

Abstract
Enzymatic polymerization provides an excellent opportunity for the conversion of renewable resources into polymeric materials in an effective and sustainable manner. A series of furan‐based copolyesters was synthesized with up to 35 kg mol−1, by using Novozyme 435 as a biocatalyst and dimethyl 2,5‐furandicarboxylate (DMFDCA), 2,5‐bis(hydroxymethyl)furan (BHMF), aliphatic linear diols, and diacid ethyl esters as monomers. The synthetic mechanism was evaluated by the variation of aliphatic linear monomers and their feed compositions. Interestingly, there was a significant decrease in the molecular weight if the aliphatic monomers were changed from diols to diacid ethyl esters. The obtained copolyesters were thoroughly characterized and compared with their polyester analogs. These findings provide a closer insight into the application of enzymatic polymerization techniques in designing sustainable high‐performance polymers.
Keywords: copolymerization, enzyme catalysis, green chemistry, oxygen heterocycles, renewable resources
Introduction
The movement towards greener alternatives in polymer science steadily grows as the drive for sustainability continues.1 The increasing concerns regarding environmental pollution and crude oil depletion and price fluctuation have pulled both academia and industry to focus more on green raw materials, chemistry, and processing.2 To this regard, the conversion of renewable resources into prevalent polymer materials through enzymatic polymerization has become a particularly irresistible path. Compared to conventional chemically‐catalyzed processes, enzymatic polymerization has been proven to be effective as a more eco‐friendly synthesis route.3 In addition to the mild reaction conditions, the high selectivity of enzymes also allows to avoid tedious protection–deprotection steps and improves the quality of the end products.3a, 4 In the past few years, enzymes have been used to synthesize a wide array of polymer classes, for instance, polyesters,5 polyamides,6 vinyl polymers,7 and polysaccharides.8 Nevertheless, compared to the conventional synthetic route, the application of enzymatic polymerization is somehow still economically limited. One interesting approach to circumvent this limitation is to design sustainable high‐performance polymers for technologically relevant applications.
In general, aromatic compounds provide rigidity to a polymer chain, owing to the inhibition of rotational flexibility.9 Polymers with rigid backbones are often characterized by their high thermal and mechanical stability and are therefore suitable for the use as high‐performance polymers.10 Among them, furan‐based polyesters are promising sustainable alternatives with great interest. In addition to their sustainability, they possess similar or even better properties than their petrol‐based counterparts. For example, poly(ethylene furanoate) (PEF) shows better barrier properties than poly(ethylene terephthalate) (PET).11
An array of different furan polyesters has been synthesized by the groups of Okada,12 Ballauff,13 and Gandini14 since the 1990s. Thereafter, various furanic–aliphatic polyesters have been reported, such as poly(ethylene furanoate) (PEF), poly(butylene furanoate) (PBF), and poly(2,3‐butylene furanoate) (P23BF).15 We recently found that enzymatic polymerization can be used to synthesize different semi‐aromatic furan‐based polyesters (Scheme 1), by using dimethyl 2,5‐furandicarboxylate (DMFDCA) or 2,5‐bis(hydroxymethyl)furan (BHMF).5d, 5f
Scheme 1.
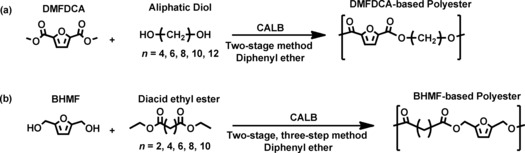
Enzymatic synthesis of semi‐aromatic furan‐based polyesters from (a) DMFDCA and aliphatic diols and (b) BHMF and diacid ethyl esters.
To further enhance the properties, the additional incorporation of aromatic content through copolymerization, in which two or more different polyester backbones are chemically linked together, can be an interesting approach. For example, Ma et al.,16 Wu et al.,17 Sousa et al.,18 and Morales‐Huerta et al.19 applied various conventional methods to synthesize furan‐based copolyesters. Recently, Morales‐Huerta et al.20 reported the enzymatic ring opening polymerization of poly(butylene 2,5‐furandicarboxylate‐co‐butylene succinate) and poly(ϵ‐caprolactone‐co‐butylene 2,5‐furandicarboxylate).
Inspired by our previous findings, we explored the enzymatic copolymerization of two carbohydrate‐sourced monomers (DMFDCA and BHMF) with aliphatic linear monomers, to prepare several semi‐aromatic copolyesters. By performing a detailed analysis of the enzymatic copolymerization, we observed the distinct activity of the enzyme towards different building blocks. We also investigated their morphologies, as well as the thermal properties of the obtained furan‐based copolyesters.
Results and Discussion
Synthesis and structural characterization
Furan‐based copolyesters were synthesized by a two‐step temperature‐varied enzymatic polymerization. The enzymatic copolymerization followed two different synthesis approaches, as depicted in Scheme 2. In the first approach, the furan‐based copolyesters were prepared by using DMFDCA, BHMF, and an aliphatic linear diol as the building blocks, whereas in the second approach, linear diacid ethyl esters were used. The number of the methylene units (n) in the dicarboxylic segments of the diacid ethyl esters is 2, 4, 6, 8, or 10, whereas the in aliphatic linear diols, n is 4, 6, 8, 10 or 12. In this study, this number is defined as the chain length of the tested aliphatic linear monomers. The obtained furan‐based copolyesters are listed in Table 1.
Scheme 2.
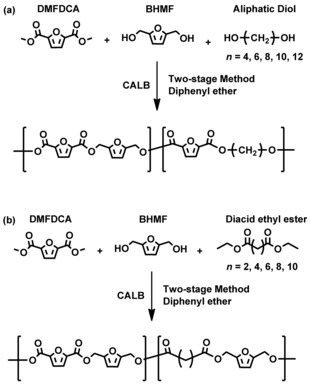
Enzymatic synthesis of furan‐based copolyesters/co‐oligoesters from (a) DMFDCA, BHMF, and aliphatic diols and (b) DMFDCA, BHMF, and diacid ethyl esters by a two‐stage method in diphenyl ether.
Table 1.
All obtained furan‐based copolyesters.
| n [a] | Copolyester | Abbreviation |
|---|---|---|
| FIRST APPROACH | ||
| 4 | poly(2,5‐furandimethylene furanoate‐co‐butylene furanoate) | P(FMF‐co‐BF) |
| 6 | poly(2,5‐furandimethylene furanoate‐co‐hexamethylene furanoate) | P(FMF‐co‐HF) |
| 8 | poly(2,5‐furandimethylene furanoate‐co‐octamethylene furanoate) | P(FMF‐co‐OF) |
| 10 | poly(2,5‐furandimethylene furanoate‐co‐decamethylene furanoate) | P(FMF‐co‐DF) |
| 12 | poly(2,5‐furandimethylene furanoate‐co‐dodecamethylene furanoate) | P(FMF‐co‐DOF) |
| SECOND APPROACH | ||
| 2 | poly(2,5‐furandimethylene furanoate‐co‐2,5‐furandimethylene succinate) | P(FMF‐co‐FMS) |
| 4 | poly(2,5‐furandimethylene furanoate‐co‐2,5‐furandimethylene adipate) | P(FMF‐co‐FMA) |
| 6 | poly(2,5‐furandimethylene furanoate‐co‐2,5‐furandimethylene suberate) | P(FMF‐co‐FMSu) |
| 8 | poly(2,5‐furandimethylene furanoate‐co‐2,5‐furandimethylene sebacate) | P(FMF‐co‐FMSe) |
| 10 | poly(2,5‐furandimethylene furanoate‐co‐2,5‐furandimethylene dodecanedioate) | P(FMF‐co‐FMD) |
[a] The number of methylene units in aliphatic linear diols (first approach) or in the dicarboxylic segments of the diacid ethyl esters (second approach).
The chemical structures and compositions of the copolyesters were confirmed by ATR‐FTIR and 1H‐NMR spectroscopy (Figure 1). The ATR‐FTIR and 1H NMR spectra of the representative furan‐based copolyesters from DMFDCA, BHMF, and diacid ethyl esters are illustrated in Figure S1 (see the Supporting Information). Detailed NMR and IR peak assignments are available in the Experimental Section. The molecular weights, yield, and the monomer feed compositions are summarized in Tables S1 and S2.
Figure 1.
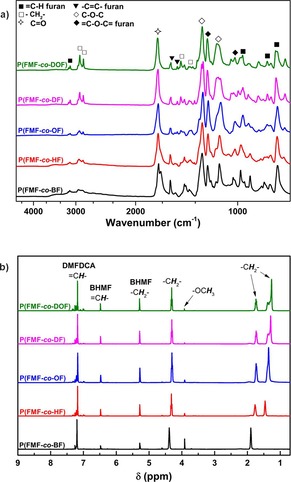
(a) ATR‐FTIR and (b) 1H NMR spectra of the representative furan‐based copolyesters from DMFDCA, BHMF, and aliphatic diols.
Influence of aliphatic linear monomers on the enzymatic copolymerization of the furan‐based copolyesters
To evaluate the influence of aliphatic linear monomers on the enzymatic synthesis of the furan‐based copolyesters, a comparative study on the degree of polymerization of the whole series of the furan‐based copolyesters was performed.
To study the effect of the chain length of aliphatic linear diols, all furan‐based copolyesters obtained from the first approach were evaluated (Figure 2 and Table S1). The results indicate that Candida antarctica lipase B (CALB) prefers longer linear diols (n=8,10 and 12) compared to shorter linear diols (n=4 and 6). If 1,8‐ODO was used, the enzymatic polymerization resulted in P(FMF‐co‐OF) with a number‐average degree of polymerization ( ) of 122 and a weight‐average degree of polymerization ( ) of 269, which was the highest amongst the tested aliphatic diols. Furan‐based copolyesters with relatively similar and values could be obtained by using 1,10‐ODO and 1,12‐DODO. Upon decreasing the chain length to 6 and 4 (1,6‐HDO and 1,4‐BDO), the and values of furan‐based copolyesters were decreased significantly. These results corroborate our previous finding on the preference of CALB on longer chain lengths of aliphatic linear diols.5d
Figure 2.
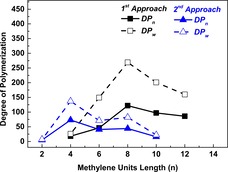
and of the furan‐based copolyesters from first and second synthetic approach against the chain length of the linear monomer. The furan‐based copolyester from the first approach obtained with a DMFDCA/BHMF/aliphatic diol feed ratio 50:12.5:37.5 and from the second approach with a DMFDCA/BHMF/diacid ethyl ester feed ratio of 12.5:50:37.5.
From the second synthetic approach, the same and trend is observed with respect to the diacid ethyl ester chain length. Furan‐based copolyesters with the highest and values of 73 and 137 were obtained by using diethyl adipate (n=4; Figure 2 and Table S2). By increasing the diacid ethyl ester length to n=6 and 8, furan‐based copolyesters with relatively similar and values were obtained. However, if diethyl succinate (n=2) and diethyl dodecanedioate (n=10) were used as the monomer, the resultant furan‐based copolyesters had only very low and values. Similar results on the effect of diacid ethyl ester/dicarboxylic acid chain length on enzymatic polymerization were reported previously.21 This result can be explained by the variable specificity of CALB towards diacid acyl esters with different chain length. This explanation is also in agreement with the study reported by McCabe and Taylor on the acyl‐binding site of CALB.22 They found that adipic acid is the preferred substrate among the tested dicarboxylic acids, which is owing to its low entropic component contribution to the enantioselectivity of CALB.
Interestingly, by changing the aliphatic monomers from aliphatic diols to diacid ethyl esters, enzymatic polymerization, in general, resulted in copolyesters with significantly lower . This can be explained by the instability of BHMF, which results in ether formation during the polymerization. As we reported previously, the high reactivity of the OH group in BHMF can lead to dehydration or reaction with ethanol to form BHMF ethers.5f Consequently, the copolyester chain propagation will be greatly limited by the formation of BHMF ethers as chain stoppers. The formation of BHMF ether was further confirmed by the presence of a small peak (≤1 wt %) at δ≈4.40 ppm in the 1H NMR spectra, as we reported previously.5f However, the substrate specificity of the enzyme also should be taken into account and this will be discussed further in later sections.
Effect of monomer feed composition on enzymatic synthesis of the furan‐based copolyesters
One fundamental issue in this work was to understand the monomer incorporation mechanism during the copolyester formation, which seemed to be governed by the enzyme catalytic activity and consequently influenced the molecular weights of the resulting furan‐based copolyesters. In this study, various molar feed compositions were used and evaluated.
We observed that as soon as we increased the 2,5‐furandimethylene furanoate (FMF) molar feed fraction to 50 %, the and of P(FMF‐co‐DOF) significantly decreased from 86 and 160 to 11 and 14, respectively. A similar trend was also observed in other copolyester series (Table 2). These results imply that the propagation mechanism is not solely limited by the formation of BHMF ethers in the system. Substrate specificity of the enzyme can also determine the enzyme catalytic activity and may also be an explanation for this. Like many enzymes, depending on the structural complementarity of transition state with the active site, CALB has the capability to catalyze diverse reactions at different efficiency ranges.23
Table 2.
Molar fraction and degree of polymerization of the furan‐based copolyesters obtained from different feed compositions of DMFDCA, BHMF, aliphatic diols, and diacid ethyl esters.
| Copolyester | Molar Fraction [%] |
|
|
|||||
|---|---|---|---|---|---|---|---|---|
| Feed[a] | Copolyester[d] | |||||||
| F FMF | F XF [b]; F FMX [c] | X FMF | X XF [e]; X FMX [f] | |||||
| FIRST APPROACH | ||||||||
| P(FMF‐co‐BF) | 25 | 75[b] | 16 | 84[e] | 18 | 26 | ||
| 50 | 50[b] | 8 | 92[e] | 13 | 13 | |||
| P(FMF‐co‐HF) | 25 | 75[b] | 23 | 77[e] | 46 | 149 | ||
| 50 | 50[b] | 15 | 85[e] | 13 | 17 | |||
| P(FMF‐co‐OF) | 25 | 75[b] | 22 | 78[e] | 122 | 269 | ||
| 50 | 50[b] | 43 | 57[e] | 24 | 43 | |||
| P(FMF‐co‐DF) | 25 | 75[b] | 22 | 78[e] | 97 | 201 | ||
| 50 | 50[b] | 18 | 82[e] | 10 | 11 | |||
| P(FMF‐co‐DOF) | 25 | 75[b] | 22 | 78[e] | 86 | 160 | ||
| 50 | 50[b] | 13 | 87[e] | 11 | 14 | |||
| SECOND APPROACH | ||||||||
| P(FMF‐co‐FMS) | 25 | 75[c] | 44 | 56[f] | 5 | 7 | ||
| 50 | 50[c] | 69 | 31[f] | 5 | 10 | |||
| P(FMF‐co‐FMA) | 25 | 75[c] | 24 | 76[f] | 73 | 137 | ||
| 50 | 50[c] | –[i] | –[f, i] | –[i] | –[i] | |||
| P(FMF‐co‐FMSu) | 25 | 75[c] | 26 | 74[f] | 41 | 71 | ||
| 50 | 50[c] | –[i] | –[f, i] | –[i] | –[i] | |||
| P(FMF‐co‐FMSe) | 25 | 75[c] | 24 | 76[f] | 44 | 82 | ||
| 50 | 50[c] | 65 | 35[f] | –[i] | –[i] | |||
| P(FMF‐co‐FMD) | 25 | 75[c] | 14 | 86[f] | 16 | 22 | ||
| 50 | 50[c] | 78 | 22[f] | 5 | 13 | |||
[a] F FMF, F XF, and F FMX represent the molar feed ratios of PFMF, PXF (in the first approach), and PFMX (in the second approach), respectively. [b] F XF. [c] F FMX. [d] X FMF, X XF, and X FMX represent the molar fractions of PFMF, PXF (in the first approach), and PFMX (in the second approach) segments in the obtained furan‐based copolyesters, respectively, determined by 1H NMR spectroscopy. [e] X XF. [f] X FMX. [g] (number‐average degree of polymerization)= [h] (weight‐average degree of polymerization)= . [i] Not determined.
A possible copolymerization mechanism of P(FMF‐co‐DOF) is depicted in Scheme 3. In this mechanism, the polymerization starts with the formation of the acyl–enzyme complex and continues with polycondensation. We propose that during the polycondensation, an intermediate product (b) forms that can inhibit the polymerization. Steric hindrance of (b) creates structure incompatibility with the enzyme active site; consequently, the polymer growth is terminated. Another possible explanation is that the OH functionality in (b) transforms into ethers, as in the case of the BHMF and eventually terminates the copolyester chain elongation. The proposed copolymerization mechanism appears to be well substantiated by the constantly lower value of the FMF molar fraction in the copolyester segment compared to the corresponding feed. Additionally, Takwa previously reported similar findings regarding the low activity of CALB towards d,d‐lactide,23 owing to the bulky conformation of the lactide if acylated. However, future studies by molecular modeling are recommended to validate the proposed reaction mechanism.
Scheme 3.
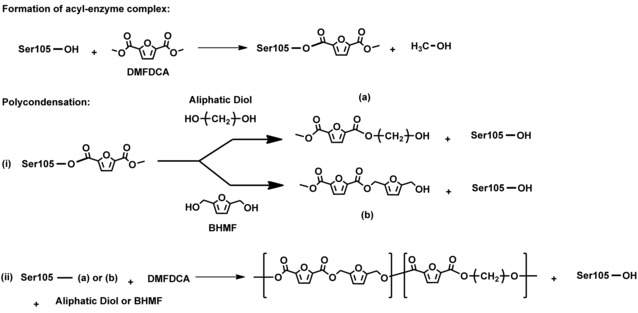
Proposed copolymerization mechanism of CALB‐catalyzed formation of P(FMF‐co‐DOF).
Thermal properties of furan‐based copolyesters
The thermal stability of the obtained furan‐based copolyesters was characterized by TGA and the representative characteristic curves for their thermal degradation behaviors are depicted in Figure 3. The values of the degradation temperatures are summarized in Table 3. The copolyesters from the first synthetic approach showed TGA traces with a two‐step degradation pattern (Figure 3 a). It consists of an initial degradation at around 230–250 °C with only approximately 10 % weight loss, followed by a secondary degradation with a maximum degradation temperature at approximately 390 °C.
Figure 3.
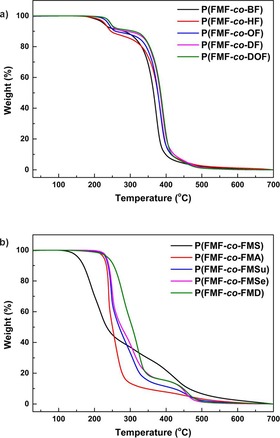
TGA traces of the obtained furan‐based copolyesters from (a) DMFDCA, BHMF, and aliphatic diols (feed ratio=50:12.5:37.5) and (b) DMFDCA, BHMF, and diacid ethyl esters (feed ratio=12.5:50:37.5).
Table 3.
Thermal properties of the obtained furan‐based copolyesters.
| Copolyester | DSC[c] | TGA[d] | ||||
|---|---|---|---|---|---|---|
|
T
g
[°C] |
T
m
[°C] |
T
c
[°C] |
T
cc
[°C] |
T
d‐10 %
[°C] |
T
d‐max
[°C] |
|
| P(FMF‐co‐BF) [a] | 15 | 142 | 83 | –[e] | 230 | 370 |
| P(FMF‐co‐HF)[a] | 12 | 120 | –[e] | –[e] | 240 | 390 |
| P(FMF‐co‐OF)[a] | 2 | 123 | 71 | 47 | 240 | 390 |
| P(FMF‐co‐DF)[a] | 6 | 90[f] | –[e] | –[e] | 250 | 390 |
| P(FMF‐co‐DOF)[a] | −2 | 88 | –[e] | 61 | 240 | 390 |
| P(FMF‐co‐FMS)[b] | −6 | –[e] | –[e] | –[e] | –[e] | 220/310/430 |
| P(FMF‐co‐FMA)[b] | −8 | –[e] | –[e] | –[e] | –[e] | 240 |
| P(FMF‐co‐FMSu)[b] | −16 | 57[f] | –[e] | –[e] | –[e] | 250/310/460 |
| P(FMF‐co‐FMSe)[b] | −19 | 62[f] | –[e] | –[e] | –[e] | 250/310/450 |
| P(FMF‐co‐FMD)[b] | −19 | 79 | 43 | 62 | –[e] | 280/320/460 |
[a] Furan‐based copolyesters from DMFDCA, BHMF, and aliphatic diol with a feed ratio of 50:12.5:37.5. [b] Furan‐based copolyesters from DMFDCA, BHMF, and diacid ethyl esters with a feed ratio of 12.5:50:37.5. [c] T g=glass transition temperature from the modulated DSC heating scan; T m=melting temperature from the second DSC heating scan; T c=crystallization temperature upon cooling; T cc=cold crystallization temperature from the second DSC heating scan. [d] T d‐10 %=decomposition temperature at 10 % weight loss; T d‐max=temperature at the maximum rate of decomposition. [e] Not detected in the tested temperature range. [f] Measured from the first DSC heating scan.
In contrast, the thermal degradation profiles of the furan‐based copolyesters from the second approach show the polymers to be less stable (Figure 3 b). Their degradation occurs in several stages and a significant weight loss was detected at temperatures around 220–280 °C. To conclude, the furan‐based copolyesters obtained from the first synthetic approach appeared to have better thermal stability, which is mainly owing to their higher molecular weights. It should be noted that, for copolyesters obtained from the first synthetic approach, the chain length of the tested aliphatic diols and molar compositions of the monomeric units have no significant influence on the decomposition temperature of the resulting copolyesters. Furthermore, the tested furan‐based copolyesters have similar degradation profiles and temperatures to their furan‐based polyester counterparts, as we reported previously.5d,5f
To evaluate the thermophysical behavior, a comparative differential scanning calorimetry (DSC) study was performed. The thermal transitions of the whole copolyester series are listed in Table 3 and representative DSC and temperature‐modulated DSC (TMDSC) traces of P(FMF‐co‐OF) is plotted in Figure 4. To enhance the visibility of the T g, we performed the TMDSC measurement.
Figure 4.
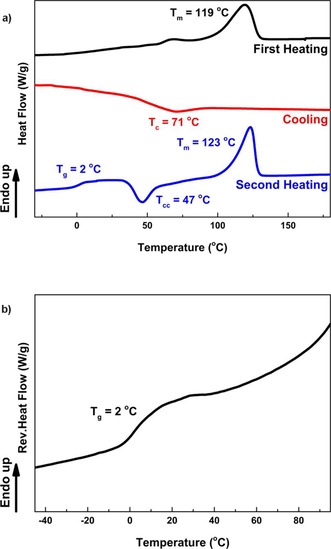
(a) DSC curves and (b) temperature‐modulated DSC (TMDSC) curves of P(FMF‐co‐OF) from 50 % DMFDCA, 12.5 % BHMF, and 37.5 % 1,8‐ODO.
For the furan‐based copolyesters obtained from the first synthetic approach, broad and multiple melting peaks are observed in all first heating cycles. A melting peak disappeared in the second heating scan of P(FMF‐co‐DF) and no crystallization was observed in the cooling cycle of P(FMF‐co‐HF) and P(FMF‐co‐DOF). In addition, a cold crystallization transition (T cc) can be observed before the melting temperature (T m) in the second heating scan of P(FMF‐co‐OF) and P(FMF‐co‐DOF). This can be explained by the polymer molecular motion and orientation, which could act as nuclei and promote the spontaneous cold crystallization. In general, we observed a constant decrease in their T g and T m values as we increased the chain length of the aliphatic linear diol. The decrease in T g value is caused by the increasing chain flexibility and mobility provided by the longer aliphatic chains. The decrease in T m value is corroborated well with our previous results of the corresponding furan‐based polyesters.5d
Except for P(FMF‐co‐FMD), no crystallization is detected in DSC traces of the furan‐based copolyester series obtained from the second synthetic approach. Owing to their low crystallization rate, no crystallization is observed, and the T m disappeared in the second heating scan of P(FMF‐co‐FMSu) and P(FMF‐co‐FMSe). For P(FMF‐co‐FMS) and P(FMF‐co‐FMA), they are amorphous materials because no melting or crystallization was observed. To verify this, we conducted further analysis by wide‐angle X‐Ray diffraction (WAXD) measurements, which is discussed below.
Crystallinity of furan‐based copolyesters
The WAXD spectra and POM images confirm the semicrystalline properties of the furan‐based copolyesters obtained from the first approach (Figure 5). As shown in Figure 5 a, P(FMF‐co‐HF), P(FMF‐co‐OF), P(FMF‐co‐DF), and P(FMF‐co‐DOF) gave rise to similar WAXD patterns, which displayed an amorphous halo at 2θ≈22° and three reflection peaks at 2θ≈10.0–13.6°, 16.8–17.9°, and 24.1–24.9°. P(FMF‐co‐BF) has three similar diffraction peaks with one additional reflection peak at 22.5°.
Figure 5.
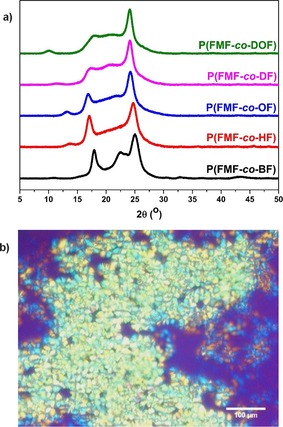
(a) Wide‐angle X‐ray diffraction (WAXD) spectra of the obtained furan‐based copolyesters from DMFDCA, BHMF and aliphatic diol with a feed ratio of 50:12.5:37.5. (b) POM image of P(FMF‐co‐DOF) obtained from 50 % DMFDCA, 25 % BHMF, and 25 % 1,8‐ODO with a feed ratio of 50:25:25.
Interestingly, we observed similar WAXD patterns in their furan‐based polyester counterparts (PBF, PHF, POF, and PDF).5d This suggested that the furan‐based copolyesters have similar crystal structures to their furan‐based polyester counterparts and that they do not change significantly as we incorporate more aromatic content in the main chain. As represented in Figure 4 b, the POM image of P(FMF‐co‐DOF) clearly showed that the product consists of birefringent spherulites with an estimated particle size of approximately 50 μm.
For the furan‐based copolyesters obtained from the second approach, distinct morphologies were observed. Analogous to their polyester counterparts, P(FMF‐co‐FMSu), P(FMF‐co‐FMSe), and P(FMF‐co‐FMD) are all semicrystalline materials. They showed a similar WAXD pattern with six reflection peaks at 2θ≈13.7–13.8°, 17.1–17.2°, 20.2–20.4°, 21.4–21.7°, 22.8–22.9°, and 23.7–23.8° (Figure S3). In contrast, WAXD spectra of P(FMF‐co‐FMS) and P(FMF‐co‐FMA) only displayed a broad halo at 2θ≈22°. This result agrees well with the DSC results, indicating that both P(FMF‐co‐FMS) and P(FMF‐co‐FMA) are indeed amorphous materials. These two copolymers, in particular, showed different morphologies with their polyester counterparts, namely PFMS and PFMA. As we incorporated FMF into the polymer main chain by copolymerization, the polymer morphology changed from semicrystalline to a completely amorphous structure.
Conclusions
We have shown that the application of enzymatic polymerization techniques can be extended to prepare a series of sustainable furan‐based copolyesters with increased content of aromatic units. Hereby, we introduce two different synthetic approaches. Using the first approach, a mixture of the furan monomers with an aliphatic diol yields a series of copolyesters with comparable high molecular weights up to =35 000 g mol−1, which is advantageous for future processability. By changing the aliphatic monomers from diols to diacid ethyl esters (second approach), a significant decrease in the molecular weight was observed. This can be explained by BHMF ether formation in the reaction system and the monomer incorporation mechanism during the copolymerization. This is further supported experimentally by the constant lower value of BHMF molar fraction in the copolyesters compared to the corresponding feed ratio, regardless of the variation in BHMF feed ratio.
The thermal stability of all copolyesters reported herein was established by TGA analysis, which indicates industrially relevant applicability. Compared to their furan‐based polyester counterparts, they possess similar decomposition profiles. DSC analysis provides insights into the thermal behavior, especially with regard to the crystallization ability of the furan‐based copolyesters. Quite interestingly, aside from P(FMF‐co‐FMS) and P(FMF‐co‐FMA), all copolyesters are semicrystalline materials. This suggests that the presence of the FMF segment in the main chain hinders the crystallinity of the furan‐based copolyesters.
One limitation of our research is that the molecular weight of the copolyesters is restricted by the incorporation of aromaticity in the backbone. Future work should focus on enhancing the molecular weight, as well as increasing the aromatic content in the polyester chain. As such, we believe that this study provides a fundamental background to design sustainable high‐performance polymers by an enzymatic pathway.
Experimental Section
Materials
Novozym 435 (N435, Candida antarctica lipase B (CALB) immobilized on acrylic resin, ≥5000 U g−1), 1,4‐butanediol (1,4‐BDO, 99 %), 1,6‐hexanediol (1,6‐HDO, 99 %), 1,8‐octanediol (1,8‐ODO, 98 %), 1,10‐decanediol (1,10‐DDO, 98 %), 1,12‐dodecanediol (1,12‐DODO, 99 %), diethyl succinate (99 %), diethyl adipate (99 %), diethyl sebacate (≥98 %), deuterated chloroform (CDCl3, 99.8 atom% D), chloroform (CHCl3, Chromasolv HPLC, ≥99.8 %, amylene stabilized) and diphenyl ether (99 %) were purchased from Sigma–Aldrich. Dimethyl 2,5‐furandicarboxylate (DMFDCA, 97 %) was purchased from Fluorochem UK. Diethyl dodecanedioate (≥95 %) was purchased from TCI Europe. Diethyl suberate (99 %) was purchased from ABCR. 2,5‐Bis(hydroxymethyl)furan (BHMF, ≥98 %) was purchased from Apollo Scientific. Chloroform (CHCl3, ChromAR HPLC, ethanol stabilized) and n‐hexane (n‐Hx, 99 %) were obtained from Macron. Absolute methanol (MeOH, AR) was obtained from Biosolve Chemicals. N435 was predried as reported previously.5b Diphenyl ether was distilled at 140 °C under reduced pressure and stored with activated 4 Å molecular sieves before use. All other chemicals were used as received.
Instrumental methods
Nuclear Magnetic Resonance (NMR) measurements were performed on a Varian VXR spectrometer (1H: 400; 13C: 300 MHz), using CDCl3 as the solvent.
Attenuated total reflectance–Fourier transform infrared (ATR‐FTIR) spectra were recorded on a Bruker VERTEX 70 spectrometer equipped with a Platinum‐ATR diamond single reflection unit. The measurement resolution was 4 cm−1 and the spectra were collected in the range of 4000–400 cm−1, with 16 scans for each sample. Atmospheric compensation and baseline correction were applied to the collected spectra using OPUS spectroscopy software (v7.0) (Bruker Optics).
Size‐exclusion chromatography (SEC) was performed on a Malvern Viscotek GPCmax equipped with triple detection, consisting of a Malvern Dual detector and Schambeck RI2912, refractive index detector. The separation was performed by utilizing two PLgel 5 μm MIXED‐C, 300 mm columns from Agilent Technologies at 35 °C. Amylene‐stabilized chloroform (CHROMASOLV, for HPLC, >99.8 %) was used as the eluent at a flow rate of 1.0 mL min−1. Data acquisition and calculations were performed using Viscotek OmniSec software version 5.0. Molecular weights were determined based on a conventional calibration curve generated from narrow dispersity polystyrene standards (Agilent and Polymer Laboratories, =645–3001 000 g mol−1). The samples were filtered over a 0.45 μm PTFE filter prior to injection.
Differential scanning calorimetry (DSC) measurements were conducted to measure the thermal transitions of the obtained furan copolyesters. The measurements were performed on a TA‐Instruments Q1000 DSC by heating–cooling–heating scans with heating–cooling rates of 10 °C min−1.
Thermogravimetric analysis (TGA) was performed on a TA‐Instruments Discovery TGA 5500. The samples were heated at a 10 °C min−1 scan rate in a nitrogen environment. Before the standard TGA measurement, the tested sample was first heated up to 100 °C and then maintained at this temperature for 30 min. to remove the remaining water and solvents in the polymer.
Wide‐angle X‐ray diffraction (WAXD) patterns of the obtained furan copolyesters were recorded on a Bruker D8 Advance diffractometer (CuKα radiation, λ=0.1542 nm) in the angular range of 5–50° (2θ) at room temperature.
Polarized optical microscopy (POM) images were observed by using a Zeiss Axiophot polarizing microscope equipped with a Sony DICC‐500 camera for image acquisition. The images were recorded by KS3000 software (Zeiss). The sample preparation was done on a Mettler Toledo FP82HT hot stage with a Mettler FP90 control panel.
General synthetic procedure for CALB‐catalyzed copolymerization with a temperature varied two‐stage method
Based on our previously reported studies,5d the following temperature varied two‐step enzymatic polymerization procedure was applied. As an example, the experimental copolymerization of DMFDCA, BHMF, and diethyl succinate was performed as follows. Predried N435 (20 wt % in relation to the total amount of the monomer) was fed into a 25 mL round bottle under a nitrogen environment. Subsequently, DMFDCA (524 mg, 2.85 mmol), BHMF (730 mg, 5.70 mmol), diethyl succinate (496 mg, 2.85 mmol), and diphenyl ether (6 mL) were added into the flask. In the first step of the reaction, the flask was magnetically stirred in an oil bath and heated to 80 °C for 2 h under a nitrogen atmosphere. Then at the second stage, the pressure was reduced stepwise to 2 mmHg while the reaction temperature was kept at 80 °C for the first 48 h. Finally, the reaction temperature was increased to 95 °C under full vacuum for the last 24 h. After that, the flask was cooled down. Chloroform (20 mL) was added to dissolve the products. N435 was filtered off by normal filtration (Folded filter type 15 Munktell 240 mm) and then washed with chloroform (3×10 mL). All the obtained solutions were then combined and concentrated by a rotary evaporator at 40 °C under a reduced pressure of 400–480 mbar. The concentrated solution was added dropwise into an excess amount of methanol (or hexane). The solution with the precipitated products were then stored for several hours at −20 °C. After that, the precipitated product was isolated by centrifugation (30 min, 4500 rpm, 4 °C in a Thermo/Heraeus Labofuge 400 R, 50 mL Greiner bio‐one, Cellstar tubes) and dried under vacuum at 40 °C for 3 days. Lastly, they were stored under vacuum at room temperature prior to analysis.
The synthesis procedure of the other copolyesters was the same as the example above, except using different monomers and feed compositions.
Furan‐based copolyesters: ATR‐FTIR: =3118–3137 (=C−H stretching vibrations of the furan ring); 2914–2954, 2848–2869 (asymmetric and symmetric C−H stretching vibrations); 1710–1729 (C=O stretching vibrations); 1573–1583, 1506–1511 (aromatic C=C bending vibrations); 1434–1471, 1371–1392 (C−H deformation and wagging vibrations); 1329 (C−H rocking vibrations); 1122–1151, 1268–1276 (asymmetric and symmetric stretching vibrations of the ester C−O−C groups);1203–1228, 1004–1031 (=C−O−C= ring vibrations, furan ring); 948–979, 798–835, 763–771 cm−1 (=C−H out‐of‐plane deformation vibrations, furan ring).
Poly(2,5‐furandimethylene furanoate‐co‐butylene furanoate) [P(FMF‐co‐BF)]: 1H NMR (400 MHz, CDCl3): δ=7.20 (2 H, s, ‐CH=, DMFDCA), 6.48 (2 H, s, ‐CH=, BHMF), 5.28 (4 H, s, ‐CO‐O‐CH 2‐, BHMF), 4.38 (4 H, m, ‐CO‐O‐CH 2‐, from 1,4‐BDO), 1.90 (4 H, m, ‐CO‐O‐CH2‐CH 2‐, from 1,4‐BDO), 4.61 (s, ‐CH 2OH, end group from BHMF), 3.92 (s, ‐O‐CH 3, end group from DMFDCA), 3.71 ppm (t, ‐CH 2‐OH, end group from 1,4‐BDO).
Poly(2,5‐furandimethylene furanoate‐co‐hexamethylene furanoate) [P(FMF‐co‐HF)]: 1H NMR (400 MHz, CDCl3): δ=7.18 (2 H, m, ‐CH=, DMFDCA), 6.48 (2 H, m, ‐CH=, BHMF), 5.28 (4 H, s, ‐CO‐O‐CH 2‐, BHMF), 4.32 (4 H, m, ‐CO‐O‐CH 2‐, from 1,6‐HDO), 1.77 (4 H, m, ‐CO‐O‐CH2‐CH 2‐, from 1,6‐HDO), 1.46 (4 H, m, ‐CO‐O‐CH2‐CH2‐CH 2‐, from 1,6‐HDO), 4.60 (s, ‐CH 2OH, end group from BHMF), 3.91 (s, ‐O‐CH 3, end group from DMFDCA), 3.64 ppm (t, ‐CH 2‐OH, end group from 1,6‐HDO).
Poly(2,5‐furandimethylene furanoate‐co‐octamethylene furanoate) [P(FMF‐co‐OF)]: 1H NMR (400 MHz, CDCl3): δ=7.18 (2 H, m, ‐CH=, DMFDCA), 6.48 (2 H, m, ‐CH=, BHMF), 5.28 (4 H, s, ‐CO‐O‐CH 2‐, BHMF), 4.30 (4 H, m, ‐CO‐O‐CH 2‐, from 1,8‐ODO), 1.74 (4 H, m, ‐CO‐O‐CH2‐CH 2‐, from 1,8‐ODO), 1.36 (8 H, m, ‐CO‐O‐CH2‐CH2‐CH 2‐, from 1,6‐HDO), 4.60 (s, ‐CH 2OH, end group from BHMF), 3.91 (s, ‐O‐CH 3, end group from DMFDCA), 3.62 ppm (t, ‐CH 2‐OH, end group from 1,8‐ODO).
Poly(2,5‐furandimethylene furanoate‐co‐decamethylene furanoate) [P(FMF‐co‐DF)]: 1H NMR (400 MHz, CDCl3): δ=7.18 (2 H, m, ‐CH=, DMFDCA), 6.48 (2 H, m, ‐CH=, BHMF), 5.29 (4 H, s, ‐CO‐O‐CH 2‐, BHMF), 4.31 (4 H, m, ‐CO‐O‐CH 2‐, from 1,10‐DDO), 1.74 (4 H, m, ‐CO‐O‐CH2‐CH 2‐, from 1,10‐DDO), 1.36 (4 H, m, ‐CH 2‐, from 1,10‐DDO), 1.29 (8 H, m, ‐CH 2‐, from 1,10‐DDO), 4.61 (s, ‐CH 2OH, end group from BHMF), 3.92 (s, ‐O‐CH 3, end group from DMFDCA), 3.63 ppm (t, ‐CH 2‐OH, end group from 1,10‐DDO).
Poly(2,5‐furandimethylene furanoate‐co‐dodecamethylene furanoate) [P(FMF‐co‐DOF)]: 1H NMR (400 MHz, CDCl3): δ=7.18 (2 H, m, ‐CH=, DMFDCA), 6.48 (2 H, m, ‐CH=, BHMF), 5.29 (4 H, s, ‐CO‐O‐CH 2‐, BHMF), 4.31 (4 H, m, ‐CO‐O‐CH 2‐, from 1,12‐DODO), 1.74 (4 H, m, ‐CO‐O‐CH2‐CH 2‐, from 1,12‐DODO), 1.38 (4 H, m, ‐CH 2‐, from 1,12‐DODO), 1.26 (12 H, m, ‐CH 2‐, from 1,12‐DODO), 4.61 (s, ‐CH 2OH, end group from BHMF), 3.92 (s, ‐O‐CH 3, end group from DMFDCA), 3.64 ppm (t, ‐CH 2‐OH, end group from 1,12‐DODO).
Poly(2,5‐furandimethylene furanoate‐co‐2,5‐furandimethylene succinate) [P(FMF‐co‐FMS)]: 1H NMR (400 MHz, CDCl3): δ=7.13 (2 H, m, ‐CH=, DMFDCA), 6.33 (2 H, m, ‐CH=, BHMF), 5.28 (4 H, s, ‐CO‐O‐CH 2‐, BHMF‐DMFDCA), 5.05 (4 H, m, ‐CO‐O‐CH 2‐, BHMF‐succinate), 2.63 (4 H, m, ‐O‐CO‐CH 2‐, succinate), 4.57 (s, ‐CH 2OH, end group from BHMF), 3.91 (s, ‐O‐CH 3, end group from DMFDCA), 4.13 (m, ‐OCH 2CH3, end group from diethyl succinate), 1.23 (t, ‐OCH2CH 3, end group from diethyl succinate), 4.45 ppm (s, ‐CH 2‐O‐CH 2‐, BHMF ether).
Poly(2,5‐furandimethylene furanoate‐co‐2,5‐furandimethylene adipate) [P(FMF‐co‐FMA)]: 1H NMR (400 MHz, CDCl3): δ=7.20 (2 H, m, ‐CH=, DMFDCA), 6.34 (2 H, m, ‐CH=, BHMF), 5.26 (4 H, s, ‐CO‐O‐CH 2‐, BHMF‐DMFDCA), 5.01 (4 H, s, ‐CO‐O‐CH 2‐, BHMF‐adipate), 2.33 (4 H, m, ‐O‐CO‐CH 2‐, adipate), 1.64 (4 H, m, ‐CH 2‐, adipate), 4.58 (s, ‐CH 2OH, end group from BHMF), 3.90 (s, ‐O‐CH 3, end group from DMFDCA), 4.11 (m, ‐OCH 2CH3, end group from diethyl adipate), 1.23 (t, ‐OCH2CH 3, end group from diethyl adipate), 4.45 ppm (s, ‐CH 2‐O‐CH 2‐, BHMF ether).
Poly(2,5‐furandimethylene furanoate‐co‐2,5‐furandimethylene suberate) [P(FMF‐co‐FMSu)]: 1H NMR (400 MHz, CDCl3): δ=7.21 (2 H, m, ‐CH=, DMFDCA), 6.34 (2 H, m, ‐CH=, BHMF), 5.27 (4 H, s, ‐CO‐O‐CH 2‐, BHMF‐DMFDCA), 5.02 (4 H, s, ‐CO‐O‐CH 2‐, BHMF‐suberate), 2.30 (4 H, m, ‐O‐CO‐CH 2‐, suberate), 1.60 (4 H, m, ‐CH 2‐, suberate), 1.30 (4 H, m, ‐CH 2‐, suberate), 4.59 (s, ‐CH 2OH, end group from BHMF), 3.91 (s, ‐O‐CH 3, end group from DMFDCA), 4.11 (m, ‐OCH 2CH3, end group from diethyl suberate), 4.46 ppm (s, ‐CH 2‐O‐CH 2‐, BHMF ether).
Poly(2,5‐furandimethylene furanoate‐co‐2,5‐furandimethylene sebacate) [P(FMF‐co‐FMSe)]: 1H NMR (400 MHz, CDCl3): δ=7.21 (2 H, m, ‐CH=, DMFDCA), 6.35 (2 H, m, ‐CH=, BHMF), 5.27 (4 H, s, ‐CO‐O‐CH 2‐, BHMF‐DMFDCA), 5.02 (4 H, s, ‐CO‐O‐CH 2‐, BHMF‐sebacate), 2.31 (4 H, m, ‐O‐CO‐CH 2‐, sebacate), 1.59 (4 H, m, ‐CH 2‐, sebacate), 1.26 (8 H, m, ‐CH 2‐, sebacate), 4.59 (s, ‐CH 2OH, end group from BHMF), 3.90 (s, ‐O‐CH 3, end group from DMFDCA), 4.11 (m, ‐OCH 2CH3, end group from diethyl sebacate), 4.47 ppm (s, ‐CH 2‐O‐CH 2‐, BHMF ether).
Poly(2,5‐furandimethylene furanoate‐co‐2,5‐furandimethylene dodecanedioate) [P(FMF‐co‐FMD)]: 1H NMR (400 MHz, CDCl3): δ=7.21 (2 H, m, ‐CH=, DMFDCA), 6.35 (2 H, m, ‐CH=, BHMF), 5.27 (4 H, s, ‐CO‐O‐CH 2‐, BHMF‐DMFDCA), 5.02 (4 H, s, ‐CO‐O‐CH 2‐, BHMF‐dodecanedioate), 2.31 (4 H, m, ‐O‐CO‐CH 2‐, dodecanedioate), 1.60 (4 H, m, ‐CH 2‐, dodecanedioate), 1.24 (12 H, m, ‐CH 2‐, dodecanedioate), 4.59 (s, ‐CH 2OH, end group from BHMF), 3.91 (s, ‐O‐CH 3, end group from DMFDCA), 4.10 (m, ‐OCH 2CH3, end group from diethyl dodecanedioate), 4.46 ppm (s, ‐CH 2‐O‐CH 2‐, BHMF ether).
Abbreviations
PEF=poly(ethylene furanoate); PET=poly(ethylene terephthalate); N435=Novozyme 435; CALB=Candida antarctica lipase B; DMFDCA=dimethyl 2,5‐furandicarboxylate; BHMF=2,5‐bis(hydroxymethyl)furan; 1,4‐BDO=1,4‐butanediol; 1,6‐HDO=1,6‐hexanediol; 1,8‐ODO=1,8‐octanediol; 1,10‐DDO=1,10‐decanediol; 1,12‐DODO=1,12‐dodecanediol; P(FMF‐co‐BF)=poly(2,5‐furandimethylene furanoate‐co‐butylene furanoate); P(FMF‐co‐HF)=poly(2,5‐furandimethylene furanoate‐co‐hexamethylene furanoate); P(FMF‐co‐OF)=poly(2,5‐furandimethylene furanoate‐co‐octamethylene furanoate); P(FMF‐co‐DF)=poly(2,5‐furandimethylene furanoate‐co‐decamethylene furanoate); P(FMF‐co‐DOF)=poly(2,5‐furandimethylene furanoate‐co‐dodecamethylene furanoate); P(FMF‐co‐FMS)=poly(2,5‐furandimethylene furanoate‐co‐2,5‐furandimethylene succinate); P(FMF‐co‐FMA)=poly(2,5‐furandimethylene furanoate‐co‐2,5‐furandimethylene adipate); P(FMF‐co‐FMSu)=poly(2,5‐furandimethylene furanoate‐co‐2,5‐furandimethylene suberate); P(FMF‐co‐FMSe)=poly(2,5‐furandimethylene furanoate‐co‐2,5‐furandimethylene sebacate); P(FMF‐co‐FMD)=poly(2,5‐furandimethylene furanoate‐co‐2,5‐furandimethylene dodecanedioate); ATR‐FTIR=attenuated total reflectance–Fourier transform infrared; SEC=size‐exclusion chromatography; DSC=differential scanning calorimetry; TGA=thermogravimetric analysis; WAXD=wide‐angle X‐ray diffraction.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
D.M. gratefully acknowledges the financial support from the Indonesian Endowment Fund for Education (Lembaga Pengelola Dana Pendidikan LPDP). We would also like to thank Dr. J. Schöbel and Y. A. Muthahari for the valuable suggestions and discussions.
D. Maniar, Y. Jiang, A. J. J. Woortman, J. van Dijken, K. Loos, ChemSusChem 2019, 12, 990.
References
- 1.
- 1a. Hillmyer M. A., Science 2017, 358, 868–870; [DOI] [PubMed] [Google Scholar]
- 1b. Zhu Y., Romain C., Williams C. K., Nature 2016, 540, 354; [DOI] [PubMed] [Google Scholar]
- 1c. Vilela C., Sousa A. F., Fonseca A. C., Serra A. C., Coelho J. F. J., Freire C. S. R., Silvestre A. J. D., Polym. Chem. 2014, 5, 3119–3141. [Google Scholar]
- 2.
- 2a. Mülhaupt R., Macromol. Chem. Phys. 2013, 214, 159–174; [Google Scholar]
- 2b. Williams C. K., Hillmyer M. A., Polym. Rev. 2008, 48, 1–10; [Google Scholar]
- 2c. Mecking S., Angew. Chem. Int. Ed. 2004, 43, 1078–1085; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 1096–1104. [Google Scholar]
- 3.
- 3a. Douka A., Vouyiouka S., Papaspyridi L.-M., Papaspyrides C. D., Prog. Polym. Sci. 2018, 79, 1–25; [Google Scholar]
- 3b. Jiang Y., Loos K., Polymers 2016, 8, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pavlidis I. V., Tzialla A. A., Enotiadis A., Stamatis H., Gournis D. in Biocatalysis in Polymer Chemistry, Wiley-VCH, 2010, pp. 35–63. [Google Scholar]
- 5.
- 5a. Muthusamy K., Lalitha K., Prasad Y. S., Thamizhanban A., Sridharan V., Maheswari C. U., Nagarajan S., ChemSusChem 2018, 11, 2453–2463; [DOI] [PubMed] [Google Scholar]
- 5b. Jiang Y., van Ekenstein G. O. R. A., Woortman A. J. J., Loos K., Macromol. Chem. Phys. 2014, 215, 2185–2197; [Google Scholar]
- 5c. Jiang Y., Woortman A., van Ekenstein G., Loos K., Biomolecules 2013, 3, 461–480; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5d. Jiang Y., Woortman A. J. J., van Ekenstein G. O. R. Alberda, Loos K., Polym. Chem. 2015, 6, 5198–5211; [Google Scholar]
- 5e. Jiang Y., Woortman A. J. J., van Ekenstein G. O. R. Alberda, Loos K., Polym. Chem. 2015, 6, 5451–5463; [Google Scholar]
- 5f. Jiang Y., Woortman A. J. J., van Ekenstein G. O. R. A., Petrovic D. M., Loos K., Biomacromolecules 2014, 15, 2482–2493. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Jiang Y., Maniar D., Woortman A. J. J., van Ekenstein G. O. R. Alberda, Loos K., Biomacromolecules 2015, 16, 3674–3685; [DOI] [PubMed] [Google Scholar]
- 6b. Jiang Y., Maniar D., Woortman A. J. J., Loos K., RSC Adv. 2016, 6, 67941–67953; [Google Scholar]
- 6c. Maniar D., Hohmann K. F., Jiang Y., Woortman A. J. J., van Dijken J., Loos K., ACS Omega 2018, 3, 7077–7085; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6d. Stavila E., van Ekenstein G. O. Alberda, Woortman A. J., Loos K., Biomacromolecules 2014, 15, 234–241; [DOI] [PubMed] [Google Scholar]
- 6e. Stavila E., Arsyi R. Z., Petrovic D. M., Loos K., Eur. Polym. J. 2013, 49, 834–842; [Google Scholar]
- 6f. Stavila E., van Ekenstein G. O. R. A., Loos K., Biomacromolecules 2013, 14, 1600–1606; [DOI] [PubMed] [Google Scholar]
- 6g.L. W. Schwab, PhD thesis, University of Groningen (Groningen), 2010.
- 7.
- 7a. Rodriguez K. J., Gajewska B., Pollard J., Pellizzoni M. M., Fodor C., Bruns N., ACS Macro Lett. 2018, 7, 1111–1119; [DOI] [PubMed] [Google Scholar]
- 7b. Hollmann F., Arends I. W. C. E., Polymers 2012, 4, 759. [Google Scholar]
- 8. Kobayashi S., Ohmae M. in Enzyme-Catalyzed Synthesis of Polymers (Eds.: S. Kobayashi, H. Ritter, D. Kaplan), Springer, Berlin, Heidelberg, 2006, pp. 159–210. [Google Scholar]
- 9. Odian G., Principles of polymerization, Wiley, 2004. [Google Scholar]
- 10. Hergenrother P. M., High Perform. Polym. 2003, 15, 3–45. [Google Scholar]
- 11.
- 11a. Burgess S. K., Karvan O., Johnson J. R., Kriegel R. M., Koros W. J., Polymer 2014, 55, 4748–4756; [Google Scholar]
- 11b. Burgess S. K., Leisen J. E., Kraftschik B. E., Mubarak C. R., Kriegel R. M., Koros W. J., Macromolecules 2014, 47, 1383–1391. [Google Scholar]
- 12. Okada M., Tachikawa K., Aoi K., J. Polym. Sci. Part A 1997, 35, 2729–2737. [Google Scholar]
- 13. Storbeck R., Ballauff M., Polymer 1993, 34, 5003–5006. [Google Scholar]
- 14.
- 14a. Khrouf A., Boufi S., El Gharbi R., Belgacem N. M., Gandini A., Polym. Bull. 1996, 37, 589–596; [Google Scholar]
- 14b. Gandini A., Silvestre A. J. D., Neto C. P., Sousa A. F., Gomes M., J. Polym. Sci. Part A 2009, 47, 295–298; [Google Scholar]
- 14c. Gharbi S., Andreolety J. P., Gandini A., Eur. Polym. J. 2000, 36, 463–472. [Google Scholar]
- 15.
- 15a. Zhu J., Cai J., Xie W., Chen P.-H., Gazzano M., Scandola M., Gross R. A., Macromolecules 2013, 46, 796–804; [Google Scholar]
- 15b. Thiyagarajan S., Vogelzang W., Knoop R. J. I., Frissen A. E., van Haveren J., van Es D. S., Green Chem. 2014, 16, 1957–1966; [Google Scholar]
- 15c. Gomes M., Gandini A., Silvestre A. J. D., Reis B., J. Polym. Sci. Part A 2011, 49, 3759–3768; [Google Scholar]
- 15d. Jiang M., Liu Q., Zhang Q., Ye C., Zhou G., J. Polym. Sci. Part A 2012, 50, 1026–1036; [Google Scholar]
- 15e. Gubbels E., Jasinska-Walc L., Koning C. E., J. Polym. Sci. Part A 2013, 51, 890–898; [Google Scholar]
- 15f. Zeng C., Seino H., Ren J., Hatanaka K., Yoshie N., Macromolecules 2013, 46, 1794–1802. [Google Scholar]
- 16. Ma J., Pang Y., Wang M., Xu J., Ma H., Nie X., J. Mater. Chem. 2012, 22, 3457–3461. [Google Scholar]
- 17. Wu L., Mincheva R., Xu Y., Raquez J. M., Dubois P., Biomacromolecules 2012, 13, 2973–2981. [DOI] [PubMed] [Google Scholar]
- 18. Sousa A. F., Matos M., Freire C. S. R., Silvestre A. J. D., Coelho J. F. J., Polymer 2013, 54, 513–519. [Google Scholar]
- 19. Morales-Huerta J. C., de Ilarduya A. Martínez, Muñoz-Guerra S., ACS Sustainable Chem. Eng. 2016, 4, 4965–4973. [Google Scholar]
- 20.
- 20a. Morales-Huerta J. C., Ciulik C. B., de Ilarduya A. M., Muñoz-Guerra S., Polym. Chem. 2017, 8, 748–760; [Google Scholar]
- 20b. Morales-Huerta J. C., Martínez de Ilarduya A., Muñoz-Guerra S., J. Polym. Sci. Part A 2018, 56, 290–299. [Google Scholar]
- 21.
- 21a. Juais D., Naves A. F., Li C., Gross R. A., Catalani L. H., Macromolecules 2010, 43, 10315–10319; [Google Scholar]
- 21b. Kanelli M., Douka A., Vouyiouka S., Papaspyrides C. D., Topakas E., Papaspyridi L.-M., Christakopoulos P., J. Appl. Polym. Sci. 2014, 131, 10.1002/app.40820. [DOI] [Google Scholar]
- 22. McCabe R. W., Taylor A., Enzyme Microb. Technol. 2004, 35, 393–398. [Google Scholar]
- 23.M. Takwa, PhD thesis, KTH Royal Institute of Technology (Sweden), 2010.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary