

Abstract
Metal‐driven self‐assembly afforded a multitude of fascinating supramolecular coordination complexes (SCCs) with applications as catalysts, host–guest, and stimuli‐responsive systems. However, the interest in the biological applications of SCCs is only starting to emerge and thorough characterization of their behavior in biological milieus is still lacking. Herein, we report on the synthesis and detailed in‐cell tracking of a Pt2L2 metallacycle. We show that our hexagonal supramolecule accumulates in cancer cell nuclei, exerting a distinctive blue fluorescence staining of chromatin resistant to UV photobleaching selectively in nucleolar G4‐rich regions. SCC co‐localizes with epitopes of the quadruplex‐specific antibody BG4 and replaces other well‐known G4 stabilizers. Moreover, the photophysical changes accompanying the metallacycle binding to G4s in solution (fluorescence quenching, absorption enhancement) also take place intracellularly, allowing its subcellular interaction tracking.
Keywords: G-quadruplex, metallacycle, platinum, SCC, subcellular localization
The thoughtful choice of transition metals and multidentate ligands allows for the generation of a number of self‐assembled supramolecular coordination complexes (SCCs) with intriguing geometries and attractive properties, including optical, sensing, and catalytic.1, 2, 3 Despite the extraordinary growth of the chemistry of SCCs, studies reporting on their potential biomedical applications are so far extremely limited. Recently, some research groups have started to exploit the cavity‐cored nature of 3D metallacages to develop innovative drug delivery systems.2, 4, 5, 6 We and others focused our efforts on the intrinsic anticancer potential of SCCs,5 devoting particular attention to those supramolecules where platinum drives the self‐assembly.7, 8, 9, 10 Pt‐ensembles can in principle be designed to mimic complex cellular regulatory motifs. Therefore, they can act through unusual modes of action potentially capable to overcome the drawbacks (for example resistance) of clinically applied PtII drugs.11 Nevertheless, apart from preliminary reports on their cytotoxic activity, detailed information about the in‐cell fate of Pt‐SCCs is still missing.
In this regard, Stang and Olenyuk reported on the intracellular localization of two emissive PtII metallacycles.12 Their compounds were proposed to accumulate in lysosomes of cancer cells producing low toxicity, yet effectively inducing tumor shrinkage in mouse xenograft models.12 Stang and his team also developed theranostic nanoconstructs incorporating platinum metallacages that displayed in vivo anticancer activity.13, 14 A further demonstration of the potential of PtII‐SCCs in biomedicine was provided by Sleiman and Mao, who discovered that PtII‐based metallacycles display fairly good ability to bind DNA G‐quadruplexes (G4s) in solution.15, 16, 17, 18 G4s are non‐canonical DNA/RNA motifs enriched in important regulatory regions,19 and regarded as emerging targets for anticancer drugs.20 They recently achieved a “star” status due to their subcellular visualization using G4‐specific antibodies.21, 22 Furthermore, two G4 binders (CX‐3543 and CX‐5461) have entered advanced clinical trials for human cancers.23, 24
In this context, we have recently reported on the promising DNA binding profiles of 4,4′‐bipyridine‐based dinuclear PtII metallacyles.25 Our SCCs, whilst not particularly cytotoxic (IC50 ≥40 μm), distinctly influence the expression of genes known to contain quadruplex motifs in their promoters, with potency and selectivity which well correlate with the size of the supramolecules.25 Herein, we report on a new hexagonal dinuclear PtII metallacycle (1, Figure 1) designed to have DNA‐binding capacity and favorable optical properties for tracking its in‐cell fate when bound to different nucleic acid structures.
Figure 1.
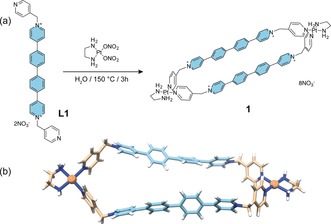
a) Synthesis of Pt‐SCC 1. b) DFT optimized structure of 1.
Over the past few years we have developed a straightforward strategy for the self‐assembly of dinuclear PtII‐based metallacycles.26 Inspired by recent works of Wasielewski and Stoddart on cyclophanes bearing isolated phenyl‐extended viologen moieties,27, 28 we argued that the ditopic pyridyl ligand L1 (Figure 1 a) would confer an adequate emission profile to the final supramolecule and overcome quenching effects typical of SCCs.29, 30, 31 The highly emissive biphenyl‐extended viologen moiety (Figure 1, in turquoise) would maintain a ligand‐centered emission profile when incorporated in the corresponding dinuclear PtII‐metallacycle 1 due to the non‐direct connectivity to the metal centers. Furthermore, the dimensions (ca. 200 Å2) and the octacationic nature of 1 are quite similar to those of our previously reported DNA/quadruplex binders,25 a fact that we envisaged would translate as well into a comparable DNA‐binding profile. Synthesis of 1 was carried out according to methodologies previously reported by us for similar SCCs (see Supporting Information for details).32
Steady‐state 3D excitation‐emission spectra (Supporting Information, Figure S1a,b) showed that 1 buffered solutions emit in the blue with a maximum at 445 nm when excited at 360 nm (an absorption band ascribed to π–π* transitions of the biphenyl moiety). The fluorescence profile of 1 is rather similar to the one of its ligand L1, whose emission maximum is slightly blue‐shifted (λ max=437 nm, Figure S1b in the Supporting Information). This, together with the observation that both 1 and L1 have emission lifetimes of about 2–3 ns (Supporting Information, Figure S1c), suggest that fluorescence is mainly dependent on the excited states of the ligand, as reported likewise for other PtII‐SCCs.12 Even though ligand L1 is more emissive in water than 1 (ϕ F=0.84 at 437 nm, Figure S2 in the Supporting Information), the metallacycle has a surprisingly good quantum yield in water (ϕ F=0.38 at 445 nm). This finding is in sharp contrast to what has been recently observed for other platinum‐based cages which display high emission only in non‐polar solvents.33 Such an aspect is particularly important for SCCs biological applications, making 1 a good candidate for tracking the cellular distribution of this class of compounds.
After proving by NMR and UV/Vis spectroscopy that 1 is stable overtime in water, buffered solutions, and cell culture media (with or without fetal bovine serum) (Supporting Information, Figure S3), we explored the interaction of the metallacycle with different DNA sequences including duplex DNA and G4 models. FRET, UV/Vis, fluorescence, and circular dichroism assays showed that 1 is able to strongly bind both duplex and quadruplex DNAs, with moderate preference for parallel G4s (see Supporting Information and Figure S4–S7 for a detailed discussion), similarly with what was observed for other Pt‐SCCs prepared by us previously.25
We then investigated the applicability of our newly synthesized Pt‐compound in biological settings, evaluating its impact on the viability of non‐transformed human keratinocytes and human lung fibroblasts (HaCaT and HLF) as well as of cancer cell models derived from different primary origins including breast cancer, osteosarcoma, melanoma, and glioblastoma (Figure 2 a and Table S1 in the Supporting Information). While HaCat cells were completely resistant against the cytotoxic impact of the metallacycle, the activity against highly proliferative HLF was comparable to malignant cell types, where 1 showed IC50 values in the range 30–45 μm after 72 h of incubation, with MCF‐7 and U2OS being the most sensitive models. Ligand L1 was slightly less active and followed the same trend (Supporting Information, Figure S8a and Table S1). These results are in good agreement with the general low cytotoxicity of similar Pt‐SCCs observed earlier by ourselves and Olenyuk.12, 25 Combination of 1 with cisplatin in MCF‐7 cells (Supporting Information, Figure S8b) gave rise to a slightly antagonistic effect suggesting that they may share mutual targets (nucleic acids) but exert a differing mode of action.
Figure 2.
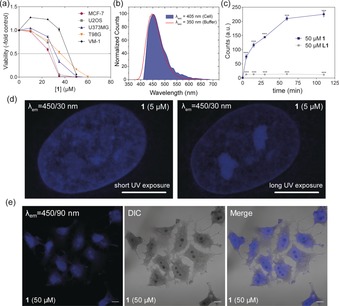
a) Cell viability assessed by MTT assay following exposure to 1 (0–50 μm). b) Normalized emission signatures of 1 in MCF‐7 cells acquired with confocal microscope (blue filled area) and in cell‐free buffer acquired with a spectrofluorometer. c) Flow cytometric analysis of U2OS cells treated with 50 μm of L1 (grey) and 1 (blue) over time (λ exc=405 nm, λ em=450/50 nm), p<0.001, 1‐way ANOVA, Bonferroni post‐test. d) Airyscan super‐resolution microscopy images of U2OS cells treated with 5 μm 1 at short‐ (10 ms) and long‐term (100 ms) exposure (λ exc=405 nm, λ em=450/30 nm) indicating selective photobleaching effects of 1. e) CLSM and DIC microscopy images acquired for PFA‐fixed MCF‐7 cells incubated for 30 min with 1 at 50 μm (λ exc=405 nm). Scale bars=10 μm.
MCF‐7 and U2OS cell sensitivity to 1 was also tested through clonogenic assay, selecting a longer incubation time (7 days). Both cell lines showed impaired colony formation upon incubation with 1 at already 15–20 μm (Supporting Information, Figure S9). Interestingly, the long‐term treatment had a stronger impact on U2OS compared to MCF‐7 cells (15 μm vs. 20 μm, respectively). Conversely, cell cycle distribution analysis revealed that after 24 h incubation there was a significant dose‐dependent cell accumulation in G0/G1 phase and clear‐cut cell death induction only in MCF‐7 cells, while U2OS cell populations presented no appreciable changes at this comparably short incubation time (Supporting Information, Figure S10).
To elucidate whether 1 indeed targets G4 regulated genes, whole genome gene expression analyses were performed on U2OS cells comparing the impact of the metallacycle with that of pyridostatin (PDS), a well‐known G4 stabilizer.21 Gene set enrichment analysis (GSEA) clearly revealed that 1 preferentially targets genes located in chromosomal regions enriched in G4 structures34 with multiple KEGG terms similarly suppressed or activated by 1 and PDS (Supporting Information, Figure S11 a–c).
Confocal laser scanning microscopy (CLSM) was used to evaluate 1 subcellular emission fingerprint. MCF‐7 cells treated with 1 revealed an intense blue fluorescence signal in the range 420–518 nm (λ exc=405 nm, Figure 2 b and S12 in the Supporting Information), showing a nearly perfect overlap with the emission band obtained in cell‐free conditions (Figure 2 b). Flow cytometry analysis of 1‐treated U2OS cells nicely confirmed CLSM results. Pt‐SCC 1 gave rise to a considerable emission in the blue portion of the spectrum (λ exc=405 nm) (Supporting Information, Figure S13a). Interestingly, 1‐associated emission decreased when cells were treated concomitantly with PDS suggesting G4s involvement.21 In contrast it was unaffected by combination with cisplatin indicating a different binding mechanism (Supporting Information, Figure S13b). In CLSM analyses as well as in FACS experiments, L1 emission was considerably lower. FACS, for example, displayed a drastic reduction of the blue fluorescence of 94 % for L1 at λ exc=405 nm (Supporting Information, Figure S13). The efficiency of 1 emission in cell can be attributed to the increased rigidity imposed by the metallacycle to the biphenyl‐extended fluorophore.28 FACS analysis of U2OS cells treated with 1 showed a significant drug accumulation within the first hour of incubation, while L1 did not share the same accumulation features (Figure 2 c). Live cell imaging indicated that exposure of U2OS cells to compound 1 at 35 μm produced an increase in fluorescence overtime (Supporting Information, Figure S14) which nicely corroborates FACS results. The emission of the ligand was in this case below the detection limit of the instrument (Supporting Information, Figure S15). Crucially, the different emissive behavior of L1 compared to 1, observed using diverse experiments and techniques, suggests a different trafficking dynamic inside the cells for the two compounds and allows ruling out possible cellular decomposition of the platinum ensemble to its free building blocks.
Drug distribution within cells was further studied by CLSM. Super‐resolution airy‐scan images of U2OS cells fixed with MeOH and incubated with 1 showed a structured blue staining of the whole nucleus confirming binding of 1 to DNA. Extended UV exposure, however, produced a photobleaching effect which was dependent on the sub‐nuclear region. Hence, at G4‐rich loci, 1 photobleaching was distinctly less efficient resulting in the labelling of nucleoli‐like substructures (Figure 2 d). This effect was corroborated by time‐lapse fluorescence microscopy (Supporting Information, Figure S16). Confocal micrographs of U2OS and MCF‐7 cells fixed with paraformaldehyde (PFA) confirmed the pan‐nuclear blue fluorescent staining induced by 1 (Figure 2 e and S17a in the Supporting Information). Interestingly, images of fixed cells exposed to 1, acquired using DIC (differential interference contrast) mode, revealed a strong contrast enhancement of nuclei and a clear‐cut “dark” staining of the nucleoli‐like substructures (see below). This unique effect is independent of the fixation method since MCF‐7 cells fixed with methanol gave the same outcome (Supporting Information, Figure S17b). Such a contrast enrichment effect was not detected when cells were incubated with L1 or DAPI, a conventional nuclear DNA stain (Supporting Information Figure S17a).
To have a first hint about possible interaction of 1 with G4s in cells, fluorescence competition assays were performed and analyzed by live cell imaging microscopy. The G4‐binders TMPyP4 and Thioflavin‐T (ThT) were used since they are also intracellularly fluorescent. In detail, TMPyP4 is a non‐selective porphyrin‐based binder displaying a red emission,35 while ThT is known for its green fluorescent, G4‐selective, light‐up effect both in solution36 and in cells.37 MCF‐7 cells incubated with the porphyrin and 1 showed an initial TMPyP4‐associated red fluorescence localized in the nuclei that turned blue overtime (Supporting Information, Figure S18a and Video S1). The same effect was obtained with ThT/1 incubation, with the ThT‐associated nucleolar green emission being replaced by the blue signals induced by 1 (Supporting Information, Figure S18b and Video S2). This outcome clearly indicated that 1 acts as competitor of both TMPyP4 and ThT for the same binding sites (duplex and G4s), showing an enhanced long‐term target affinity. It is worth noting that live cell imaging was affected by considerable cell death after treatment with 1, probably based on phototoxicity or synergistic effects when combined with TMPyP4 and ThT. We verified the latter hypothesis through viability assays, demonstrating that co‐incubations of 1 with ThT (or PDS) produced strong synergistic cytotoxicity in MCF‐7 and U2OS cells (Supporting Information, Figure S18c).
A competition assay was also performed in cells fixed after ThT exposure. Metallacycle 1 blocked the G4‐selective green fluorescence staining of nucleoli by ThT (Figure 3 a and S19a in the Supporting Information), comparable to recent reports for PDS.37 Similarly, 1 completely displaced the green nucleoli marker SYTO®RNASelect (SYTO, Figure 3 b), while it did not interact with mitochondrial labelling by MitoTracker Red (MR, Figure S19b in the Supporting Information).
Figure 3.
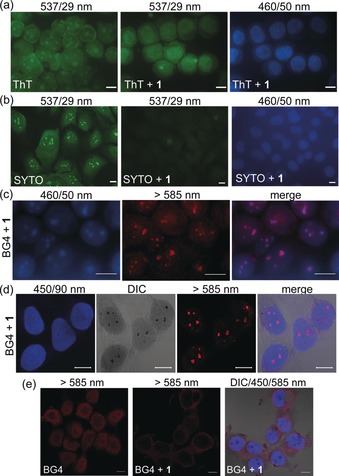
a) Fluorescence microscopy images of MeOH fixed MCF‐7 cells, stained with ThT (5 μm) with and without 1 (5 μm) as indicated. Metallacycle 1 prevented ThT G4‐specfic nucleolar signals. b) Fluorescence microscopy images of U2OS cells stained with SYTO (500 nm), then fixed with MeOH and exposed to 1 (5 μm). c) Fluorescence microscopy images of MeOH‐fixed MCF‐7 cells co‐stained with BG4 and 1 (order: BG4, 1) after photobleaching. d,e) CLSM images of MeOH‐fixed MCF‐7 cells co‐stained with BG4 and 1 (order: BG4, 1) e) PFA‐fixed MCF‐7 cells co‐stained with 1 and BG4 (order: 1, BG4). Scale bars=10 μm.
To further evaluate that cellular distribution of 1 could be governed also by the presence of G4s besides duplex DNA, a series of immunofluorescence studies using the quadruplex specific antibody BG421, 22 was performed (Figure 3 c–e). Fluorescence microscopy images of MeOH‐fixed MCF‐7 cells were acquired after BG4 immunofluorescence and subsequent 1 staining at extended UV exposure(photobleaching). Figure 3 c depicts the clear and distinct cellular co‐localization of BG4‐associated red G4‐rich foci with photobleaching‐resistant 1 fluorescence, both enriched in nucleolar regions.
We then repeated BG4 co‐localization experiments, taking advantage of the clear‐cut “dark” staining of sub‐nuclear structure observed previously for compound 1. CLSM images of MeOH fixed MCF‐7 cells (Figure 3 d) proves that, also in this case, 1 co‐localized with the anti‐G4 immunostaining by BG4. In detail, our Pt‐SCC labelled the whole nucleus with a blue fluorescence and rendered nuclear substructures visible in the DIC channel as dark stains. BG4 revealed red signals exactly in the same regions (λ exc=555 nm) as depicted from representative pixel intensity plots of fluorescence and DIC images (Supporting Information, Figure S20). It should be noted that BG4 epitope distribution is known to be dependent on the fixation method used. Recent studies reported that the antibody labels preferentially nucleoli when MeOH is used for fixation.38 Hence, our experiments indicate that Pt‐metallacycle 1 actually targets G4‐rich nucleolar structures. The co‐labelling assays were repeated both changing either the incubation order or the fixation method. When MCF‐7 cells were fixed with MeOH, incubated with 1 and then with the antibody, a loss of BG4 staining in the nucleoli was produced (Supporting Information, Figure S21a). The micrographs showed instead a weak overall red labelling within the cytoplasm. The outcome was similar when living cells were pre‐treated with 1, then fixed and finally labelled with BG4 (Supporting Information, Figure S21b). Additionally, comparable displacement effects were obtained when MCF‐7 cells were fixed with PFA. Incubation with BG4 produced the classic nuclear red punctate staining pattern (Figure 3 e and Figure S22 in the Supporting Information) as reported by Biffi et al.21, 22 When PFA‐fixed cells were pre‐treated with 1, the BG4 punctate pattern got diffused, appearing enriched in the cytoplasm, demonstrating that metallacycle 1 disrupted BG4 ability to stain G4s in the nuclear region. Eventually, co‐incubation of 1 and BG4 rendered comparable results at the level of individual chromosomes, obtaining the same effect (Supporting Information, Figure S23d).
Overall, these experiments clearly indicate that 1, once inside the nucleus (and the nucleoli), interacts with nucleic acids and interferes with the G4 epitope recognition by BG4.
Besides its cellular co‐localization with BG4 and its emissive properties, our Pt‐metallacycle showed the unique ability to provide well‐contrasted images of cells in transmission (monochrome) DIC mode. The brightfield channel is often ignored in drug accumulation studies for several reasons (poor contrast, difficult localization of cell borders, faint nuclei and others).39 Nevertheless, we were particularly intrigued by the clear‐cut and intense dark staining of nucleoli provided by 1, which resulted to be easily reproducible and independent of the fixation method and cell line used.
Considering the complex optical mechanism at the basis of DIC microscopy,40 we hypothesized that the dark nucleolar “staining” could arise from absorption or refraction (or both) of the light by the Pt‐metallacycle within the sub‐nuclear structures, associated to a simultaneous quenching (and photobleaching resistance) of its blue fluorescence. The UV/Vis spectrum of 1 in solution showed a band centred at 355 nm with a tail ending at around 430 nm (Figure 4 a). Interestingly, the absorbance at 355 nm was significantly red‐shifted when aliquots of pre‐folded parallel quadruplex were added, resulting in a considerable increase in absorption at 405 nm (Figure 4 a). This effect was noticeably less pronounced when the same experiment was performed using a double‐stranded DNA model (Supporting Information, Figure S5b). At the same time, a strong fluorescence quenching was observed after interaction of 1 with the same parallel G4 in solution (Figure 4 b). Crucially, the quenching was less pronounced after the interaction with ds‐DNA, as confirmed by the binding and the Stern–Volmer constants lower in two and one orders of magnitudes, respectively (Supporting Information, Figure S6).
Figure 4.
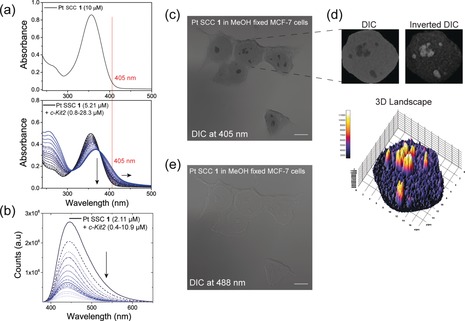
a) UV/Vis absorption spectrum of 1 alone (up) and in combination with increasing amount of c‐Kit2 G4 solution (down). b) Fluorescence titration of 1 with c‐Kit2 G4. c) Image of MCF‐7 cells fixed with MeOH and incubated with 1 acquired using DIC mode at 405 nm. d) Differences in absorption within subnuclear regions of a representative 1‐treated nucleus derived from c) visualized by three‐dimensional surface plotting of single pixel intensities of the inverted DIC image. e) Image of MCF‐7 cells fixed with MeOH and incubated with 1 acquired using DIC mode at 488 nm. Scale bars=10 μm.
Furthermore, MCF‐7 cells fixed with MeOH and incubated with 1 afforded cell images with dramatically enhanced contrast in the DIC setting with 405 nm excitation (where the metallacycle‐G4 adduct preferentially absorbs light, Figure 4 a), depicting prominently visible nucleoli (Figure 4 c,d). When changing the excitation wavelength to 488 nm, just faint cell contours typical for DIC remained visible (Figure 4 e). Of note, nucleoli staining was not observed when fixed MCF‐7 cells were incubated with another Pt‐SCC, selected among our toolbox of metallacycles because of its transparency at 405 nm (Supporting Information, Figure S24). Likewise, staining of fixed MCF‐7 cells with DAPI resulted in the expected blue chromatin staining without any contrast enhancement in the DIC setting. While nucleoli remained invisible in absorption imaging, they appeared darker in the blue fluorescence channel based on the well‐known binding of DAPI to AT‐rich regions (Supporting Information, Figure S25).
These considerations strongly suggest that the dark staining is produced by absorption of 1 interacting with G4 structures within the nucleoli in association with initial distinct fluorescence quenching.
In summary, we thoroughly described the intracellular fate of a Pt‐SCC. Based on its fluorescence properties, we show, for the first time for a Pt‐based SCC, that 1 is taken up by cancer cells and accumulates in the nucleus. Furthermore, the particular alterations of the metallacycle optical features when interacting with G4 as compared to duplex DNA offered a unique tool for its subcellular interaction profiling. Compound 1 preferentially targets nucleolar structures, co‐localizes with a G4‐specific antibody and displaces well‐known G4 stabilizers. Our in‐solution and cellular results, together with the information that nucleoli are particularly rich in G4 forming sequences,38, 41, 42, 43 strongly support that these noncanonical nucleic acid structures are also the final target of the Pt‐SCC. Quarfloxin (CX‐3543), the first‐in‐class G4‐binder in clinical trials, is believed to exert its anticancer activity through a similar mechanism, that is, precisely by accumulation in nucleoli.23 To conclude, platinum‐based SCCs are an extremely versatile class of compounds with the potential to mimic recognition properties of complex biological systems. This manuscript provides evidence that nucleic acids, and in particular G‐quadruplex structures, can be key cellular targets of platinum‐driven supramolecular coordination compounds. Our findings open new avenues in the development of anticancer platinum‐based SCCs with promising and defined mechanisms of action.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary
Acknowledgements
A.T. has received funding from the Mahlke–Obermann Stiftung and the European Union's Seventh Framework Programme (grant agreement no. 609431). C.D. is a recipient of a DOC Fellowship of the Austrian Academy of Sciences (24660). C.P. is grateful to the Ministerio de Economía, Industria y Competitividad and FEDER (CTQ2016‐75629‐P). A.T. is grateful to Prof. L. Salassa for his support and advice.
O. Domarco, C. Kieler, C. Pirker, C. Dinhof, B. Englinger, J. M. Reisecker, G. Timelthaler, M. D. García, C. Peinador, B. K. Keppler, W. Berger, A. Terenzi, Angew. Chem. Int. Ed. 2019, 58, 8007.
Contributor Information
Prof. Carlos Peinador, Email: carlos.peinador@udc.es.
Prof. Walter Berger, Email: walter.berger@meduniwien.ac.at.
Dr. Alessio Terenzi, Email: aterenzi@dipc.org.
References
- 1. Chakrabarty R., Mukherjee P. S., Stang P. J., Chem. Rev. 2011, 111, 6810–6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cook T. R., Stang P. J., Chem. Rev. 2015, 115, 7001–7045. [DOI] [PubMed] [Google Scholar]
- 3. McConnell A. J., Wood C. S., Neelakandan P. P., Nitschke J. R., Chem. Rev. 2015, 115, 7729–7793. [DOI] [PubMed] [Google Scholar]
- 4. Casini A., Woods B., Wenzel M., Inorg. Chem. 2017, 56, 14715–14729. [DOI] [PubMed] [Google Scholar]
- 5. Cook T. R., Vajpayee V., Lee M. H., Stang P. J., Chi K. W., Acc. Chem. Res. 2013, 46, 2464–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Therrien B., Süss-Fink G., Govindaswamy P., Renfrew A. K., Dyson P. J., Angew. Chem. Int. Ed. 2008, 47, 3773–3776; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 3833–3836. [Google Scholar]
- 7. Ahmedova A., Momekova D., Yamashina M., Shestakova P., Momekov G., Akita M., Yoshizawa M., Chem. Asian J. 2016, 11, 474–477. [DOI] [PubMed] [Google Scholar]
- 8. Kaiser F., Schmidt A., Heydenreuter W., Altmann P. J., Casini A., Sieber S. A., Kühn F. E., Eur. J. Inorg. Chem. 2016, 5181–5181. [Google Scholar]
- 9. Terenzi A., Ducani C., Blanco V., Zerzankova L., Westendorf A. F., Peinador C., Quintela J. M., Bednarski P. J., Barone G., Hannon M. J., Chem. Eur. J. 2012, 18, 10983–10990. [DOI] [PubMed] [Google Scholar]
- 10. Mounir M., Lorenzo J., Ferrer M., Prieto M. J., Rossell O., Avilès F. X., Moreno V., J. Inorg. Biochem. 2007, 101, 660–666. [DOI] [PubMed] [Google Scholar]
- 11. Englinger B., Pirker C., Heffeter P., Terenzi A., Kowol C. R., Keppler B. K., Berger W., Chem. Rev. 2019, 119, 1519–1624. [DOI] [PubMed] [Google Scholar]
- 12. Grishagin I. V., Pollock J. B., Kushal S., Cook T. R., Stang P. J., Olenyuk B. Z., Proc. Natl. Acad. Sci. USA 2014, 111, 18448–18453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yu G., Cook T. R., Li Y., Yan X., Wu D., Shao L., Shen J., Tang G., Huang F., Chen X., et al., Proc. Natl. Acad. Sci. 2016, 113, 13720–13725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yu G., Zhang M., Saha M. L., Mao Z., Chen J., Yao Y., Zhou Z., Liu Y., Gao C., Huang F., et al., J. Am. Chem. Soc. 2017, 139, 15940–15949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kieltyka R., Englebienne P., Fakhoury J., Autexier C., Moitessier N., Sleiman H. F., J. Am. Chem. Soc. 2008, 130, 10040–10041. [DOI] [PubMed] [Google Scholar]
- 16. Zheng X.-H., Chen H.-Y., Tong M.-L., Ji L.-N., Mao Z.-W., Chem. Commun. 2012, 48, 7607–7609. [DOI] [PubMed] [Google Scholar]
- 17. Zheng X.-H., Zhong Y.-F., Tan C.-P., Ji L.-N., Mao Z.-W., Dalton Trans. 2012, 41, 11807–11812. [DOI] [PubMed] [Google Scholar]
- 18. Garci A., Castor K. J., Fakhoury J., Do J. L., Di Trani J., Chidchob P., Stein R. S., Mittermaier A. K., Friščić T., Sleiman H., J. Am. Chem. Soc. 2017, 139, 16913–16922. [DOI] [PubMed] [Google Scholar]
- 19. Hänsel-Hertsch R., Di Antonio M., Balasubramanian S., Nat. Rev. Mol. Cell Biol. 2017, 18, 279–284. [DOI] [PubMed] [Google Scholar]
- 20. Neidle S., Nat. Rev. Chem. 2017, 1, 0041. [Google Scholar]
- 21. Biffi G., Tannahill D., McCafferty J., Balasubramanian S., Nat. Chem. 2013, 5, 182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Biffi G., Di Antonio M., Tannahill D., Balasubramanian S., Nat. Chem. 2014, 6, 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Drygin D., Siddiqui-Jain A., O'Brien S., Schwaebe M., Lin A., Bliesath J., Ho C. B., Proffitt C., Trent K., Whitten J. P., et al., Cancer Res. 2009, 69, 7653–7661. [DOI] [PubMed] [Google Scholar]
- 24. Xu H., Di Antonio M., McKinney S., Mathew V., Ho B., O'Neil N. J., Dos Santos N., Silvester J., Wei V., Garcia J., et al., Nat. Commun. 2017, 8, 14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Domarco O., Lötsch D., Schreiber J., Dinhof C., Van Schoonhoven S., García M. D., Peinador C., Keppler B. K., Berger W., Terenzi A., Dalton Trans. 2017, 46, 329–332. [DOI] [PubMed] [Google Scholar]
- 26. García M. D., Alvariño C., López-Vidal E. M., Rama T., Peinador C., Quintela J. M., Inorg. Chim. Acta 2014, 417, 27–37. [Google Scholar]
- 27. Wu Y., Zhou J., Phelan B. T., Mauck C. M., Stoddart J. F., Young R. M., Wasielewski M. R., J. Am. Chem. Soc. 2017, 139, 14265–14276. [DOI] [PubMed] [Google Scholar]
- 28. Roy I., Bobbala S., Zhou J., Nguyen M. T., Nalluri S. K. M., Wu Y., Ferris D. P., Scott E. A., Wasielewski M. R., Stoddart J. F., J. Am. Chem. Soc. 2018, 140, 7206–7212. [DOI] [PubMed] [Google Scholar]
- 29. Blanco V., García M. D., Terenzi A., Pía E., Fernández-Mato A., Peinador C., Quintela J. M., Chem. Eur. J. 2010, 16, 12373–12380. [DOI] [PubMed] [Google Scholar]
- 30. Blanco V., García M. D., Peinador C., Quintela J. M., Chem. Sci. 2011, 2, 2407–2416. [Google Scholar]
- 31. Alvariño C., Terenzi A., Blanco V., García M. D., Peinador C., Quintela J. M., Dalton Trans. 2012, 41, 11992. [DOI] [PubMed] [Google Scholar]
- 32. López-Vidal E. M., Blanco V., García M. D., Peinador C., Quintela J. M., Org. Lett. 2012, 14, 580–583. [DOI] [PubMed] [Google Scholar]
- 33. Yan X., Cook T. R., Wang P., Huang F., Stang P. J., Nat. Chem. 2015, 7, 342–348. [DOI] [PubMed] [Google Scholar]
- 34. Lam E. Y. N., Beraldi D., Tannahill D., Balasubramanian S., Nat. Commun. 2013, 4, 1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Henderson A., Wu Y., Huang Y. C., Chavez E. A., Platt J., Johnson F. B., Brosh R. M., Sen D., Lansdorp P. M., Nucleic Acids Res. 2014, 42, 860–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mohanty J., Barooah N., Dhamodharan V., Harikrishna S., Pradeepkumar P. I., Bhasikuttan A. C., J. Am. Chem. Soc. 2013, 135, 367–376. [DOI] [PubMed] [Google Scholar]
- 37. Zhang S., Sun H., Chen H., Li Q., Guan A., Wang L., Shi Y., Xu S., Liu M., Tang Y., Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1101–1106. [DOI] [PubMed] [Google Scholar]
- 38. Laguerre A., Wong J. M. Y., Monchaud D., Sci. Rep. 2016, 6, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Selinummi J., Ruusuvuori P., Podolsky I., Ozinsky A., Gold E., Yli-Harja O., Aderem A., Shmulevich I., PLoS One 2009, 4, e7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang G., Fang N. in Imaging and Spectroscopic Analysis of Living Cells (Ed.: P. M. Conn), Academic Press, New York, 2012, pp. 83–108. [Google Scholar]
- 41. Rodriguez R., Miller K. M., Forment J. V., Bradshaw C. R., Nikan M., Britton S., Oelschlaegel T., Xhemalce B., Balasubramanian S., Jackson S. P., Nat. Chem. Biol. 2012, 8, 301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lefebvre J., Guetta C., Poyer F., Mahuteau-Betzer F., Teulade-Fichou M. P., Angew. Chem. Int. Ed. 2017, 56, 11365–11369; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11523–11527. [Google Scholar]
- 43. Doria F., Nadai M., Zuffo M., Perrone R., Freccero M., Richter S. N., Chem. Commun. 2017, 53, 2268–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary