmacologic analysis of drug-receptor interaction, 2 nd ed., Raven Press, New York, 1993. [Google Scholar]
- 16. Gómez-Santacana X., Pittolo S., Rovira X., Lopez M., Zussy C., Dalton J. A., Faucherre A., Jopling C., Pin J. P., Ciruela F., Goudet C., Giraldo J., Gorostiza P., Llebaria A., ACS Cent. Sci. 2017, 3, 81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zussy C., Gomez-Santacana X., Rovira X., De Bundel D., Ferrazzo S., Bosch D., Asede D., Malhaire F., Acher F., Giraldo J., Valjent E., Ehrlich I., Ferraguti F., Pin J. P., Llebaria A., Goudet C., Mol. Psychiatry 2018, 23, 509–520. [DOI] [PubMed] [Google Scholar]
- 18. Williams R. H., Chee M. J., Kroeger D., Ferrari L. L., Maratos-Flier E., Scammell T. E., Arrigoni E., J. Neurosci. 2014, 34, 6023–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary