

Abstract
Despite recent advances in the use of immunotherapy, only a minority of small cell lung cancer (SCLC) patients respond to immune checkpoint blockade (ICB). Here, we show that targeting DNA damage response (DDR) proteins, poly ADP-ribose polymerase (PARP) and checkpoint kinase 1 (CHK1) significantly increased protein and surface expression of PD-L1. PARP or CHK1 inhibition remarkably potentiated the anti-tumor effect of PD-L1 blockade and augmented cytotoxic T-cell infiltration in multiple immunocompetent SCLC in vivo models. CD8 depletion reversed the anti-tumor effect demonstrating the role of CD8+ T-cells in combined DDR-PD-L1 blockade in SCLC. We further demonstrate that DDR inhibition activated the STING/TBK1/IRF3 innate immune pathway, leading to increased levels of chemokines such as CXCL10 and CCL5 that induced activation and function of cytotoxic T-lymphocytes. Knockdown of cGAS and STING successfully reversed the anti-tumor effect of combined inhibition DDR and PD-L1. Our results define previously unrecognized innate immune pathway-mediated immunomodulatory functions of DDR proteins and provide a rationale for combining PARP/CHK1 inhibitors and immunotherapies in SCLC.
Keywords: SCLC, DNA damage response, immune checkpoint blockade, STING pathway
Introduction
In the past several years, immunotherapies that harness or enhance a patient’s own immune system to target and kill cancer cells have been developed (1,2). The most commonly used therapeutic approaches use antibodies against inhibitory signaling molecules expressed on tumor and immune cells. Common targets include the immune checkpoints programmed death-1 (PD-1), PD-1 ligand (PD-L1), and cytotoxic T-lymphocyte associated protein 4 (CTLA4) (3). Since the first FDA approval of ipilimumab (anti-CTLA4) for melanoma, several such immune checkpoint blockade (ICB) agents have been approved for patients with advanced cancers, including non-small cell lung cancer (NSCLC) (4–6).
Although ICB agents are very promising, their activity varies across different cancer types, and there is increasing evidence of primary and adaptive resistance to ICB in multiple cancer types (7). Thus, efforts are underway to develop new therapeutic strategies with novel drug combinations to enhance the antitumor efficacy of ICB. High mutational burden has been reported as a potential predictor of response to immunotherapy (8). However, despite having one of the highest mutational burdens, small cell lung cancer (SCLC) is often accompanied by relatively high immunosuppression with low levels of T-cell infiltration and reduced antigen presentation (9–11). Consistent with this, clinical trials investigating PD-1 or PD-L1 blockade in patients with SCLC have shown low overall response rates (12,13).
Recent studies have demonstrated the potential of targeting the DNA damage response (DDR) pathway as a therapeutic strategy for SCLC (14–19), including drugs targeting poly (ADP) ribose polymerase (PARP) and checkpoint kinase 1 (CHK1). Several DDR inhibitors have been developed and are now either approved for the treatment of other cancers (e.g., PARP inhibitors) or are in clinical trials (20). Although best known for its functions in repairing DNA damage and controlling the cell cycle, the DDR pathway has also been shown to be involved in the antitumor immune response (21). For example, the PARP inhibitor olaparib was recently reported to show a synergistic effect with PD-L1 blockade in triple-negative breast cancer in preclinical models (22). However, little is currently known regarding the mechanistic interactions between DDR targeting and response to ICB, as well as the immune-activating properties of DDR targeting.
In the current study, we identified a role of DDR targeting through CHK1 and PARP inhibition in modulating T-cell action via regulation of the innate immune response pathway. We found that pharmacologic inhibition of CHK1 or PARP increased levels of tumor-infiltrating T-lymphocytes and synergized with anti-PD-L1 therapy in multiple SCLC models. Taken together, our results elucidate a mechanism of action for DDR inhibitors in antitumor immunity and suggest that treatment with DDR inhibitors may increase the effectiveness of ICB in SCLC patients.
Results:
DDR inhibition enhances PD-L1 expression in vitro and in vivo.
To determine the effect of DDR targeting on PD-L1 expression in SCLC models, we treated a panel of human SCLC cell lines with either a CHK1 inhibitor (prexasertib-300nM) or a PARP inhibitor (olaparib-1μM) for 72 hours and analyzed protein expression by Reverse Phase Protein Array (RPPA), immunoblot and flow cytometry. DDR targeting significantly increased the total level of PD-L1 protein in all cell lines tested with the greatest PD-L1 fold change (up to 5 fold) with prexasertib and an appreciable PD-L1 increase (up to 3 fold) with olaparib treatment as detected by RPPA (p<0.05, Fig 1A). The RPPA result was validated by immunoblot analysis, which further demonstrated PD-L1 upregulation upon DDR targeting in a time-dependent manner (Fig 1B). As PD-L1 must be expressed on the cell surface for successful targeting, we then assayed cell surface PD-L1 expression by fluorescence-activated cell sorting (FACS). Cell surface PD-L1 levels significantly increased following treatment with either prexasertib or olaparib in a time dependent manner in both human (Fig 1C) and murine (RPP/mTmG) (Fig 1D) SCLC cell lines (p<0.05 for all).
Figure 1: DDR inhibition enhances PD-L1 expression in vitro and in vivo and enhances antitumor response of anti-PD-L1 antibody in SCLC.
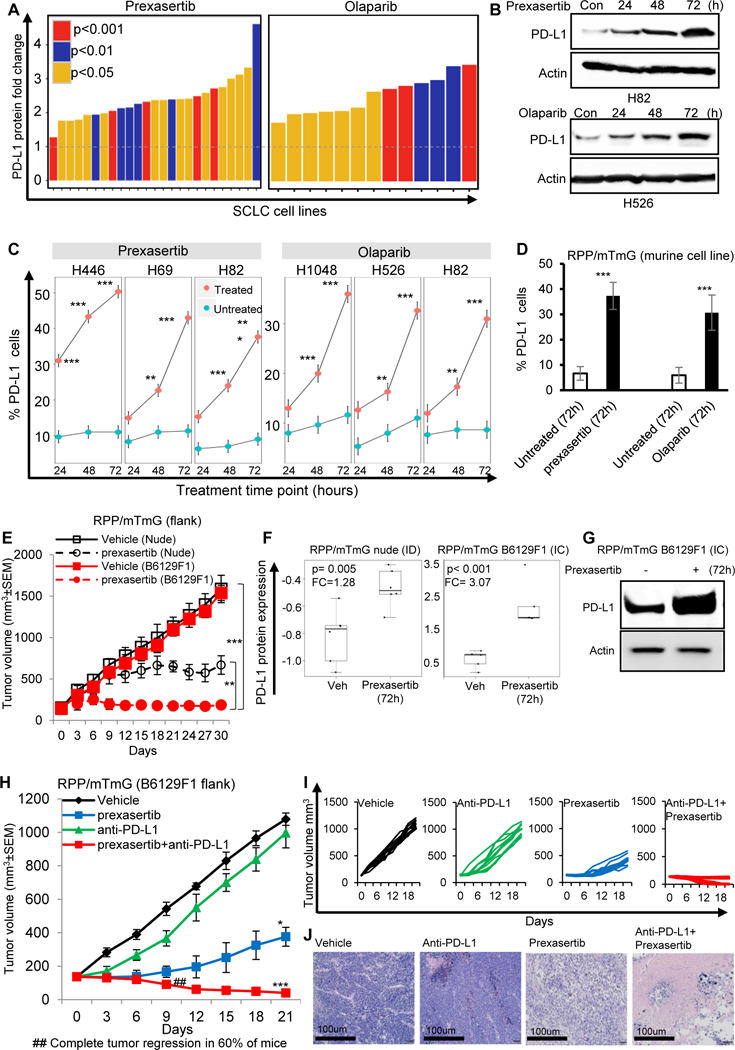
(A-D) DDR inhibition by targeting with small molecule inhibitors of CHK1 (prexasertib), and PARP (olaparib) enhances the PD-L1 protein expression as measured by RPPA (A) and immunoblot analysis (B); and increases PD-L1 surface expression, as measured by flow cytometry in human (C) and murine (D) SCLC cell lines.
(E) Tumor growth curve of immunocompetent B6129F1 (red lines) model and immunocompromised nude (black lines) SCLC RPP/mTmG (flank) models treated with CHK1 inhibitor, prexasertib (12mg/kg, BID, 2 out of 7 days) for 30 days. Prexasertib showed enhanced anti-tumor efficacy in immunocompetent model (TC=0.13; p<0.0001) as compared to immunocompromised model (T/C=0.47; p<0.01).
(F) Prexasertib treatment enhanced PD-L1 protein expression in SCLC tumors, with improved enhancement of PD-L1 expression in immunocompetent (IC) RPP/mTmG B6129F1 model (FC=3.07; p<0.001) as compared to immunodeficient (ID) RPP/mTmG nude model (FC=1.28; p=0.005).
(G) Immunoblot analysis confirms higher PD-L1 protein expression post-prexasertib treatment in immunocompetent (IC) RPP/mTmG B6129F1 model.
(H-I) Tumor growth curves +/− SEM (H) and for each RPP/mTmG B6129F1 mouse (I) from vehicle (black, n=10, median tumor volume=1110mm3), prexasertib alone (10mg/kg, 2 out of 7 days, BID) (blue, n=10, median tumor volume=410mm3), anti-PD-L1 alone (300μg, 1 out of 7 days, ip) (green, n=10, median tumor volume=1020mm3) and prexasertib+anti-PD-L1 (red, n=10, median tumor volume=40mm3).
(J) Representative H&E of the tumor sections from vehicle, prexasertib alone, anti-PD-Ll alone and combination treated group. Scale bar 100μm. All data represent at least three independent experiments. Means ± SEM are plotted. In all panels- *p<0.05, **p<0.01, ***p<0.0001, ns-not significant.
To confirm that PD-L1 upregulation is specifically due to inhibition of CHK1 or PARP and not an off target effect of the inhibitors, we knocked down (KD) CHEK1 or PARP in multiple SCLC cell lines. Consistent with pharmacologic inhibition, PD-L1 expression was substantially higher in CHEK1 knockdown (Fig S1A) or PARP knockdown (Fig S1B) cells compared with the scrambled control. PD-L1 upregulation upon CHK1 targeting was further confirmed by treating cells with a second CHK1 inhibitor (LY2603618) in SCLC cell lines (Fig S1C).
Olaparib and prexasertib-induced cytogenetic stress was evaluated by using a micronuclei (MN) assay and represented as MN-frequency, as demonstrated in Fig S1D and S1E. Treatment of SCLC cell lines H69, H446 and RPP/mTmG with prexasertib (1μM) or olaparib (10μM) for 24 hours led to significant (p<0.001) increase in MN frequency in treated samples. Representative micrographs using DAPI have been provided in Fig S1D and number MN/1000 cells (H69, H446 and RPP/mTmG) summarized in Fig S1E.
Given that PD-L1 expression was significantly enhanced following CHK1 inhibition (CHK1i), we hypothesized that CHK1i may induce an immune response in addition to direct anti-tumor effects in SCLC in vivo models and that CHK1i would be more effective in the immunocompetent setting. To test this possibility, we compared the effect of a low dose of prexasertib (12 mg/kg, BID, 2 out of 7 days, i.e., total 48 mg/kg/week), previously shown to cause growth delay but not tumor regression (18), on flank tumors grown in immunocompromised (nude) versus immune competent (B6129F1) mice. For these experiments, we used murine RPP/mTmG cells derived from a genetically engineered SCLC mouse with conditional loss of Trp53, p130, and Rb1 (RPP) (23,24). The prexasertib-induced delay in tumor growth in the immune competent (B6129F1) model was significantly greater as compared to the immune compromised (nude) model (p<0.001), demonstrating the efficacy of CHK1 targeting in the context of an intact immune system (Fig 1E). Prexasertib treatment induced PD-L1 protein expression in both the immune deficient (ID) and immune competent (IC) in vivo model. However a greater degree of PD-L1 upregulation was seen in the IC model (FC=3.07) as compared to ID model (FC= 1.28) (Fig. 1F). The enhancement of PD-L1 expression in the IC model was further confirmed by immunoblot (Fig. 1G).
Next we sought to determine whether CHK1i may induce changes in tumor infiltrating immune cells in the lung microenvironment. We treated an immunocompetent model of SCLC, 4 months after Ad-CMV Cre intubation, bearing appreciable spontaneous RPP lung tumors (at least 50 tumors/lung), (23,24), with prexasertib at a dose that was previously shown to reduce tumor growth (10 mg/kg, BID) (18). As predicted, prexasertib treatment increased PD-L1 protein expression in this model in a time-dependent manner (Fig S1F). At day 7, prexasertib-treated lung tumors had significantly more CD3+ total T cells (Fig S1G) and CD8+ cytotoxic T cells (Fig S1H), but decreased CD4+ helper T cells (Fig S1I), PD1+/TIM3+ exhausted T cells (Fig S1J), and CD62L+ naive T cells (Fig S1K) with a corresponding enhancement of CD44+ memory/effector T cell (Fig S1L) infiltration as compared to vehicle treated tumors. The induction of PD-L1 expression, rapid tumor regression, and intratumoral immune infiltration following CHK1 targeting support a direct role for DDR modulation in regulating the immune microenvironment in these SCLC models.
CHK1i augments anti-PD-L1 antibody-induced antitumor immunity.
As CHK1i alone is not sufficient to eradicate tumors despite reduced tumor growth, increased T-cell infiltration, and abrogated T-cell exhaustion in vivo, we next evaluated the ability of CHK1i to sensitize tumors to PD-L1 blockade. We treated B6129F1 immunocompetent flank RPP/mTmG tumor bearing mice with prexasertib (10mg/kg, 2/7days, BID, Day 1 and 2, i.e., total 40mg/kg/week) and/or anti-PD-L1 (300μg, 1/7days, Day 3) for 3 weeks (n = 10 per group) (Fig S2A). Mice treated with anti-PD-L1 alone showed no anti-tumor response in this model and were sacrificed due to excessive tumor burden within three weeks. However, within 1 week, remarkable tumor regression was observed in the combination treated group (Fig 1H, 1I and 1J).
Of the ten mice treated with the combination of prexasertib and anti-PD-Ll, six had a complete response (100% reduction).
Tumors were resected at Day 21 (when available) and analyzed by multicolor flow cytometry for the changes in tumor infiltrating lymphocytes (TILs) (Fig S2B). Combination treatment with CHKli and PD-L1 blockade significantly increased CD3+ total T-cell infiltration (Fig 2A and 2B) (p<0.0001), CD8+ cytotoxic T-cell infiltration (Fig 2C and 2D) (p<0.0001). Additionally, the single agent prexasertib treatment increased the CD44+ memory/effector T-cell population, which was further enhanced in the combination treatment (p<0.001) (Fig 2E and 2F). Prexasertib+anti-PD-L1 treatment furthermore reduced the CD62L+ naïve T-cell population (p<0.001) (Fig 2E, 2F and 2G). Prexasertib treatment alone or combination with PD-L1 blockade decreased CD4+ helper T-cell infiltration (p<0.001) (Fig S2C), the PD1+/TIM3+ exhausted CD8+ T cell population (p<0.001) (Fig S2D) and CD25+/FOXP3+ regulatory T-cell infiltration (p<0.001) (Fig. S2E).
Figure 2: Analysis of immune infiltrates of tumors after CHK1i.
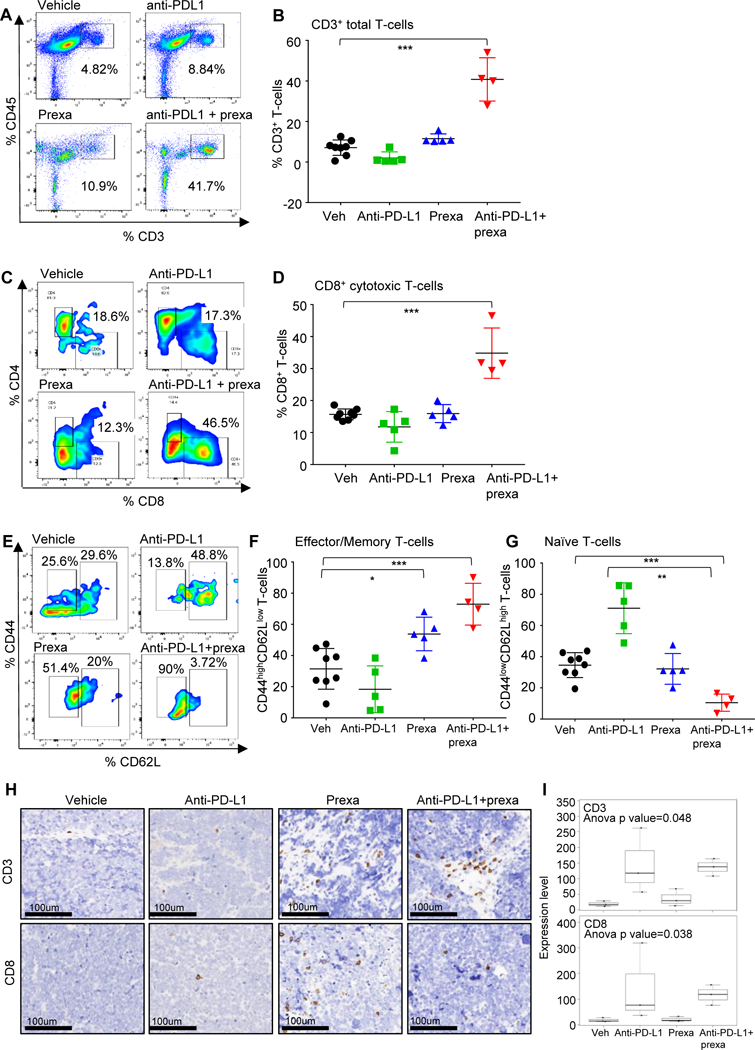
(A-H) SCLC tumors in Fig 1H were harvested at Day 21 and the immune profiling was analyzed by FACS at the endpoint, the representative plots and cumulative data for all the tumors is shown. FACS analysis of CD3+CD45+ Total T-cells (A-B), CD3+CD45+CD8+ cytotoxic T-cells (C-D), effector memory CD8 T cells: CD45+CD3+CD8+CD44highCD62Llow (E-F) and naïve T-cells CD45+CD3+CD8+CD44low CD62Lhigh (E, G) from the endpoint primary tumors. The statistical summary is shown with ANOVA test. ns, no significance; *, p < 0.05; **, p < 0.001; ***, p < 0.0001.
(H-I) The CD3 and CD8 IHC staining were performed in tumors from the resected tumors at Day 21 (from Fig 1H). Representative images of staining intensity are shown (H). The staining intensity and percentage of positive cells were analyzed and used to generate an H-score for each sample that passed quality control. Samples were stratified as CD3/8+ (+1, +2, +3), CD3/8- (0 and lower). The percent of each expression pattern of CD3 and CD8 IHC staining was summarized and shown in the bar chart (I).
CD3 and CD8 IHC staining and scoring were performed on the resected tumors (from Fig 1H). The IHC data confirms the flow cytometry observations with higher CD8 staining intensity and percentage of CD8+cells in the prexasertib treated group as compared to vehicle or anti-PD-L1 alone and further enhancement of this population in the anti-PD-L1+prexasertib treatment tumors (p<0.05) (Fig 2H and 2I). Therefore, CHK1 targeting by prexasertib significantly augments the anti-tumor immune response of anti-PD-L1 and causes cytotoxic T-cell infiltration and activation in an immunocompetent in vivo model of SCLC.
CD8+ T-cells are required for anti-tumor immunity induced by CHK1i with or without anti-PD-L1 blockade.
As CHK1 plus PD-L1 blockade resulted in complete tumor regression in 60% of the animals and FACS profiling of TILs from Day 21 resected tumors revealed that prexasertib+anti-PD-Ll treatment induced CD8+ but not CD4+ T-cell infiltration, we hypothesized that the observed responses were mediated through CD8+ immune cell populations. To test whether intratumoral CD8+ T-cell suppression would reverse the anti-tumor effect of this combination, tumor-bearing mice (RPP/mTmG-flank) were treated with prexasertib +/− anti-PD-Ll antibody in the presence of either control IgG or anti-CD8 antibody to immunodeplete CD8+ T-cells (Fig S3A). CD8 depletion slightly enhanced tumor growth in the vehicle and PD-L1 antibody treated groups (Fig 3A). In tumors treated with single agent prexasertib or combined treatment of prexasertib and anti-PD-L1, we observed that depletion of CD8+ cytotoxic T-cells significantly decreased the degree of tumor shrinkage relative to the control arms (p<0.0001) (Fig 3A and S3B).
Figure 3: CD8+ T-cells are required for anti-tumor immunity induced by CHK1i with or without anti-PD-L1 blockade.
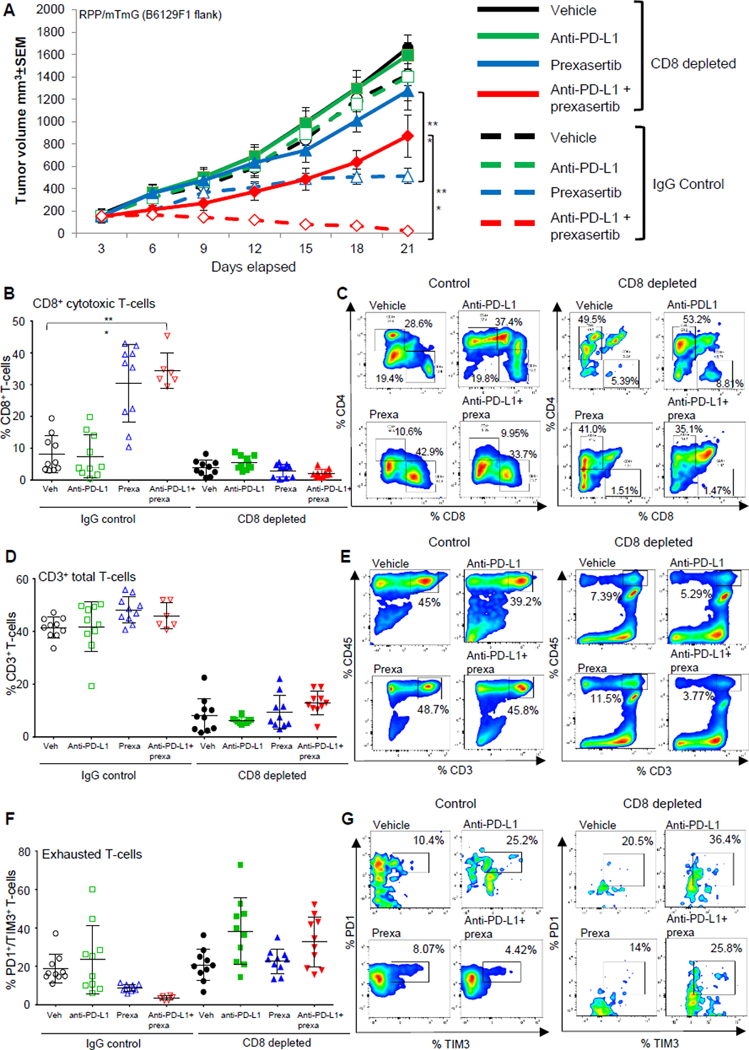
(A) Tumor growth curves +/−SEM from vehicle, prexasertib alone (10mg/kg, 2 out of 7 days, BID), anti-PD-L1 alone (300μg, 1 out of 7 days) and prexasertib+anti-PD-L1 treatment groups in RPP B6 mice in IgG control and CD8-depleted (anti-CD8, 200μg, 2 out of 7 days) groups.
(B-C) CD8+ T cells measured by flow cytometric analysis in single-cell suspensions prepared from tumors (n=10) in CD8-depleted groups as compared to IgG control groups. The analysis was independently repeated at least three times. t-test, p<0.0001.
(D-H) SCLC tumors in Fig 3A were harvested at Day 21 and the immune profiling was analyzed by FACS at the endpoint, the representative plots and cumulative data for all the tumors is shown. FACS analysis of CD3+CD45+ Total T-cells (D-E), exhausted CD8 T cells: CD45+CD3+CD8+PD-1+TIM3+ (F-G) from the endpoint primary tumors. The statistical summary is shown with ANOVA test. ns, no significance; *, p < 0.05; **, p < 0.001; ***, p < 0.0001.
Flow cytometry confirmed successful intratumoral CD8 depletion in tumors from all treatment arms (p<0.0001) (Fig 3B and 3C). In the CD8 depleted models, we did not observe significant changes in the CD3+ total T-cell (Fig 3D and 3E) or CD4+ helper T-cell infiltration following combination treatment as compared to the vehicle (Fig 3C and S3C). However, CD8 depletion was associated with a higher percentage of exhausted T-cells (PD1+/TIM3+) in animals treated with the combination, as compared to the CD8 intact group (Fig 3F and 3G). CD8 depletion also did not cause any significant changes in the naïve (CD44medium/CD62Lhigh) or memory/effector (CD44high/CD62Llow) T-cells (Fig S3D and S3E). Thus, we conclude that CHK1i greatly potentiates the effects of PD-L1 blockade in vivo through a CD8+ T-cell induced anti-tumor immune response.
PARP inhibition augments anti-PD-L1 antibody-induced antitumor immunity.
Based on the findings above, we next tested whether targeting a second important DNA repair protein, PARP, could enhance the anti-tumor immunity of anti-PD-Ll antibody, similar to the effect we observed with CHKli. We have previously shown that PARP is overexpressed in SCLC tumors (14), that PARP inhibition has activity in preclinical models (15,25) and recent clinical trials have demonstrated clinical benefit from PARP inhibitors in some SCLC patients (16,19). As described above, we observed a significant increase in PD-L1 levels following PARP inhibition or knockdown in SCLC cell lines in culture (Fig 1A, 1B and 1C).
To test the effect of PARP targeting on response to ICB and the immune microenvironment, we treated an immunocompetent SCLC model (RPP) with an FDA approved PARP inhibitor (olaparib, 50 mg/kg, 5 out of 7 days) and/or anti-PD-L1 (10mg/kg, 3 out of 7 days) (Fig S4A). Single agent olaparib treatment had no significant anti-tumor activity and consistent with the previous observation, anti-PD-L1 alone had no anti-tumor effect in these models (Fig 4A). However, we observed striking tumor regressions in animals treated with the combination of olaparib and anti-PD-L1. All animals had a complete tumor regression as early as Day 7 and the effect was sustained until Day 80 (Fig 4A, 4B). Consistent with these findings, the overall survival of the olaparib+anti-PD-L1 treated group was significantly higher than the IgG, anti-PD-L1 or olaparib treated groups (p < 0.001; Fig 4B).
Figure 4: PARP inhibition augments anti-PD-L1 antibody-induced antitumor immunity.
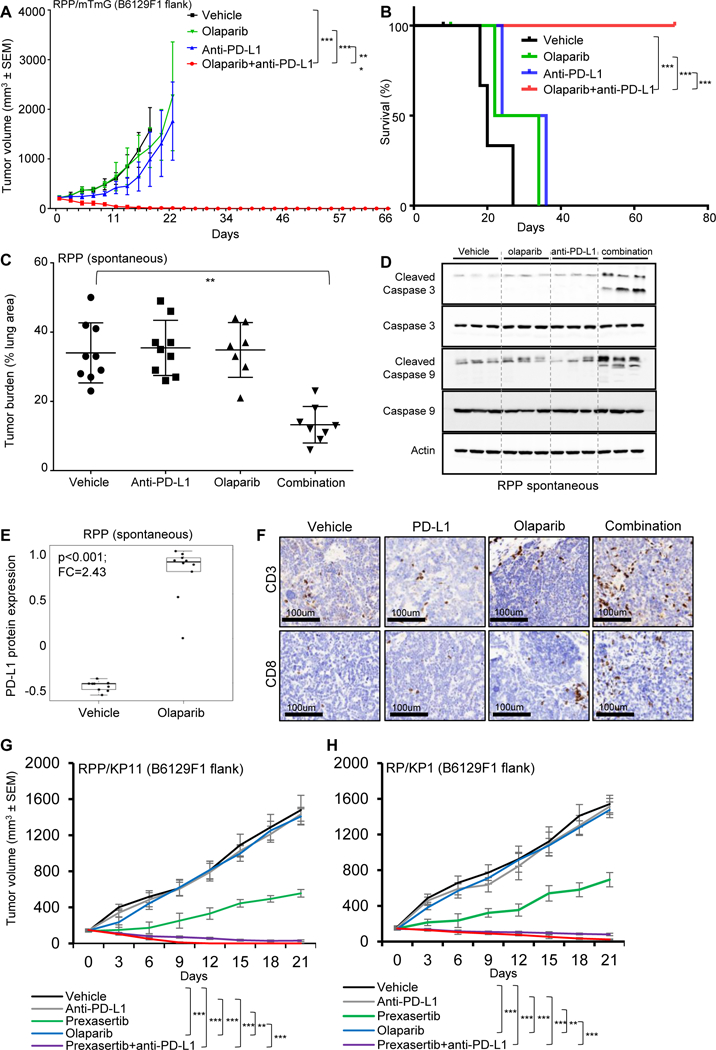
(A-B) B6129F1 mice were injected with murine RPP derived from SCLC in a genetically engineered mice with conditional loss of Trp53, p130, and Rb1. Tumor volume changes (means +/− standard error of the mean [SEM; error bars]) (A) and survival of mice (B) treated with IgG (10mg/kg, 3 out of 7 days), anti-PD-L1 (10mg/kg. 3 out of 7 days), olaparib (50mg/kg, 5 out of 7 days) and the combination (n=5 per group) up to 80 days. For the survival curve the p value was established by the Mantel-Cox test.
(C) Quantification of tumor burden as a percentage of lung area in Trp53/Rb1/p130-knockout mice treated with vehicle IgG, olaparib (50 mg/kg, 5 out of 7 days), anti-PD-L1 (300μg, 1 out of 7 days) or combination (one random lung section quantified per mouse).
(D) Immunoblot analysis of tumors treated with vehicle, olaparib, anti-PD-Ll and combination resected at Day 21. Analysis performed for a panel of apoptosis markers, pro and cleaved caspase 3 and 9. Actin was used a loading control.
(E) Olaparib treatment enhanced PD-L1 protein expression in RPP spontaneous tumors as measured by RPPA analysis (FC=2.43; p<0.001).
(F) The CD3 and CD8 IHC staining were performed in tumors from the resected tumors from Fig 4C. Representative images of staining intensity are shown. The staining intensity and percentage of positive cells were analyzed and used to generate an H-score for each sample that passed quality control. Samples were stratified as CD3/8+ (+1, +2, +3), CD3/8- (0 and lower).
(G-H) B6129F1 mice were injected with an additional cell line derived from RPP model (KP11) and from RP model (derived from SCLC in a genetically engineered mice with conditional loss of Trp53, and Rb1), KP1. Tumor volume changes (means +/− standard error of the mean [SEM; error bars]) of KP11 (G) and KP1 (H) treated with IgG, anti-PD-L1 (300pg, 1/7), olaparib (50mg/kg, 5 out of 7 days), prexasertib (10mg/kg, 2 out of 7 days, BID), combination of olaparib and anti-PD-L1 and combination of prexasertib and anti-PD-L1 (n=6 per group) treated for 21 days.
Due to total tumor regression in the olaparib+anti-PD-L1 treated group in the immunocompetent RPP flank model (Fig 4A and 4B), there were no resectable tumors for downstream analyses. Thus, we tested a lower dose of olaparib and collected tumors from all treatment arms on day 21 to determine changes in the immune microenvironment. In this experiment, we treated pre-established RPP flank tumors in immunocompetent B6129F1 mice with olaparib (50mg/kg, day 1–4 out of 7 days) and/or anti-PD-L1 (300μg, 1 out of 7 days) for 3 weeks (Fig S4B). As expected, neither single-agent olaparib nor anti-PD-Ll antibody demonstrated significant anti-tumor benefit as compared to IgG vehicle control (Fig S4C and S4D). However, even at a lower dose, the combination treatment led to significant delay in tumor growth (Fig S4C and S4D). Following olaparib treatment, we again observed a significant increase in tumor PD-L1 protein expression by RPPA analysis (p<0.0001, FC=1.47) (Fig S4E) which was confirmed by immunoblot analysis (Fig S4F).
To investigate the effects of this combination in the endogenous lung microenvironment, we tested olaparib and anti-PD-Ll in the spontaneous RPP GEMM mode of SCLC. We further performed histologic analysis of the pre-treatment tumors at 4 months after Ad-CMV-Cre administration to ascertain tumor burden. As expected from prior experience with this model, we observed appreciable tumor burden at this time by analysis of H&E-stained sections (Fig S5A). The mice were randomized into groups based on their baseline tumor burden (as measured by luciferase imaging represented in Fig S5B) to ensure comparable tumor burdens between treatment groups. Based on prior experience with this model (18), we treated spontaneous RPP tumor-bearing mice with olaparib (50mg/kg, day 1–4 out of 7 days) and/or anti-PD-L1 (300μg, 1 out of 7 days) for 3 weeks (treatment schema-Fig S4B) starting about four months after administration with Ad-CMV-Cre, when all mice had more than 50 SCLC tumors growing in their lungs. When tumor burden was quantified after three weeks of treatment (Fig 4C), single agent olaparib or anti-PD-L1 did not cause any appreciable change in the tumor burden as compared to the IgG control mice. However, olaparib+anti-PD-L1-treated tumors occupied a significantly smaller fraction of the total lung area and were fewer in number, suggestive of tumor regression (Fig 4C; p<0.001). We further performed an immunoblot analysis of tumor lysates resected from the lungs for a panel of apoptosis markers (cleaved caspase 3 and 9) to investigate tumor cell killing in the animals treated with the combination regimen. In agreement with the changes in tumor volume, we observed no appreciable change in cleaved caspase 3 or 9 post-olaparib or anti-PD-Ll single agent treatments (Fig 4D). However, there was a noticeable increase in the expression of cleaved caspase 3 and 9 in the tumors treated with combination of olaparib and anti-PD-Ll, thus confirming tumor cell killing with this treatment (Fig 4D). Moreover, olaparib treatment appreciably increased PD-L1 protein expression compared with vehicle-treated animals (p<0.001) (Fig 4E). IHC staining of lung sections demonstrated no change in CD8 positive cells in the olaparib treated group as compared to vehicle or anti-PD-Ll alone. However, we observed a significantly higher CD8 staining intensity and percentage of CD8+ cells in anti-PD-Ll+olaparib treated group (Fig 4F).
Immune profiling by flow cytometry of resected lung tumors showed no change in the CD3+ total T-cells, CD4+ helper T-cells or CD8+ cytotoxic T-cell infiltration upon single agent olaparib or anti-PD-Ll treatment (Fig S5C-S5G). However, there was a significant increase in the CD3+ total T-cells and CD8+ cytotoxic T-cell and decrease in the CD4+ helper T-cell infiltration in the olaparib+anti-PD-Ll treatment group (Fig S5C-S5G). Interestingly, we observed an increase in the PDl+/TIM3+ exhausted T-cells and CD25+/FOXP3+ T-regulatory cells upon treatment with single agent olaparib or anti-PD-Ll antibody (Fig S5H-S5K). However, consistent with the anti-tumor response, treatment with olaparib+anti-PD-Ll antibody decreased the percentage of tumor infiltrating PDl+/TIM3+ exhausted CD8 T-cells and CD25+/FOXP3+ CD4 T-regulatory cells (Fig S5H-S5K ).
We further performed immune profiling of flank tumors shown in Fig S4C and S4D to see whether consistent changes were induced versus the endogenous lung tumors. Single agent olaparib or anti-PD-Ll antibody did not change CD3+ total T-cell or CD8+cytotoxic T-cell and only minimally decreased CD4+helper T-cell infiltration as compared to IgG control (Fig S6A-S6E). However, treatment with the combination of olaparib and anti-PD-Ll led to a significant increase in the CD3+ total T-cell or CD8+cytotoxic T-cell, and decrease CD4+helper T-cell infiltration as compared to IgG control (Fig S6A-S6E). Furthermore, we observed either single agent olaparib or anti-PD-Ll increased CD25+FOXP3+ CD4 T-regulatory and PD+/TIM3+ CD8 exhausted T-cell infiltration with a significant decrease in these populations upon combined inhibition with olaparib and anti-PDLl (Fig S6F-S6I).
Thus, in summary, while PARP targeting by olaparib alone does not significantly change CD8+cytototoxic T-cell infiltration, combined inhibition of PARP and PD-L1 caused remarkable tumor regressions and increased infiltration of CD8+ cytotoxic T-cells and decrease in the exhausted and regulatory T-cells.
To further confirm that the synergistic anti-tumor effect of combined DDR-PD-L1 targeting is not model specific, we investigated the effect of this combination in additional SCLC models. For this, we selected an additional RPP model (KP11 cell line) and a Trp53f/f, Rb1f/f (p53−/−/Rb1−/−) double knockout RP model (KP1 cell line). We treated B6129F1 immunocompetent flank RPP/KP11 (Fig 4G) and RP/KP1 (Fig 4H) tumor bearing mice with prexasertib (10mg/kg, 2/7days, BID, Day 1 and 2) or olaparib (50 mg/kg, 5 out of 7 days) with or without anti-PD-L1 (300μg, 1/7days) for 3 weeks (n = 5 per group). As expected anti-PD-L1 has no anti-tumor benefit in either KP11 or KP1 model (Fig 4G and 4H). Similar to the effect observed in the RPP/mTmG model, we saw no change in tumor growth with olaparib treatment in either KP11 or KP1 model (Fig 4G and 4H). Single agent prexasertib, however, showed significant delay in tumor growth in both models (Fig 4G and 4H). Notably, in agreement with previous models, combined targeting of CHK1 or PARP and PD-L1 led to remarkable anti-tumor effect in these models. In KP11, by Day 21, we observed complete tumor regression in 2 out of 5 animals when treated with prexasertib + PD-L1 antibody; and complete regression in 5 out of 5 animals when treated with olaparib + PD-L1 antibody. In KP1, by Day 21, we observed complete tumor regression in 1 out of 5 animals when treated with prexasertib + PD-L1 antibody; and complete regression in 3 out of 5 animals when treated with olaparib + PD-L1 antibody. These observations clearly demonstrated that the synergistic anti-tumor benefit of combined DDR and PD-L1 blockade is not model-specific in SCLC.
Anti-tumor immune response post-DDR targeting is mediated via STING-TBK1-IRF3 pathway in SCLC.
Based on the rapid anti-tumor immune response we observed following DDR inhibitor treatment, we hypothesized that the immune modulation induced by DDR targeting may occur, at least in part, through activation of the innate immune system. Previous reports have shown that DNA damage that arises from cytotoxic agents can activate the STING pathway, an innate immune pathway activated by cytoplasmic DNA (26–30). Cyclic guanosine monophosphate (GMP)–adenosine monophosphate (AMP) synthase (cGAS) is a DNA sensor that triggers innate immune responses through production of the second messenger cyclic GMP-AMP (cGAMP), which binds and activates the adaptor protein STING (26–30). cGAMP binding to STING triggers phosphorylation of IRF3 via TBK1, and IRF3 then translocates to the nucleus to trigger transcription of inflammatory genes.
To test whether the STING pathway was activated in SCLC models in response to the DDR inhibitors prexasertib and olaparib (31), we treated SCLC cells lines and tumors with prexasertib and olaparib and assessed STING pathway activation by genomic and proteomic assays. We observed the presence of cytosolic DNA post-treatment with prexasertib and olaparib which is indicative of DNA damage (Fig. S7A). We next found that treatment with prexasertib for 24 and 72 hours led to a time dependent activation of the STING pathway including pSTING_S366, pTBK1_S172, cGAS, and pIRF3_S396 in multiple human SCLC cell lines (H82, H69 and H841) (Fig 5A). STING pathway activation was also observed in flank murine RPP tumor samples collected on Days 5, 14 and 21 of prexasertib treatment (Fig. 5A). A similar STING pathway activation was observed upon olaparib treatment in multiple SCLC cell lines (H82, H526, H1048) where PARP targeting enhanced expression of pSTING_S366, pTBK1_S172, cGAS, and pIRF3_S396 in a time-dependent manner (Fig. 5B). Activation of the STING pathway was also observed in olaparib-treated murine RPP tumors collected from flank RPP and spontaneous lung RPP models 21 days post treatment (Fig. 5B). In contrast, when SCLC cell lines were treated with a drug that does not induce DDR, paclitaxel (PTX) (10 and 20μM for 48 hours) we did not observe any change in the expression of PD-L1 or any of the main STING pathway proteins (pSTING_S366 and cGAS) (Fig. S7B). Phospho-histone H3 was assessed to demonstrate ongoing cell division (Fig S7B). These data indicate that, while DDR inhibition causes release of cytosolic DNA which further triggers STING pathway activation, no such effect is observed with a drug that does not induce DDR.
Figure 5: Anti-tumor immune response post-DDR targeting is mediated via STING-TBK1-IRF3 pathway in SCLC.
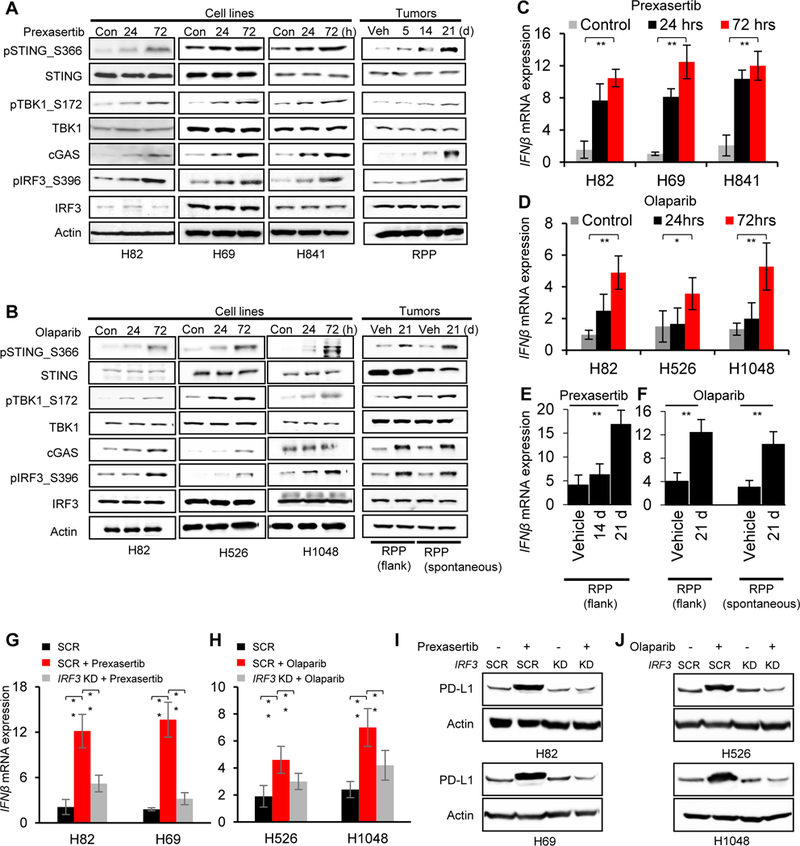
(A-B) Immunoblots of markers in the STING pathway including total and phospho STING (S366), total and phospho TBK1 (S172), cGAS, total and phospho IRF3 (S396) in lysates collected from SCLC cell lines and tumors treated with prexasertib (A) or olaparib (B). Actin served as a loading control.
(C-D) Quantitative PCR (qPCR) measurement of IFNβ mRNA expression in SCLC cell lines 24 and 72 hours after prexasertib (C) and olaparib (D) treatment.
(E-F) Quantitative PCR (qPCR) measurement of IFNβ mRNA expression in SCLC tumor models after treatment with prexasertib in RPP flank model (E) and olaparib in RPP flank and spontaneous (F) SCLC in vivo models.
(G-H) qPCR measurement of IFNβ mRNA expression 72 hours after treatment with prexasertib (G) and olaparib (H). IRF3 expression was knocked down 24 hours prior to drug treatment using siRNA targeting IRF3. A scrambled siRNA control (SCR) is included. All data representative of mean ± SD and p values represented as * p<0.05, **p<0.001, ***p<0.0001.
(I-J) Immunoblot of PD-L1 in SCLC cells treated with prexasertib (I) and olaparib (J) (72 hours). IRF3 expression was knocked down in all cells 24 hours prior to drug treatment using siRNA targeting IRF3. A scrambled siRNA control (SCR) is included.
IRF3 has been previously identified as a transcription factor for Type I interferon genes, particularly IFNβ (32,33). Since DDR targeting led to the activation of IRF3, we predicted that the increase in IRF3 levels post-DDR targeting would enhance the mRNA expression of IFNβ. Prexasertib and olaparib treatment caused significant increases in the mRNA expression of IFNβ in multiple SCLC cell lines in a time-dependent manner (Fig 5C and 5D). Similar enhancement of IFNβ expression upon treatment with prexasertib (Fig. 5E) or olaparib (Fig. 5F) was observed in SCLC RPP-mTmG in vivo tumors. In contrast, treatment with PTX (10, 20 μM for 48 hours) did not cause any appreciable change in the mRNA expression of IFNβ (Fig S7C), further demonstrating that the STING-mediated activation of Type I interferon is DDR targeting mediated. Moreover, prexasertib and olaparib treatment caused significant increases in the mRNA expression of IFNβ in RP-KP1 and RPP-KP11 tumors at Day 21 (Fig S7D). Notably, siRNA-mediated IRF3 knockdown, as confirmed by immunoblot analysis (Fig S7E), abrogated the prexasertib (Fig 5G) or olaparib (Fig 5H) –mediated upregulation of IFNβ mRNA expression. Because the Type I interferon pathway can regulate PD-L1 expression, we next investigated the role of IRF3 on PD-L1 expression. Interestingly, IRF3 knockdown abrogated the increase in PD-L1 protein expression post-prexasertib (Fig 5I) or -olaparib (Fig 5J) treatment in SCLC models, supporting a direct role of IRF3-mediated type 1 interferon genes in regulating PD-L1 in SCLC.
Next, we aimed to determine the extent to which the synergistic anti-tumor effect of combined DDR-PD-L1 blockade is mediated through tumor-cell cGAS-mediated STING activation. To accomplish this, we depleted either cGAS or STING in SCLC RPP/mTmG cells. The knockdown efficiency was determined by western blot analysis (Fig. S7F-cGAS; Fig. S7G-STING). The cGAS or STING-depleted tumor cells were then injected into immune competent mice and treated with either prexasertib (10mg/kg, BID, 2/7 days) or olaparib (50mg/kg, 5/7 days) with or without PD-L1 (300μg, 1/7 days) blockade (n=5/group). In tumors harboring knockdown of cGAS or STING, we observed a significantly decreased degree of tumor shrinkage relative to control arms (Fig. 6A and 6B). Contrary to parental control (Con) and scrambled control (SCR) tumors, all cGAS and STING KD tumors progressed even with combined CHK1/PARP and PD-L1 blockade (p<0.001), confirming the vital role of the cGAS/STING pathway in DDR-mediated anti-tumor response in SCLC models. We collected tumors from animals treated with either vehicle or olaparib at the end of the three weeks and investigated the expression of cGAS and STING in control vs knockdown tumors. As predicted, we observed an increase in cGAS and no detectable changes in total STING post-olaparib treatment in control cells (Fig. 6C and 6D). Furthermore, we observed undetectable levels of cGAS and STING pre- and post-olaparib treatment in cGAS and STING knockdown tumors respectively (Fig. 6C and 6D).
Figure 6: Knockdown of cGAS and STING reverses the anti-tumor effect of combined DDR and PD-L1 blockade in vivo:
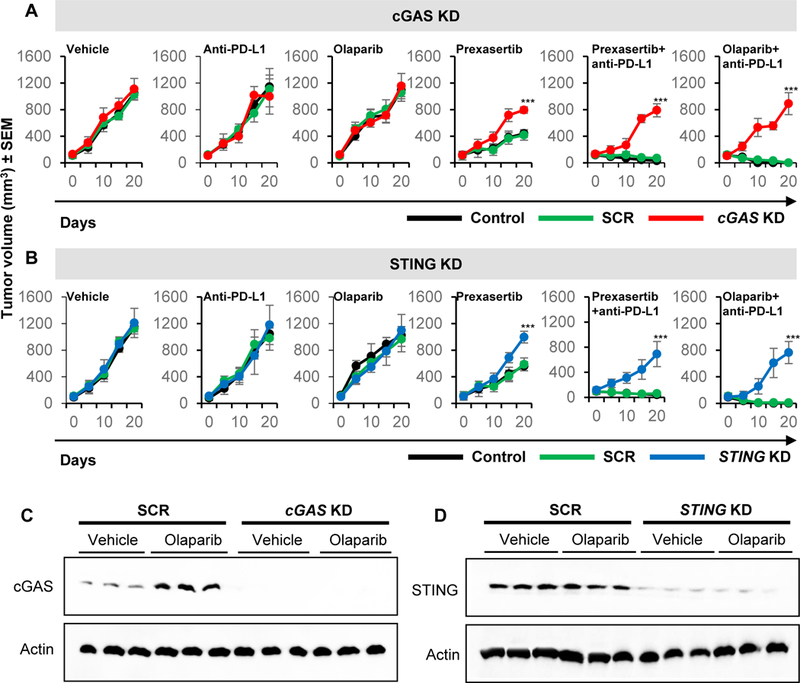
(A-B) Tumor growth curves +/−SEM from vehicle, prexasertib alone (10mg/kg, 2 out of 7 days, BID), olaparib alone (50mg/kg, 5 out of 7 days), anti-PD-L1 alone (300μg, 1 out of 7 days), prexasertib+anti-PD-L1 and olaparib+anti-PD-L1 treatment groups in B6129 mice with RPP/mTmG parental control (Con), scrambled (SCR), cGAS knockdown (A) and STING knockdown (B) groups.
(C-D) Immunoblot analysis for cGAS and STING of tumors resected at the end of treatment (Day 21) from SCR or cGAS and STING knockdown models treated with either vehicle or olaparib. Actin used as loading control.
IRF3, a major effector of the STING/TBK1 signaling pathway, has been reported to regulate the expression of chemokines such as CXCL10 and CCL5. Since, CXCL10 and CCL5 are key mediators for the chemotaxis of CD8+ T lymphocytes, and CXCL10 and CCL5 overexpression is associated with the presence of CD8+ T lymphocytes in melanoma, gastric, and colorectal cancers (34,35), we next investigated the previously unexplored link between DDR targeting and chemokine expression in SCLC. Prexasertib and olaparib treatment caused significant upregulation of CXCL10 and CCL5 mRNA expression in multiple SCLC cell line (Fig. 7A) and tumor (Fig. 7B) models (p<0.001 for all). siRNA-mediated knockdown of STING (Fig. S7H) resulted in significantly reduced expression of the CXCL10 (Fig. 7C) and CCL5 (Fig. 7D) post-prexasertib or olaparib treatment in multiple SCLC models.
Figure 7: Role of STING pathway in DDR-targeting dependent chemokine expression:
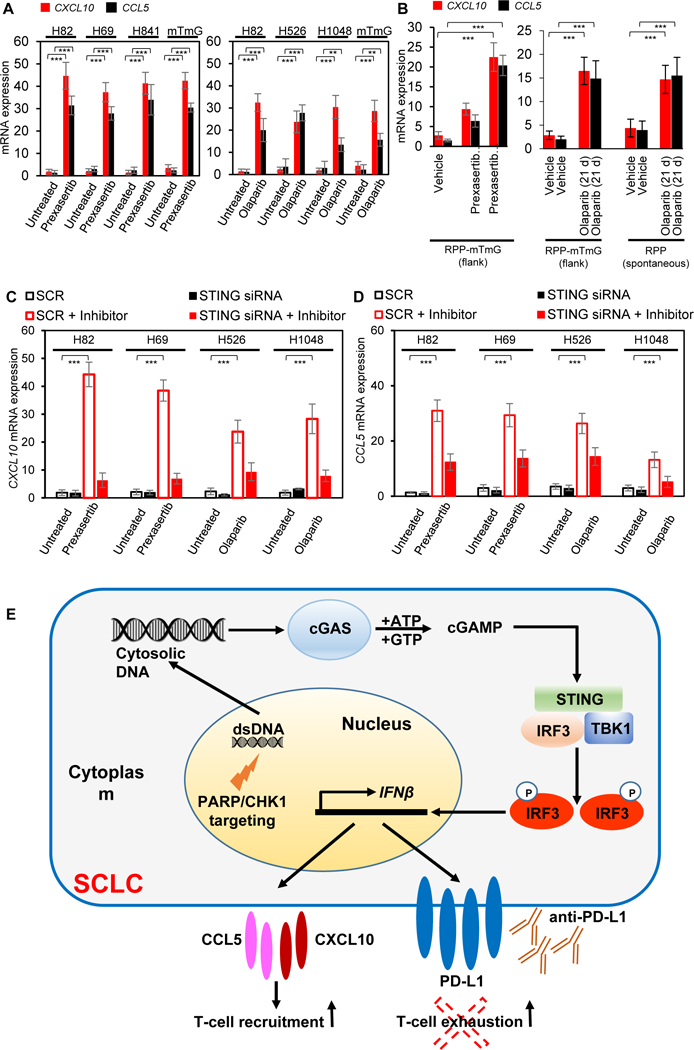
(A-B) RT-qPCR measurement of chemokines CXCL10 (red) and CCL5 (black) mRNA extracted from SCLC cell lines treated with prexasertib (left panel) and olaparib (right panel) for 72 hours (A) and tumors treated with prexasertib (RPP flank-left panel) and olaparib (RPP flank and spontaneous model- right panel) after 1 cycle of treatment (B).
(C-D) qPCR measurement of CXCL10 (C) and CCL5 (D) mRNA 72 hours following knockdown of STING using siRNA in SCLC cell lines normalized to a non-targeting scrambled control (SCR) post-prexasertib and olaparib (inhibitor) treatment.
(E) Model for STING pathway activation in response to DDR targeting in SCLC. In the proposed model, targeting the DDR proteins PARP and CHK1 with the small molecule inhibitors prexasertib and olaparib leads to cytosolic DNA in SCLC models. The cytosolic DNA is then recognized by cGAS, which leads to activation of the STING/TBK1/IRF3 pathway. IRF activation leads to increased expression of IFNβ and enhanced expression of the chemokines CXCL10 and CCL5. STING pathway activation and increased chemokine expression lead to increased PD-L1 expression and T-cell recruitment in SCLC models. Finally, DDR co-targeting leads to enhanced antitumor immunity in SCLC models.
In summary, we observed that DDR targeting by CHK1 or PARP inhibition induces DNA damage and activates the STING-TBK1-IRF3 pathway in SCLC models leading to the induction of PD-L1 expression and Type 1 interferon IFNβ. Furthermore, DDR targeting leads to the higher expression of chemokines CXCL10 and CCL5 in SCLC, dependent on STING pathway activation. This activation leads to the recruitment of CD8+ T lymphocytes and antitumor immunity, as summarized in our working model (Fig 7E).
Discussion
DDR inhibition and ICB are both therapeutic strategies under pre-clinical and clinical development for SCLC patients. Here we report a previously unexplored role of DDR pathway targeting in regulating anti-tumor immune response in SCLC models, specifically, that inhibition of DDR proteins such as CHK1 and PARP potentiates the anti-tumor immune response of PD-L1 blockade through T-cell-mediated effects. This effect is mediated through activation of the STING/TBK1/IRF3 innate immune response pathway, which ultimately enhances expression of the Type 1 interferon gene IFNβ and down-stream chemokines such as CXCL10 and CCL5 to induce activation and function of cytotoxic T lymphocytes. When combined with ICB, by anti-PD-L1 antibody, DDR targeting demonstrates significant anti-tumor effect, suggesting that these combinations may be valuable clinically to overcome primary and adaptive resistance to ICB in SCLC.
Despite significant enthusiasm for immunotherapy approaches in lung cancer, only a minority of SCLC patients respond to anti-PD-1 monotherapy or anti-PD-1/anti-CTLA4 combination (approximately 10% with monotherapy and 23% with the combination) (36,37). Despite having one of the highest mutational burdens among solid tumors, SCLC paradoxically shows lower expression of PD-L1 and relatively immunosuppressed phenotypes with low levels of infiltrating T-cells and reduced antigen presentation (38). Recent clinical data illustrating that tumors with defective DDR, such as MSI-high or MMR-deficient tumors, predicts improved response to anti-PD-1 therapy supports the hypothesis that the addition of a DDR inhibitor to anti-PD-1 therapy may significantly enhance response rates and outcomes (39). Furthermore, some recent reports have demonstrated PD-L1 upregulation by DNA damage (40). However, it is not known to what extent pharmacologic inhibitors of DDR targets may enhance ICB response or the mechanism through which this occurs.
SCLC has a ubiquitous loss in two key regulators of the DNA damage and cell cycle checkpoint pathway, TP53 and RB1, which leads to high genomic instability and replication stress (20,41). Therefore, we hypothesized that targeting this inherent DNA repair vulnerability with DDR inhibitors to promote an immune response may be an effective strategy to achieve a quick and sustained anti-tumor immune response by potentiating the effect of a combination ICB regimen. Because the response with DDR targeting occurs quickly, we hypothesized that in SCLC the activation of the innate immune response machinery may lead to DDR-mediated immune activation.
Previous studies have reported the presence of cytosolic DNA post-S phase damage and additional evidence suggests that cells may actively export DNA fragments from the nucleus possibly to prevent misincorporation into genomic DNA (42). In turn, the presence of cytosolic DNA in the absence of efficient DNA damage repair triggers cGAS-mediated innate immune response. Here, we demonstrated increased cytosolic DNA following treatment with either DDR inhibitor (prexasertib and olaparib) in vitro. We further observed that targeting CHK1 or PARP leads to the activation of the STING/cGAS/TBK1 pathway, thus upregulating PD-L1 expression. IRF3 knockdown studies confirmed the role of the transcription factor in the DDR targetingmediated anti-tumor immune response and PD-L1 regulation in SCLC. We further observed upregulation of IFNβ after DDR-targeting in SCLC, which could be an indirect mechanism of regulating PD-L1 levels in these tumors. Moreover, in our models we demonstrated significant increase in the expression of the chemokines CXCL10 and CCL5 post-DDR-inhibitor treatment. The abrogation of the expression after STING knockdown indicates the role of these chemokines in STING-TBK1-RF3 mediated anti-tumor immune response. It is not known to what extent increased tumor mutation burden induced by DDR targeting may also contribute to enhancing ICB response. However, based on our observations here with tumor shrinkage starting soon after treatment (often seen within days), the rapid innate response – rather than changes in neoantigens – seems likely to be a more important contributor at least in the initial response. Also since the mouse tumor have previously been shown to have low mutation burden (41) future studies to monitor the effects of DDR inhibition in SCLC patients will be valuable to provide further insights of the effect of DDR-mediated STING activation and immune response.
In summary, the effects of CHK1 and PARP on DNA damage repair and cell cycle progression lead to T-cell recruitment and enhanced effector cell function in SCLC tumors, mediated by the activation of the innate immune response pathway STING/TBK1/IRF3 and increased IFNβ. Activation of the STING-mediated pathway is responsible for chemokine production in response to DNA damage in vitro thereby resulting in increased immunogenicity of the otherwise immunosuppressed tumors. The pathway also leads to the upregulation of PD-L1 expression. Expression of PD-L1 is associated with tumors deficient in DNA damage repair and we, for the first time, provide rationale for investigating the role of immunotherapy in the context of DDR targeting in SCLC. Moreover, our results demonstrating the remarkable efficacy of the combination of PD-L1 blockade with PARP or CHK1 inhibition provide a strong scientific rationale for combining these modalities in clinical trials for SCLC patients. Further studies will be necessary to carefully interrogate the contribution, if any, of the neoantigen load and other immune cell populations, such as NK cells (43) during DDR-mediated T-cell activation. Because prexasertib, olaparib and other PARP inhibitors are already in clinical trials for SCLC, we expect that this hypothesis has the potential for rapid translation into the clinic.
Materials and Methods
Cell lines and characterization
SCLC human-derived cell lines were provided by Dr. John Minna (The University of Texas Southwestern Medical Center, Dallas, TX) or obtained from American Type Culture Collection (ATCC) (between the years 2011–2015).
The GEMM-derived SCLC cell lines were established from a genetically engineered mouse model, either derived from a double knockout Trp53f/f, Rb1f/f (p53−/−/Rb1−/−)(RP-KP1) model or a triple-knockout model of SCLC, which closely mimics the human disease and has a Trp53f/f Rb12f/f Rb1f/f (p53−/−/p130−/−/Rb1−/−) (RPP/mTmG; RPP/KP11) allelic genotype with a ROSA26R reporter. Complete cell line information is provided in Table S1.
All cell lines were tested and authenticated by Short Tandem Repeat Profiling (DNA Fingerprinting) within 6 months of the study and routinely tested for Mycoplasma species before any experiments were performed.
Chemical compounds
LY2603618 was obtained from Selleckchem.com. Prexasertib and olaparib were manufactured by MD Anderson’s Institute for Applied Chemical Science. All compounds were dissolved in dimethyl sulfoxide for in vitro treatments.
DNA fingerprinting to confirm cell line identity
DNA was isolated using a QIAamp DNA mini kit from 5–6 × 106 cells was isolated (Qiagen, Valencia, CA) following the manufacturer’s protocol. The isolated DNA was eluted in elution buffer (100μl) (Buffer AE, Qiagen). The concentration of the DNA and it purity was measured by Nanodrop. Cell line authentication was done by using DNA (50ng) for DNA fingerprint analysis of short tandem repeat profiling (PowerPlex 1.2, Promega, Madison, WI).
Fingerprinting results for each cell line were compared to reference fingerprints provided by Dr. Minna or the ATCC.
Mice
For the syngeneic mouse model, six-week-old immunocompetent female B6129F1 (Taconic) were used. These animals were maintained in accordance with the Institutional Animal Care and Use Committee (IACUC) of The University of Texas MD Anderson Cancer Center and the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
The spontaneous GEMM mice were maintained according to practices prescribed by the National Institutes of Health at the Stanford Research Animal Facility, which is accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care. The RPP conditional knockout mouse model induced by intratracheal administration of Ad-CMV-Cre for SCLC has been described previously (24).
Establishment of syngeneic flank tumors and studies in B6129F1 mice
The mouse SCLC cell line was derived from tumors isolated from RPP mice (RPP-mTmG; RPP-KP11 cell lines) or RP mice (RP-KP1). For subcutaneous injections, 0.5×106 mouse SCLC (mTmG) cells were injected into one flank of each mouse with Matrigel (1:1, BD Biosciences).
Treatment schedule of SCLC in vivo models
Mice with mouse SCLC tumors received one of the following treatments: 1) vehicle; 2) anti-PD-L1 (9G2; BioXcell) (300μg), once per week; 3) prexasertib (10 mg/kg twice daily), 2 consecutive days per week; or 4) combination of anti-PD-L1 (300μg) once per week and prexasertib (10 mg/kg twice daily) 2 consecutive days per week.
For the PARP inhibitor experiments, mice received one of the following treatments: 1) vehicle; 2) olaparib (50 mg/kg daily) 4 times per week; 3) anti-PD-Ll (300μg), once per week; or 4) combination of olaparib and anti-PD-Ll.
Tumors were collected for single cell preparation for flow cytometry, snap-frozen in liquid nitrogen for protein isolation or fixed in 4% paraformaldehyde in phosphate-buffered saline at 4oC and processed for paraffin histologic analysis. Sections of paraffin-embedded tissues (4p,m) were stained with hematoxylin and eosin and collected for immunohistochemistry (IHC).
Development of spontaneous SCLC tumors and dosing schedule
Mice were maintained according to practices prescribed by the National Institutes of Health at the Stanford Research Animal Facility, which is accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care. The RPP triple-knockout mouse model for SCLC has been previously described. The mice were bred onto a mixed genetic background composed of C57BL/6, 129/SvJ, and 129/SvOla. Tumors were induced in young adult mice by intratracheal instillation of 4 × 107 plaque-forming units of adenovirus expressing Cre recombinase (Ad-Cre; Baylor College of Medicine, Houston, TX).
To determine baseline amount of tumors before treatment mice were injected with luciferin (Biosynth), anesthetized with isoflurane (VetOne), and dorsal hair shaved between the head and stomach. The mice were imaged for luciferase luminescence using an IVIS CCD camera. The mice were separated into treatment groups to ensure all cohorts had comparable levels of luminescence, which is proportional to the amount of tumors in the mouse.
Treatment was initiated 4 months after Ad-Cre delivery, at a time when infected mice have developed more than 50 independent lesions. The CHK1 inhibitor, prexasertib (10 mg/kg, BID) was injected subcutaneously at the nape of the neck every 12 hours for 1, 3 and 3 three days in a row, for one week (i.e., 10 mg/kg, twice daily, days 1, 2 and 3 of a 7-day cycle, for one cycle). For olaparib, the mice were treated either with olaparib (50 mg/kg, 4/7) and/or anti-PD-L1 (300μg, 1/7). Control mice were injected with IgG control. The lung tumors were collected and processed for single cell isolation.
Real-time polymerase chain reaction (PCR)
Real-time PCR was done using SYBR Select Master Mix (Life Technologies, cat# 4472908) according to the manufacturer’s protocol. The primers were purchased from Sigma (USA); the details of the primers are given in Table S2. Triplicate PCR reactions were run on ABI (7500 Fast Real Time PCR System) according to the manufacturer’s instructions. The comparative Ct method using the average 2ΔΔCT value for each set of triplicates was used, and the average of the biological replicates was calculated. Negative controls were included for every primer set, and GAPDH was used as the positive control.
Flow cytometry
Single-cell suspensions were prepared and stained according to standard protocols for flow cytometry with antibodies listed in Table S3. For intracellular staining, cells were fixed and permeabilized with BD Cytofix/Cytoperm (BD Biosciences). The data were acquired on a Fortessa or Calibur platform (BD Biosciences) and analyzed with FlowJo software (version 7.6; Tree Star). For analyzing the abundance and the function of CD4+ or CD8+ TILs, single-cell suspensions were prepared from tumors and inguinal lymph nodes and stained; the staining of inguinal lymph node cells was used as the reference of lymphocyte gating, then CD3+ cells were gated, and then CD4+ or CD8+ population was analyzed.
Supplementary Material
Statement of Relevance
Our results define previously unrecognized immunomodulatory functions of DDR inhibitors and suggest that adding PARP or CHK1 inhibitors to immune checkpoint blockade may enhance treatment efficacy in SCLC patients. Furthermore, our study supports a role of innate immune STING pathway in DDR-mediated anti-tumor immunity in SCLC.
Acknowledgements:
The authors wish to thank Dr. Erica Goodoff (Department of Scientific Publications, MD Anderson) and Dr. Lara Carolina Alvarez de Lacerda Landry for their excellent assistance with editing the manuscript and the figures; Dr. Emily Roarty for scientific and administrative support.
Financial Support: This work was supported by: The NIH/NCI award U01-CA213273 (JS, JVH, LAB); NIH/NCI award 1-R01-CA207295 (LAB), and the Lung Cancer Research Foundation (TS); NIH/NCI CCSG P30-CA016672 (shRNA and ORFeome Core, Boinformatics Shared Resource); The University of Texas-Southwestern and MD Anderson Cancer Center Lung SPORE (5 P50 CA070907); through generous philanthropic contributions to The University of Texas MD Anderson Lung Cancer Moon Shot Program (JVH, JW LAB); The MD Anderson Cancer Center Small Cell Lung Cancer Working Group and Abell Hangar Foundation Distinguished Professor Endowment (BSG), MD Anderson Cancer Center Physician Scientist Award (LAB); Lee Clark Fellowship of The University of Texas MD Anderson Cancer Center, supported by the Jeane F. Shelby Scholarship Fund (LAB); an NCI Cancer Clinical Investigator Team Leadership Award (P30 CA016672) (LAB); The Rexanna Foundation for Fighting Lung Cancer (JVH, LAB); The University of Texas Lung Specialized Programs of Research Excellence (SPORE) grant (P50CA70907) (IIW); Elsa U. Pardee Foundation grant (LC) and P50CA070907–18 (UT Lung SPORE CEP) (LC).
J.V.H serves on the advisory committees of AstraZeneca, Boehringer Ingelheim, Exelixis, Genentech, GSK, Guardant Health, Hengrui, Lilly, Novartis, Spectrum, EMD Serono, and Synta; received research support from AstraZeneca, Bayer, GlaxoSmithKline, Spectrum; receives royalties and licensing fees from Spectrum.
L.A.B. serves on scientific advisory boards for AstraZeneca, AbbVie, GenMab, BergenBio, Pharma Mar, SA, and has research support from AbbVie, AstraZeneca, GenMab, Tolero Pharmaceuticals.
D.L.G. serves on scientific advisory committees for Astrazeneca, GlaxoSmithKline, Sanofi and Janssen and has research support from Janssen, Takeda, and Astrazeneca.
I.I.W serves in advisory boards for Genentech/Roche, Bristol-Myers Squibb, Astra Zeneca/Medimmune, HTG Molecular, Merck, GlaxoSmithKline, and MSD; and receives research support from Genentech, HTG Molecular, DepArray, Merck, Bristol-Myers Squibb, Medimmune, Adaptive, Adaptimmune, EMD Serono, Pfizer, Takeda, Amgen, Karus, Johnson & Johnson, Bayer, and 4D.
J.S. has research support from Abbvie/StemCentrx.
Footnotes
For details on cell culture, transient knockdown of CHEK1, PARP, IRF3, TMEM173 (STING), stable knockdown of STING and MB21D1 (cGAS), RNA isolation, reverse transcription, primer information, preparation of protein lysates, western blot analysis, western blot antibodies, reverse-phase protein array (RPPA), method for tumor growth assessment, in vivo CD8 depletion, flow cytometry antibodies, histologic analysis, micronuclei assay and statistical methods-see supplementary materials and methods.
Conflict of Interest:
Data and materials availability: All data available in the main text or the supplementary materials.
References:
- 1.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science (New York, NY) 2018;359(6382):1350–5 doi 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharma P, Allison JP. The future of immune checkpoint therapy. Science (New York, NY) 2015;348(6230):56–61 doi 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 3.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer cell 2015;27(4):450–61 doi 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferris RL, Blumenschein G Jr., Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. The New England journal of medicine 2016;375(19):1856–67 doi 10.1056/NEJMoa1602252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. The New England journal of medicine 2016;375(19):1823–33 doi 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 6.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. The New England journal of medicine 2015;372(4):320–30 doi 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 7.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017;168(4):707–23 doi 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hellmann MD, Callahan MK, Awad MM, Calvo E, Ascierto PA, Atmaca A, et al. Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination with Ipilimumab in Small-Cell Lung Cancer. Cancer cell 2018;33(5):853–61.e4 doi 10.1016/j.ccell.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng Y, Li H, Liu Y, Ma L, Liu X, Liu Y, et al. Distribution and clinical significance of CTLA4, PD-1 and PD-L1 in peripheral blood of patients with small-cell lung cancer. Journal of Clinical Oncology 2015;33(15_suppl):7574-doi 10.1200/jco.2015.33.15_suppl.7574. [DOI] [Google Scholar]
- 10.Ishii H, Azuma K, Kawahara A, Yamada K, Imamura Y, Tokito T, et al. Significance of programmed cell death-ligand 1 expression and its association with survival in patients with small cell lung cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 2015;10(3):426–30 doi 10.1097/jto.0000000000000414. [DOI] [PubMed] [Google Scholar]
- 11.Yamane H, Isozaki H, Takeyama M, Ochi N, Kudo K, Honda Y, et al. Programmed cell death protein 1 and programmed death-ligand 1 are expressed on the surface of some small-cell lung cancer lines. American journal of cancer research 2015;5(4):1553–7. [PMC free article] [PubMed] [Google Scholar]
- 12.Paglialunga L, Salih Z, Ricciuti B, Califano R . Immune checkpoint blockade in small cell lung cancer: is there a light at the end of the tunnel? ESMO Open 2016;1(4) doi 10.1136/esmoopen-2015-000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Majem M, Rudin CM. Small-cell lung cancer in the era of immunotherapy. Translational Lung Cancer Research 2017;6(Suppl 1):S67–S70 doi 10.21037/tlcr.2017.10.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Byers LA, Wang J, Nilsson MB, Fujimoto J, Saintigny P, Yordy J, et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer discovery 2012;2(9):798–811 doi 10.1158/2159-8290.cd-12-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cardnell RJ, Feng Y, Diao L, Fan YH, Masrorpour F, Wang J, et al. Proteomic markers of DNA repair and PI3K pathway activation predict response to the PARP inhibitor BMN 673 in small cell lung cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2013;19(22):6322–8 doi 10.1158/1078-0432.ccr-13-1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Bono J, Ramanathan RK, Mina L, Chugh R, Glaspy J, Rafii S, et al. Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA½ Mutations and Selected Sporadic Cancers. Cancer discovery 2017;7(6):620–9 doi 10.1158/2159-8290.cd-16-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sen T, Tong P, Diao L, Li L, Fan Y, Hoff J, et al. Targeting AXL and mTOR Pathway Overcomes Primary and Acquired Resistance to WEE1 Inhibition in Small-Cell Lung Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2017;23(20):6239–53 doi 10.1158/1078-0432.ccr-17-1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sen T, Tong P, Stewart CA, Cristea S, Valliani A, Shames DS, et al. CHK1 Inhibition in Small-Cell Lung Cancer Produces Single-Agent Activity in Biomarker-Defined Disease Subsets and Combination Activity with Cisplatin or Olaparib. Cancer research 2017;77(14):3870–84 doi 10.1158/0008-5472.can-16-3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pietanza MC, Waqar SN, Krug LM, Dowlati A, Hann CL, Chiappori A, et al. Randomized, DoubleBlind, Phase II Study of Temozolomide in Combination With Either Veliparib or Placebo in Patients With Relapsed-Sensitive or Refractory Small-Cell Lung Cancer. Journal of Clinical Oncology;0(0):JCO.2018.77.7672 doi 10.1200/jco.2018.77.7672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sen T, Gay CM, Byers LA. Targeting DNA damage repair in small cell lung cancer and the biomarker landscape. Translational Lung Cancer Research 2018;7(1):50–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chatzinikolaou G, Karakasilioti I, Garinis GA. DNA damage and innate immunity: links and tradeoffs. Trends in immunology 2014;35(9):429–35 doi 10.1016/j.it.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 22.Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clinical cancer research : an official journal of the American Association for Cancer Research 2017;23(14):3711–20 doi 10.1158/1078-0432.ccr-16-3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jahchan NS, Dudley JT, Mazur PK, Flores N, Yang D, Palmerton A, et al. A drug repositioning approach identifies tricyclic antidepressants as inhibitors of small cell lung cancer and other neuroendocrine tumors. Cancer discovery 2013;3(12):1364–77 doi 10.1158/2159-8290.cd-13-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schaffer BE, Park KS, Yiu G, Conklin JF, Lin C, Burkhart DL, et al. Loss of p130 accelerates tumor development in a mouse model for human small-cell lung carcinoma. Cancer research 2010;70(10):3877–83 doi 10.1158/0008-5472.can-09-4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cardnell RJ, Feng Y, Mukherjee S, Diao L, Tong P, Stewart CA, et al. Activation of the PI3K/mTOR Pathway following PARP Inhibition in Small Cell Lung Cancer. PLoS ONE 2016;11(4):e0152584 doi 10.1371/journal.pone.0152584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li T, Chen ZJ. The cGAS–cGAMP–STING pathway connects DNA damage to inflammation, senescence, and cancer. The Journal of Experimental Medicine 2018. doi 10.1084/jem.20180139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Corrales L, McWhirter SM, Dubensky TW Jr., Gajewski TF The host STING pathway at the interface of cancer and immunity. The Journal of clinical investigation 2016;126(7):2404–11 doi 10.1172/JCI86892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature 2011;478(7370):515–8 doi 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009;461(7265):788–92 doi 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science (New York, NY) 2013;339(6121):786–91 doi 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brzostek-Racine S, Gordon C, Van Scoy S, Reich NC. The DNA damage response induces IFN. Journal of immunology (Baltimore, Md : 1950) 2011;187(10):5336–45 doi 10.4049/jimmunol.1100040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barber GN. STING: infection, inflammation and cancer. Nature reviews Immunology 2015;15(12):760–70 doi 10.1038/nri3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS–STING pathway of cytosolic DNA sensing. Nature Immunology 2016;17:1142 doi 10.1038/ni.3558. [DOI] [PubMed] [Google Scholar]
- 34.Groom JR, Luster AD. CXCR3 in T cell function. Experimental cell research 2011;317(5):620–31 doi 10.1016/j.yexcr.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kunz M, Toksoy A, Goebeler M, Engelhardt E, Brocker E, Gillitzer R. Strong expression of the lymphoattractant C-X-C chemokine Mig is associated with heavy infiltration of T cells in human malignant melanoma. The Journal of pathology 1999;189(4):552–8 doi 10.1002/(sici)1096-9896(199912)189:4<552::aid-path469>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 36.Antonia SJ, Lopez-Martin JA, Bendell J, Ott PA, Taylor M, Eder JP, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase ½ trial. The Lancet Oncology 2016;17(7):883–95 doi 10.1016/s1470-2045(16)30098-5. [DOI] [PubMed] [Google Scholar]
- 37.Ott PA, Elez E, Hiret S, Kim DW, Morosky A, Saraf S, et al. Pembrolizumab in Patients With Extensive-Stage Small-Cell Lung Cancer: Results From the Phase Ib KEYNOTE-028 Study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2017;35(34):3823–9 doi 10.1200/jco.2017.72.5069. [DOI] [PubMed] [Google Scholar]
- 38.Wang W, Hodkinson P, McLaren F, MacKinnon A, Wallace W, Howie S, et al. Small cell lung cancer tumour cells induce regulatory T lymphocytes, and patient survival correlates negatively with FOXP3+ cells in tumour infiltrate. International journal of cancer Journal international du cancer 2012;131(6):E928–37 doi 10.1002/ijc.27613. [DOI] [PubMed] [Google Scholar]
- 39.Vranić S Microsatellite instability status predicts response to anti-PD-1/PD-L1 therapy regardless the histotype: A comment on recent advances. Bosnian Journal of Basic Medical Sciences 2017;17(3):274–5 doi 10.17305/bjbms.2017.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sato H, Niimi A, Yasuhara T, Permata TBM, Hagiwara Y, Isono M, et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nature communications 2017;8(1):1751 doi 10.1038/s41467-017-01883-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015;524(7563):47–53 doi 10.1038/nature14664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parkes EE, Walker SM, Taggart LE, McCabe N, Knight LA, Wilkinson R, et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J Natl Cancer Inst 2017; 109(1) doi 10.1093/jnci/djw199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Y, Li Y, Liu S, Adeegbe DO, Christensen CL, Quinn MM, et al. NK cells mediate synergistic antitumor effects of combined inhibition of HDAC6 and BET in a SCLC preclinical model. Cancer research 2018. doi 10.1158/0008-5472.can-18-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.