

Abstract
Toxoplasma gondii is a successful protozoan parasite that cycles between definitive felid hosts and a broad range of intermediate hosts, including rodents and humans. Within intermediate hosts, this obligate intracellular parasite invades the small intestine, inducing an inflammatory response. Toxoplasma infects infiltrating immune cells, using them to spread systemically and reach tissues amenable to chronic infection. An intact immune system is necessary to control life-long chronic infection. Chronic infection is characterized by formation of parasite cysts, which are necessary for survival through the gastrointestinal tract of the next host. Thus, Toxoplasma must evade sterilizing immunity, but still rely on the host's immune response for survival and transmission. To do this, Toxoplasma exploits a central cost-benefit tradeoff in immunity: the need to escalate inflammation for pathogen clearance vs. the need to limit inflammation-induced bystander damage. What are the consequences of sustained inflammation on host biology? Many studies have focused on aspects of the immune response that directly target Toxoplasma growth and survival, commonly referred to as “resistance mechanisms.” However, it is becoming clear that a parallel arm of the immune response has evolved to mitigate damage caused by the parasite directly (for example, egress-induced cell death) or bystander damage due to the inflammatory response (for example, reactive nitrogen species, degranulation). These so-called “disease tolerance” mechanisms promote tissue function and host survival without directly targeting the pathogen. Here we review changes to host metabolism, tissue structure, and immune function that point to disease tolerance mechanisms during Toxoplasma infection. We explore the impact tolerance programs have on the health of the host and parasite biology.
Keywords: tolerance, Toxoplasma gondii, resistance, cachexia, parasite, innate immunity, chronic infection
Introduction: Disease Tolerance vs. Resistance
A successful immune response requires two distinct, but complementary components: “resistance mechanisms” and “tolerance mechanisms.” Resistance (or restriction) mechanisms directly target pathogens to limit microbial replication or dissemination. Examples of resistance mechanisms include antimicrobial peptides, complement, and degranulation by neutrophils or cytotoxic T cells. Tolerance mechanisms target host cell biology to improve tissue integrity and function in the setting of damage caused by the pathogen or the inflammatory response. Examples include extracellular matrix remodeling, the DNA damage response, antioxidant production, and shifts in cell metabolism (Ayres and Schneider, 2009, 2012; Medzhitov et al., 2012; Soares et al., 2014). Although tolerance mechanisms are often induced by and intimately related to an immune response, many disease tolerance programs are carried out by non-immune (non-hematopoietic) cell types. In fact, disease tolerance mechanisms were first described in plants, which lack a distinct cellular immune system, where they have been associated with tissue integrity, growth, and reproductive capacity (Caldwell et al., 1958).
Historically, immunologists have used the word “tolerance” to describe two processes that limit lymphocyte autoreactivity. In central tolerance, B or T lymphocytes with an affinity for self-antigens are deleted in the bone marrow or thymus before entering circulation. In peripheral tolerance, auto-reactive lymphocytes that arise later in development are deleted or rendered unresponsive to antigen (anergic) in peripheral tissues or in lymph nodes. Regulatory T cells (Tregs) play an important role in peripheral tolerance. Central and peripheral lymphocyte tolerance are included under the broader umbrella of so-called “disease tolerance” strategies because they are mechanisms of limiting inflammation and preventing auto-immunity but do not directly influence the pathogen. In this review, we refer to the broader definition of “disease tolerance” as mechanisms that support host fitness and survival during infection, not by directly targeting pathogen biology, but by shifting homeostasis to maintain tissue function during infection and inflammation (Martins et al., 2019).
It is important to note that tolerance biology, like resistance mechanisms, represent a shift from normal homeostasis. The costs of overactive resistance mechanisms are well-established. Examples include bystander damage from reactive oxygen species, delayed wound healing, and recognition of autoantigens like DNA- or RNA-binding proteins. The negative effects of excessive tolerance programs are comparatively under-appreciated, but include mutations due to imperfect DNA repair, fibrotic wound healing and, potentially, metabolic disorders like Type II diabetes. We can assume that long-term reliance on tolerance mechanisms must be maladaptive, otherwise they would be selected for homeostatic function. This is an important consideration, as tolerance programs are receiving attention as therapeutic targets to interrupt the maladaptive consequences of acute inflammation (for example, sepsis or influenza infection) without increasing susceptibility to pathogens. Host survival during infection depends having both tolerance and resistance mechanisms, otherwise the host will succumb either to pathogen overgrowth or lethal immunopathology. As discussed in the following sections, it is also important to consider how promoting tolerance biology may benefit the life cycle of pathogens, particularly those that have evolved a strategy for long-term persistence within a host (Figure 1).
Figure 1.
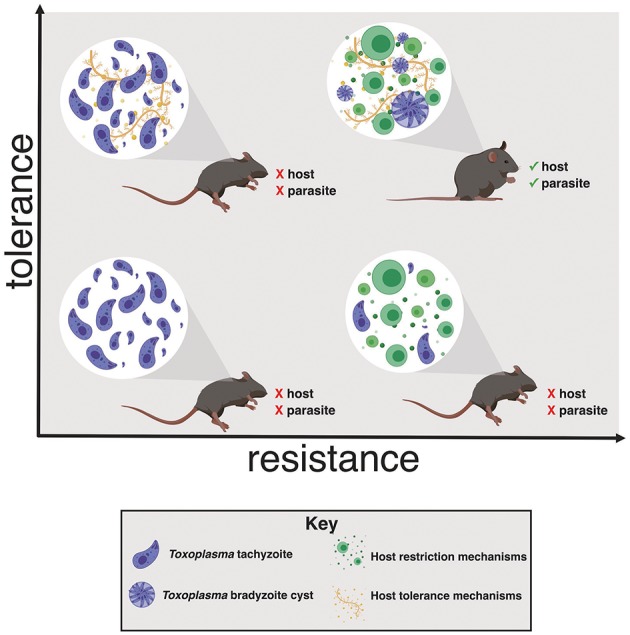
Toxoplasma gondii induces host tolerance and resistance mechanisms in successful infections. In the absence of a strong restrictive immune response the host succumbs to Toxoplasma overgrowth early in infection (left quadrants). Without tolerance mechanisms the host is susceptible to inflammation-induced pathology, even if parasite replication effectively restricted (lower right quadrant). Both resistance and tolerance mechanisms are necessary for host survival (upper right quadrant). This balance also benefits Toxoplasma by ensuring that the host survives long enough to enable bradyzoite cyst differentiation, a requirement for transmission to another host.
Reinterpreting the Immune Response to Toxoplasma in the Framework of Disease Tolerance
Toxoplasma gondii is an obligate intracellular protozoan parasite that establishes life-long infection in a wide range of warm-blooded intermediate hosts, including rodents and humans. Three major haplotypes, types I–III, dominate Europe and North America which differ in virulence by several logs. Feline definitive hosts support Toxoplasma sexual recombination and shed millions of environmentally stable, highly infectious oocysts in feces. This confers a tremendous benefit for the parasite in terms of genetic diversity and dissemination potential, which suggests that rodents may be an important intermediate host for Toxoplasma given the predator-prey relationship between the two host organisms.
Intermediate hosts are infected by ingesting Toxoplasma oocysts or tissue cysts, the so-called bradyzoite form, from a previously infected intermediate host. Toxoplasma invades the small intestine, converting to the rapidly dividing tachyzoite form and inducing inflammation (Dubey et al., 1998). This process activates an immune response that is critical to the parasite's life cycle in two major ways. First, Toxoplasma infects and replicates within infiltrating immune cells (Courret et al., 2006). The parasite uses immune cells to disseminate systemically, reaching tissue sites that support chronic infection including the brain, muscle, and other tissues (Bierly et al., 2008). Second, chronic infection is defined by parasites shifting to a bradyzoite transcriptional program and clearance of most tachyzoites. This involves synthesis of a saccharide-rich parasite cyst wall which is necessary for the parasite to survive transit through the gastrointestinal tract of the next host (Bierly et al., 2008; Gregg et al., 2013). Thus, without a robust immune response, the parasite kills the host before this shift occurs and the opportunity for transmission is limited.
In the mouse, Toxoplasma is recognized by innate immune sensors including Toll-like receptors, and the NLRP1 and NLRP3 inflammasomes. Mice deficient in tlr11 or its signaling adaptor myd88 fail to control parasite replication after intraperitoneal (i.p.) infection (Yarovinsky et al., 2008). Similarly, mice lacking the inflammasome effector caspases-1 and -11 die of parasite overgrowth early in chronic infection (Ewald et al., 2014; Gorfu et al., 2014). Disabling the IFN-γ-regulated GTPase (IRG) system, comprised of GTPases which identify modified intracellular membranes and target them for degradation, leads to rapid parasite expansion and host death in inbred laboratory mice in a manner that depends on parasite genetics. Type II Toxoplasma, the most frequently isolated type in Europe and North America, expresses alleles of the effector proteins ROP5 and ROP18 which make it susceptible to IRG-mediated clearance. Hypervirulent type I and hypovirulent type III Toxoplasma express alleles of rop5 and rop18 that inactivate IRGs at the parasite vacuole improving intracellular replication in many inbred strains of mice (Reese et al., 2011; Etheridge et al., 2014). However, some wild-derived mouse strains are able to restrict growth of hypervirulent type I parasites in an IRG-dependent manner, suggesting that the IRG restriction pathway has imparted an important selective pressure on both host and parasite evolution (Lilue et al., 2013).
Innate immune recognition of Toxoplasma promotes IL-12 release, NK cell-mediated IFN-γ production and a Th1 polarized adaptive immune response (Sher et al., 1993; Hunter et al., 1994). The cellular mechanisms of these responses and the importance of CD8+ cytotoxic T cell response for host survival have been reviewed extensively elsewhere (Dupont et al., 2012; Sasai et al., 2018). Importantly, many cytokines and effector molecules central to this response have been shown to be necessary for parasite resistance. Specifically, mice deficient in IL-12 (Yap et al., 2000), IFN-γ (Suzuki et al., 1988; Deckert-Schlüter et al., 1996), IL-6 (Jebbari et al., 1998), TNF-α (Schlüter et al., 2003), iNOS (Scharton-Kersten et al., 1997), or their receptors die from parasite overgrowth in acute infection or early chronic infection. Host haplotype also plays a dominant role in parasite resistance. BALB/c mice are resistant to infection and express the H-2Ld MHC haplotype, which presents an immunodominant peptide from the dense granule protein GRA6 (Blanchard et al., 2008). In contrast, C57BL/6 mice have the H2b haplotype which presents ROP5, a lower abundance rhoptry protein that elicits a weak CD8+ T cell response, resulting in worse parasite restriction (Grover et al., 2014).
A number of immune cell-intrinsic pathways have been identified that mitigate Th1-mediated immunopathology during Toxoplasma infection which are relevant to disease tolerance (Table 1). At acute infection, mice deficient in IL-10 had a similar liver parasite burden as wildtype mice, but died from a cytokine storm of IL-12, TNF-α, and IFN-γ (Gazzinelli et al., 1996; Neyer et al., 1997). This phenotype could be reversed by depleting CD4+ T cells (Gazzinelli et al., 1996), or by depleting IFN-γ (Suzuki et al., 2000). More recently, Jankovic et al. showed that T-bet+Foxp3− Th1 cells were a major source of IL-10 during chronic i.p. infection with 20 ME49 cysts. These IL-10-producing cells were required to limit fatal immunopathology during both acute and chronic infection (Jankovic et al., 2007). In 2005, Wilson et al. found that lethal Toxoplasma encephalitis in chronically infected IL-10−/− mice is not due to higher parasite loads, implicating IL-10 as a tolerance effector in the central nervous system (CNS) during Toxoplasma infection (Wilson et al., 2005). Similarly, mice lacking IL-4 were more susceptible to Toxoplasma infection despite having slightly reduced cyst burdens and microglial nodules in the brain at chronic infection (Roberts et al., 1996). These data suggest that tolerance mechanisms induced during infection are critical to the survival of host. Although the mechanistic basis for the effect of IL-4 is currently unclear, it has been well-studied in other systems. One example is the rodent helminth Nippostrongylus brasiliensis. N. brasiliensis burrows through the skin then migrates to the lung where it is coughed up and swallowed to complete its life cycle in the gastrointestinal tract. N. brasiliensis chitin promotes IL-4, driving alveolar macrophages to limit lung pathology by producing insulin-like growth factor 1, resistin-like molecule-α and arginase-1 (Chen et al., 2012). These effectors dampen inflammation and promote the fibroblast wound repair response (Knipper et al., 2015). Importantly, the parasite's egg-laying stage occurs in the gut, after trans-lung migration. There may be significant selective pressure for N. brasiliensis to activate tolerance programs that promote wound healing and host survival so that the parasite has sufficient time for transmission.
Table 1.
Summary of literature describing immunoregulatory pathways during Toxoplasma gondii infection.
| Observation | Reference |
|---|---|
| IL-10−/− mice die of acute encephalitis during Toxoplasma infection, associated with elevated cytokine levels, but not with enhanced parasite proliferation | Gazzinelli et al., 1996 |
| IL-4−/− mice succumb during acute Toxoplasma infection despite having fewer circulating parasites, but chronic IL-4 suppresses the restriction response, leading to increased pathogen burden and pathology | Roberts et al., 1996 |
| Mice deficient in 5-lipoxygenase (the enzyme responsible for generating lipoxin A4) succumb to otherwise sublethal doses of Toxoplasma, have elevated circulating IL-12, IFN-y, and TNF-a, and have increased T cell infiltration, despite harboring lower cyst burdens than WT mice | Aliberti et al., 2002 |
| Mice lacking the IL-27 receptor have enhanced mortality and CD4+ mediated tissue damage, independently of parasite burden during oral Toxoplasma infection | Villarino et al., 2003 |
| Chronic IL-10 is required to limit fatal immunopathology mediated by CD4+ T cells in the brain during Toxoplasma infection | Wilson et al., 2005 |
| IL-10 is required to limit fatal immunopathology at acute and chronic Toxoplasma infection. In this context, it is primarily produced by T-bet+Foxp3− Th1 cells | Jankovic et al., 2007 |
| IL-27 signaling promotes a subset of Tregs that limit intestinal pathology and improve host survival during oral Toxoplasma infection | Hall et al., 2012 |
| Lamina propria-infiltrating Ly6Chi monocytes release IL-10 and PGE2 that limit neutrophil-induced tissue pathology in oral Toxoplasma infection | Grainger et al., 2013 |
| Gut infiltrating neutrophils create multicellular casts to prevent bacterial translocation, improving host survival independently of parasite burden | Molloy et al., 2013 |
In addition to their well-characterized roles in pathogen clearance and adaptive immune response activation during Toxoplasma infection, innate immune cells also play a critical role in mitigating tissue pathology. A 2013 study from Yasmine Belkaid's laboratory found that lamina propria-infiltrating neutrophils generated “casts” containing cells and extracellular DNA over regions of small intestine damaged by per oral (p.o.) Toxoplasma infection. These casts which appeared to limited bacterial translocation into host circulation. Depleting neutrophils with an α-Gr-1 antibody increased mortality without increasing parasite burden, indicating that the immune-commensal axis also plays an important role in disease tolerance during Toxoplasma infection (Molloy et al., 2013). During p.o. Toxoplasma infection, lamina propria-infiltrating Ly6Chi monocytes were also shown to release IL-10 and prostaglandin E2 (PGE2) which limited neutrophil-induced pathology. Administration of a PGE2 analog during infection was sufficient to reduce intestinal tissue pathology and immune infiltration in the absence of monocytes without affecting parasite burden (Grainger et al., 2013).
T cells are important players in disease tolerance mechanisms during Toxoplasma infection. 5-lipoxygenase is the rate-limiting enzyme in the synthesis of lipoxin A4, an eicosanoid mediator of the resolution phase of an inflammatory response (Colgan et al., 1993). The importance of this pathway in disease tolerance is underscored by the observation that 5-lipoxygenase deficient mice succumbed to i.p. infection with 20 ME49 cysts by 35 days post-infection, despite harboring significantly fewer brain cysts than wild type mice. Death was associated with encephalitis, increased T cell infiltration into the brain and elevated circulating IL-12, IFN-γ, and TNF-α (Aliberti et al., 2002). IL-27 is part of the IL-12 family of JAK-STAT signaling cytokines with an emerging role in tolerance to Toxoplasma infection. WSX-1 knockout mice (which lack the IL-27 receptor) i.p. infected with 20 ME49 cysts succumbed by 13 days post-infection. No change in parasite burden was detected between knockout and wild type mice in peritoneal lavage fluid at 7 days post-infection, but knockout mice had elevated circulating IL-12, IFN-γ, increased inflammation and necrosis in the liver and lungs, and hyperactive and hyperproliferative CD4+ T cells in the spleen. Survival was partially rescued when CD4+ T cells were depleted (Villarino et al., 2003). Subsequently, the same group found that IL-27 limited production of IL-2 by activated CD4+ T cells, implicating IL-27 receptor as a negative regulator of T cell responses during Toxoplasma infection (Villarino et al., 2006). This conclusion was also supported by a 2012 study from the Hunter Laboratory where mice were p.o. infected with 100 ME49 cysts or i.p. infected with 20 cysts. IL-27 promoted development of Tregs at primary sites of infection, the small intestine or peritoneal cavity, respectively. IL-27−/− mice succumbed to acute infection, which could be partially rescued by adoptive transfer of Treg cells (Hall et al., 2012). Although this result is consistent with a tolerance strategy, parasite burden was not directly measured.
IL-2 is also important for Treg biology in Toxoplasma infection. Using p.o. infection, several groups have shown a transient, but significant loss of Treg function in the small intestine (Oldenhove et al., 2009; Benson et al., 2012). Addition of IL-2 promoted Treg survival and prevented liver pathology, consistent with a role in disease tolerance in the liver. However, IL-2 treatment also resulted in a higher brain cyst burden (Oldenhove et al., 2009). Similarly, Benson et al. found that IL-2 treated mice had more Tregs and significantly higher brain cyst burdens causing lethality (Benson et al., 2012). Whether the inability to restrict cerebral Toxoplasma infection in these studies was due to Treg-mediated suppression of effector T cells or a direct effect of IL-2 on naïve or effector T cell activity is unclear. However, directly targeting effector T cell responses has been shown to reduce tissue damage in a mouse model of ocular toxoplasmosis. Specifically, in the retina both infiltrating antigen-presenting cells (MHCII expressing) and tissue resident retinal cells expressed PD-L1, the ligand for the T cell inhibitory receptor PD-1. PD-L1 expressing retinal cells were able to suppress splenic T cell proliferation ex vivo using a co-culture system with Toxoplasma-antigen loaded dendritic cells (Charles et al., 2010). These results suggest that retinal cells may be able to moderate local immune responses and reduce tissue damage by directly suppressing T cell activation in the eye. Cumulatively, these studies underscore the importance of targeting the T cell response to limit tissue damage and restrict parasite growth.
Host Metabolic Dysregulation in Toxoplasma Infection
There is a growing appreciation that immune responses are intimately linked with shifts in metabolic homeostasis. This is a critical arm of pathogen resistance, for example, T cell activation requires a glycolytic burst; anorexia during infection mobilizes glycogen and lipid stores to support gluconeogenesis for the immune system; metabolic shifts are also used to sequester trace metals or nutrients to limit pathogen growth, often referred to as nutritional immunity (Hood and Skaar, 2012; Núñez et al., 2018). Shifts in nutrient utilization are also important mediators of disease tolerance. For example, diet restriction improves fruit fly survival during S. typhimurium infection without affecting bacterial load (Ayres and Schneider, 2009).
There is a growing body of literature showing metabolic shifts in the host during acute Toxoplasma infection. However, a role for these shifts in disease tolerance has not been addressed specifically. Using Swiss-Webster mice infected p.o. with 8 cysts of the human-derived Type II strain BGD-1, one group reported reduced circulating cholesterol and HDL at 14 days post infection (Milovanović et al., 2017). Untargeted proteomic analysis of sera isolated from BALB/c mice infected per orally with 10 Type II Pru cysts showed increased circulating amino acids and reduced choline levels in the first 21 days of infection (Zhou et al., 2016). The same group observed an increase in choline-derived phosphatidylcholine and phosphatidylethanolamine in the brains of infected mice (Zhou et al., 2015). Choline is an essential dietary nutrient as the precursor of phosphatidylcholine and the neurotransmitter acetylcholine. Phosphatidylcholine is a major component of cell membranes and is necessary for the packaging and export of very-low-density lipoproteins (VLDL) from the liver (Corbin and Zeisel, 2012). Together, these studies suggest that Toxoplasma infected mice shift toward amino acid and fat metabolism and away from glycolytic metabolism during Toxoplasma infection. This is consistent with a well-described, although transient, period of anorexia during acute infection in Toxoplasma infected mice which would induce such programs (Arsenijevic et al., 1997; Weiss and Dubey, 2009; Jin et al., 2017; Hatter et al., 2018). In mice infected with L. monocytogenes, anorexia-induced ketogenesis protected tissues from oxidative stress, whereas glucose supplementation increased mortality. Interestingly, the opposite effect was observed with influenza infection, suggesting that the effectiveness of tolerance programs, like restriction mechanims, depend on the pathogen (Wang et al., 2016). Similar experiments to look at the role of glycolytic and beta-oxidative host metabolism in Toxoplasma will be necessary to determine if anorexia respresents a host tolerance strategy or if the parasite capatilizes on altered host metabolism for infection.
It is well-established that scavenging host lipids is necessary for Toxoplasma survival in vivo and in vitro. For example, most of the cholesterol in Toxoplasma is derived from scavenged host LDL (Bisanz et al., 2006). Metabolic labeling with 14C-acetate showed that Toxoplasma must scavenge fatty acid precursors from its host to synthesize its full range of lipids (Sehgal et al., 2005). Recent studies indicate that Toxoplasma competes with the host cell for lipids at the level of lipid droplet recruitment, mitochondrial interaction, and vesicular transport for intracellular survival (Hu et al., 2017; Nolan et al., 2017; Pernas et al., 2018). Further studies are warranted to determine whether host metabolic shifts that mobilize lipid stores at a systemic (rather than a cellular) level benefit Toxoplasma. By contrast, there is growing evidence that altered metabolism has a long-term negative impact on the host during Toxoplasma infection. A series of studies by Arsenijevic et al. demonstrated that mice orally infected with 10 Me49 cysts undergo anorexia-associated hypermetabolism during acute infection. Nearly half of the infected cohort could not regain weight as they progressed to chronic infection. These “non-gainers” harbored a higher parasite load than mice that regained weight and sustained the hypermetabolic phenotype along with elevated circulating inflammatory cytokines (Arsenijevic et al., 1997, 2001). A subsequent study showed that non-gainers challenged with LPS had a more severe inflammatory response, worse pathology, and a longer rebound period than infected mice that gained weight (Arsenijevic et al., 1998). Related, a study by Kugler et al. showed that Toxoplasma infection led to long-term defects in thymus and lymph node structure, hindering naïve T cell responses to subsequent viral challenge (Kugler et al., 2016). More recently, the Wohlfert lab showed that oral infection with 5 Me49 cysts causes acute weight loss in mice and inability to regain weight as chronic infection progresses (Jin et al., 2017). Surprisingly, muscle inflammation and myositis were driven by Tregs, a population classically associated with tolerance, suggesting that sustained tolerance programs may negatively impact the host. In 2018 Hatter et al. similarly reported that C57BL/6J mice orally infected with Me49 cysts develop chronic cachexia defined by lean muscle and fat wasting and chronic elevation of circulating innate cytokines. Although cachectic mice recovered from acute ileitis, dysbiosis in the intestinal commensal microbiota population was sustained (Hatter et al., 2018). Loss of total gut commensal diversity and a shift toward “pathobiotic” Gram negative species is well-established in acute Toxoplasma infection (Heimesaat et al., 2006; Benson et al., 2009; Molloy et al., 2013; Hatter et al., 2018). Although the precise composition of outgrowth species is animal colony-dependent, several groups have reported E. coli outgrowth (Benson et al., 2009; Raetz et al., 2012; Molloy et al., 2013; Fonseca et al., 2015). This is interesting in the context of the Zhao Lab metabolomics data because these bacteria are major metabolizers of the choline derivative ethanolamine (Garsin, 2012). It is highly plausible that shifts in commensals influence nutrient availability, in addition to playing a better-described role in influencing host immunity during Toxoplasma infection
Toxoplasma interactions with major organs involved in nutrient regulation may have important implications for host metabolism as well. The liver coordinates dietary nutrient uptake (bile acid recycling), availability and storage (fat and glycogen), and detoxifies the blood. Liver resident macrophages called Kupffer cells screen incoming blood for pathogens and intestinal microbes that leak from the gut during intestinal inflammation, including during Toxoplasma infection (Molloy et al., 2013). Toxoplasma has been detected in the livers of mice at acute infection using bioluminescence, histology, and PCR in a number of studies using Type II strains (Silva et al., 2002; Di Cristina et al., 2008; Zhou et al., 2015; He et al., 2016a,b). In the first week of infection, Toxoplasma has been observed replicating in hepatocytes near regions of inflammatory infiltrate and focal necrosis in both Swiss-Webster and BALB/c mice (Atmaca et al., 2013; Bottari et al., 2014). Interestingly, Atmaca et al. also reported an expansion of hepatic stellate cells during Toxoplasma infection (Atmaca et al., 2013). During inflammation, hepatic stellate cells differentiate into myofibroblasts and produce extracellular matrix which is consistent with the induction of a tissue remodeling tolerance program during Toxoplasma infection. However, an important caveat with these studies is that they were performed with lethal doses of the hypervirulent Type I strain RH raising the question of relevance to hepatic infection with strains that are more commonly found in mice or humans. Using clinically isolated Type II strains, Toxoplasma cysts were found in the livers of infected Swiss-Webster mice as late as 33 weeks post-infection, indicating that the liver may be a reservoir for chronic infection (Autier et al., 2018). This is consistent with clinical reports of Toxoplasma-negative transplant recipients who have developed toxoplasmosis after receiving livers from sera positive donors (Assi et al., 2007; Galván-Ramírez et al., 2018). While there are no studies directly implicating Toxoplasma in the development of liver disease, approximately 30% of patients with chronic liver disease test sera-positive for Toxoplasma B1 compared to 6% in control populations (El-Sayed et al., 2016). These Toxoplasma-infected patients had significantly elevated circulating ALT and AST, clinical markers of liver damage, compared to uninfected patients with liver disease. Although these data do not imply causality, they are consistent with the interpretation that chronic Toxoplasma infection may negatively impact host fitness in diseases of co-occurrence.
Toxoplasma infection may also change the metabolic landscape of the liver. In a recent liver proteomics study, BALB/c mice intraperitoneally infected with 200 type II PYS tachyzoites had reduced signatures of fatty-acid oxidation proteins and an upregulation of cell death, inflammatory, and stress response pathways at 6 days post-infection (He et al., 2016b). A parallel liver transcriptomics study reported down-regulation of gene families involved in lipid metabolism, cholesterol and bile synthesis, and amino acid metabolism, with an increase in inflammatory transcripts (He et al., 2016a). Together, these studies provide evidence that Toxoplasma occupies a liver niche in acute and chronic infection and may directly contribute to shifts in liver metabolic homeostasis.
Adipose tissue depots are another important site of calorie storage and immune regulation. Fat tissues have long been described as an anti-inflammatory environment that become inflamed in diseases of the adipose tissue, including obesity and diabetes. As better tools have become available to survey low pathogen loads in tissue, researchers have begun to appreciate the frequency of microbial translocation to and persistence in this nutrient-rich environment. M. tuberculosis, T. brucei, and facultative pathogen strains of E. coli have been detected in this niche, indicating that it serves as a reservoir of infection for many pathogens (Neyrolles et al., 2006; Schieber et al., 2015; Trindade et al., 2016). More recently, Toxoplasma has been reported in the visceral fat following intraperitoneal infection and oral infection by bioluminescence assay and PCR (Di Cristina et al., 2008). Interestingly, using a stage-specific luciferase reporter system, di Cristina et al. showed that parasites in visceral fat expressed GFP driven by the SRS9 promoter, a gene product enriched during bradyzoite differentiation (Di Cristina et al., 2008). Future studies will be necessary to determine if fat is a long-term reservoir for Toxoplasma, and whether colonization of the fat is related to host metabolic shifts.
Conclusions
Toxoplasma gondii has evolved sophisticated mechanisms to evade sterilizing immunity, yet activating a robust immune response is necessary to ensure host survival long enough for Toxoplasma encystation and transmission (Bohne et al., 1993, 1994). Disease tolerance programs are adaptations to cell biology and metabolism that allow tissues to function in the harsh environment of an inflammatory response (Tzelepis et al., 2018). However, tolerance adaptations must come at a cost to the host, otherwise they would be selected for homeostatic use. The literature reviewed here are consistent with a model where tolerance programs initiated in acute Toxoplasma infection, including immune-microbiota interactions, T cell-mediated responses and metabolic shifts fail to return to homeostasis in chronic infection. Emerging studies suggest that these shifts in homeostasis have sustained negative consequences for the host, including muscle wasting, and impaired responses to secondary immune stimuli (Arsenijevic et al., 1997; Kugler et al., 2016; Jin et al., 2017; Hatter et al., 2018). Whether or not these shifts in homeostasis confer a benefit to the parasite is an open question. Compromising rodent fitness in these ways would likely enhance the opportunity for predation by felines, the parasite's definitive host. Passage through a cat is extremely advantageous for Toxoplasma because feline hosts facilitate genetic diversity and range expansion. In this way, Toxoplasma may benefit from promoting tolerance programs that ensure host survival during the acute phase of infection, during the tachyzoite to bradyzoite transition, but ultimately impair host fitness in the long term.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was supported by NIH NIAID K22 AI116727-01 (SE) and T32 AI7496-23 (SM).
References
- Aliberti J., Serhan C., Sher A. (2002). Parasite-induced lipoxin A 4 is an endogenous regulator of IL-12 production and immunopathology in Toxoplasma gondii Infection. J. Exp. Med. 196, 1253–1262. 10.1084/jem.20021183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsenijevic D., Bilbao F. D., Giannakopoulos P., Girardier L., Samec S., Richard D. (2001). A role for interferon-gamma in the hypermetabolic response to murine toxoplasmosis. Eur. Cytokine Netw. 12, 518–527. [PubMed] [Google Scholar]
- Arsenijevic D., Girardier L., Seydoux J., Chang H. R., Dulloo A. G. (1997). Altered energy balance and cytokine gene expression in a murine model of chronic infection with Toxoplasma gondii. Am. J. Physiol. 272(Pt. 1), E908–E917. 10.1152/ajpendo.1997.272.5.E908 [DOI] [PubMed] [Google Scholar]
- Arsenijevic D., Girardier L., Seydoux J., Pechere J. C., Garcia I., Lucas R., et al. (1998). Metabolic-cytokine responses to a second immunological challenge with LPS in mice with T. gondii infection. Am. J. Physiol. 274(Pt. 1), E439–E445. 10.1152/ajpendo.1998.274.3.E439 [DOI] [PubMed] [Google Scholar]
- Assi M. A., Rosenblatt J. E., Marshall W. (2007). Donor-transmitted toxoplasmosis in liver transplant recipients: a case report and literature review. Transpl. Infect. Dis. 9, 132–136. 10.1111/j.1399-3062.2006.00187.x [DOI] [PubMed] [Google Scholar]
- Atmaca H., Gazyagci A., Canpolat S., Kul O. (2013). Hepatic stellate cells increase in Toxoplasma gondii infection in mice. Parasites Vectors 6:135. 10.1186/1756-3305-6-135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autier B., Dion S., Robert-gangneux F. (2018). The liver as an organ at risk for Toxoplasma transmission during transplantation: myth or reality? J. Clin. Pathol. 71, 763–766. 10.1136/jclinpath-2018-205066 [DOI] [PubMed] [Google Scholar]
- Ayres J. S., Schneider D. S. (2009). The role of anorexia in resistance and tolerance to infections in Drosophila. PLoS Biol. 7:e1000150. 10.1371/journal.pbio.1000150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayres J. S., Schneider D. S. (2012). Tolerance of infections. Annu. Rev. Immunol. 30, 271–294. 10.1146/annurev-immunol-020711-075030 [DOI] [PubMed] [Google Scholar]
- Benson A., Murray S., Divakar P., Burnaevskiy N., Pifer R., Forman J., et al. (2012). Microbial infection-induced expansion of effector T cells overcomes the suppressive effects of regulatory T cells via an IL-2 deprivation mechanism. J. Immunol. 188, 800–810. 10.4049/JIMMUNOL.1100769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson A., Pifer R., Behrendt C. L., Hooper L. V., Yarovinsky F. (2009). Gut commensal bacteria direct a protective immune response against Toxoplasma gondii. Cell Host Microbe 6, 187–196. 10.1016/j.chom.2009.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierly A. L., Shufesky W. J., Sukhumavasi W., Morelli A. E., Denkers E. Y. (2008). Dendritic cells expressing plasmacytoid marker PDCA-1 are trojan horses during Toxoplasma gondii infection. J. Immunol. 181, 8485–8491. 10.4049/jimmunol.181.12.8485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisanz C., Bastien O., Grando D., Jouhet J., Maréchal E., Cesbron-Delauw M.-F. (2006). Toxoplasma gondii acyl-lipid metabolism: de novo synthesis from apicoplast-generated fatty acids versus scavenging of host cell precursors. Biochem. J. 394, 197–205. 10.1042/BJ20050609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard N., Gonzalez F., Schaeffer M., Joncker N. T., Cheng T., Shastri A. J., et al. (2008). Immunodominant, protective response to the parasite Toxoplasma gondii requires antigen processing in the endoplasmic reticulum. Nat. Immunol. 9, 937–944. 10.1038/ni.1629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohne W., Heesemann J., Gross U. (1993). Induction of bradyzoite-specific Toxoplasma gondii antigens in gamma interferon-treated mouse macrophages. Infect. Immunity 61, 1141–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohne W., Heesemann J., Gross U. (1994). Reduced replication of Toxoplasma gondii is necessary for induction of bradyzoite-specific antigens: a possible role for nitric oxide in triggering stage conversion. Infect. Immunity 62, 1761–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottari N. B., Tonin A. A., Fighera R., Flores M. M., França R. T., Camillo G., et al. (2014). Experimental parasitology Neospora caninum and Toxoplasma gondii: relationship between hepatic lesions, cytological and biochemical analysis of the cavitary liquid during the acute phase of the diseases in experimental models. Exp. Parasitol. 136, 68–73. 10.1016/j.exppara.2013.11.004 [DOI] [PubMed] [Google Scholar]
- Caldwell R. M., Schafer J. F., Compton L. E., Patterson F. L. (1958). Tolerance to cereal leaf rusts. Science 128, 714–715. 10.1126/science.128.3326.714 [DOI] [PubMed] [Google Scholar]
- Charles E., Joshi S., Ash J. D., Fox B. A., Farris A. D., Bzik D. J., et al. (2010). CD4 T-cell suppression by cells from Toxoplasma gondii-infected retinas is mediated by surface protein PD-L1. Infect. Immunity 78, 3484–3492. 10.1128/IAI.00117-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F., Liu Z., Wu W., Rozo C., Bowdridge S., Millman A., et al. (2012). An essential role for T H 2-type responses in limiting acute tissue damage during experimental helminth infection. Nat. Med. 18, 262–268. 10.1038/nm.2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgan S. P., Serhan C. N., Parkos C. A., Delp-Archer C., Madara J. L. (1993). Lipoxin A4 modulates transmigration of human neutrophils across intestinal epithelial monolayers. J. Clin. Invest. 92, 75–82. 10.1172/JCI116601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin K. D., Zeisel S. H. (2012). Choline metabolism provides novel insights into nonalcoholic fatty liver disease and its progression. Curr. Opin. Gastroenterol. 28, 159–165. 10.1097/MOG.0b013e32834e7b4b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courret N., Darche S., Sonigo P., Milon G., Buzoni-Gâtel D., Tardieux I. (2006). CD11c- and CD11b-expressing mouse leukocytes transport single Toxoplasma gondii tachyzoites to the brain. Blood 107, 309–316. 10.1182/blood-2005-02-0666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckert-Schlüter M., Rang A., Weiner D., Huang S., Wiestler O. D., Hof H., et al. (1996). Interferon-gamma receptor-deficiency renders mice highly susceptible to toxoplasmosis by decreased macrophage activation. Lab. Investig. 75, 827–841. [PubMed] [Google Scholar]
- Di Cristina M., Marocco D., Galizi R., Proietti C., Spaccapelo R., Crisanti A. (2008). Temporal and spatial distribution of Toxoplasma gondii differentiation into Bradyzoites and tissue cyst formation in vivo. Infect. Immun. 76, 3491–3501. 10.1128/IAI.00254-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey J. P., Lindsay D. S., Speer C. A. (1998). Structures of Toxoplasma gondii tachyzoites, bradyzoites, and sporozoites and biology and development of tissue cysts. Clin. Microbiol. Rev. 11, 267–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont C. D., Christian D. A., Hunter C. A. (2012). Immune response and immunopathology during toxoplasmosis. Semin. Immunopathol. 34, 793–813. 10.1007/s00281-012-0339-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sayed N. M., Ramadan M. E., Ramadan M. E. (2016). Toxoplasma gondii infection and chronic liver diseases: evidence of an association. Trop. Med. Infect. Dis. 1, 1–8. 10.3390/tropicalmed1010007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etheridge R. D., Alaganan A., Tang K., Lou H. J., Turk B. E., Sibley L. D. (2014). The Toxoplasma pseudokinase ROP5 forms complexes with ROP18 and ROP17 kinases that synergize to control acute virulence in mice. Cell Host Microbe. 15, 537–550. 10.1016/j.chom.2014.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald S. E., Chavarria-Smith J., Boothroyd J. C. (2014). NLRP1 is an inflammasome sensor for Toxoplasma gondii. Infect. Immun. 82, 460–468. 10.1128/IAI.01170-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca D. M., Hand T. W., Han S. J., Gerner M. Y., Glatman Zaretsky A. G., Byrd A. L., et al. (2015). Microbiota-dependent sequelae of acute infection compromise tissue-specific immunity. Cell 163, 354–366. 10.1016/j.cell.2015.08.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galván-Ramírez M. L., Sánchez-Orozco L. V., Gutiérrez-Maldonado A. F., Rodriguez Pérez L. R. (2018). Does Toxoplasma gondii infection impact liver transplantation outcomes ? A systematic review. J. Med. Microbiol. 67, 499–506. 10.1099/jmm.0.000694 [DOI] [PubMed] [Google Scholar]
- Garsin D. A. (2012). Ethanolamine: a signal to commence a host-associated lifestyle? MBio 3, 172–174. 10.1128/mbio.00172-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazzinelli R. T., Wysocka M., Hieny S., Scharton-Kersten T., Cheever A., Kühn R., et al. (1996). In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF-alpha. J. Immunol. 157, 798–805. [PubMed] [Google Scholar]
- Gorfu G., Cirelli K. M., Melo M. B., Mayer-Barber K., Crown D., Koller B. H., et al. (2014). Dual role for inflammasome sensors NLRP1 and NLRP3 in murine resistance to Toxoplasma gondii. MBio 5, 1–12. 10.1128/mBio.01117-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainger J. R., Wohlfert E. A., Fuss I. J., Bouladoux N., Askenase M. H., Legrand F., et al. (2013). Inflammatory monocytes regulate pathologic responses to commensals during acute gastrointestinal infection. Nat. Med. 19, 713–721. 10.1038/nm.3189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg B., Taylor B. C., John B., Tait-Wojno E. D., Girgis N. M., Miller N., et al. (2013). Replication and distribution of Toxoplasma gondii in the small intestine after oral infection with tissue cysts. Infect. Immun. 81, 1635–1643. 10.1128/IAI.01126-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover H. S., Chu H. H., Kelly F. D., Yang S. J., Reese M. L., Blanchard N., et al. (2014). Article impact of regulated secretion on antiparasitic CD8 T cell responses. Cell Rep. 7, 1716–1728. 10.1016/j.celrep.2014.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A. O., Beiting D. P., Tato C., John B., Oldenhove G., Lombana C. G., et al. (2012). The cytokines interleukin 27 and interferon-γ promote distinct Treg cell populations required to limit infection-induced pathology. Immunity 37, 511–523. 10.1016/j.immuni.2012.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatter J. A., Kouche Y. M., Melchor S. J., Ng K., Bouley D. M., Boothroyd J. C., et al. (2018). Toxoplasma gondii infection triggers chronic cachexia and sustained commensal dysbiosis in mice. PLoS ONE 13:e0204895. 10.1371/journal.pone.0204895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J. J., Ma J., Elsheikha H. M., Song H. Q., Huang S. Y., Zhu X. Q. (2016a). Transcriptomic analysis of mouse liver reveals a potential hepato-enteric pathogenic mechanism in acute Toxoplasma gondii infection. Parasites Vectors 9:427. 10.1186/s13071-016-1716-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J. J., Ma J., Elsheikha H. M., Song H. Q., Zhou D. H., Zhu X. Q. (2016b). Proteomic profiling of mouse liver following acute toxoplasma gondii infection. PLoS ONE 11:e0152022. 10.1371/journal.pone.0152022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimesaat M. M., Bereswill S., Fischer A., Fuchs D., Struck D., Niebergall J., et al. (2006). Gram-negative bacteria aggravate murine small intestinal Th1-type immunopathology following oral infection with Toxoplasma gondii. J. Immunol. 177, 8785–8795. 10.4049/jimmunol.177.12.8785 [DOI] [PubMed] [Google Scholar]
- Hood M. I., Skaar E. P. (2012). Nutritional immunity: transition metals at the pathogen-host interface. Nat. Rev. Microbiol. 10, 525–537. 10.1038/nrmicro2836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X., Binns D., Reese M. L. (2017). The coccidian parasites Toxoplasma and Neospora dysregulate mammalian lipid droplet biogenesis. J. Biol. Chem. 292, 11009–11020. 10.1074/jbc.M116.768176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter C. A., Subauste C. S., Cleave V. H. V. A. N., Remington J. S. (1994). Production of gamma interferon by natural killer cells from Toxoplasma gondii-infected SCID mice : regulation by necrosis factor alpha. Infect. Immun. 62, 2818–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankovic D., Kullberg M. C., Feng C. G., Goldszmid R. S., Collazo C. M., Wilson M., et al. (2007). Conventional T-bet+Foxp3− Th1 cells are the major source of host-protective regulatory IL-10 during intracellular protozoan infection. J. Exp. Med. 204, 273–283. 10.1084/JEM.20062175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jebbari H., Roberts C. W., Ferguson D. J., Bluethmann H., Alexander J. (1998). A protective role for IL-6 during early infection with Toxoplasma gondii. Parasite Immunol. 20, 231–239. [DOI] [PubMed] [Google Scholar]
- Jin R. M., Blair S. J., Warunek J., Reid R., Blader I. J., Wohlfert E. A., et al. (2017). Regulatory T cells promote myositis and muscle damage in Toxoplasma gondii infection. J. Immunol. 198, 352–362. 10.4049/jimmunol.1600914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipper J. A., Willenborg S., Hammerschmidt M., Allen J. E., Eming S. A., Knipper J. A., et al. (2015). Interleukin-4 receptor a signaling in myeloid cells controls collagen fibril assembly in skin repair article interleukin-4 receptor a signaling in myeloid cells controls collagen fibril assembly in skin repair. Immunity 43, 803–816. 10.1016/j.immuni.2015.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugler D. G., Flomerfelt F. A., Costa D. L., Laky K., Kamenyeva O., Mittelstadt P. R., et al. (2016). Systemic toxoplasma infection triggers a long-term defect in the generation and function of naive T lymphocytes. 213, 3041–3056. 10.1084/jem.20151636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilue J., Müller U. B., Steinfeldt T., Howard J. C. (2013). Reciprocal virulence and resistance polymorphism in the relationship between Toxoplasma gondii and the house mouse. Elife 2:e01298. 10.7554/eLife.01298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins R., Carlos A. R., Braza F., Thompson J. A., Amador-Bastos P., Ramos S., et al. (2019). Disease tolerance as an inherent component of immunity. Annu. Rev. Immunol. 13, 405–437. 10.1146/annurev-immunol-042718 [DOI] [PubMed] [Google Scholar]
- Medzhitov R., Schneider D. S., Soares M. P. (2012). Disease tolerance as a defense strategy. Science 335, 936–941. 10.1126/science.1214935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milovanović I., Busarčević M., Trbovich A., Ivović V., Uzelac A., Djurković-Djaković O. (2017). Evidence for host genetic regulation of altered lipid metabolism in experimental toxoplasmosis supported with gene data mining results. PLoS ONE 12:e0176700. 10.1371/journal.pone.0176700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy M. J., Grainger J. R., Bouladoux N., Hand T. W., Koo L. Y., Naik S., et al. (2013). Intraluminal containment of commensal outgrowth in the gut during infection-induced dysbiosis. Cell Host Microbe 14, 318–328. 10.1016/j.chom.2013.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyer L. E., Grunig G., Fort M., Remington J. S., Rennick D., Hunter C. A. (1997). Role of interleukin-10 in regulation of T-cell-dependent and T-cell-independent mechanisms of resistance to Toxoplasma gondii. Infect. Immunity 65, 1675–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyrolles O., Hernández-Pando R., Pietri-Rouxel F., Fornès P., Tailleux L., Payán J. A. B., et al. (2006). Is adipose tissue a place for Mycobacterium tuberculosis persistence? PLoS ONE 1:e43. 10.1371/journal.pone.0000043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan S. J., Romano J. D., Coppens I., Mellman I., Gartner F., Provenzano M. (2017). Host lipid droplets: an important source of lipids salvaged by the intracellular parasite Toxoplasma gondii. PLoS Pathog. 13:e1006362. 10.1371/journal.ppat.1006362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Núñez G., Sakamoto K., Soares M. P. (2018). Innate nutritional immunity. J. Immunol. 201, 11–18. 10.4049/JIMMUNOL.1800325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenhove G., Bouladoux N., Wohlfert E. A., Hall J. A., Chou D., Dos santos L., et al. (2009). Decrease of Foxp3+ treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity 31, 772–786. 10.1016/j.immuni.2009.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernas L., Bean C., Boothroyd J. C., Scorrano L. (2018). Mitochondria restrict growth of the intracellular parasite Toxoplasma gondii by limiting its uptake of fatty acids. Cell Metab. 27, 886–897.e4. 10.1016/j.cmet.2018.02.018 [DOI] [PubMed] [Google Scholar]
- Raetz M., Hwang S., Wilhelm C. L., Kirkland D., Benson A., Sturge C. R., et al. (2012). Parasite-induced TH1 cells and intestinal dysbiosis cooperate in IFN-γ-dependent elimination of Paneth cells. Nat. Immunol. 14, 136–142. 10.1038/ni.2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese M. L., Zeiner G. M., Saeij J. P. J., Boothroyd J. C., Boyle J. P. (2011). Polymorphic family of injected pseudokinases is paramount in Toxoplasma virulence. Proc. Natl. Acad. Sci. USA. 108, 9625–9630. 10.1073/pnas.1015980108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts C. W., Ferguson D. J. P., Jebbari H., Satoskar A., Bluethmann H., Alexander J. (1996). Different roles for interleukin-4 during the Course of Toxoplasma gondii infection. Infect. Immun. 64, 897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasai M., Pradipta A., Yamamoto M. (2018). Host immune responses to Toxoplasma gondii. Int. Immunol. 30, 113–119. 10.1093/intimm/dxy004 [DOI] [PubMed] [Google Scholar]
- Scharton-Kersten T. M., Yap G., Magram J., Sher A. (1997). Inducible nitric oxide is essential for host control of persistent but not acute infection with the intracellular pathogen Toxoplasma gondii. J. Exp. Med. 185, 1261–1273. 10.1084/jem.185.7.1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieber A. M. P., Lee Y. M., Chang M. W., Leblanc M., Collins B., Downes M., et al. (2015). Disease tolerance mediated by microbiome E. coli involves inflammasome and IGF-1 signaling. Science 350, 558–563. 10.1126/science.aac6468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlüter D., Kwok L.-Y., Lütjen S., Soltek S., Hoffmann S., Körner H., et al. (2003). Both lymphotoxin-alpha and TNF are crucial for control of Toxoplasma gondii in the central nervous system. J. Immunol. 170, 6172–6182. 10.4049/JIMMUNOL.170.12.6172 [DOI] [PubMed] [Google Scholar]
- Sehgal A., Bettiol S., Pypaert M., Wenk M. R., Kaasch A., Blader I. J., et al. (2005). Peculiarities of host cholesterol transport to the unique intracellular vacuole containing Toxoplasma. Traffic 6, 1125–1141. 10.1111/j.1600-0854.2005.00348.x [DOI] [PubMed] [Google Scholar]
- Sher A., Oswald I. P., Hieny S., Gazzinelli R. T. (1993). Toxoplasma gondii induces a T-independent IFN-gamma response in natural killer cells that requires both adherent accessory cells and tumor necrosis factor-alpha. J. Immunol. 150, 3982–3989. [PubMed] [Google Scholar]
- Silva N. M., Rodrigues C. V., Santoro M. M., Reis L. F. L., Alvarez-leite J. I., Gazzinelli R. T., et al. (2002). Expression of indoleamine 2, 3-dioxygenase, tryptophan degradation, and kynurenine formation during in vivo infection with Toxoplasma gondii: induction by endogenous gamma interferon and requirement of interferon regulatory factor 1. Infect. Immun. 70, 859–868. 10.1128/IAI.70.2.859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares M. P., Gozzelino R., Weis S. (2014). Tissue damage control in disease tolerance. Trends Immunol. 35, 483–494. 10.1016/J.IT.2014.08.001 [DOI] [PubMed] [Google Scholar]
- Suzuki Y., Orellana M. A., Schreiber R. D., Remington J. S. (1988). Interferon-gamma: the major mediator of resistance against Toxoplasma gondii. Science 240, 516–518. [DOI] [PubMed] [Google Scholar]
- Suzuki Y., Sher A., Yap G., Park D., Neyer E., Liesenfeld O., et al. (2000). IL-10 is required for prevention of necrosis in the small intestine and mortality in both genetically resistant BALB/c and susceptible C57BL/6 mice following peroral infection with Toxoplasma gondii. J. Immunol. 164, 5375–5382. 10.4049/jimmunol.164.10.5375 [DOI] [PubMed] [Google Scholar]
- Trindade S., Rijo-Ferreira F., Carvalho T., Pinto-Neves D., Guegan F., Aresta-Branco F., et al. (2016). Trypanosoma brucei parasites occupy and functionally adapt to the adipose tissue in mice. Cell Host Microbe 19, 837–848. 10.1016/j.chom.2016.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzelepis F., Blagih J., Khan N., Gillard J., Mendonca L., Roy D. G., et al. (2018). Mitochondrial cyclophilin D regulates T cell metabolic responses and disease tolerance to tuberculosis. Sci. Immunol. 3:eaar4135. 10.1126/sciimmunol.aar4135 [DOI] [PubMed] [Google Scholar]
- Villarino A., Hibbert L., Lieberman L., Wilson E., Mak T., Yoshida H., et al. (2003). The IL-27R (WSX-1) is required to suppress T cell hyperactivity during infection. Immunity 19, 645–655. 10.1016/S1074-7613(03)00300-5 [DOI] [PubMed] [Google Scholar]
- Villarino A. V., Stumhofer J. S., Saris C. J. M., Kastelein R. A., de Sauvage F. J., Hunter C. A. (2006). IL-27 limits IL-2 production during Th1 differentiation. J. Immunol. 176, 237–247. 10.4049/JIMMUNOL.176.1.237 [DOI] [PubMed] [Google Scholar]
- Wang A., Huen S. C., Luan H. H., Yu S., Zhang C., Gallezot J.-D., et al. (2016). Opposing effects of fasting metabolism on tissue tolerance in bacterial and viral inflammation. Cell 166, 1512–1525.e12. 10.1016/j.cell.2016.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss L. M., Dubey J. P. (2009). Toxoplasmosis: a history of clinical observations. Int. J. Parasitol. 39, 895–901. 10.1016/j.ijpara.2009.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson E. H., Wille-Reece U., Dzierszinski F., Hunter C. A. (2005). A critical role for IL-10 in limiting inflammation during toxoplasmic encephalitis. J. Neuroimmunol. 165, 63–74. 10.1016/J.JNEUROIM.2005.04.018 [DOI] [PubMed] [Google Scholar]
- Yap G., Pesin M., Sher A. (2000). Cutting edge: IL-12 is required for the maintenance of IFN-gamma production in T cells mediating chronic resistance to the intracellular pathogen, Toxoplasma gondii. J. Immunol. 165, 628–631. 10.4049/jimmunol.165.2.628 [DOI] [PubMed] [Google Scholar]
- Yarovinsky F., Hieny S., Sher A. (2008). Recognition of Toxoplasma gondii by TLR11 prevents parasite-induced immunopathology. J. Immunol. 181, 8478–8484. 10.4049/jimmunol.181.12.8478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C.-X., Zhou D.-H., Elsheikha H. M., Liu G.-X., Suo X., Zhu X.-Q. (2015). Global metabolomic profiling of mice brains following experimental infection with the cyst-forming Toxoplasma gondii. PLoS ONE 10:e0139635. 10.1371/journal.pone.0139635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C.-X., Zhou D.-H., Elsheikha H. M., Zhao Y., Suo X., Zhu X.-Q. (2016). Metabolomic profiling of mice serum during toxoplasmosis progression using liquid chromatography-mass spectrometry. Sci. Rep. 6:19557. 10.1038/srep19557 [DOI] [PMC free article] [PubMed] [Google Scholar]