

Abstract
A new method for the one‐step C−H amination of xanthene and thioxanthene with sulfonamides is reported, without the need for any metal catalyst. A benzoquinone was employed as a hydride (or two‐electron and one‐proton) acceptor. Moreover, a previously unknown and uncatalyzed reaction between iminoiodanes and xanthene, thioxanthene and dihydroacridines (9,10‐dihydro‐9‐heteroanthracenes or dihydroheteroanthracenes) is disclosed. The reactions proceed through hydride transfer from the heteroarene substrate to the iminoiodane or benzoquinone, followed by conjugate addition of the sulfonamide to the oxidized heteroaromatic compounds. These findings may have important mechanistic implications for metal‐catalyzed C−H amination processes involving nitrene transfer from iminoiodanes to dihydroheteroanthracenes. Due to the weak C−H bond, xanthene is an often‐employed substrate in mechanistic studies of C−H amination reactions, which are generally proposed to proceed via metal‐catalyzed nitrene insertion, especially for reactions involving nitrene or imido complexes that are less reactive (i.e., less strongly oxidizing). However, these substrates clearly undergo non‐catalyzed (proton‐coupled) redox coupling with amines, thus providing alternative pathways to the widely assumed metal‐catalyzed pathways.
Keywords: benzoquinone, C−H amination, dihydroheteroanthracene, hydride transfer, iminoiodane
Introduction
The development of new synthetic methods for the synthesis of (secondary) amines is a constantly evolving field, due to the ever increasing demand for nitrogen containing compounds in for example, pharmaceuticals and agrochemicals.1 Direct (sp3) C−H amination via metal‐nitrene intermediates has received increasing attention in the last two decades, as no pre‐functionalization of the hydrocarbon substrates is required.2, 3, 4, 5 Key developments are the use of activated6, 7, 8 and non‐activated organic azides,9, 10, 11, 12, 13, 14, 15 Haloamine‐T16, 17 and (in situ generated) iminoiodanes (PhI=NR) as nitrene precursors (Figure 1).18, 19, 20, 21, 22, 23 Transition‐metal complexes have proven to be excellent catalysts for these amination reactions, and the commonly accepted mechanism comprises the formation of a reactive metal‐nitrene intermediate, followed by stepwise hydrogen atom abstraction and radical recombination or concerted insertion of the nitrene into the C−H bond.3, 5, 24 In addition, organocatalysts that are also capable of nitrene transfer have been reported.25, 26
Figure 1.
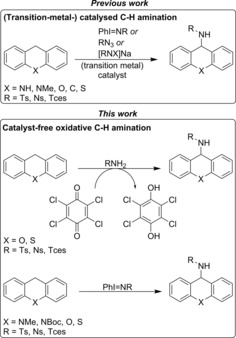
Comparison between previously reported (transition‐metal‐) catalyzed amination of C−H bonds and the catalyst‐free protocols presented in this work.
Our group is interested in the formation and characterization of new metal–nitrene complexes from known nitrene precursors and ideally directly from primary amines. As others, we adopted the reasoning that successful nitrene transfer could be dictated by the relative bond dissociation free energy (BDFE) of the C−H bond, with a lower BDFE expectedly resulting in faster nitrene insertion.25, 27, 28, 29, 30, 31, 32, 33 Dihydroheteroanthracenes (xanthene, thioxanthene, and dihydroacridine derivatives) have low C−H bond dissociation energies (BDE) in the range of 74–81 kcal mol−1.34 Therefore, dihydroheteroanthracenes are often assumed to be suitable model substrates to test for basic C−H amination activity, even for relatively non‐reactive nitrene intermediates.35 Especially xanthene is a commonly used substrate to investigate reaction kinetics of such reactions.28, 29, 30, 31, 33
In the course of our investigations, we initially reasoned in a similar manner. However, much to our surprise, we observed that sulfonamides are able to react with xanthene and thioxanthene in the presence of a benzoquinone derivative as a sacrificial oxidant and base, without the need for a (transition‐metal) catalyst. Even more interestingly, we also observed that dihydroheteroanthracenes react with commonly used iminoiodanes to afford the corresponding amination product in the absence of any catalyst (Figure 1). To the best of our knowledge, this background reaction has not been reported in literature. In this contribution, we disclose the details of catalyst‐free amination reactions of dihydroheteroanthracenes. The obtained insights are of considerable interest for researchers interested in (transition‐metal‐) catalyzed nitrene transfer, considering that we describe hitherto unknown, uncatalyzed background reactions and report a new mechanism for amination of dihydroheteroanthracenes that is very different from the generally accepted (metal) catalyzed nitrene transfer processes.
Results and Discussion
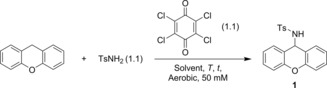
During our efforts to develop new (transition‐) metal catalyzed sp3 C−H amination strategies directly from amines, we stumbled across the uncatalyzed amination of xanthene with p‐toluenesulfonamide (TsNH2) in the presence of tetrachloro‐p‐benzoquinone (chloranil) as an oxidant. We decided to optimize the reaction conditions of this reaction (Table 1) to shed new light on this unexpected reaction. The C−H aminated product 1 was obtained in 22–48 % yield after 20 hours at 30 °C in solvents most commonly used in nitrene transfer reactions (entries 1–4). Decreasing the reaction time to 5 hours resulted in a lower yield, whereas increasing the reaction temperature to 60 °C afforded 1 in 43 % yield in benzene (entries 5 and 6). Performing the reaction at 60 °C for a longer time (20 hours) in benzene led to the formation of 1 in 72 % or 83 % in the presence and absence of light, respectively (entries 7 and 8). The conditions in entry 8 proved to be the optimal reaction conditions. For practical purposes we employed the conditions in entry 7 for further screening (vide infra).36 Dilution of the total concentration from 50 to 25 mm, increasing the amount of chloranil or performing the reaction under an argon atmosphere did not improve the yield (see Table S1 in the Supporting Information). It is worth mentioning that the reaction can be performed without drying the solvent and that the only by‐product is xanthone (3–7 %).37
Table 1.
Optimization of the reaction conditions for the amination of xanthene with TsNH2 and chloranil.
| ||||
|---|---|---|---|---|
| Entry | Solvent | T [oC] | t [h] | Yield [%][a] |
| 1 | C6H6 | 30 | 20 | 26 |
| 2 | PhCH3 | 30 | 20 | 22 |
| 3 | MeCN | 30 | 20 | 23 |
| 4 | CH2Cl2 | 30 | 20 | 48 |
| 5 | C6H6 | 30 | 5 | 18 |
| 6 | C6H6 | 60 | 5 | 43 |
| 7 | C6H6 | 60 | 20 | 72 |
| 8 [b] | C6H6 | 60 | 20 | 83 |
[a] Based on 1H NMR integration by using 1,3,5‐tris‐(tert‐butyl)benzene as the internal standard. [b] Performed in the absence of light.
With the optimized conditions in hand, we screened various p‐benzoquinone derivatives for their reactivity in the amination of xanthene by TsNH2 (Scheme 1). The mildly oxidizing parent p‐benzoquinone did not lead to conversion of xanthene. However, the use of 2‐chloro‐p‐benzoquinone, 2,6‐dichloro‐p‐benzoquinone, chloranil or 2,3‐dichloro‐5,6‐dicyano‐p‐benzoquinone (DDQ) afforded 1 in 17, 34, 72 and 81 %, respectively. The trends in the yield of 1 nicely correlate with the reported 1 e− and 2 e−/1 H+ reduction potentials of the corresponding benzoquinones.38 The more oxidizing quinones lead to higher yields, therefore indicating that oxidation of one of the substrates is involved in the reaction mechanism. DDQ, the strongest oxidant employed, is capable of oxidizing 1 to the corresponding imine, which was detected as a side product (10 % yield). Moreover, when using this oxidant, 9 % of xanthone was formed. Other quinones showed the same correlation between yield and redox potential, but afforded larger amounts of (unidentified) side products (Table S2 in the Supporting Information).
Scheme 1.
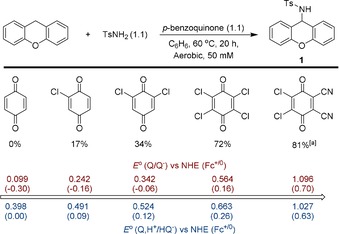
Screening of various p‐benzoquinones for the synthesis of 1 from xanthene and TsNH2. Yields based on 1H NMR integration by using 1,3,5‐tris‐(tert‐butyl)benzene as the internal standard. [a] 10 % oxidation of 1 to the imine and 9 % xanthone observed. Potentials for the 1 e− (Q/Q−) and 2 e−/1 H+ (Q,H+/HQ−) couples versus NHE (NHE=normal hydrogen electrode) in water.38 Potentials versus Fc+/0 are estimated by a correction of −0.40 V versus NHE.39
A small, but representative substrate scope of different sulfonamides and dihydroheteroanthracenes was explored, as shown in Scheme 2. TsNH2 and 2,2,2‐trichloroethoxysulfonamide (TcesNH2) form 1 (72 %) and 2 (67 %) in comparable yields. However, the use of p‐nitrobenzenesulfonamide (NsNH2) yields only 15 % of 3 with a considerable amount of xanthone. This is most likely caused by the reduced nucleophilicity of NsNH2 compared to TsNH2 and TcesNH2, thus leading to higher yields of reaction products stemming from reaction with H2O (which is present in the solvent, vide infra). Performing the reaction in thoroughly dried benzene led to reduced xanthone formation (2 %), but does not increase the yield of 3. 9,10‐Dihydroanthracene did not afford the desired product 4, but generated anthracene in 22 % yield. Similarly, various 9,10‐dihydroacridine derivatives did not afford the desired products 5‐H, 5‐Me or 5‐Boc; 9,10‐dihydroacridine was quantitatively converted to acridine, N‐methyldihydroacridine was oxidized to unidentified products and N‐Boc‐9,10‐dihydroacridine was not converted. However, thioxanthene reacted in a similar manner as xanthene, producing product 6 in 37 % yield. Other hydrocarbon substrates with weak C−H bonds, for example ethylbenzene and cyclohexadiene, did not afford the desired aminated products (Table S3 in the Supporting Information).
Scheme 2.
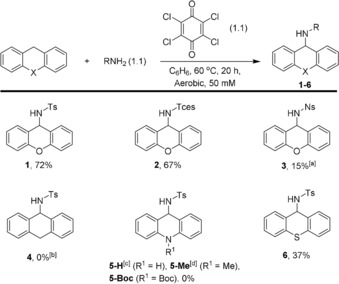
Substrate scope with different sulfonamides and dihydroheteroanthracenes. R=Ts, Tces or Ns. Yields based on 1H NMR integration by using 1,3,5‐tris‐(tert‐butyl)benzene or 1,3,5‐trimethoxybenzene as the internal standard. [a] 28 % xanthone formation and 2 % xanthone formation in anhydrous solvent. [b] 22 % anthracene formation. [c] Quantitative conversion to acridine. [d] Quantitative conversion of substrate, no conversion of TsNH2.
Tetrachloro‐p‐hydroquinone formation was observed by 1H NMR spectroscopy for all reactions in which the dihydroheteroanthracene was converted. We also obtained crystals of tetrachloro‐p‐hydroquinone from the reaction mixture, and single‐crystal X‐ray diffraction analysis of these crystals confirmed the formation of the aromatic hydroquinone (Figure S25 in the Supporting Information). This, in combination with the observed oxidation of the dihydroheteroanthracenes and the absence of reaction between chloranil and TsNH2, proves that chloranil is acting as a proton and electron acceptor in the oxidative amination process. Moreover, in the absence of a sulfonamide, the only formed product is xanthone.37 To rule out the possible involvement of xanthone in the formation of 1 through nucleophilic reaction of the sulfonamide with the carbonyl moiety, we also tested xanthone as the substrate under the same reaction conditions. However, neither in the presence or absence of chloranil we observed formation of any product. We therefore rule out that xanthone is involved in the formation of the C−H aminated product 1. Moreover, we exclude the involvement of oxygen‐sensitive free‐radical species that result from single‐electron transfer, as the conversion and yield do not change in the presence or absence of oxygen (vide supra).
Surprised by these results, we wondered if these reactions could perhaps proceed through two‐electron oxidation and deprotonation of the dihydroheteroanthracenes followed by nucleophilic attack of the sulfonamide. We therefore investigated the redox potentials of the dihydroheteroanthracenes with cyclic voltammetry and differential pulse voltammetry (Figures S26 and S27 in the Supporting Information). All observed electrochemical oxidations were found to be irreversible, with the potentials varying from +0.32 V (dihydroacridine) to +1.28 V (dihydroanthracene) versus Fc+/0, see Figure 2. These potentials seem to be too high for outer‐sphere single‐electron transfer from the dihydroheteroanthracene to chloranil in the absence of a proton donor (E o 1/2 = −0.43 V versus Fc+/0 in CH2Cl2, see Figure S29 in the Supporting Information).
Figure 2.
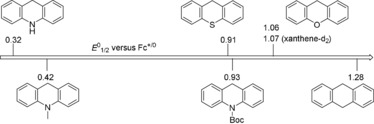
E o 1/2 versus Fc+/0 in CH2Cl2 for various dihydroheteroanthracenes, obtained from DPV measurements in a three‐electrode cell with a glassy carbon working electrode, Pt auxiliary electrode and leak‐free Ag/AgCl 3.0 m KCl reference electrode.
However, as an alternative, the reaction could proceed through a hydride‐transfer step from the dihydroheteroanthracenes to chloranil, followed by conjugate addition of the sulfonamide to the cationic heteroanthracenium derivative. The two‐electron oxidation and deprotonation of xanthene, thioxanthene and N‐methyldihydroacridine has previously been studied by electrochemical or combined radiolytic and photochemical oxidation and hydride transfer to triphenylmethyl perchlorate (Scheme 3 a,b).40, 41, 42
Scheme 3.
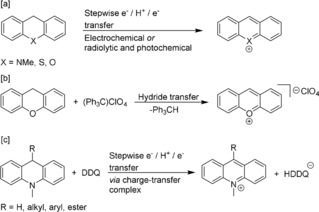
[a] Electrochemical or combined radiolytic and photochemical stepwise two‐electron and one‐proton transfer from dihydroheteroanthracenes.40, 41 [b] Chemical hydride transfer of xanthene to (Ph3C)ClO4.42 [c] Stepwise two‐electron and one‐proton transfer from 9‐substituted 10‐methyl‐9,10‐dihydroacridines to DDQ.43
The hydride‐transfer reactions were shown to proceed through sequential electron‐proton‐electron transfer to form the heteroanthracenium ions. Moreover, hydride transfer from different 9‐substituted 10‐methyl‐9,10‐dihydroacridines (0.41<E o ox<0.52 V versus Fc+/0) to DDQ (E o 1/2=+0.70 V versus Fc+/0) has been studied in detail and was shown to proceed via a charge‐transfer complex, followed by stepwise electron‐proton‐electron transfer within the charge‐transfer complex (Scheme 3 c).43 Interestingly, it has been shown that the initial electron‐transfer step is in equilibrium and the proton transfer is rate determining. Moreover, the separately prepared xanthylium ion (obtained by hydride transfer of xanthene to triphenylmethyl perchlorate) was recently indeed shown to react with 4‐(cyclohepta‐2,4,5‐trien‐1‐yl)aniline and pyrimidin‐2‐amine to produce aminated products.42, 44 These data and observations suggest that the reactions in Scheme 2 might indeed also proceed through initial hydride transfer.
To test our hypothesis that chloranil might act as a hydride acceptor, we performed an intermolecular kinetic isotope competition study with xanthene, xanthene‐d 2, TsNH2 and chloranil. This led to a kinetic isotope effect (KIE) of 2.6, clearly indicating that a proton or hydride‐transfer step is involved in the rate‐determining step or in a pre‐equilibrium leading to the rate‐determining step. To obtain more insight into this step, we monitored the reaction under standard conditions in the absence of sulfonamide. We were unable to detect the formation of the xanthylium cation by 1H NMR spectroscopy. However, under aerobic conditions larger amounts of oxidized products (xanthone and xanthydrol) were observed than those obtained when the reaction was performed under argon. This indicates that hydride transfer from xanthene to chloranil is in a thermodynamically unfavorable equilibrium with the xanthylium ion and the 2,3,5,6‐tetrachloro‐4‐hydroxyphenoxyl anion. However, in the presence of a nucleophile (H2O or sulfonamide) the xanthylium cation can react to form the aminated or hydrated products (vide supra).
Therefore, we propose that chloranil acts as a hydride (or one‐proton and two‐electron) acceptor for dihydroheteroanthracene oxidation to form the heteroanthracenium ion and the 2,3,5,6‐tetrachloro‐4‐hydroxy‐phenoxyl anion (Figure 3). Subsequent conjugate addition of the sulfonamide to the heteroanthracenium ion leads to product formation. The products 4 and 5 are not formed, probably because the oxidized substrates from anthracene and acridine are not electrophilic enough to react with the weakly nucleophilic TsNH2. A control reaction with acridine as the substrate indeed confirmed this (Scheme S4 in the Supporting Information).
Figure 3.
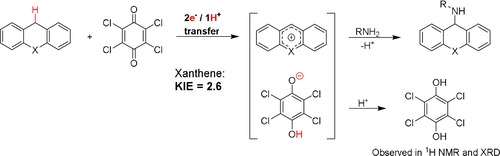
Proposed reaction mechanism for the amination reaction with chloranil and sulfonamides. X=O or S, R=Ts, Tces or Ns. Intermolecular KIE (2.6) for the formation of 1.
Intrigued by the results obtained by using the combination of chloranil as the hydride (or two‐electron and one‐proton) acceptor and sulfonamides as the nitrogen source in the amination of xanthene and thioxanthene, we wondered whether the oxidant and nitrogen‐group donor could also be combined in a single reagent. We therefore decided to investigate whether the use of hypervalent iodine reagents, such as PhINTs, could be used as amide‐delivering oxidants in the absence of a transition‐metal catalyst. PhINTs is a common nitrene precursor for C−H amination and alkene aziridination reactions in combination with various transition‐metal catalysts, but the free iminoiodane is considered to be non‐reactive towards hydrocarbons.45 The hypervalent iodine oxidant can be synthesized separately or formed in situ from TsNH2 and di‐(pivaloyloxy)iodobenzene [PhI(OPiv)2]. To the best of our knowledge, there is no report on the direct (non‐catalyzed) use of PhINTs for net C−H amination. However, the use of an in situ generated hypervalent iodine reagent from PhI and mCPBA has been recently reported for a dehydrogenative C−H imination reaction with benzylic anilines.46
However, much to our surprise the reaction between xanthene and PhINTs cleanly afforded 1 in 62 % yield after 60 minutes and 68 % yield (49 % yield with in situ generated PhINTs) after 20 hours (see Figure 4 and Scheme 4). Performing the reaction at lower temperatures afforded 1 in 23 % yield after 3.5 hours at 27 °C, or 21 % after 1.5 hours at 40 °C. At 60 °C, the reaction almost reached full conversion after 1 hour. Using PhINTces and PhINNs afforded 2 and 3 after 20 hours in comparable yields as observed for the amination reaction described in Scheme 2. Dihydroanthracene did not afford 4 and reactions with ethylbenzene and 1,4‐cyclohexadiene also did not lead to the desired products. However, 5‐Me (46 %), 5‐Boc (40 %), and 6 (47 %) were obtained from the corresponding dihydroacridines and thioxanthene after 1 hour. Interestingly, the yield of 5‐Me is higher after 1 hour than after 20 hours, suggesting that the product is over‐oxidized under these reaction conditions.47 Consistent with these observations, hydride abstraction reactions from substrates similar to 5‐Me have been described (vide supra and Scheme 3 c). In general (except for 5‐Me) the highest yields were obtained with pre‐formed iminoiodane.
Figure 4.
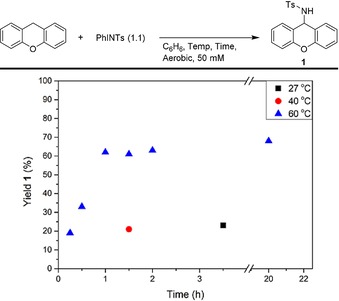
Formation of 1 from xanthene and PhINTs at different temperatures and reaction times. Yields based on 1H NMR integration by using 1,3,5‐tris‐(tert‐butyl)benzene as the internal standard.
Scheme 4.
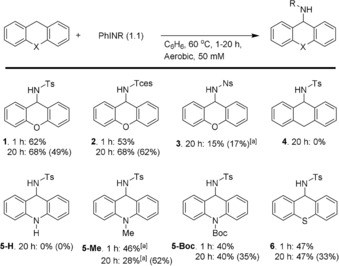
C−H Amination of dihydroheteroanthracenes by PhINR (R=Ts, Tces, Ns). Yields in parentheses concern reactions using in situ generated PhINR, generated from RNH2 and PhI(OPiv)2 in the presence of MgO. Yields based on 1H NMR integration by using 1,3,5‐tris‐(tert‐butyl)benzene or 1,3,5‐trimethoxybenzene as an internal standard. [a] Unidentified by‐products formed.
Mechanistic insight was obtained from an intermolecular competition experiment between xanthene and xanthene‐d 2 in the reaction with PhINTs, which gave a KIE of 2.1. Analogous to the 2 e−/1 H+ transfer described above, this suggests that proton or hydride transfer is involved in, or before, the rate‐determining step. Moreover, the reaction of dihydroacridine with PhINTs, which did not afford the desired product 5‐H, gave quantitative conversion to acridine, iodobenzene, and TsNH2. Hydride (or two‐electron and one‐proton) transfer from the substrate to the iminoiodane is thus a feasible process. For acridine the reaction stops at this point, whereas for the substrates that afford the respective desired product this step is followed by conjugate addition of the sulfonamide to the oxidized substrate.
Based on the above combined data, we propose the reaction mechanism shown in Scheme 5. The mechanism is supported by DFT calculations at the B3LYP/def2‐TZVP/disp3 level of theory with implicit solvation in benzene (COSMO), a method that typically affords reliable energies for charged intermediates.38 Endergonic hydride transfer from xanthene to the nitrogen atom in PhINTs (ΔG o=+12.6 kcal mol−1 at 298 K) affords intermediate B through transition state TS (ΔG ≠ = +17.7 kcal mol−1). Simultaneously with the hydride transfer, heterolytic cleavage of the I−N bond is observed, as the bond elongates from 1.999 Å (A) to 2.447 Å (TS). The I−N bond is completely cleaved in B, in which iodobenzene and the anionic tosylamide remain as a close‐contact pair. Slightly exergonic breaking of this close contact pair affords the negatively charged tosylamido and positively charged xanthylium ion (C, ΔG o=+10.1 kcal mol−1). Product D is formed by a virtually barrierless conjugate addition in an overall highly exergonic reaction (ΔG o=−62.1 kcal mol−1). The use of dihydroacridine as the substrate is believed to follow the same mechanism until intermediate C, after which the tosylamide deprotonates the cationic N‐protonated‐acridinium cation to afford acridine and TsNH2, as was experimentally observed.
Scheme 5.
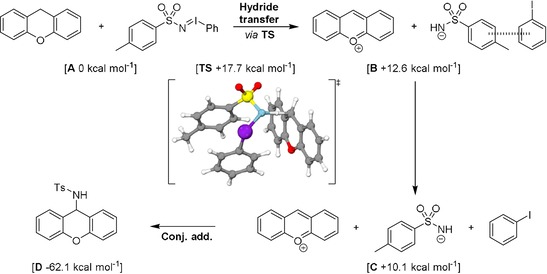
Proposed mechanism for the C−H amination of xanthene with PhINTs. Energies in ΔG o at 298 K calculated with DFT at the B3LYP/def2‐TZVP/disp3 m4‐grid/COSMO(benzene) level of theory. Graphical representation of TS generated with IboView. Grey=C, white=H, purple=I, yellow=S, red=O, blue=N.
Conclusions
To conclude, we have shown that xanthene and thioxanthene can be aminated at the bridgehead sp3 C−H position by using chloranil (or a related benzoquinone) as the oxidant and with sulfonamides as the nitrogen donor. The benzoquinone acts as a hydride (or two‐electron and one‐proton) acceptor and the amination step proceeds through conjugate addition of a sulfonamide to the formed heteroanthracenium ion. We have also demonstrated that often‐employed iminoiodanes can react in an uncatalyzed manner with xanthene, thioxanthene and dihydroacridines to afford the sp3 C−H aminated products. The key mechanistic step is a hydride transfer from the dihydroheteroanthracene to the iminoiodane, followed by conjugate addition of an anionic sulfonamido intermediate to the thus formed heteroanthracenium cation. This finding is relevant for the chemical community interested in (the mechanisms of) nitrene‐transfer catalysis, because it describes a previously unknown background reaction that may compete with the postulated catalytic cycles. We would therefore like to emphasize that this uncatalyzed process should be carefully considered when using xanthene‐like substrates in mechanistic studies of catalytic nitrene‐transfer reactions.
Experimental Section
General procedure for the oxidative amination with chloranil
A 4.0 mL vial was charged with TsNH2, NsNH2 or TcesNH2 (0.11 mmol, 1.1 equiv), the dihydroheteroanthracene (0.10 mmol, 1.0 equiv), chloranil (0.11 mmol, 1.1 equiv) and benzene (2.0 mL). The resulting suspension was stirred, with a closed cap, under aerobic conditions at 60 °C for 20 h. After cooling to room temperature and concentration under reduced pressure, the yield was determined by 1H NMR spectroscopy, using 1,3,5‐tris‐(tertbutyl)benzene or 1,3,5‐trimethoxybenzene as the internal standard.
General procedure for the oxidative amination with PhINR
A 4.0 mL vial was charged with the PhINTs, PhINNs or PhINTces (0.11 mmol, 1.1 equiv), the dihydroheteroanthracene (0.10 mmol, 1.0 equiv) and benzene (2.0 mL). The resulting suspension was stirred, with a closed cap, under aerobic conditions at 60 °C for 20 h (or 1 hour, if specified). After cooling to room temperature and concentration under reduced pressure, the yield was determined by 1H NMR spectroscopy, using 1,3,5‐tris‐(tertbutyl)benzene or 1,3,5‐trimethoxybenzene as the internal standard.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support from The Netherlands Organization for Scientific Research (NWO TOP‐Grant 716.015.001) is gratefully acknowledged. Ed Zuidinga is thanked for MS measurements.
N. P. van Leest, L. Grooten, J. I. van der Vlugt, B. de Bruin, Chem. Eur. J. 2019, 25, 5987.
References
- 1. Hili R., Yudin A. K., Nat. Chem. Biol. 2006, 2, 284–287. [DOI] [PubMed] [Google Scholar]
- 2. Hazelard D., Nocquet P.-A., Compain P., Org. Chem. Front. 2017, 4, 2500–2521. [Google Scholar]
- 3. Kuijpers P. F., van der Vlugt J. I., Schneider S., de Bruin B., Chem. Eur. J. 2017, 23, 13819–13829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chu J. C. K., Rovis T., Angew. Chem. Int. Ed. 2018, 57, 62–101; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 64–105. [Google Scholar]
- 5. Park Y., Kim Y., Chang S., Chem. Rev. 2017, 117, 9247–9301. [DOI] [PubMed] [Google Scholar]
- 6. Driver T. G., Org. Biomol. Chem. 2010, 8, 3831–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Intrieri D., Zardi P., Caselli A., Gallo E., Chem. Commun. 2014, 50, 11440–11453. [DOI] [PubMed] [Google Scholar]
- 8. Shin K., Kim H., Chang S., Acc. Chem. Res. 2015, 48, 1040–1052. [DOI] [PubMed] [Google Scholar]
- 9. Hennessy E. T., Betley T. A., Science 2013, 340, 591–595. [DOI] [PubMed] [Google Scholar]
- 10. Kuijpers P. F., Tiekink M. J., Breukelaar W. B., Broere D. L. J., van Leest N. P., van der Vlugt J. I., Reek J. N. H., de Bruin B., Chem. Eur. J. 2017, 23, 7945–7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bagh B., Broere D. L. J., Sinha V., Kuijpers P. F., van Leest N. P., de Bruin B., Demeshko S., Siegler M. A., van der Vlugt J. I., J. Am. Chem. Soc. 2017, 139, 5117–5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Broere D. L. J., de Bruin B., Reek J. N. H., Lutz M., Dechert S., van der Vlugt J. I., J. Am. Chem. Soc. 2014, 136, 11574–11577. [DOI] [PubMed] [Google Scholar]
- 13. Broere D. L. J., van Leest N. P., de Bruin B., Siegler M. A., van der Vlugt J. I., Inorg. Chem. 2016, 55, 8603–8611. [DOI] [PubMed] [Google Scholar]
- 14. Thacker N. C., Lin Z., Zhang T., Gilhula J. C., Abney C. W., Lin W., J. Am. Chem. Soc. 2016, 138, 3501–3509. [DOI] [PubMed] [Google Scholar]
- 15. Zardi P., Intrieti D., Caselli A., Gallo E., J. Organomet. Chem. 2012, 716, 269–274. [Google Scholar]
- 16. Albone D. P., Aujla P. S., Challenger S., Derrick A. M., J. Org. Chem. 1998, 63, 9569–9571. [Google Scholar]
- 17. Albone D. P., Challenger S., Derrick A. M., Fillery S. M., Irwin J. L., Parsons C. M., Takada H., Taylor P. C., Wilson D. J., Org. Biomol. Chem. 2005, 3, 107–111. [DOI] [PubMed] [Google Scholar]
- 18. Fiori K. W., Du Bois J., J. Am. Chem. Soc. 2007, 129, 562–568. [DOI] [PubMed] [Google Scholar]
- 19. Du Bois J., Org. Process Res. Dev. 2011, 15, 758–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fructos M. R., Trofimenko S., Díaz-Requejo M. M., Pérez P. J., J. Am. Chem. Soc. 2006, 128, 11784–11791. [DOI] [PubMed] [Google Scholar]
- 21. Li Z., Capretto D. A., Rahaman R., He C., Angew. Chem. Int. Ed. 2007, 46, 5184–5186; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 5276–5278. [Google Scholar]
- 22. Chang J. W. W., Chan P. W. H., Angew. Chem. Int. Ed. 2008, 47, 1138–1140; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 1154–1156. [Google Scholar]
- 23. Wang L., Agnew D. W., Yu X., Figueroa J. S., Cohen S. M., Angew. Chem. Int. Ed. 2018, 57, 511–515; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 520–524. [Google Scholar]
- 24. Goswami M., Lyaskovskyy V., Domingos S. R., Buma W. J., Woutersen S., Troeppner O., Ivanović-Burmazović I., Lu H., Cui X., Zhang X. P., Reijerse E. J., DeBeer S., van Schooneveld M. M., Pfaff F. F., Ray K., de Bruin B., J. Am. Chem. Soc. 2015, 137, 5468–5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Combee L. A., Raya B., Wang D., Hilinski M. K., Chem. Sci. 2018, 9, 935–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lamar A. A., Nicholas K. M., J. Org. Chem. 2010, 75, 7644–7650. [DOI] [PubMed] [Google Scholar]
- 27. Liu Y., Guan X., Wong E. L., Liu P., Huang J., Che C.-M., J. Am. Chem. Soc. 2013, 135, 7194–7204. [DOI] [PubMed] [Google Scholar]
- 28. Hong S., Lu X., Lee Y.-M., Seo M. S., Ohta T., Ogura T., Clémancey M., Maldivi P., Latour J.-M., Sarangi R., Nam W., J. Am. Chem. Soc. 2017, 139, 14372–14375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fujita D., Sugimoto H., Shiota Y., Morimoto Y., Yoshizawa K., Itoh S., Chem. Commun. 2017, 53, 4849–4852. [DOI] [PubMed] [Google Scholar]
- 30. Hong S., Sutherlin K. D., Vardhaman A. K., Yan J. J., Park S., Lee Y. M., Jang S., Lu X., Ohta T., Ogura T., Solomon M. E. L., Nam W., J. Am. Chem. Soc. 2017, 139, 8800–8803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Leung S. K.-Y., Tsui W.-M., Huang J.-S., Che C.-M., Liang J.-L., Zhu N., J. Am. Chem. Soc. 2005, 127, 16629–16640. [DOI] [PubMed] [Google Scholar]
- 32. Bryant J. R., Mayer J. M., J. Am. Chem. Soc. 2003, 125, 10351–10361. [DOI] [PubMed] [Google Scholar]
- 33. Liang J.-L., Yuan S.-X., Huang J.-S., Che C.-M., J. Org. Chem. 2004, 69, 3610–3619. [DOI] [PubMed] [Google Scholar]
- 34. Lou Y.-R., Comprehensive Handbook of Chemical Bond Energies, CRC, Boca Raton, 2007. [Google Scholar]
- 35.An often used argument is: if the nitrene intermediate does not react with xanthene, nitrene-insertion reactions with hydrocarbon substrates having stronger C−H bonds can likely be ruled out as well.
- 36.Considering that most laboratories usually perform reactions in the presence of light and that the difference in presence and absence of light is only small, we reasoned that the conditions in entry 7 are more widely applicable.
- 37.Xanthone is most likely formed in a side reaction involving the oxidized xanthene cation with water. See Figures S34 and S35 in the Supporting Information.
- 38. Huynh M. T., Anson C. W., Cavell A. C., Stahl S. S., Hammes-Schiffer S., J. Am. Chem. Soc. 2016, 138, 15903–15910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Connelly N. G., Geiger W. E., Chem. Rev. 1996, 96, 877–910. [DOI] [PubMed] [Google Scholar]
- 40. Marcinek A., Rogowski J., Adamus J., Gȩbicki J., Platz M. S., J. Phys. Chem. 1996, 100, 13539–13543. [Google Scholar]
- 41. Ben Salah N., Mhalla F. M., J. Electroanal. Chem. 2000, 485, 42–48. [Google Scholar]
- 42. Bonthrone W., Reid D. H., J. Chem. Soc. 1959, 2773–2779. [Google Scholar]
- 43. Fukuzumi S., Ohkubo K., Tokuda Y., Suenobu T., J. Am. Chem. Soc. 2000, 122, 4286–4294. [Google Scholar]
- 44. Yunnikova L. P., Esenbaeva V. V., Russ. J. Org. Chem. 2018, 54, 1018–1022. [Google Scholar]
- 45. Darses B., Rodrigues R., Neuville L., Mazurais M., Dauban P., Chem. Commun. 2017, 53, 493–508. [DOI] [PubMed] [Google Scholar]
- 46. Bose A., Maiti S., Sau S., Mal P., Chem. Commun. 2019, 55, 2066–2069. [DOI] [PubMed] [Google Scholar]
- 47.For this particular substrate, a number of side products are formed of which only the ketone could be unequivocally identified. After 1 hour 9 % ketone of 5-Me was observed, while after 20 hours 15 % ketone was detected (Figures S21 and S22 in the Supporting Information).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary