

Abstract
In this article, we expand upon the catalytic hydrothiolation of 1,3-dienes to afford either allylic or homoallylic sulfides with high regiocontrol. Mechanistic studies support a pathway where regioselectivity is dictated by the choice of counter-ion associated with the Rh-center. Non-coordinating counter-ions, such as SbF6−, allow for η4-diene coordination to Rh-complexes and result in allylic sulfides. In contrast, coordinating counter-ions, such as Cl−, favor neutral Rh-complexes where the diene binds η2 to afford homoallylic sulfides. We propose mechanisms that rationalize a fractional dependence on thiol for the 1,2-Markovnikov hydrothiolation while accounting for an inverse dependence on thiol in the 3,4-anti-Markovnikov pathway. Through the hydrothiolation of an essential oil (β-farnesene), we achieve the first enantioselective synthesis of (−)-agelasidine A.
Graphical Abstract
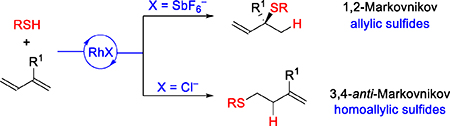
Introduction
Given the value of organosulfur compounds as metabolites and medicines,1 synthetic chemists strive to develop versatile methods for accessing these motifs.2 Both allylic and homoallylic sulfides, as well as their respective derivatives (e.g., sulfones and thioesters), comprise natural products and analogs with a wide range of bioactivities (Figure 1A).3 The hydrothiolation of olefins and dienes represents an atom-economical strategy4 for constructing C–S bonds.5,6 Despite its high atomeconomy, hydrothiolation remains an unexploited strategy for the synthesis of complex targets and further development is warranted. Breit demonstrated an enantioselective hydrothiolation of allenes to generate allylic sulfides via Rh-catalysis (Figure 1B).7a, 7b Using Au-catalysis, the He group achieved the hydrothiolation of 1,3-dienes to access allylic sulfides, with excellent 3,4-Markovnikov selectivity, albeit as racemic mixtures (Figure 1C).7c Our laboratory recently communicated the first enantioselective 1,2-Markovnikov hydrothiolation of 1,3-dienes to generate allylic sulfides (Figure 1C).8 Although hydrothiolations have been developed to access allylic sulfides, selective access to the homoallylic isomer has been elusive.
Figure 1.

Inspiration for the regiodivergent hydrothiolation of 1,3-dienes.
In this article, to expand the power of diene hydrothiolation, we focused on elucidating the mechanism for the 1,2-Markovnikov hydrothiolation. In theory, the addition of a thiol to an unsymmetrical diene (e.g., 2-phenyl-1,3-diene), can afford up to 11 isomers.9 Yet, the use of cationic Rh and a bidentate phosphine ligand afforded secondary and tertiary sulfide motifs with excellent regioselectivity and enantioselectivity. By studying the mechanism, we determine the fundamental steps that govern regiocontrol. Guided by these insights, we then focused on developing a complementary hydrothiolation to provide access to the homoallylic sulfide (Figure 1C). While regiodivergent hydrothiolation of dienes has not previously been reported, Hull demonstrated regiodivergent hydrothiolations of allylic amines by choice of ligand on Rh.10 Regiodivergent hydrosilylation of 1,3-dienes has been reported by Ritter through the use of an Fe versus Pt catalyst.11 In this study, we enable access to homoallylic sulfides by simply changing the counter ion that coordinates to Rh from non-coordinating (SbF6−) to coordinating (Cl−). The scope and mechanism of this new hydrothiolation of 1,3-dienes is presented. Within this article, we also showcase hydrothiolation in the first enantioselective synthesis of (−)-agelasidine A, a natural product that bears a chiral tertiary sulfide derived motif (Figure 1A).
Results and Discussion
In our previous studies, we observed that different 1,3-diene substitution patterns require the use of different ligand families for optimal results (Figure 2).8 By using this empirical guide, one can identify either the desired product or the commodity diene of choice to functionalize. For cyclic, 1-substituted, 1,2-disubstituted, and 2,3-disubstituted dienes, we found that the BINAP ligand family is best for furnishing enantioenriched allylic sulfides. Whereas 2-substituted dienes require the use of the Josiphos ligand family. The Garphos ligand scaffold provides good yields and enantioselectivities for 1,3-butadiene. In an effort to better understand the catalyst design and its effects on the hydrofunctionalizations of dienes, we interrogated the hydrothiolation mechanism to elucidate the factors that affect selectivity.
Figure 2.

Empirical guide for optimal ligand choice for 1,2-Markovnikov hydrothiolation. Results previously published.8
Mechanism of 1,2-Markovnikov hydrothiolation of 1,3-dienes
On the basis of both literature precedence and the following mechanistic studies, we propose the 1,2-Markovnikov hydrothiolation mechanism depicted in Figure 3. Ligand exchange between 1,5-cyclooctadiene (cod) with a bidentate phosphine ligand, thiol 1, and diene 2 generates intermediate I. In the turnover limiting step, oxidative addition results in formation of a η4-diene coordinated Rh–H intermediate II.12 Subsequent 1,4-insertion of the diene into the Rh–H furnishes Rh-π-allyl intermediate III.13 Intermediate III undergoes reductive elimination to provide IV, where product 3 remains coordinated to Rh. Ligand exchange of product 3 with thiol 1 and diene 2 regenerates the Rh catalyst I.
Figure 3.

Proposed mechanism of 1,2-Markovnikov hydrothiolation of 1,3-dienes.
For the model system, we chose to study the mechanism using an achiral ligand, Xantphos, because we previously observed that it is an effective ligand for the transformation.8 This bidentate ligand bears a coordinating oxygen atom that can act as a hemilabile ligand.14 Our initial mechanistic studies used thiophenol (1a) and myrcene (2a) to explore the kinetic profile of the transformation. We found a first-order dependence on the catalyst and a zeroth-order dependence on diene 2a, which is consistent with a mechanism where the Rh complex I is saturated with diene 2, or diene 2 coordination occurs after the turnover limiting step (Figure 3). We found that thiophenol (1a) can participate in two reaction pathways: desired hydrothiolation (path a) or dimerization (path b).15 Thiophenol (1a) dimerization increases proportionally with its concentration. When adding bis(4-methoxyphenyl) disulfide to a mixture of thiophenol with myrcene under the standard conditions, we observe crossover products, which suggests that thiol dimerization (path b) is reversible (see SI, Figure S4). In accordance with these competing pathways, we observe a fractional-order dependence (0.4) on thiophenol (1a).
We also performed deuterium-labeling experiments to further probe the mechanism (Figure 4). When subjecting deuterated thiophenol (d-1a) and myrcene (2a) to the standard conditions, we found that the recovered diene starting material 2a exhibits no deuterium incorporation (eq 1). This lack of scrambling supports our proposal that Rh–H insertion is an irreversible step in the catalytic cycle. However, we observed deuterium scrambling in the allylic sulfide product d-3aa. To examine the origin of this deuterium incorporation, we subjected a non-deuterated product 3aa to a mixture of deuterated thiophenol (d-1a), Rh(cod)2SbF6, and Xantphos (eq 2). We detected similar deuterium incorporation only in the terminal olefin moiety of d-3aa′. Collectively, these results suggest that deuterium scrambling in product 3aa occurs from a pathway external to the catalytic cycle. We hypothesize that intermediate IV can undergo oxidative addition to an equivalent of thiol to form complex V (Figure 3). Subsequent reversible Rh–H insertion into the terminal olefin results in the deuterium scrambling observed in d-3aa.
Figure 4.

Deuterium-labeling studies for the 1,2-Markovnikov hydrothiolation of 1,3-dienes.
Next, we studied key steps of the hydrothiolation by NMR spectroscopy. First, we monitored a mixture of thiophenol (1a), Rh(cod)2SbF6 (10 mol%), and Xantphos (10 mol%) in DCE-d4 by 1H NMR analysis. A resonance at −13.5 ppm was observed in less than ten minutes at room temperature in the 1H NMR spectrum, which is consistent with previously reported values for Rh−H complexes.16 This observation suggests that a Rh−H is rapidly generated from the catalyst precursor Rh(cod)2SbF6 in the presence of Xantphos and thiophenol (1a). While observation of a Rh−hydride does not necessitate its involvement in catalysis, we found that this hydride species is consumed when treated with an equivalent of diene (myrcene, 2a). In this stoichiometric experiment, we observe formation of a new Rh-complex with non-equivalent phosphine resonances in the 31P NMR spectrum at −40 °C [a pair of doublet of doublet signals (δ = 26.6 ppm, JRh−p = 174 Hz, JP−P = 8 Hz; δ = 16.0 ppm, JRh−p = 115 Hz, JP−P = 8 Hz)]. When we subjected the product 3aa to a mixture of Rh(cod)2SbF6 and Xantphos in DCE-d4, we observed the same species by 31P NMR spectroscopy. Based on these results, we label Rh intermediate V as the resting state in the catalytic cycle (Figure 3).
To investigate the turnover limiting step, we carried out several kinetic experiments. First, a H/D kinetic isotope effect (KIE) experiment with thiophenol (1a) and deuterated thiophenol (d-1a) was performed. The initial rate constants were determined in parallel, and we observed a primary KIE (kH/kD = 2.8, Figure 5). Second, a Hammett plot was constructed, using various p-substituted thiophenols, to determine if there was a rate dependence on the electronic character of the thiol 1 partner (Figure 6). A relatively small ρ value (−0.22 ± 0.02) is observed with more electron-rich thiophenols undergoing hydrothiolation slightly faster. We hypothesize that the thiol initially coordinates to Rh to provide a transient species (see I, Figure 3), which then undergoes insertion of the Rh into the S–H bond to form the Rh–H species (see II). Electron-rich thiols can accelerate this process by stabilizing positive charge build up on the Rh-center during the transition state for oxidative addition.
Figure 5.

KIE from two parallel reactions using initial rates (1,2-Markovnikov).
Figure 6.

Hammett plot (log k/kH = mσ+ + b (m = −0.22 ± 0.02; b = 0.03 ± 0.01).
Based on these mechanistic studies, we reason that the elementary steps from intermediate II to IV account for the observed regioselectivity (Figure 3). Hydrometallation occurs with the bulky Rh-center preferentially adding to the less sterically encumbered terminal position (C4). This net 1,4-insertion ultimately yields the Rh-πc-allyl intermediate III. Reductive elimination of III at the more-substituted position to form the branched product is preferred, which is consistent with other Rh-catalyzed alkyne, allene, and diene hydrofunctionalizations.17
When intermediate II bears a chiral thiolate ligand, the configuration appears to have little/no influence on the stereochemical outcome. Our initial report included an example of a chiral cysteine-derived thiol undergoing hydrothiolation to selectively give one diastereomer, depending on which enantiomer of the bisphosphine ligand was used (Figure 7, entry A).8 To elaborate on this observation, we investigated chiral secondary thiols, where the chiral information is closer to the Rh-center. Hydrothiolation occurs with high reactivity (3cb and 3db, 83–92% yield, entry B and C), regioselectivity (>20:1 rr), and diastereoselectivity (>20:1 dr) when using chiral secondary thiols (1c and 1d). These results demonstrate complete catalyst control when forging the C–S bond. Thus, chiral secondary thiols can be transformed to sulfides in a diastereodivergent fashion. With a better understanding of the 1,2-Markovnikov hydrothiolation mechanism, we set out to apply this asymmetric hydrothiolation methodology to the total synthesis of a natural product.
Figure 7.

Catalyst-controlled diastereoselective 1,2-Markovnikov hydrothiolation.
Total synthesis of (−)-agelasidine A
(−)-Agelasidine A (4), an antifungal and antimicrobial agent isolated from marine sponges of the genus Agelas,3b has previously been synthesized as a racemate from farnesol. Ichikawa reported two different methods for the installation of the key tertiary sulfide moiety of (±)-agelasidine A; a [2,3]-sigmatropic rearrangement or hetero-Claisen rearrangement have been used to construct the C–S bond and access (±)-4 in three and eight steps, respectively.18
We focused on intercepting an enantioenriched variant of sulfone 5, which was previously elaborated to (±)-4 in Ichikawa’s synthesis (Figure 8). To achieve this goal, we focused on coupling β-farnesene (2c), which is a renewable feedstock found in many essential oils,19 and 2-mercaptoethyl acetate (1e). Referencing our hydrothiolation guide (Figure 2), the Josiphos ligand scaffold is the most promising choice for achieving high reactivity and selectivity because 2c is a 2-substituted 1,3-diene. In line with this guide, we found that β-farnesene (2c) can be coupled with 1e to give the tertiary sulfide 3ec in 78% yield with high enantioselectivity (>99:1 er) when using a Josiphos ligand (R = Cy, Figure 2). Various methods have been developed to chemoselectively oxidize sulfides to the corresponding sulfones.20 We observed high reactivity (77% yield) when using catalytic (NH4)6Mo7O244H2O and H2O2 to oxidize sulfide 3ec to sulfone 5.20i Following Ichikawa’s report, we found that enantioenriched sulfone 5 could be transformed to (−)-agelasidine A (4, 67% yield) in the presence of excess guanidine.18c Collectively, our approach requires only three steps from commercially available β-farnesene (2c) to afford enantioenriched (−)-4 in 40% overall yield. We anticipate that this methodology will be applicable to other natural products and synthetic targets bearing C–S bonds.21
Figure 8.

Enantioselective synthesis of (−)-agelasidine A. Reagents and conditions: (a) Rh(cod)2SbF6 (5 mol%), Josiphos (5 mol%, R = Cy, Figure 2), DCE, 30 °C, 15 h. (b) (NH,)6Mo7O24–4H2O (10 mol%), H2O2, MeOH, rt, 4 h. (c) NaH, EtOH, NH2C(=NH)NH2 · HCl, 1,4-dioxane, rt, 12 h.
Development of 3,4-anti-Markovnikov hydrothiolation of 1,3-dienes
Based on the 1,2-Markovnikov mechanism depicted in Figure 3, we reasoned that it would be possible to access other hydrothiolation regioisomers by tuning the ligands22 and/or counter-ions on Rh. Previous reports have demonstrated that coordination modes of 1,3-dienes to a metal center can switch the observed regioselectivity of transition-metal catalyzed hydrofunctionalizations. For example, Ritter and coworkers found that η4-diene coordination provides 1,4-addition products,11a whereas η2-diene coordination gives 3,4-anti-Markovnikov hydrosilylation products.11b We envisioned using this concept to design a regiodivergent hydrothiolation of 1,3-dienes by switching from η4- to η2-diene binding. As shown in Figure 9, cationic Rh-precatalysts prefer η4-diene binding due to the presence of two open coordination sites. In contrast, a neutral Rh species would prefer η2-diene binding due to the availability of only one coordination site. Subsequent 1,2-insertion would lead to an intermediate B in which Rh adds to the less sterically hindered terminal position. Reductive elimination of this Rh-alkyl species B would yield homoallylic sulfides 6.
Figure 9.

Proposed counter-ion controlled regiodivergent hydrothiolations
To begin our study, we chose isoprene (2d), a petroleum feedstock, and thiophenol (1a) as model substrates (Figure 10). Since 6ad is achiral, we focused on identifying an achiral ligand for the 3,4-anti-Markovnikov hydrothiolation. In the early stages of 1,2-Markovnikov hydrothiolation development, we found that Xantphos is a viable choice for the ligand. Indeed, with a combination of Rh(cod)2SbF6 and Xantphos, the expected tertiary allylic sulfide 3ad could be synthesized in 83% yield and >20:1 rr (entry 1). In stark contrast, when using the neutral [Rh(cod)Cl]2 as a catalyst precursor, the homoallylic sulfide 6ad was obtained in 74% yield and 1:>20 rr (entry 2). These results suggest that regioselectivity is controlled by the counter-ion on Rh (SbF6− vs Cl−), which is in line with our proposal (η4- vs η2-diene coordination).23 Switching the counterion to I− or MeO− lowers the reactivity (18% and 49% yield, respectively, entries 3 and 4) while maintaining high regioselectivity (1:>20 rr). With further tuning, we found that [Rh(C2H4)2Cl]2 and bidentate phosphine ligand dppe furnishes 6ad in 94% yield with 1:>20 rr in 3 hours (entry 5). Furthermore, with this catalyst, we can lower the loading to 0.1 mol% and synthesize 6ad on gram-scale (1.3 g) in 74% yield with 1:>20 rr.
Figure 10.

Rh-precatalyst leads to a switch in regioselectivity
a Reaction conditions: 1a (0.1 mmol), 2d (0.5 mmol), [Rh] (5 mol%), ligand (5 mol%), DCE (0.2 mL), 15 h. Isolated yield. Regioselectivity ratio (rr) is the ratio of f 3ad to 6ad, which is determined by 1H NMR analysis of reaction mixture. b Using dppe as ligand, 3 h.
With these optimal conditions, we examined the coupling of fifteen different thiols with isoprene (2d) to generate the corresponding homoallylic sulfides (Table 1A). High reactivity (6bd–6sd, 54–95% yield) and regioselectivity (>20:1 rr) are obtained with both aromatic and aliphatic thiol partners. This method is also compatible with heteroarene (6nd, 6od), imide (6sd), amide (6bd), and ester (6bd) functionalities.
Table 1.
3,4-anti-Markovnikov Hydrothiolation of 1,3-Dienes.a
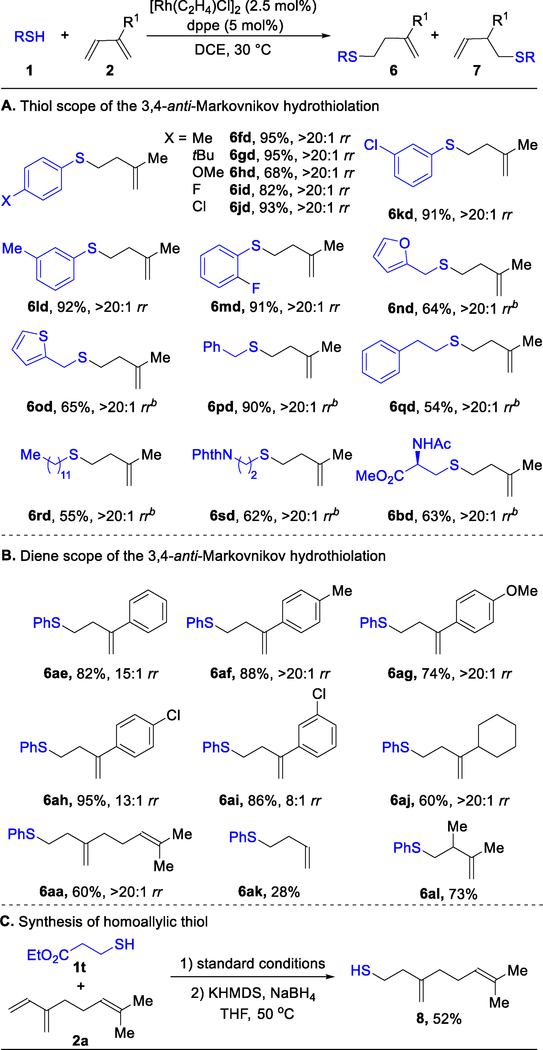 |
Reaction conditions: 1 (0.2 mmol), 2 (0.4 mmol), [Rh(C2H4)2Cl]2 (2.5 mol%), dppe (5 mol%), DCE (0.4 mL), 3 h. Isolated yield. Regioselectivity ratio (rr) is the ratio of 6 to 7, which is determined by 1H NMR analysis of reaction mixture. b Using [Rh(cod)Cl]2 (2.5 mol%), Xantphos (5 mol%) and 3,5-dimethylbenzoic acid (50 mol%), 12 h.
Next, we investigated the scope of the 1,3-diene partner in the 3,4-anti-Markovnikov hydrothiolation using thiophenol (1a) as a model thiol partner (Table 1B). Both aromatic and aliphatic 2-substituted 1,3-dienes are converted to the sulfide products (6aa–6aj) in high yields (60–95%). The electronics of the 2-aryl ring on the 1,3-diene has a noticeable effect on the regioselectivity of the transformation. Electron-rich 1,3-dienes (6af, 6ag, >20:1 rr) yield higher regioselectivity than electron-poor 1,3-dienes (6ah, 6ai, 13:1 and 8:1 rr, respectively). 3,4-Anti-Markovnikov hydrothiolation of butadiene (2k) provides the corresponding homoallylic sulfide (6ak) in 28% yield. A 2,3-disubstituted diene 2l also transforms well to the homoallylic sulfide 6al (73% yield). Moreover, myrcene (2a) could be converted to the corresponding homoallylic thiol 8 via a formal addition of H2S, which consisted of 3,4-anti-Markovnikov hydrothiolation followed by deprotection (Table 1C).24
Mechanism of 3,4-anti-Markovnikov hydrothiolation of 1,3-dienes
Based on kinetic studies and NMR experiments, we propose the mechanism shown in Figure 11A for the 3,4-anti-Markovnikov hydrothiolation of 1,3-dienes. Oxidative addition of thiol 1 with Rh provides intermediate II’. Two equivalents of thiol 1 can then associate to furnish the resting state III’. A similar off-cycle resting state with an Ir(III)–H complex bearing a six-membered ring formed from two hydrogen bonds to ethanol has been reported.25 Intermediate III’ displays a hydride resonance at −15.8 ppm with symmetrical phosphines [doublet (δ = 52.2 ppm, JRh–p = 94 Hz)]. Moreover, a negative half-order dependence on thiol 1 supports the proposed side pathway.
Figure 11.

Mechanistic studies of 3,4-anti-Markovnikov hydrothiolation of 1,3-dienes.
In contrast to the η4-diene binding exhibited in II (Figure 3), we propose that the less substituted olefin coordinates to intermediate II’ to form η2-diene coordinated Rh complex IV’. This diene binding mode is due to the presence of one coordination site and could be the foundation for the switch in regioselectivity. Insertion of the 1,3-diene into the Rh−H bond then provides the less sterically encumbered intermediate V’. The observed primary KIE (kH/kD = 1.9, Figure 11B) supports that either oxidative addition to the S–H bond or diene insertion into the Rh−H is turnover limiting. Given the first-order rate dependence on diene 2e and catalyst (Figure 11A), as well as the results of deuterium incorporation into the allylic position of d-6ae (Figure 11C), we postulate diene insertion is the turnover limiting step. Rh-π-allyl V’ then undergoes reductive elimination to yield intermediate VI’, which can perform a ligand exchange of product 6 with thiol 1 to regenerate Rh catalyst I’.
Conclusion
Hydrothiolation of 1,3-dienes provides an efficient and straightforward way to construct primary, secondary and tertiary C–S centers. A concise total synthesis of (−)-agelasidine A (4) exemplifies the facile use of this methodology in a synthetic setting. Allylic and homoallylic sulfides can be synthesized in a regiodivergent manner with the choice of the Rh-precatalyst. Mechanistic investigations shed light on the origin of the high regioselectivity observed for both hydrothiolations. Future efforts will focus on the development of a unified mechanistic approach for accessing different regioisomers of 1,3-diene hydrofunctionalizations.
Supplementary Material
ACKNOWLEDGMENT
Funding provided by UC Irvine, the National Institutes of Health (R35GM127071) and the National Science Foundation (CHE-1465263). Shao-Zhen Nie thanks Liaocheng University for a scholarship. Faben A. Cruz is grateful for an NSF Graduate Research Fellowship. We thank Dr. Peter Dornan (Amgen) and Jan Riedel for helpful discussion.
Footnotes
The authors declare no competing financial interests.
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures and spectral data for all new compounds (PDF)
REFERENCES
- (1).For reviews, see:Ilardi EA; Vitaku E; Njardarson JT Data-Mining for Sulfur and Fluorine: An Evaluation of Pharmaceuticals To Reveal Opportunities for Drug Design and Discovery. J. Med. Chem 2014, 57, 2832–2842.Feng M; Tang B; Liang SH; Jiang X Sulfur Containing Scaffolds in Drugs: Synthesis and Application in Medicinal Chemistry. Curr. Top. Med. Chem 2016, 16, 1200–1216.Scott KA; Njardarson JT Analysis of US FDA-Approved Drugs Containing Sulfur Atoms. Top. Curr. Chem 2018, 376, 5.
- (2).For reviews on C–S bond formation, see:Kondo T; Mitsudo T-A Metal-Catalyzed Carbon-Sulfur Bond Formation. Chem. Rev 2000, 100, 3205–3220.Arisawa M; Yamaguchi M Transition-metal-catalyzed synthesis of organosulfur compounds. Pure Appl. Chem 2008, 80, 993–1003.Chauhan P; Mahajan S; Enders D Organocatalytic Carbon-Sulfur Bond-Forming Reactions. Chem. Rev 2014, 114, 8807–8864.Shen C; Zhang P; Sun Q; Bai S; Hor TSA; Liu X Recent advances in C–S bond formation via C–H bond functionalization and decarboxylation. Chem. Soc. Rev 2015, 44, 291–314.Yu J-S; Huang H-M; Ding P-G; Hu X-S; Zhou F; Zhou J Catalytic Enantioselective Construction of Sulfur-Containing Tetrasubstituted Carbon Stereocenters. ACS Catal. 2016, 6, 5319–5344.Qiao A; Jiang X Recent developments in sulfur-carbon bond formation reaction involving thiosulfates. Org. Biomol. Chem 2017, 15, 1942–1946. For a review on enzymatic C–S bond formation, see:Dunbar KL; Scharf DH; Litomska A; Hertweck C Enzymatic Carbon-Sulfur Bond Formation in Natural Product Biosynthesis. Chem. Rev 2017, 117, 5521–5577.
- (3).(a) Bernström K; Hammarström S A novel leukotriene formed by transpeptidation of leukotriene E. Biochem. Biophys. Res. Commun 1982, 109, 800–804. [DOI] [PubMed] [Google Scholar]; (b) Nakamura H; Wu H; Kobayashi J; Ohizumi Y Agelasidine-A, a novel sesquiterpene possessing antispasmodic activity from the okinawa sea sponge Agelassp. Tetrahedron Lett. 1983, 24, 4105–4108. [Google Scholar]; (c) Sumiyoshi H; Wargovich MJ Chemoprevention of 1,2-Dimethylhydrazine-induced Colon Cancer in Mice by Naturally Occurring Organosulfur Compounds. Cancer Res. 1990, 50, 5084–5087. [PubMed] [Google Scholar]; (d) Arora A; Siddiqui IA; Shukla Y Modulation of p53 in 7,12-dimethylbenz[a]anthracene–induced skin tumors by diallyl sulfide in Swiss albino mice. Mol. Cancer Ther 2004, 3, 1459–1466. [PubMed] [Google Scholar]; (e) Arunkumar A; Vijayababu MR; Venkataraman P; Senthilkumar K; Arunakaran J Chemoprevention of Rat Prostate Carcinogenesis by Diallyl Disulfide, an Organosulfur Compound of Garlic. Biol. Pharm. Bull 2006, 29, 375–379. [DOI] [PubMed] [Google Scholar]; (f) Taori K; Paul VJ; Luesch H Structure and Activity of Largazole, a Potent Antiproliferative Agent from the Floridian Marine Cyanobacterium Symploca sp. J. Am. Chem. Soc 2008, 130, 1806–1807. [DOI] [PubMed] [Google Scholar]
- (4).Trost BM The atom economy--a search for synthetic efficiency. Science 1991, 254, 1471–1477. [DOI] [PubMed] [Google Scholar]
- (5).For select hydrothiolations of alkenes, see:Cabrero-Antonino JR; LeyvaPerez A; Corma A Iron-Catalysed Markovnikov Hydrothiolation of Styrenes. Adv. Synth. Catal 2012, 354, 678–687.Tamai T; Ogawa A Regioselective Hydrothiolation of Alkenes Bearing Heteroatoms with Thiols Catalyzed by Palladium Diacetate. J. Org. Chem 2014, 79, 5028–5035.Tamai T; Fujiwara K; Higashimae S; Nomoto A; Ogawa A Gold-Catalyzed Anti-Markovnikov Selective Hydrothiolation of Unactivated Alkenes. Org. Lett 2016, 18, 2114–2117.Yi H; Song C; Li Y; Pao C-W; Lee J-F; Lei A Single-Electron Transfer between CuX2 and Thiols Determined by Extended X-Ray Absorption Fine Structure Analysis: Application in Markovnikov-Type Hydrothiolation of Styrenes. Chem. Eur. J 2016, 22, 18331–18334.Mosaferi E; Ripsman D; Stephan DW The air-stable carbocation salt [(MeOC6H4)CPh2][BF4] in Lewis acid catalyzed hydrothiolation of alkenes. Chem. Commun 2016, 52, 8291–8293.Teders M; Henkel C; Anhäuser L; Strieth-Kalthoff F; Gómez-Suárez A; Kleinmans R; Kahnt A; Rentmeister A; Guldi D; Glorius F The energy-transfer-enabled biocompatible disulfide–ene reaction. Nat. Chem 2018, 10, 981–988.Kristensen SK; Laursen SLR; Taarning E; Skrydstrup T Ex Situ Formation of Methanethiol: Application in the Gold(I)-Promoted Anti-Markovnikov Hydrothiolation of Olefins. Angew. Chem., Int. Ed 2018, 57, 13887–13891. For review, see:Castarlenas R; Giuseppe AD; Pérez-Torrente JJ; Oro LA The Emergence of Transition-Metal-Mediated Hydrothiolation of Unsaturated Carbon-Carbon Bonds: A Mechanistic Outlook. Angew. Chem., Int. Ed 2013, 52, 211–222.
- (6).For select hydrofunctionalizations of 1,3-dienes, see:Zbieg JR; Yamaguchi E; Mclnturff EL; Krische MJ Enantioselective C–H Crotylation of Primary Alcohols via Hydrohydroxyalkylation of Butadiene. Science 2012, 336, 324–327.Chen Q-A; Kim DK; Dong VM Regioselective Hydroacylation of 1,3-Dienes by Cobalt Catalysis. J. Am. Chem. Soc 2014, 136, 3772–3775.Saini V; O’Dair M; Sigman MS Synthesis of Highly Functionalized Tri- and Tetrasubstituted Alkenes via Pd-Catalyzed 1,2-Hydrovinylation of Terminal 1,3-Dienes. J. Am. Chem. Soc 2015, 137, 608–611.Marcum JS; Roberts CC; Manan RS; Cervarich TN; Meek SJ Chiral Pincer Carbodicarbene Ligands for Enantioselective Rhodium-Catalyzed Hydroarylation of Terminal and Internal 1,3-Dienes with Indoles. J. Am. Chem. Soc 2017, 139, 15580–15583.Jing SM; Balasanthiran V; Pagar V; Gallucci JC; RajanBabu TV Catalytic Enantioselective Hetero-dimerization of Acrylates and 1,3-Dienes. J. Am. Chem. Soc 2017, 139, 18034–18043.Yang X-H; Lu A; Dong VM Intermolecular Hydroamination of 1,3-Dienes To Generate Homoallylic Amines. J. Am. Chem. Soc 2017, 139, 14049–14052.Gui Y-Y; Hu N; Chen X-W; Liao L-L; Ju T; Ye JH; Zhang Z; Li J; Yu D-G Highly Regio- and Enantioselective Copper-Catalyzed Reductive Hydroxymethylation of Styrenes and 1,3-Dienes with CO2. J. Am. Chem. Soc 2017, 139, 17011–17014.Thullen SM; Rovis T A Mild Hydroaminoalkylation of Conjugated Dienes Using a Unified Cobalt and Photoredox Catalytic System. J. Am. Chem. Soc 2017, 139, 15504–15508.Adamson NJ; Hull E; Malcolmson SJ Enantioselective Intermolecular Addition of Aliphatic Amines to Acyclic Dienes with a Pd-PHOX Catalyst. J. Am. Chem. Soc 2017, 139, 7180–7183.Adamson NJ; Wilbur KCE; Malcolmson SJ Enantioselective Intermolecular Pd-Catalyzed Hydroalkylation of Acyclic 1,3-Dienes with Activated Pronucleophiles. J. Am. Chem. Soc 2018, 140, 2761–2764.Schmidt VA; Kennedy CR; Bezdek MJ; Chirik PJ Selective [1,4]-Hydrovinylation of 1,3-Dienes with Unactivated Olefins Enabled by Iron Diimine Catalysts. J. Am. Chem. Soc 2018, 140, 3443–3453Cheng L; Li M-M; Xiao L-J; Xie J-H; Zhou Q-L Nickel(0)-Catalyzed Hydroalkylation of 1,3-Dienes with Simple Ketones. J. Am. Chem. Soc 2018, 140, 11627–11630.
- (7).(a) Pritzius AB; Breit B Asymmetric Rhodium-Catalyzed Addition of Thiols to Allenes: Synthesis of Branched Allylic Thioethers and Sulfones. Angew. Chem., Int. Ed 2015, 54, 3121–3125. [DOI] [PubMed] [Google Scholar]; (b) Pritzius AB; Breit B Z-Selective Hydrothiolation of Racemic 1,3-Disubstituted Allenes: An Atom-Economic Rhodium-Catalyzed Dynamic Kinetic Resolution. Angew. Chem., Int. Ed 2015, 54, 15818–15822. [DOI] [PubMed] [Google Scholar]; (c) Brouwer C; Rahaman R; He C Gold(I)-Mediated Hydrothiolation of Conjugated Olefins. Synlett 2007, 2007, 1785–1789. [Google Scholar]
- (8).Yang X-H; Davison RT; Dong VM Catalytic Hydrothiolation: Regio- and Enantioselective Coupling of Thiols and Dienes. J. Am. Chem. Soc 2018, 140, 10443–10446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).See Supporting Information for details
- (10).Kennemur JL; Kortman GD; Hull KL Rhodium-Catalyzed Regiodivergent Hydrothiolation of Allyl Amines and Imines. J. Am. Chem. Soc 2016, 138, 11914–11919. [DOI] [PubMed] [Google Scholar]
- (11).(a) Wu JY; Stanzl BN; Ritter T A Strategy for the Synthesis of Well-Defined Iron Catalysts and Application to Regioselective Diene Hydrosilylation. J. Am. Chem. Soc 2010, 132, 13214–13216. [DOI] [PubMed] [Google Scholar]; (b) Parker SE; Borgel J; Ritter T 1,2-Selective Hydrosilylation of Conjugated Dienes. J. Am. Chem. Soc 2014, 136, 4857–4860. [DOI] [PubMed] [Google Scholar]
- (12).For examples of oxidative addition of thiols with Rh-catalysts, see:Ogawa A; Ikeda T; Kimura K; Hirao T Highly Regio- and Stereocontrolled Synthesis of Vinyl Sulfides via Transition-Metal-Catalyzed Hydrothiolation of Alkynes with Thiols. J. Am. Chem. Soc 1999, 121, 5108–5114.Shoai S; Bichler P; Kang B; Buckley H; Love JA Catalytic Alkyne Hydrothiolation with Alkanethiols using Wilkinson’s Catalyst. Organometallics 2007, 26, 5778–5781.Han L; Li Y; Liu T Theoretical investigation of the impact of ligands on the regiodivergent Rh-catalyzed hydrothiolation of allyl amines. Dalton Trans. 2018, 47, 150–158, and ref. 10
- (13).For the same sterically preferred 1,4-insertion into a Ir-H, see:Nguyen KD; Herkommer D; Krische MJ Enantioselective Formation of All-Carbon Quaternary Centers via C–H Functionalization of Methanol: Iridium-Catalyzed Diene Hydrohydroxymethylation. J. Am. Chem. Soc 2016, 138, 14210–14213.
- (14).Xantphos is a hemilabile ligand that can coordinate through the oxygen atom, see:Ren P; Pike SD; Pernik I; Weller AS; Willis MC Rh–POP Pincer Xantphos Complexes for C–S and C–H Activation. Implications for Carbothiolation Catalysis. Organometallics 2015, 34, 711–723.
- (15).For the formation of a disulfide under iron-catalyzed alkene hydrothiolation, see: ref. 5a
- (16).For the resonance for Rh–H, see:Di Giuseppe A; Castarlenas R; Perez-Torrente JJ; Crucianelli M; Polo V; Sancho R; Lahoz FJ; Oro LA Ligand-Controlled Regioselectivity in the Hydrothiolation of Alkynes by Rhodium N-Heterocyclic Carbene Catalysts. J. Am. Chem. Soc 2012, 134, 8171–8183; and ref. 10 and 12a.
- (17).For reviews, see:Koschker P; Breit B Branching Out: Rhodium-Catalyzed Allylation with Alkynes and Allenes. Acc. Chem. Res. 2016, 49, 1524–1536.Haydl AM; Breit B; Liang T; Krische MJ Alkynes as Electrophilic or Nucleophilic Allylmetal Precursors in Transition-Metal Catalysis. Angew. Chem., Int. Ed 2017, 56, 11312–11325. For select papers, see:Chen Q-A; Chen Z; Dong VM Rhodium-Catalyzed Enantioselective Hydroamination of Alkynes with Indolines. J. Am. Chem. Soc 2015, 137, 8392–8395.Yang X-H; Dong VM Rhodium-Catalyzed Hydrofunctionalization: Enantioselective Coupling of Indolines and 1,3-Dienes. J. Am. Chem. Soc 2017, 139, 1774–1777.
- (18).(a) Ichikawa Y First synthesis of agelasidine A. Tetrahedron Lett. 1988, 29, 4957–4958. [Google Scholar]; (b) Ichikawa Y; Kashiwagi T; Urano N A biomimetic synthesis of agelasidine A. J. Chem. Soc., Chem. Commun 1989, 987–988. [Google Scholar]; (c) Ichikawa Y; Kashiwagi T; Urano N Biomimetic synthesis of agelasidine A. J. Chem. Soc., Perkin Trans 1, 1992, 1497–1500. [Google Scholar]
- (19).Simionatto E; Porto C; Stuker CZ; Dalcol II; da Silva UF Chemical composition and antimicrobial activity of the essential oil from Aeolanthus suaveolens Mart. ex Spreng. Quim. Nova 2007, 30, 1923–1925. [Google Scholar]
- (20).(a) Priebe W; Grynkiewicz G A facile and selective oxidation of sulfides to sulfones. Tetrahedron Lett. 1991, 32, 7353–7356. [Google Scholar]; (b) Su W An efficient method for the oxidation of sulfides to sulfones. Tetrahedron Lett. 1994, 35, 4955–4958. [Google Scholar]; (c) Beckerbauer R; Smart BE Oxidation of Electron-Deficient Sulfides to Sulfones Using HOF·UH3CN. J. Org. Chem 1995, 60, 6186–6187. [Google Scholar]; (d) Barton DHR; Li W; Smith JA Binuclear manganese complexes as catalysts in the selective and efficient oxidation of sulfides to sulfones. Tetrahedron Lett. 1998, 39, 7055–7058. [Google Scholar]; (e) Choi S; Yang JD; Ji M; Choi H; Kee M; Ahn KH; Byeon SH; Baik W; Koo S Selective Oxidation of Allylic Sulfides by Hydrogen Peroxide with the T rirutile-type Solid Oxide Catalyst LiNbMoO6. J. Org. Chem 2001, 66, 8192–8198. [DOI] [PubMed] [Google Scholar]; (f) Xu L; Cheng J; Trudell ML Chromium(VI) Oxide Catalyzed Oxidation of Sulfides to Sulfones with Periodic Acid. J. Org. Chem 2003, 68, 5388–5391. [DOI] [PubMed] [Google Scholar]; (g) Shaabani A; Teimouri F; Lee DG Ion Exchange Catalysis in Oxidation of Organic Compounds with KMnO4. Synth. Commun 2003, 33, 1057–1065. [Google Scholar]; (h) Baciocchi E; Gerini MF; Lapi A Synthesis of Sulfoxides by the Hydrogen Peroxide Induced Oxidation of Sulfides Catalyzed by Iron Tetrakis(pentafluorophenyl)porphyrin: Scope and Chemoselectivity. J. Org. Chem 2004, 69, 3586–3589. [DOI] [PubMed] [Google Scholar]; (i) Jeyakumar K; Chakravarthy DR; Chand DK Simple and efficient method for the oxidation of sulfides to sulfones using hydrogen peroxide and a Mo(VI) based catalyst. Catal. Commun 2009, 10, 1948–1951. [Google Scholar]
- (21).Paige Stout E; Yu LC; Molinski TF Antifungal Diterpene Alkaloids from the Caribbean Sponge Agelas citrina: Unified Configurational Assignments of Agelasidines and Agelasines. Eur. J. Org. Chem 2012, 5131–5135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).For a regiodivergent hydrothiolation controlled by ligand choice, see: ref. 10 and ref. 16.
- (23).For coordinating ability of anions to transition-metals, see:Diaz-Torres R; Alvarez S Coordinating ability of anions and solvents towards transition metals and lanthanides. Dalton Trans. 2011, 40, 10742–10750.
- (24).Jin L; Wang J; Dong G Palladium-Catalyzed γ-C(sp3)–H Arylation of Thiols by a Detachable Protecting/Directing Group. Angew. Chem, Int. Ed 2018, 57, 12352–12355. [DOI] [PubMed] [Google Scholar]
- (25).Wang Y; Huang Z; Leng X; Zhu H; Liu G; Huang Z Transfer Hydrogenation of Alkenes Using Ethanol Catalyzed by a NCP Pincer Iridium Complex: Scope and Mechanism. J. Am. Chem. Soc 2018, 140, 4417–4429. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.