

Abstract
The direct generation of aldehydes from carboxylic acids is often a challenging synthetic task but undoubtedly attractive in view of abundant supply of such feedstocks from nature. Though long known, biocatalytic carboxylate reductions are at an early stage of development, presumably because of their co‐factor requirement. To establish an alternative to whole‐cell‐based carboxylate reductions which are limited by side reactions, we developed an in vitro multi‐enzyme system that allows for quantitative reductions of various carboxylic acids with full recycling of all cofactors and prevention of undesired over‐reductions. Regeneration of adenosine 5′‐triphosphate is achieved through the simultaneous action of polyphosphate kinases from Meiothermus ruber and Sinorhizobium meliloti and β‐nicotinamide adenine dinucleotide 2′‐phosphate is reduced by a glucose dehydrogenase. Under these conditions and in the presence of the carboxylate reductases from Neurospora crassa or Nocardia iowensis, various aromatic, heterocyclic and aliphatic carboxylic acids were quantitatively reduced to the respective aldehydes.
Keywords: aldehyde, carboxylate reductase, enzyme cascade, in vitro ATP generation, multi-enzyme catalysis
Carboxylic acids represent a class of compounds that are produced in large quantities in nature in the form of fatty acids, citric acid, amino acids and many more. Consequently, they serve as attractive starting materials to promote the implementation of bio‐based pathways in chemistry and related sciences. Their applications as free acids or—after transformation to derivatives with unchanged oxidation state—as corresponding esters and amides are well established. By contrast, selective reductions of the carboxylate group are often not amenable in a direct way towards aldehydes. The most frequent way to obtain aldehydes from carboxylic acids is to perform a sequence consisting of ester formation, reduction to the alcohol and selective re‐oxidation to the desired product. Poor atom economy and the use of reagents like LiAlH4 for the reduction,1 and chromine(VI) reagents, Dess–Martin periodinane or Swern conditions for the re‐oxidation2 raise concerns when considering the compatibility with functional groups and applications on larger scales. However, apart from chemical methods, there is a biocatalytic alternative available through the application of carboxylate reductases (CARs).3 This class of enzymes was first described decades ago,4 and is gaining increased attention within the last few years. Carboxylate reductases exhibit the intrinsic advantage of processing reactions via chemoselective and mild activation under aqueous conditions. CARs leave various functional groups and unsaturated moieties untouched. These reductases are comprised of three domains and possess the desirable feature of directly reducing carboxylic acids into aldehydes without the need for any external pre‐activation. Mechanistically, the carboxylate is activated first in an adenylation domain of the CAR as a mixed anhydride of the carboxylic acid and adenosine monophosphate. Next, the carboxylate is transferred to a 4′‐phosphopantetheinyl residue in the respective binding domain, thereby forming an enzyme‐bound thioester. Finally, NADPH‐dependent reduction in the reductase domain generates the corresponding aldehyde.5 The overall reduction of one equivalent of carboxylic acid consumes equimolar amounts of NADPH and ATP and produces AMP, NADP+, pyrophosphate, and aldehyde. Although NADPH recycling is well established, ATP recycling systems are still at the proof‐of‐concept level.6 Because of the obvious complexity associated with the simultaneous recycling of NADPH and ATP, the only economic way was to utilize CARs embedded in living microbial cells and exploit their cellular metabolism for co‐factor regeneration. Processes involving microbial cells have many advantages, but they also have drawbacks such as competing metabolic pathways, kinetic restrictions due to physical barriers or aldehyde‐related toxicity to the biological system.7 Host background activities can annihilate perfect aldehyde yields by detoxification mechanisms, leading for example, to undesired over‐reduction.8 In vitro systems on the other hand require the imitation of all relevant reaction steps by applying appropriate enzymes.9 They provide advantages in testing and optimizing the reaction conditions, ideally allowing reducing—if not eliminating—side reactions of the starting materials and aldehyde products. On the other hand, possible incompatibilities with reagents required to regenerate the co‐factors and pH shifts during the reaction course that would have played no role in a whole‐cell transformation need to be considered.
Herein, we focused on the selective preparation of aldehydes as final product in a cell‐free system. To make this process viable for synthesis, the key is to implement a balanced, catalytic cofactor recycling system for the activation and reduction of thermodynamically nearly inert carboxylate moieties.
In initial test series, we identified polyphosphate kinases (PPK)10 as the optimal enzymes to perform the generation of ATP from AMP in view of the reaction system to be realized (Scheme 1).11 Full regeneration of ATP was found most efficient when combining two PPK enzymes, that is, MrPPK and SmPPK from Meiothermus ruber 12 and Sinorhizobium meliloti,13 respectively, with an excess of MrPPK to obtain similar conversion rates in each of the two phosphorylation steps (Supporting information Table S3). Mg2+ ions are essential to adenylate forming enzymes14 such as PPKs and CARs but their complexation or precipitation by several phosphate species in the system must be assumed, necessitating a determination of the optimal Mg2+ input. Therefore, the formation of ATP from AMP was monitored at a defined polyphosphate (polyP) concentration by varying the Mg2+ concentration. Additionally, six different buffer systems were investigated with the aim to stabilize a pH of 7.5—a value compatible to all enzymes and chemical components present in the designed in vitro carboxylate reduction.
Scheme 1.
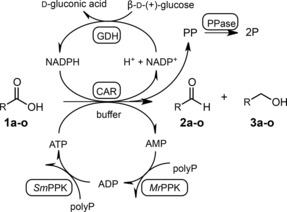
Reaction Scheme for the in vitro reduction of carboxylic acids with full recycling of all cofactors. CAR=carboxylate reductase, PPK=polyphosphate kinase, GDH=glucose dehydrogenase, PolyP=polyphosphate, PP=pyrophosphate, PPase=pyrophosphatase, P=ortho‐phosphate. For compound names see Table 2.
In line with reports in the literature,15 we found increasing conversions with increasing concentrations of MgCl2 (Figure 1 and Supporting Information Table S4). Beyond about 30 mm Mg2+, which corresponds to half the concentration of monophosphate units in polyP, no significant further increase of ATP formation was found. Consequently, this ratio was used for all following investigations. Notably, at 30 mm and slightly higher concentrations, no precipitation of magnesium phosphates leading to undesired loss of the free divalent ions was visible at pH 7.5. Tris‐HCl was found as overall best and phosphate buffer the least suitable for ATP recycling.
Figure 1.

Regeneration of ATP from AMP (2.0 mm) in the presence of MrPPK (20 μg mL−1) and SmPPK (4 μg mL−1) at pH 7.5, 30 °C in buffers (100 mm) containing NaCl (50 mm) and sodium polyP (medium chain length: 25, 60 mm related to monophosphate units). The buffer systems are as follows: white bar: HEPES, grey shaded: imidazole‐HCl, hatched: MOPS, horizontal lines: PIPES, black: potassium phosphate and vertical lines: Tris‐HCl. An average standard deviation of <5 % conversion was observed.
One of the challenges in developing a multi‐enzyme system is the identification of the operational window that is dependent on the requirements of each reaction factor (i.e., pH, temperature, and buffer preference of each enzyme, solubility and inhibition, etc.). An important aspect to be considered is potential cross inhibition of CAR enzymes by substrates required for the cofactor regeneration, especially the different phosphate species.16
To evaluate the effect of polyP, piperonylic acid 1 a reduction by Neurospora crassa carboxylate reductase (NcCAR) was studied.17 All conversions in reactions without polyP were similar after 2 h at 30 °C and almost independent of the buffer type used (Table 1). Adding sodium polyP (100 mm; Merck type 1.065.291.000, medium chain length n=25) to identical reaction mixtures caused a significant drop of all conversions. This effect was especially strong in case of phosphate buffer. We rationalized this observation by the enforced complexation of Mg2+ ions by polyP which, in addition to the ortho‐phosphate (P) in the buffer lowers the amount of freely accessible divalent ions. In the case of all other buffers tested, this reduction in activity is substantially lower and especially imidazole‐HCl and HEPES seemed to be most suitable for the carboxylate reduction with concurrent cofactor regeneration.
Table 1.
Influence of polyphosphate and different buffer systems on NcCAR‐catalyzed carboxylate reductions of piperonylic acid 1 a (5 mm).
| Buffer[a] | Conversion no polyP [% ][b] |
Conversion 100 mm polyP [%][c] |
|---|---|---|
| HEPES | 95 | 60 |
| Imidazole‐HCl | 94 | 64 |
| MOPS | 94 | 48 |
| PIPES | 89 | 42 |
| K‐phosphate | 94 | 14 |
| Tris‐HCl | 94 | 39 |
[a] 100 mm buffer containing 50 mm NaCl. [b] Conversion to piperonal 2 a after 2 h at 30 °C in the presence of NADPH (7.5 mm), ATP (10.0 mm), MgCl2 (10 mm) and NcCAR (50 μg mL−1). [c] as described in [b] supplemented with sodium polyP (100 mm).
Having investigated in vitro ATP recycling, we went on to select a suitable reduction system for NADP+. Ideally, this system operates well at pH 7.5, tolerates moderate to high salt concentrations and forms only non‐interfering co‐products. In view of these requirements, the glucose dehydrogenase (GDH)‐based NADPH formation was selected. Initial experiments with GDH showed substantial over‐reductions to the corresponding alcohols 3 catalyzed by host protein activities in the tested commercial GDH preparations. Eventually, the most active GDH was used at limiting amounts in the complex reaction system (Scheme 1) to keep the steady‐state concentration of NADPH low. This measure minimized the formation of alcohols 3 to negligible amounts in most of the investigated cases, allowing to exploit the full selectivity advantage of CARs to form pure aldehydes.
Assembling both cofactor regeneration reactions and the carboxylate reduction in vitro, the system was tested with a number of carboxylic acid substrates (Table 2). Pyrophosphate (PP) inhibits CARs but this can be circumvented by adding a pyrophosphate‐hydrolyzing enzyme, pyrophosphatase (PPase) as shown previously.16, 17, 18 Expecting inhibitory effects of PP in our investigations, PPase was added to standard reactions.
Table 2.
Reduction of several carboxylic acids 1 to their corresponding aldehydes 2 in vitro after 18 h at 30 °C.
| Example | Carboxylic acid 1 [a] | C [%] NcCAR[b] |
C [%] NiCAR[b] |
|---|---|---|---|
| a | Piperonylic acid | >99 | >99 |
| b | Benzoic acid | >99 | >99 |
| c | 2‐Methoxy‐benzoic acid | 21[c] | 66 |
| d | 3‐Methoxy‐benzoic acid | 77 | >99 |
| e | 4‐Methoxy‐benzoic acid | >99 | >99 |
| f | 2‐Chloro‐benzoic acid | 48 | 65 |
| g | 3‐Chloro‐benzoic acid | 55 | >99 |
| h | 4‐Chloro‐benzoic acid | 48 | >99 |
| i | 3‐Nitro‐benzoic acid | 16[c] | >99[c] |
| j | 2‐Thiophenecarboxylic acid | >99 | >99 |
| k | trans‐Cinnamic acid | 41[d] | 34[d] |
| l | 3‐Phenyl‐propionic acid | 30 | 99 |
| m | Hexanoic acid | 20 | >99 |
| n | Octanoic acid | 0 | 80 |
| o | Dodecanoic acid | 0 | 2 |
[a] Reaction conditions: 10 mm carboxylic acid, ATP (1.0 mm), NADPH (0.5 mm), NcCAR or NiCAR (50 μg mL−1), MrPPK (100 μg mL−1), SmPPK (40 μg mL−1), PPase (25 μg mL−1), GDH (0.2 U mL−1), 100 mm imidazole‐HCl buffer pH 7.5, β‐d‐glucose (100 mm), MgCl2 (70 mm), sodium polyP (140 mm related to ortho‐phosphate units) and 30 °C, [b] Determined by rp‐HPLC analysis; <2 % alcohol formed. [c] 5–8 % of alcohol formed. [d] Minor amounts of aldehyde‐derived byproducts were formed via β‐hydration and aldol reactions.
Under the conditions used, reactions in the presence of Nocardia iowensis CAR (NiCAR)19 showed the overall best performance in converting a set of diverse carboxylic acids 1 a–o comprising of substituted benzoic, heterocyclic, α,β‐unsaturated and aliphatic carboxylic acids. Apart from o‐substituted benzoic, cinnamic and aliphatic acids bearing longer chains than C8 were fully or almost fully converted to the corresponding aldehydes. In general, the amounts of alcohols formed were below 2 % in most cases (Table 2). NcCAR provided complete conversions only with a few carboxylic derivatives under these conditions (1 a, b, e and j). One reason for lower conversions with NcCAR in this system may be the reaction pH, which better overlaps with the optimal pH‐range of NiCAR. In this setup, no significant difference between the outcome of reactions of 1 a with or without PPase was observed as determined by kinetic and endpoint comparisons. We hypothesize the high Mg2+ concentration being the main factor for complexing PP and thereby its inhibitory effect on CAR is alleviated, again emphasizing on the critical role of Mg2+ concentrations in this context.
To estimate the limits for ATP regeneration under the general reaction conditions, further investigations with varying polyP concentrations were made. As outlined before, polyP should be used at lowest possible concentrations to minimize adverse effects on the Mg2+ supply and, consequently, on the conversion in CAR reactions. ATP recycling starting from AMP requires two molecules of ortho‐phosphate which are added stepwise by PPKs from polyP sources, thereby defining the minimum amount of active phosphate needed. PPKs recently reported to be also able to transfer more than one phosphate unit in a single event were not considered for use in this work.20 In addition to the selection of PPKs, shorter polyphosphate chains are expected to be no efficient donors15 and therefore, an appropriate excess of polyP must be provided.
Figure 2 clearly shows that a minimum amount of 40 mm polyP (calculation based on ortho‐phosphate units) is required to reach complete reduction of 10 mm of 1 a in vitro. Overall, almost 50 % of the polyP (Merck type no. 1.065.291.000) can act as active phosphate donor when using a combination of MrPPK and SmPPK for ATP regeneration. In other words, polyphosphate chains longer than 12 on average can serve as donor.12b To enable a full conversion of the carboxylic acid, an at least five‐fold excess of this specific polyP should be used in the ATP recycling system. This observation of a practically restricted use of shorter chain polyphosphates may partially explain incomplete conversions obtained in an earlier study when sodium polyphosphate crystals (Aldrich cat # 305 553) with an average chain length of 17, according to the manufacturers product specification, were applied in a reduction of 10 mm of 4‐hydroxybutanoic acid.11a In this reaction system, different CARs were tested with the formate/formate dehydrogenase (FDH) system for NADPH regeneration as well as two PPKs for ATP regeneration and a pyrophosphatase.
Figure 2.

In vitro reduction of 1 a (10 mm) to 2 a+3 a (minor amount) in the presence of different concentration of polyP. Reaction conditions: ATP (1.0 mm), NADPH (0.5 mm), NcCAR (50 μg mL−1), MrPPK (100 μg mL−1), SmPPK (40 μg mL−1), PPase (25 μg mL−1), GDH (0.2 U mL−1), 100 mm imidazole‐HCl buffer pH 7.5, 30 °C, β‐d‐glucose (100 mm), MgCl2 (70 mm) and sodium polyP (medium chain length: 25, 0–100 mm related to monophosphate units). Circles: overall conversion of 1 a, squares: formally consumed fraction of polyP calculated based on the amount of polyP and ATP initially added and the assumption that polyP is only consumed in the regeneration of ATP.
The optimized reaction conditions (Supporting Information section 1.4.4 and 1.4.5) for in vitro reduction of carboxylic acids were applied at increased substrate concentration. Taking advantage of the protocol already designed to convert 20 mm carboxylic acid at the 1 mL scale without any need to increase the enzyme and reagent concentrations, 25 mm of piperonylic acid 1 a were reduced close to completion to piperonal 2 a in a 20 mL reaction (Scheme 2). To obtain full conversion at increased carboxylic acid concentrations in the shortest time possible, the replacement of imidazole by MOPS buffer at the same pH and the concomitant use of PPase were crucial.
Scheme 2.

Reaction Scheme for the in vitro reduction of piperonylic acid 1 a (25 mm) in the presence of NADP+ (0.5 mm), ATP (1.0 mm), MgCl2 (70 mm), polyP (140 mm) and β‐d‐glucose (150 mm) at 30 °C.
In contrast to the reactions at 10 mm carboxylic acid concentrations, the preparative scale variant required continuous control and adjustment of the pH to keep within the optimal range of 7.0–7.5. Moreover, with a target time of one day for full conversion, it was possible to reduce the enzyme amounts per volumetric unit in the preparative scale reaction but still keep the performance maintained.
In summary, we have developed a cell‐free system for the direct reduction of carboxylic acids to aldehydes, reaching full conversion for the majority of investigated cases. Using three compatible enzymes, the concurrent formation of ATP and NADPH with only catalytic amounts of these expensive cofactors fuels enzymatic reduction of carboxylic acids to up to 100 % aldehyde. With the protocol described herein, we show in vitro aldehyde synthesis in an aqueous system, which mimics the opportunities provided by a whole‐cell reduction but avoids their inherent disadvantages in applications.
Experimental Section
General
ATP was obtained from Carl Roth (PN HN35.2, Lot 347262238; 98.1 % purity, 8.6 % water content). ADP and AMP were obtained from Roche Diagnostics. NADP+ and NADPH were purchased from Carl Roth and MgCl2 from Merck. HPLC‐MS grade acetonitrile was purchased from J.T. Baker/Avantor Performance Materials (Deventer, The Netherlands) and VWR (Vienna, Austria). Sodium polyphosphate was obtained from Merck (Darmstadt, Germany; order no 1.065.291.000, Lot K46879329603; medium chain length n=25) and concentrations mentioned in this work are based on ortho‐phosphate units. Glucose dehydrogenase GDH‐105 was obtained from Codexis (Redwood City, CA, USA). All other chemicals were obtained from Sigma–Aldrich/Fluka or Carl Roth and used without further purification. Enzymes were prepared as described in the supporting information. Small scale reactions were performed on a Thermomixer comfort (Eppendorf) in 1.5 mL‐polypropylene tubes (Eppendorf, Hamburg, Germany). Flash column chromatography was performed on silica gel 0.035–0.070 mm, 60 Å (product no. 240360300; Acros Organics, Geel, Belgium). 1H and 13C NMR spectra were recorded on a Bruker AVANCE III 300 spectrometer (1H: 300.36 MHz; 13C: 75.53 MHz) and chemical shifts are referenced to residual protonated solvent signals as internal standard.
Preparative scale synthesis of piperonal 2 a
To MOPS buffer pH 7.70 (5.0 mL, 400 mm, 100 mm final concentration in reaction) in a 50 mL‐polypropylene tube (CELLSTAR®, Greiner Bio‐One GmbH, Kremsmünster, Austria) was added water (10.83 mL), piperonylic acid 1 a (83.1 mg, 500 μmol, 2.00 mL of a 250 mm stock solution in 250 mm NaOH), NADP+ (8.5 mg, 10.0 μmol), ATP (12.3 mg, 20.0 μmol), MgCl2 (133.3 mg, 1.40 mmol), β‐d‐glucose (541 mg, 3.00 mmol), sodium polyphosphate (288 mg, 2.80 mmol based on ortho‐phosphate units) and bromothymol blue solution (50 μL, 8.0 mm in 20 mm potassium phosphate buffer pH 7.50). The reaction was started by adding sequentially GDH‐105 (8.0 μL, 8 U, 1000 U mL−1 in 20 mm in potassium phosphate buffer pH 7.50), NiCAR (429 μL, 1.50 mg), MrPPK (151 μL, 1.00 mg), SmPPK (116 μL, 0.40 mg), EcPPase (18.5 μL, 0.20 mg) and then gently stirred with a floating stirring bar under temperature control at 30 °C. Through the stepwise addition of NaOH (900 μL in total, 1.8 mmol, 2.0 m in water), the pH was kept between 7.0 and 7.5, as visualized by the bromothymol blue color. After 24 h, the conversion had reached more than 90 % as checked by rp‐HPLC analysis and the product was extracted with ethyl acetate (3×20 mL). The combined extracts were dried over sodium sulfate, then concentrated under reduced pressure and finally separated using flash chromatography [12.0 g silica gel, 18×1.3 cm column size, cyclohexane/ethyl acetate=3:1 (v/v)]. All fractions containing the product 2 a (R f=0.51; cyclohexane/ethyl acetate 3:1 (v/v)) were pooled and concentrated under reduced pressure. The yield was 57.0 mg (76 %) of 2 a as colorless solid. M.p. 37–38 °C (commercial 2 a, m.p. 38 °C). 1H‐NMR (300 MHz, CDCl3): δ=6.08 (2 H, s, ‐CH2‐), 6.93 (1 H, d, 3 J(H,H)=7.9 Hz, C3‐H), 7.33 (1 H, d, 4 J(H,H)=1.3 Hz, C6‐H), 7.41 ppm (1 H, dd, 3 J(H,H)=7.9 Hz, 4 J(H,H)=1.3 Hz, C4‐H). 13C‐NMR (75 MHz, CDCl3): δ=102.3, 107.1, 108.5, 128.8, 132.1, 148.9, 153.3, 190.4 ppm.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work has been supported by the Federal Ministry of Science, Research and Economy (BMWFW), the Federal Ministry of Traffic, Innovation and Technology (bmvit), the Styrian Business Promotion Agency SFG, the Standortagentur Tirol, the Government of Lower Austria and Business Agency Vienna through the COMET‐Funding Program managed by the Austrian Research Promotion Agency FFG. A.S. received funding from the Austrian Research Promotion Agency FFG (FEM‐Tech stipend, grant no. 870897). We thank Jun. Prof. Jennifer N. Andexer for generously providing the genes coding for polyphosphate kinases and Prof. Bernd Nidetzky for pyrophosphatase.
G. A. Strohmeier, I. C. Eiteljörg, A. Schwarz, M. Winkler, Chem. Eur. J. 2019, 25, 6119.
References
- 1. Seyden-Penne J., Reductions by the Alumino- and Borohydrides in Organic Synthesis m, 2nd ed., Wiley-VCH, Weinheim, 1997. [Google Scholar]
- 2. Tojo G., Fernández M. I., Oxidation of Alcohols to Aldehydes and Ketones: A Guide to Current Common Practice, Springer, New York, 2006. [Google Scholar]
- 3.
- 3a. Qu G., Guo J., Yang D., Sun Z., Green Chem. 2018, 20, 777–792; [Google Scholar]
- 3b. Winkler M., Curr. Opin. Chem. Biol. 2018, 43, 23–29; [DOI] [PubMed] [Google Scholar]
- 3c. Lamm A. S., Venkitasubramanian P., Rosazza J. P. N., in Science of Synthesis: Biocatalysis in Organic Synthesis Vol. 2 (Eds.: K. Faber, W.-D. Fessner, N. J. Turner), Thieme, 2015, pp. 459–478; [Google Scholar]
- 3d. Napora-Wijata K., Strohmeier G. A., Winkler M., Biotechnol. J. 2014, 9, 822–843. [DOI] [PubMed] [Google Scholar]
- 4. Gross G. G., Eur. J. Biochem. 1972, 31, 585–592. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Stolterfoht H., Schwendenwein D., Sensen C. W., Rudroff F., Winkler M., J. Biotechnol. 2017, 257, 222–232; [DOI] [PubMed] [Google Scholar]
- 5b. Venkitasubramanian P., Daniels L., Rosazza J. P. N., J. Biol. Chem. 2007, 282, 478–485. [DOI] [PubMed] [Google Scholar]
- 6. Andexer J. N., Richter M., ChemBioChem 2015, 16, 380–386. [DOI] [PubMed] [Google Scholar]
- 7. Kunjapur A. M., Prather K. L. J., Appl. Environ. Microbiol. 2015, 81, 1892–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Venkitasubramanian P., Daniels L., Das S., Lamm A. S., Rosazza J. P. N., Enzyme Microb. Technol. 2008, 42, 130–137. [DOI] [PubMed] [Google Scholar]
- 9.Recent applications for aldehyde and amine formation:
- 9a. Ramsden J. I., Heath R. S., Derrington S. R., Montgomery S. L., Mangas-Sanchez J., Mulholland K. R., Turner N. J., J. Am. Chem. Soc. 2019, 141, 1201–1206; [DOI] [PubMed] [Google Scholar]
- 9b. Thompson M. P., Derrington S. R., Heath R. S., Porter J. L., Mangas-Sanchez J., Devine P. N., Truppo M. D., Turner N. J., Tetrahedron 2019, 75, 327–334. [Google Scholar]
- 10. Kornberg A., Rao N. N., Ault-Riché D., Annu. Rev. Biochem. 1999, 68, 89–125. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Khusnutdinova A. N., Flick R., Popovic A., Brown G., Tchigvintsev A., Nocek B., Correia K., Joo J. C., Mahadevan R., Yakunin A. F., Biotechnol. J. 2017, 12, 1600751; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Wang J., Zheng C., Zhang T., Liu Y., Cheng Z., Liu D., Ying H., Niu H., Biotechnol. Lett. 2017, 39, 1875–1881; [DOI] [PubMed] [Google Scholar]
- 11c. Liu S., Li Y., Zhu J., Process Biochem. 2016, 51, 1458–1463; [Google Scholar]
- 11d. Achbergerová L., Nahálka J., Microb. Cell Fact. 2011, 10, 63; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11e. Shiba T., Itoh H., Kameda A., Kobayashi K., Kawazoe Y., Noguchi T., J. Bacteriol. 2005, 187, 1859–1865; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11f. Kornberg A., J. Bacteriol. 1995, 177, 491–496; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11g. Murata K., Uchida T., Kato J., Chibata I., Agric. Biol. Chem. 1988, 52, 1471–1477. [Google Scholar]
- 12.
- 12a. Parnell A. E., Mordhorst S., Kemper F., Giurrandino M., Prince J. P., Schwarzer N. J., Hofer A., Wohlwend D., Jessen H. J., Gerhardt S., Einsle O., Oyston P. C. F., Andexer J. N., Roach P. L., Proc. Natl. Acad. Sci. USA 2018, 115, 3350–3355; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Kulmer S. T., Gutmann A., Lemmerer M., Nidetzky B., Adv. Synth. Catal. 2017, 359, 292–301. [Google Scholar]
- 13. Mordhorst S., Siegrist J., Müller M., Richter M., Andexer J. N., Angew. Chem. Int. Ed. 2017, 56, 4037–4041; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4095–4099. [Google Scholar]
- 14. Schmelz S., Naismith J. H., Curr. Opin. Struct. Biol. 2009, 19, 666–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Motomura K., Hirota R., Okada M., Ikeda T., Ishida T., Kuroda A., Appl. Environ. Microbiol. 2014, 80, 2602–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Finnigan W., Thomas A., Cromar H., Gough B., Snajdrova R., Adams J. P., Littlechild J. A., Harmer N. J., ChemCatChem 2017, 9, 1005–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schwendenwein D., Fiume G., Weber H., Rudroff F., Winkler M., Adv. Synth. Catal. 2016, 358, 3414–3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kunjapur A. M., Cervantes B., Prather K. L. J., Biochem. Eng. J. 2016, 109, 19–27. [Google Scholar]
- 19. He A., Li T., Daniels L., Fotheringham I., Rosazza J. P. N., Appl. Environ. Microbiol. 2004, 70, 1874–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mordhorst S., Singh J., Mohr M., Hinkelmann R., Keppler M., Jessen H. J., Andexer J., ChemBioChem 2019, DOI: 10.1002/cbic.201800704. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary