

Abstract
The 3D shape of glycosyl oxocarbenium ions determines their stability and reactivity and the stereochemical course of SN1 reactions taking place on these reactive intermediates is dictated by the conformation of these species. The nature and configuration of functional groups on the carbohydrate ring affect the stability of glycosyl oxocarbenium ions and control the overall shape of the cations. We herein map the stereoelectronic substituent effects of the C2‐azide, C2‐fluoride and C4‐carboxylic acid ester on the stability and reactivity of the complete suite of diastereoisomeric furanoses by using a combined computational and experimental approach. Surprisingly, all furanosyl donors studied react in a highly stereoselective manner to provide the 1,2‐cis products, except for the reactions in the xylose series. The 1,2‐cis selectivity for the ribo‐, arabino‐ and lyxo‐configured furanosides can be traced back to the lowest‐energy 3 E or E 3 conformers of the intermediate oxocarbenium ions. The lack of selectivity for the xylosyl donors is related to the occurrence of oxocarbenium ions adopting other conformations.
Keywords: conformation analysis, energy landscape maps, glycosylation, oxocarbenium ions, substituent effect
Introduction
Stereoelectonic effects dictate the shape and behaviour of molecules. Understanding and harnessing these effects enables the conception of effective and stereoselective synthetic chemistry.1 Carbohydrates are densely decorated molecules bearing a variety of different functional groups in numerous configurational and stereochemical constellations.2, 3 The decoration pattern of carbohydrates plays an all‐important role in manipulations/transformations of the functional groups in the assembly of carbohydrate building blocks as well as in the union of two carbohydrates in a glycosylation reaction. During a glycosylation reaction a donor glycoside is generally activated to give an electrophilic species bearing significant oxocarbenium ion character.4 Although steric effects are often decisive in determining the overall shape of a neutral molecule, in charged molecules electronic effects become more important and they may in fact outweigh steric effects. For example, protonated iminosugars, that is, carbohydrates having the endocyclic oxygen replaced by a nitrogen, may change their conformation to place their ring substituents in a sterically unfavourable (pseudo)‐axial orientation to stabilise the positive charge on the ring nitrogen.5, 6, 7, 8, 9, 10 In line with these stereoelectronic effects, glycosyl donors that feature an “axial‐rich” substitution pattern, are generally more reactive than glycosyl donors equipped with equatorially disposed functional groups.11, 12, 13 However, it is extremely challenging to understand—let alone predict—what the overall effect of multiple ring substituents is on the reactivity of a particular glycosyl donor and as a result the effect on the stereoselectivity in a glycosylation reaction. Based on a computational strategy of Rhoad and co‐workers,14 we have recently introduced a method to determine the conformational behaviour of furanosyl oxocarbenium ions.15, 16, 17 By calculating the relative energy of a large number of fixed furanosyl oxocarbenium ion conformers and mapping these in energy contour plots we could determine, which conformations played an important role during furanosylation reactions and we were able to relate the population of the different conformational states to the stereoselectivity of the reactions. The introduced conformational energy landscape mapping method provided detailed insight into how the ring substituents—as stand‐alone entities but also collectively—influenced the shape, stability and reactivity of the furanosyl oxocarbenium ions. In this initial study, only ether substituents were assessed. We here present an in‐depth study on the stereoelectronic substituent effects of different functional groups that are all highly relevant in oligosaccharide synthesis. Understanding these effects will enable the development of effective glycosylation methodologies and aid in the interpretation of the outcome of glycosylation reactions. We have studied the effect of C2‐fluoride and C2‐azide substituents, as well as C4‐carboxylic acid ester groups, as these functionalities are commonly employed in the assembly of fluorinated, cis‐linked glycosamine‐containing or glycuronic acid‐featuring oligosaccharides, respectively.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25
Herein, we describe the synthesis of a panel of twelve structurally varying furanosyl imidate donors, comprising all possible pentofuranosyl diastereoisomers (Figure 1, 1–12), their glycosylation properties were studied by experimental chemical glycosylations as well as by a computational investigation on the reactive intermediates active during the glycosylation and responsible for the stereoselective outcome of the reaction, that is, the furanosyl oxocarbenium ions.
Figure 1.
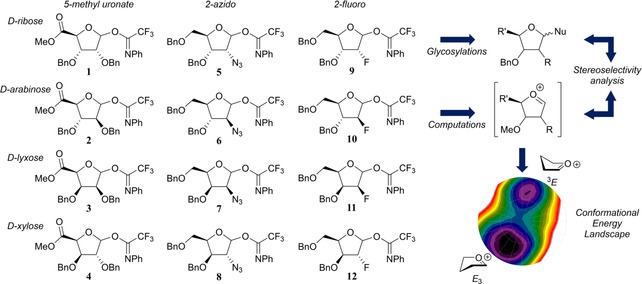
Target donors 1–12 and the subsequent stereoselectivity investigation by glycosylations and computational analysis (Bn=benzyl).
Results and Discussion
Synthesis
The set of d‐ribo‐, d‐arabino‐, d‐lyxo‐ and d‐xylo‐configured furanosyl donors 1–12 (Figure 1) that was needed for this study was prepared as depicted in Scheme 1. All donors studied here were equipped with an N‐phenyl trifluoroacetimidate anomeric leaving group.26 The uronic acid methyl esters 17–20 were obtained from their parent methyl furanosides 13–16 27, 28, 29, 30 by a straightforward TEMPO/BAIB oxidation procedure of the primary alcohols, followed by methylation with MeI and K2CO3 (Scheme 1 A).31 Aqueous TFA‐mediated hydrolysis of the anomeric methyl group and installation of the trifluoro‐N‐phenyl imidate group with Cs2CO3 proceeded uneventfully to give donors 1–4.
Scheme 1.
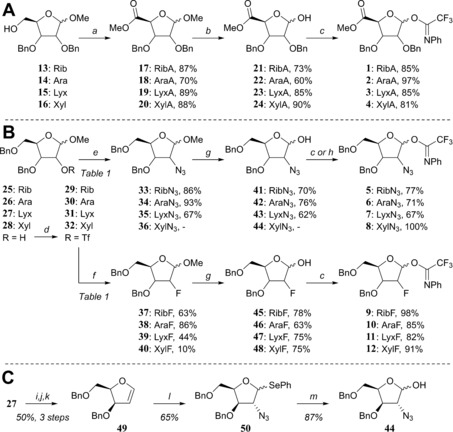
a) i) 2,2,6,6‐Tetramethylpiperidine N‐oxyl (TEMPO), diacetoxyiodobenzene (BAIB), dichloromethane, H2O; ii) MeI, K2CO3, DMF; b) trifluoroacetic acid (TFA)/H2O (9/1); c) 2,2,2‐trifluoro‐N‐phenylacetimidoyl chloride, Cs2CO3, acetone, H2O; d) trifluoromethanesulfonic anhydride (Tf2O), pyridine, dichloromethane; e) NaN3, DMF, see Table 1; f) tetrabutylammonium fluoride (TBAF), THF; or CsF, tert‐amyl alcohol, see Table 1; g) HCOOH, H2O; h) 2,2,2‐trifluoro‐N‐phenylacetimidoyl chloride, 1,5‐diazabicyclo[5.4.0]undec‐5‐ene (DBU), dichloromethane; i) TFA, H2O, THF; j) 4‐dimethylaminopyridine (DMAP), diisopropylethylamine (DIPEA), thiophosgene, dichloromethane; k) 1,3‐dimethyl‐2‐phenyl‐1,3,2‐diazaphospholidine, toluene; l) N‐(phenylseleno)phthalimide, azidotrimethylsilane (TMSN3), TBAF, dichloromethane; m) N‐iodosuccinimide (NIS), H2O, acetone, THF.
For the functionalisation on C2 we first investigated the inversion of the C2‐triflates 29–32, generated from the corresponding C2‐alcohols 25–28 32, 33, 34, 35 with a suitable azide or fluoride nucleophile (Scheme 1 B).36 Inversion of the ribosyl C2‐OTf group in 29 by using an excess of NaN3 in DMF at 80 °C proceeded smoothly to give the 2‐azidoarabinoside 34 in high yield (see Table 1, entry 1, conditions A). The inversion of 29 by using tetrabutylammonium fluoride as the source of the fluoride nucleophile in THF at ambient temperature gave 38 in good yield (71 %, Table 1, entry 2, conditions B). This yield could be further improved to 86 % by employing CsF in tert‐amyl alcohol at 90 °C (Table 1, entry 3, conditions C).37
Table 1.
Synthesis of C2‐modified methyl glycosides 33–40 through C2‐triflate inversion.
| Entry | Triflate | Cond.[a] | Substitution product |
Yield [%] |
Side products (yield [%]) |
|---|---|---|---|---|---|
| from d‐ribo‐ to d‐arabino‐configured | |||||
| 1 | 29 | A (N3) | 34 | 93 | – |
| 2 | 29 | B (TBAF) | 38 | 71 | – |
| 3 | 29 | C (CsF) | 38 | 86 | – |
| from d‐arabino‐ to d‐ribo‐configured | |||||
| 4 | 30 | A | 33 | 86[b] | 51 [b] |
| 5 | 30 | B | 37, β only | 42 | 30α (17) |
| 6 | 30 | C | 37 | 63 | 52 f (17) |
| from d‐xylo‐ to d‐lyxo‐configured | |||||
| 7 | 32α | A[c] | 35α | 67 | 53α (12), 55 (7) |
| 8 | 32α | B[d] | 39α | 44 | 28 (17) |
| 9 | 32α | C[e] | 39α | – | 54α (57), 55 (21) |
| 10 | 32β | A[c] | 35β | – | 53β, (30) |
| 11 | 32β | B[d] | 39β | – | 54β (18), 56[ g] |
| 12 | 32β | C | 39β | – | 54β (47), 40β (10) |
| from d‐lyxo‐ to d‐xylo‐configured | |||||
| 13 | 31 | A,B,C | 36/40 | – | 56[ g] |
[a] Reagents and conditions: A) 0.2 m solution in DMF, NaN3 (5 equiv), 80 °C, 2 h; B) 0.2 m solution in THF, TBAF (2.5 equiv), 0–20 °C, overnight; C) 0.35 m solution in tert‐amyl alcohol, CsF (4 equiv), 90 °C, overnight. [b] Combined yield of 33 and 51 as a 4:1 mixture. [c] Overnight. [d] 70 °C, 5 h for entry 8, overnight for entry 11. [e] 110 °C overnight. [f] α/β=88:12. [g] Yield not determined.
When the arabinoside C2‐triflate 30 (a mixture of anomers) was treated under conditions A to install the C2 azide group and provide 33, a mixture of products resulted consisting of the desired C2‐azide 33 and the anomeric azide 51 (Figure 2 A, 86 %, 33/51=4:1, Table 1, entry 4). The stereospecific formation of the β‐azide 51 (Figure 2 A) can be explained by the generation of a transient methyl oxiranium ion intermediate, that is substituted in an SN2‐like fashion by the azide anion on the anomeric centre (see Figure 2 B, path A).38
Figure 2.
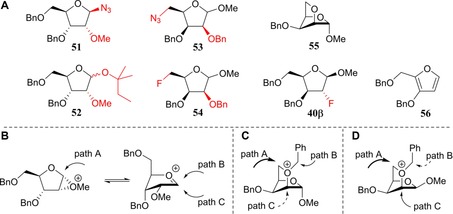
A) Observed side products 51–56 and 40β. B) Proposed reaction pathways for the formation of 51 and 52. C) Proposed reaction pathways for the formation of 53α and 54α (path A) as well as 55 (path B). D) Proposed reaction pathways for the formation of 53β and 54β (path A) as well as 40β (path C).
The fluoride substitution on 30 also comes along with side reactions. When 30 was subjected to conditions B by using TBAF, only the β‐anomer of 30 reacted to provide 37, leaving the α‐anomer untouched (Table 1, entry 5).32 At higher temperatures, by using CsF (conditions C), both anomers reacted to provide the corresponding C2‐fluorides. However, reaction of the α‐anomer of 30 also provided the anomeric tert‐amyl product 52 (Figure 2 A), resulting from a migration of the anomeric methoxide by substitution of the C2‐triflate and subsequent solvolysis of the formed oxocarbenium ion (Figure 2 B, paths A, B and/or C). The weaker nucleophilicity of tert‐amyl alcohol with respect to the azide anion likely leads to more SN1 character in the substitution reaction of the methyl oxiranium ion and generation of an anomeric mixture of 52, where the azide stereospecifically provided the β‐azide 51.
The xylosyl C2‐alcohols 28α and 28β could be readily separated and their C2‐triflates 32α and 32β could therefore be individually investigated in the substitution reactions (Scheme 1 B, Table 1, entries 7–12). The inversion of the α‐anomer 32α with NaN3 provided the 2‐azidolyxoside 35α (67 %, Table 1, entry 7), alongside two side products, that is, the 5‐azidolyxoside 53α and the bicycle 55 (Figure 2 A), which were formed in 12 and 7 % yield, respectively. The generation of these side products stems from the participation of the primary C5‐OBn group, which is capable of substituting the C2‐OTf group. Nucleophilic attack at C5 provides 53α, whereas substitution at the benzylic position generates the bicycle 55 (see Figure 2 C, paths A and B). When the substitution of the C2‐triflate 32α was tried under conditions B to furnish the desired C2‐fluoro lyxose 39α (Table 1, entry 8), no conversion was observed and therefore, the reaction was heated to 70 °C. Under these conditions, the 2‐fluorolyxoside 39α was formed in 44 % yield, whereas alcohol 28 was regenerated through hydrolysis of the triflate. Application of conditions C (Table 1, entry 9) only resulted in the formation of products originating from C5‐OBn participation: the 5‐fluoroxyloside 54α and the bicycle 55 (Figure 2 A and D) were obtained in 57 and 21 % yield, respectively.
Inversion of the C2‐OTf group of the β‐xyloside 32β with either the azide or fluoride nucleophiles did not lead to any desired inversion products (Table 1, entries 10–12). Conditions A only provided the C5‐azido product 53β (Figure 2 A), whereas conditions B led to the formation of 54β, through the participation of the C5‐OBn group (Figure 2 C, path A). Elimination to give furan 56 was also observed under conditions B (Table 1, entry 11).39 Interestingly, the use of CsF (conditions C, Table 1, entry 12) provided, besides the side product 54β, the 2‐fluoroxyloside 40β in low yield, apparently through a double‐displacement mechanism (Figure 2 D, path C). Generation of product 40β through this route proved advantageous because its generation from lyxo‐triflate 31 was ineffective (see below).
All conditions examined to transform lyxo‐triflate 31 to one of the inverted products (i.e., 36/40) were ineffective (Table 1, entry 13) and furan 56 was formed exclusively within minutes. We therefore took a different approach to generate the 2‐azidoxyloside (Scheme 1 C). Thus, glycal 49 40 was functionalised by azidophenylselenation with TMSN3 and N‐(phenylseleno)phthalimide to give the desired 2‐azidoxyloside 50 with good diastereoselectivity (xylo/lyxo 9:1).41, 42 Oxidative hydrolysis of the selenophenyl group by aqueous NIS then gave lactol 44.
Acidolysis of the anomeric methyl ethers 33–35 and 37–40 by using aqueous formic acid provided the other lactols 41–43 and 45–48, respectively (Scheme 1 B). Finally, all eight lactols were transformed into the corresponding N‐phenyl trifluoracetimidates 5–12 to complete the set of donor furanosides.
Glycosylations
With the complete set of functionalised furanosyl imidate donors 1–12 in hand, the stereoselectivity of the glycosylation reactions by using allyltrimethylsilane (allyl‐TMS) or [D]triethylsilane ([D]TES) as acceptors, were examined.43 Allyl‐TMS and [D]TES are poor nucleophiles and are ideal acceptors to study the SN1 reaction pathways of the glycosylations at hand.15, 16, 44, 45, 46 The results of these glycosylations together with results obtained previously for the tri‐O‐benzyl series (i.e., donors 57–60) are listed in Table 2. As previously described, the reactions in the tri‐O‐benzyl series proceed with good to excellent 1,2‐cis selectivity for all four configurations.15 The ribo‐, arabino‐ and lyxo‐configured donors 57, 58 and 59 provided exclusively the 1,2‐cis‐substitution products, whereas the xylose donor 60 gave the anomeric deuterium α and β products in a 85:15 α/β ratio (Table 2, entry 1, 5, 9 and 13). Strikingly, the 1,2‐cis selectivity in the glycosylation reactions was also observed for the reactions of the C2‐ and C5‐modified furanosyl donors. All reactions performed with the ribose donors 1, 5 and 9 (Table 2, entries 2–4), the arabinose donors 2, 6 and 10 (Table 2, entries 6–8) and the lyxose donors 3, 7 and 11 (Table 2, entries 10–12) proceeded with excellent 1,2‐cis stereoselectivity. The reactions of the xylose donors 4, 8 and 12 (Table 2, entries 14–16) proceed with poorer stereoselectivity. The 2‐azidoxyloside donor 8 gives a 85:15 mixture of anomers (product 72), which is in line with the outcome of the reaction of the corresponding tri‐O‐benzyl donor 60 (Table 2, entries 13 and 15). The 2‐fluoroxyloside 76 is formed in a 70:30 α/β ratio (Table 2, entry 16) and the uronic acid xyloside donor 4 provided the least selective reaction giving roughly equal amounts of both the α and the β product (68, Table 2, entry 14). The reactions of the xylosyl donors also provided significant quantities of side products. In all reactions, the anomeric N‐phenyl‐trifluoracetamides (78–80, Figure 3) were formed. Although these kind of side products are well known for (pyranosyl) trichloroacetimidate donors,47 to our knowledge they have never been reported for N‐phenyl trifluoroacetimidates. In the reaction of the xyluronic acid ester 4, the tricyclic compound 81 (Figure 3) was also formed, originating from an intramolecular electrophilic aromatic substitution reaction of the C2‐O‐benzyl group.
Table 2.
Glycosylation reactions of donors 1–12 and 57–60 with the acceptors [D]TES and allyl‐TMS.[a]
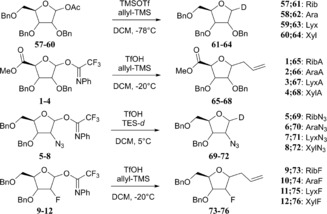
| |||||
|---|---|---|---|---|---|
| Entry | Donor | Acceptor | Product | 1,2‐cis/1,2‐trans (α/β) | Yield [%] |
| d‐ribo‐configured | |||||
| 1 | 57 | [D]TES | 61 | >98:2 | 50[b] |
| 2 | 1 | allyl‐TMS | 65 | >98:2 | 79 |
| 3 | 5 | [D]TES | 69 | >98:2 | 68 |
| 4 | 9 | allyl‐TMS | 73 | >98:2 | 76 |
| d‐arabino‐configured | |||||
| 5 | 58 | [D]TES | 62 | <2:98 | 62[b] |
| 6 | 2 | allyl‐TMS | 66 | 5:95 | 76 |
| 7 | 6 | [D]TES | 70 | <2:98 | 57 |
| 8 | 10 | allyl‐TMS | 74 | <2:98 | 79 |
| d‐lyxo‐configured | |||||
| 9 | 59 | [D]TES | 63 | <2:98 | 100[b] |
| 10 | 3 | allyl‐TMS | 67 | <2:98 | 76 |
| 11 | 7 | [D]TES | 71 | <2:98 | 59 |
| 12 | 11 | allyl‐TMS | 75 | <2:98 | 90 |
| d‐xylo‐configured | |||||
| 13 | 60 | [D]TES | 64 | 85:15 | 40[b] |
| 14 | 4 | allyl‐TMS | 68 | 45:55 | 57[c] |
| 15 | 8 | [D]TES | 72 | 85:15 | 68[c] |
| 16 | 12 | allyl‐TMS | 76 | 70:30 | 62 |
Figure 3.
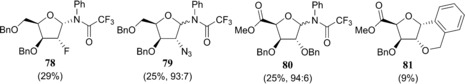
Structures 78–81 identified as side products in the glycosylation reactions of xylosyl imidate donors. Percentages obtained from the crude 1H NMR.
Overall it can be concluded that—quite surprisingly—the nature of the substituents on the furanosyl donors has relatively little effect on the stereochemical outcome of the glycosylation reactions.
Computations
To rationalise the stereochemical outcome of the glycosylations described above, we next assessed the structure of the oxocarbenium ions involved. Woerpel and co‐workers have devised an empirical model to rationalise the stereoselectivity observed in addition reactions to furanosyl oxocarbenium ions. This model takes the two most relevant structures of the ions, that is, the E 3 and 3 E conformers, into account and describes that these will be preferentially attacked form the “inside” of the envelope (see Figure 4 A).32, 51, 52, 53, 54 Thus, the former ion is stereoselectively attacked from the bottom face, whereas the latter is substituted on the top face and the population of both conformational states determines the overall stereoselectivity. The stereoelectronic effects of the ring substituents dictate the relative stability of the conformers and the stabilising/destabilising spatial positions are graphically presented in Figure 4 B for the four tri‐O‐benzylfuranosyl oxocarbenium ions. The oxocarbenium ion conformers are most stable when the positive charge at the anomeric centre is stabilised by C2−H hyperconjugation, and by placing the alkoxy substituents at C3 and C5 in an orientation that brings the lone pairs of the oxygen atoms closest to the anomeric centre. In the ribose oxocarbenium ion all substituents can work in concert to stabilise the cation, whereas in the other three ions the stabilising effect of the substituents cannot be matched. For these ions it is difficult to predict what the net effect of the combination of the substituents is and therefore, we have developed, based on the initial work of Rhoad and co‐workers,14 a computational method that maps the relative energy of all possible conformations as a function of their shape. By plotting the energy of the conformers on the pseudo‐rotational circle, which is used to graphically represent all possible five‐ring geometries (Figure 4 C),55 conformational energy landscape (CEL) maps are created that provide detailed insight into the overall stabilising/destabilising effects of the ring substituents in every possible conformation and configuration (see the Supporting Information for the full computational method).15 These maps can account for the stereoselectivity of addition reactions to fully or partially substituted furanosyl oxocarbenium ions, and the method thus provides an excellent tool to assess the stereoelectronic effects of the functional groups on the ring as a function of their electronic nature and spatial orientation.
Figure 4.
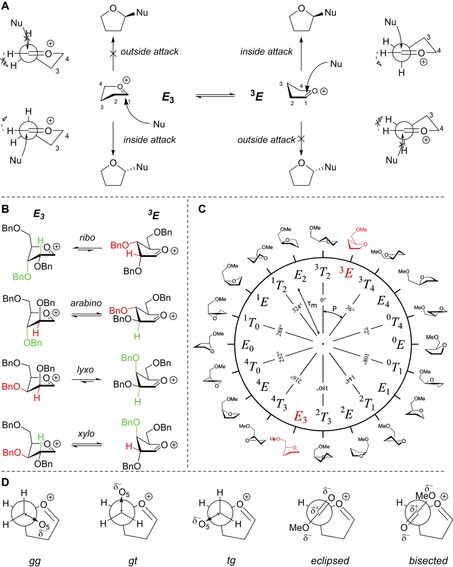
A) The two‐conformer model, visualising the preferential nucleophilic attack from the inside face. Important rotations are denoted by dashed arrows. B) The two principal conformations of the two‐conformer model (E 3–3 E) shown for every carbohydrate configuration, examples taken as their tri‐O‐benzyl‐protected form. The colours indicate the relative preferential orientations for H2 and O3: green is relatively stabilising whereas red is relatively destabilising. C) Pseudo‐rotational circle showing the twenty canonical furanoside structures, with phase‐angles (P) and puckering amplitudes (τ m). D) Possible C4−C5 rotamers: gg, gt and tg for the C5‐OMe oxocarbenium ions, and two rotamers, eclipsed and bisected, for the C4‐CO2Me oxocarbenium ions.
We therefore adopted this method here, to probe the effect of the C2 and C5 modifications on the stability of the oxocarbenium ion conformers and we have calculated the relative energy of the C4‐CO2Me, C2‐N3 and C2‐F furanosyl ions as a function of their shape to deliver the CEL maps shown in Figure 5. To generate these maps the benzyl ethers in the substrates used in the experiments described above, have been replaced for methyl ethers (see Figure 5, 82–97), to minimise computational costs.56 For the C2‐N3 and C2‐F ions, three maps were generated for each of the C4−C5 gg, gt and tg rotamers (Figure 4 D), and these were combined to provide the overall CEL map shown in Figure 5. A similar approach was taken for the bisected and eclipsed structures of the C4‐CO2Me oxocarbenium ions. The most important conformations for each ion are given next to the CEL map of each oxocarbenium ion in Figure 5 (see the Supporting Information for the corresponding energies).
Figure 5.
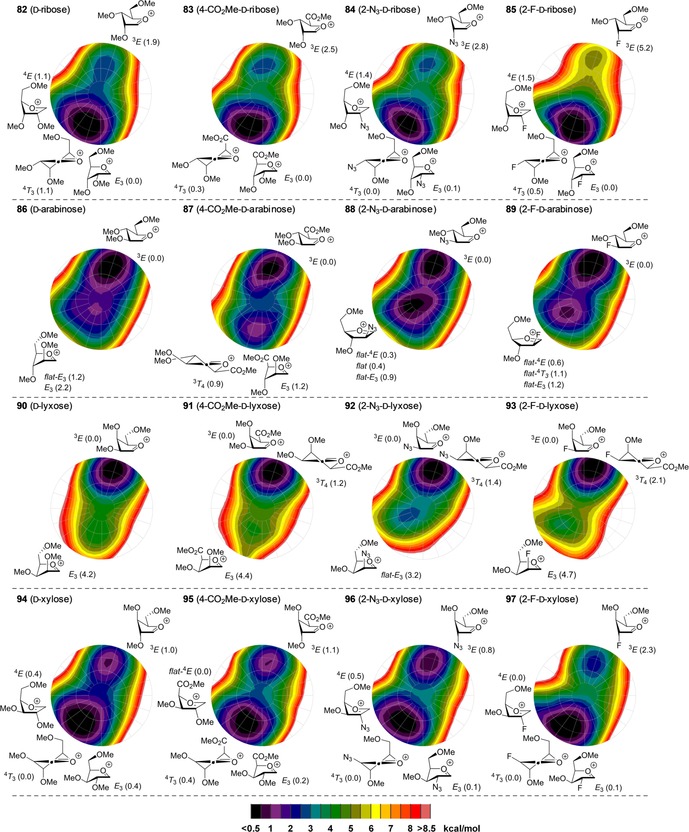
Conformational energy landscape maps for the four diastereoisomeric pentofuranosides and their C5 and C2 modifications. Energies are expressed as Δ in [kcal mol−1].
From the CEL maps of the ribo‐configured furanosyl oxocarbenium ions 82–85 (Figure 5, top row) it becomes clear that the overall shape of the energy landscape is comparable for all four ions, with the energy minima centred on the E 3 conformation. This indicates that a fluoride or azide at C2 is best positioned in a pseudo‐equatorial orientation to allow for stabilisation of the ion by hyperconjugation of the C2−H bond, which is in line with the effect of a C2‐ether functionality.8, 15 From the CEL map of the C2‐F ion it does become apparent that there is a stronger tendency of the fluorine atom to occupy a pseudo‐equatorial orientation. The 3 E conformer of 85 is 5.2 kcal mol−1 higher in energy than the lowest‐energy E 3 conformer, whereas this difference is only 1.9 kcal mol−1 for 82 and around 2.5 kcal mol−1 for 83 and 84. The preference of the C4‐CO2Me to take up an axial orientation becomes apparent from the CEL map of ion 83. Interestingly, there is only a marginal difference between the eclipsed and bisected orientation of the carboxylic acid ester and both orientations seem to be equally capable of stabilising the electron‐depleted anomeric centre when the C4‐CO2Me takes up a pseudo‐axial orientation (see Supporting Information). By using the lowest‐energy E 3 conformers as product‐forming intermediates, the formation of the 1,2‐cis products can be readily accounted for by using the inside attack model for all of the examined ribofuranosides.
The CEL maps for the arabino‐configured furanosyl oxocarbenium ions 86–89 (Figure 5, second row) also show great similarity, with each map showing an energy minimum around the 3 E conformation. Thus, the hyperconjugative stabilisation of the C2−H bond, in combination with a sterically favourable pseudo‐equatorial orientation for all substituents seems decisive for these ions. Inside attack on the 3 E conformers leads to the formation of the 1,2‐cis products as found experimentally. Of note, the CEL map of the C2‐N3 does show a second energy minimum for the 4 T 3/4 E conformer with minimal ring puckering. From the stereochemical outcome of the glycosylation reactions it seems that this conformer does not play a major role in the addition reaction. This could indicate that attack on this almost flat conformer is significantly less favourable than the inside attack of the 3 E envelope, which leads to the favourable C1−C2 staggered product.
The CEL maps of the lyxo‐configured oxocarbenium ions 90–93 (Figure 5, third row) show a single energy minimum on the 3 E side of the CEL maps and the difference in energy between these structures and the other conformers appears to be even larger than the energy differences observed for the ribosyl oxocarbenium ions. This can be understood by realising that the E 3 envelope not only loses the stabilising interactions of the C2 and C3 substituents, present in the 3 E conformer, but also experiences severe 1,3‐diaxial interactions between the C2 and C4 groups, especially for the electronically most favourable gg rotamer. Again, the CEL maps show great similarity for all substitution patterns, indicating analogous behaviour of the lyxofuranosides in the glycosylation reaction. This is indeed borne out in the experimental glycosylations that all proceed in a completely stereoselective fashion to provide the all‐cis products.
Finally, the xylo‐configured oxocarbenium ions 94–97 (Figure 5, fourth row) were assessed. Again, the CEL maps of the differently functionalised xylosides appear to be rather similar. Two minima are apparent on either side of the CEL maps. In the low‐energy E 3‐like structures the C5‐OMe groups are positioned in a gg orientation to stabilise the electron‐depleted anomeric centre, whereas in the low‐energy 3 E‐like structures, on the other side of the CEL map, the C5‐OMe takes up a gt orientation. Notably, the energy minima located on the south side of the CEL maps are relatively broad and not only encompass E 3‐like conformations but also 4 T 3 structures, and perhaps more striking, the 4 E‐like conformers. This latter conformer is in fact the lowest energy species for the 2‐fluoroxyloside 97 and the xylosyl uronate 95, which are the two least selective species (see Table 2). This conformation lacks the stabilising effect of the O3 electron lone pairs as well as hyperconjugative stabilisation by the C2−H bond. Instead, the driving stabilisation now appears to be the interaction of the C5‐O‐methyl or C5 carbonyl group, which is positioned over the ring in an eclipsed conformation, with the anomeric centre. In the 4 E conformation, the steric interactions between C5 and the substituents at C3 and the C2−H bond are reduced when compared to the sterically unfavourable situation in the E 3 conformer. The established broad energy minima may be at the basis for the poor stereoselectivity observed in the condensations of the xylosyl donors as attack of the 4 E conformers may occur from both sides of the ring.
Conclusions
In summary, we have disclosed synthetic routes to access all diastereoisomeric C2‐azido and C2‐fluoro furanosides as well as all furanosyl uronic acid esters. In total, a set of twelve differently functionalised furanosyl donors has been synthesised and these have been glycosylated with allyltrimethylsilane and [D]triethylsilane to establish the stereoselectivity of these donors in SN1‐type glycosylation reactions. An exclusive 1,2‐cis selectivity was observed for all ribo‐, arabino‐ and lyxo‐configured donors, despite the structural modifications made on the C2‐ and C5‐positions. The 2‐azido and 2‐fluoro xylose donors were moderately 1,2‐cis selective, whereas the xyluronic acid donor reacted in a non‐stereoselective manner. The experimental results have been complemented by computational studies, generating conformational energy landscape (CEL) maps for the intermediate oxocarbenium ions. These maps have shown that the stereoelectronic effects of the C2 and C5 modifications are, across the board, similar to a C2‐ether substituent. These groups therefore have a similar effect on the stereochemical outcome of glycosylation reactions taking place through an SN1‐like mechanism and the lowest‐energy oxocarbenium ion conformers, revealed by the CEL maps, have, in combination with the inside attack model, provide a suitable explanation for the experimentally observed cis stereoselectivity. The maps have revealed that for most of the studied furanosyl oxocarbenium ions the canonical 3 E and E 3 envelopes represent the lowest‐energy structures. However, for the xylosyl oxocarbenium ions other low‐energy structures can be found, taking up 4 T 3 and 4 E conformations. The occurrence of these structures coincides with a relatively poor selectivity in the addition reactions. For these ions it appears that the “two‐conformer model” falls short in providing an adequate explanation to account for the (lack of) stereoselectivity and that more oxocarbenium ion conformations have to be taken into account as product‐forming intermediates. Further insight into the structure of glycosyl oxocarbenium ions and the trajectories of nucleophiles that attack these will lead to a better understanding of the SN1 side of the glycosylation reaction mechanism continuum and this can eventually pave the way to a new stereoselective glycosylation methodology.57
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the Netherlands Organization for Scientific Research (NWO) for financial support and the use of supercomputer facilities (SURFsara and the Lisa system) and we kindly acknowledge Mark Somers for technical support.
S. van der Vorm, T. Hansen, E. R. van Rijssel, R. Dekkers, J. M. Madern, H. S. Overkleeft, D. V. Filippov, G. A. van der Marel, J. D. C. Codée, Chem. Eur. J. 2019, 25, 7149.
References
- 1. Deslongchamps P., Stereoelectronic Effects in Organic Chemistry, Pergamon, Oxford, 1983. [Google Scholar]
- 2. Sinnott M., Carbohydrate Chemistry and Biochemistry: Structure and Mechanism, RSC, London, 2007. [Google Scholar]
- 3. Taha H. A., Richards M. R., Lowary T. L., Chem. Rev. 2013, 113, 1851–1876. [DOI] [PubMed] [Google Scholar]
- 4. Adero P. O., Amarasekara H., Wen P., Bohé L., Crich D., Chem. Rev. 2018, 118, 8242–8284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van Rijssel E. R., Janssen A. P. A., Males A., Davies G. J., van der Marel G. A., Overkleeft H. S., Codée J. D. C., ChemBioChem 2017, 18, 1297–1304. [DOI] [PubMed] [Google Scholar]
- 6. Pedersen C. M., Bols M., Org. Biomol. Chem. 2017, 15, 1164–1173. [DOI] [PubMed] [Google Scholar]
- 7. Ingemar Olsen J., Sauer S. P. A., Pedersen C. M., Bols M., Org. Biomol. Chem. 2015, 13, 3116–3121. [DOI] [PubMed] [Google Scholar]
- 8. Smith D. M., Woerpel K. A., Org. Biomol. Chem. 2006, 4, 1195–1201. [DOI] [PubMed] [Google Scholar]
- 9. Jensen H. H., Bols M., Acc. Chem. Res. 2006, 39, 259–265. [DOI] [PubMed] [Google Scholar]
- 10. Jensen H. H., Lyngbye L., Jensen A., Bols M., Chem. Eur. J. 2002, 8, 1218–1226. [DOI] [PubMed] [Google Scholar]
- 11. Woods R. J., Andrews C. W., Bowen J. P., J. Am. Chem. Soc. 1992, 114, 859–864. [Google Scholar]
- 12. Hagen B., van der Vorm S., Hansen T., van der Marel G. A., Codée J. D. C. in Selective Glycosylations: Synthetic Methods and Catalysts, Wiley-VCH, Weinheim, 2017, pp. 1–28. [Google Scholar]
- 13.For selected examples on donor reactivities, see:
- 13a. Reactivity Tuning in Oligosaccharide Assembly (Eds.: B. Fraser-Reid, J. C. López), Springer, Heidelberg, 2011, pp. 1–29; [Google Scholar]
- 13b. Grice P., Ley S. V., Pietruszka J., Priepke H. W. M., Walther E. P. E., Synlett 1995, 781–784; [Google Scholar]
- 13c. Douglas N. L., Ley S. V., Lücking U., Warriner S. L., J. Chem. Soc. Perkin Trans. 1 1998, 51–66; [Google Scholar]
- 13d. Zhang Z., Ollmann I. R., Ye X.-S., Wischnat R., Baasov T., Wong C.-H., J. Am. Chem. Soc. 1999, 121, 734–753; [Google Scholar]
- 13e. Mong K.-K. T., Wong C.-H., Angew. Chem. Int. Ed. 2002, 21, 4087–4090; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 4261–4264; [Google Scholar]
- 13f. Pedersen C. M., Marinescu L. G., Bols M., Chem. Commun. 2008, 2465–2467; [DOI] [PubMed] [Google Scholar]
- 13g. Heuckendorff M., Pedersen C. M., Bols M., J. Org. Chem. 2013, 78, 7234–7248. [DOI] [PubMed] [Google Scholar]
- 14. Rhoad J. S., Cagg B. A., Carver P. W., J. Phys. Chem. A 2010, 114, 5180–5186. [DOI] [PubMed] [Google Scholar]
- 15. van Rijssel E. R., van Delft P., Lodder G., Overkleeft H. S., van der Marel G. A., Filippov D. V., Codée J. D. C., Angew. Chem. Int. Ed. 2014, 53, 10381–10385; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10549–10553. [Google Scholar]
- 16. Madern J. M., Hansen T., van Rijssel E. R., Kistemaker H. A. V., van der Vorm S., Overkleeft H. S., van der Marel G. A., Filippov D. V., Codée J. D. C., J. Org. Chem. 2019, 84, 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van Rijssel E. R., van Delft P., van Marle D. V., Bijvoets S. M., Lodder G., Overkleeft H. S., van der Marel G. A., Filippov D. V., Codée J. D. C., J. Org. Chem. 2015, 80, 4553–4565. [DOI] [PubMed] [Google Scholar]
- 18. Kim J.-H., Yu J., Alexander V., Choi J. H., Song J., Lee H. W., Kim H. O., Choi J., Lee S. K., Jeong L. S., Eur. J. Med. Chem. 2014, 83, 208–225. [DOI] [PubMed] [Google Scholar]
- 19. Evdokimov N. M., Clark P. M., Flores G., Chai T., Faull K. F., Phelps M. E., Witte O. N., Jung M. E., J. Med. Chem. 2015, 58, 5538–5547. [DOI] [PubMed] [Google Scholar]
- 20. Weinstein E. A., Ordonez A. A., DeMarco V. P., Murawski A. M., Pokkali S., MacDonald E. M., Klunk M., Mease R. C., Pomper M. G., Jain S. K., Sci. Transl. Med. 2014, 6, 259ra146–259ra146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Richards M. R., Lowary T. L., ChemBioChem 2009, 10, 1920–1938. [DOI] [PubMed] [Google Scholar]
- 22. Lowary T. L., Acc. Chem. Res. 2016, 49, 1379–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van den Bos L. J., Codée J. D. C., Litjens R. E. J. N., Dinkelaar J., Overkleeft H. S., van der Marel G. A., Eur. J. Org. Chem. 2007, 3963–3976. [DOI] [PubMed] [Google Scholar]
- 24. Lindberg B. in Advances in Carbohydrate Chemistry and Biochemistry (Eds.: R. S. Tipson, D. Horton), Academic Press, Cambridge, 1990, pp. 279–318. [DOI] [PubMed] [Google Scholar]
- 25. Codée J. D. C., Christina A. E., Walvoort M. T. C., Overkleeft H. S., van der Marel G. A. in Reactivity Tuning in Oligosaccharide Assembly (Eds.: B. Fraser-Reid, J. Cristóbal López), Springer, Heidelberg, 2011, pp. 253–289. [Google Scholar]
- 26.In a set of preliminary glycosylations the anomeric acetates of the C2- and C5-modified glycosides were discarded as donors because they were unreactive even at room temperature, by nature of their electron-withdrawing substituents. The tri-O-benzyl furanoside donors were converted to trifluoro-N-phenylacetimidates and reacted at −78 °C with allyltrimethylsilane. Analysis of the crude product samples by 1H NMR spectroscopy revealed the same stereochemistry as reported in Table 1, acetyl donors 57–60, deuterated products 61–64 A sample of the unknown 1-allyl-2,3,5-tri-O-benzyl 1-deoxy-lyxofuranoside is reported in the Supporting Information (77).
- 27. Wender P. A., Bi F. C., Buschmann N., Gosselin F., Kan C., Kee J.-M., Ohmura H., Org. Lett. 2006, 8, 5373–5376. [DOI] [PubMed] [Google Scholar]
- 28. Cuzzupe A. N., Di Florio R., Rizzacasa M. A., J. Org. Chem. 2002, 67, 4392–4398. [DOI] [PubMed] [Google Scholar]
- 29. Veeneman G. H., Gomes L. J. F., van Boom J. H., Tetrahedron 1989, 45, 7433–7448. [Google Scholar]
- 30. Li K.-Y., Jiang J., Witte M. D., Kallemeijn W. W., van den Elst H., Wong C.-S., Chander S. D., Hoogendoorn S., Beenakker T. J. M., Codée J. D. C., Aerts Gijs J. M. F. G., van der Marel A., Overkleeft H. S., Eur. J. Org. Chem. 2014, 6030–6043. [Google Scholar]
- 31. van den Bos L. J., Codée J. D. C., van der Toorn J. C., Boltje T. J., van Boom J. H., Overkleeft H. S., van der Marel G. A., Org. Lett. 2004, 6, 2165–2168. [DOI] [PubMed] [Google Scholar]
- 32. Larsen C. H., Ridgway B. H., Shaw J. T., Smith D. M., Woerpel K. A., J. Am. Chem. Soc. 2005, 127, 10879–10884. [DOI] [PubMed] [Google Scholar]
- 33. Jia C., Zhang Y., Zhang L.-H., Sinaÿ P., Sollogoub M., Carbohydr. Res. 2006, 341, 2135–2144. [DOI] [PubMed] [Google Scholar]
- 34. Popsavin V., Grabež S., Stojanović B., Popsavin M., Pejanović V., Miljković D., Carbohydr. Res. 1999, 321, 110–115. [Google Scholar]
- 35. Chakraborty T. K., Koley D., Ravi R., Krishnakumari V., Nagaraj R., Chand Kunwar A., J. Org. Chem. 2008, 73, 8731–8744. [DOI] [PubMed] [Google Scholar]
- 36. Hale K. J., Hough L., Manaviazar S., Calabrese A., Org. Lett. 2015, 17, 1738–1741. [DOI] [PubMed] [Google Scholar]
- 37. Soueidan O.-M., Trayner B. J., Grant T. N., Henderson J. R., Wuest F., West F. G., Cheeseman C. I., Org. Biomol. Chem. 2015, 13, 6511–6521. [DOI] [PubMed] [Google Scholar]
- 38. Nicolaou K. C., Ladduwahetty T., Randall J. L., Chucholowski A., J. Am. Chem. Soc. 1986, 108, 2466–2467. [DOI] [PubMed] [Google Scholar]
- 39. Su T.-L., Klein R. S., Fox J. J., J. Org. Chem. 1981, 46, 1790–1792. [Google Scholar]
- 40. Diaz R. R., Rodríguez Melgarejo C., Cubero I. I., Plaza López-Espinosa M. T., Carbohydr. Res. 1997, 300, 375–380. [Google Scholar]
- 41. Hernández R., León E. I., Moreno P., Riesco-Fagundo C., Suárez E., J. Org. Chem. 2004, 69, 8437–8444. [DOI] [PubMed] [Google Scholar]
- 42. Czernecki S., Ayadi E., Randriamandimby D., J. Org. Chem. 1994, 59, 8256–8260. [Google Scholar]
- 43.Allylations with allyltrimethylsilane were preferred over reductions with [D]triethylsilane, as the allyl glycosides were easier to purify, gave higher yields and their anomeric configuration was easier to identify.
- 44. van der Vorm S., Hansen T., Overkleeft H. S., van der Marel G. A., Codee J. D. C., Chem. Sci. 2017, 8, 1867–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Krumper J. R., Salamant W. A., Woerpel K. A., Org. Lett. 2008, 10, 4907–4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Krumper J. R., Salamant W. A., Woerpel K. A., J. Org. Chem. 2009, 74, 8039–8050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Christensen H. M., Oscarson S., Jensen H. H., Carbohydr. Res. 2015, 408, 51–95. [DOI] [PubMed] [Google Scholar]
- 48. Napolitano J. G., Gavín J. A., García C., Norte M., Fernández J. J., Hernández Daranas A., Chem. Eur. J. 2011, 17, 6338–6347. [DOI] [PubMed] [Google Scholar]
- 49. Koźmiński W., Nanz D., J. Magn. Reson. 1997, 124, 383–392. [Google Scholar]
- 50. Marquez B. L., Gerwick W. H., Williamson R. T., Magn. Reson. Chem. 2001, 39, 499–530. [Google Scholar]
- 51. Shaw J. T., Woerpel K. A., J. Org. Chem. 1997, 62, 6706–6707. [DOI] [PubMed] [Google Scholar]
- 52. Shaw J. T., Woerpel K. A., Tetrahedron 1999, 55, 8747–8756. [Google Scholar]
- 53. Larsen C. H., Ridgway B. H., Shaw J. T., Woerpel K. A., J. Am. Chem. Soc. 1999, 121, 12208–12209. [Google Scholar]
- 54. Bear T. J., Shaw J. T., Woerpel K. A., J. Org. Chem. 2002, 67, 2056–2064. [DOI] [PubMed] [Google Scholar]
- 55. Altona C., Sundaralingam M., J. Am. Chem. Soc. 1972, 94, 8205–8212. [DOI] [PubMed] [Google Scholar]
- 56.Benzyl ethers and methyl ethers behave similarly in glycosylation leading to similar stereoselectivity. See, for example: Beaver M. G., Woerpel K. A., J. Org. Chem. 2010, 75, 1107–1118 and Reference [15].20108907 [Google Scholar]
- 57. Zhang L., Shen K., Taha H. A., Lowary T. L., J. Org. Chem. 2018, 83, 7659–7671. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary