

Abstract
There is increasing awareness that in addition to the metabolic crisis of diabetic ketoacidosis (DKA) caused by severe insulin deficiency, the immune inflammatory response is likely an active multicomponent participant in both the acute and chronic insults of this medical crisis, with strong evidence of activation for both the cytokine and complement system. Recent studies report that the matrix metalloproteinase enzymes and their inhibitors are systemically activated in young Type 1 diabetes mellitus (T1D) patients during DKA and speculate on their involvement in blood-brain barrier (BBB) disruption. Based on our previous studies, we address the question if matrix metalloproteinase 9 (MMP9) is expressed in the brain in the fatal brain edema (BE) of DKA. Our data show significant expression of MMP9 on the cells present in brain intravascular areas. The presence of MMP9 in intravascular cells and that of MMP+ cells seen passing the BBB indicates a possible role in tight junction protein disruption of the BBB, possibly leading to neurological complications including BE. We have also shown that MMP9 is expressed on neurons in the hippocampal areas of both BE/DKA cases investigated, while expression of tissue inhibitor of metalloproteinases 1 (TIMP1) was reduced in the same areas. We can speculate that intraneuronal MMP9 can be a sign of neurodegeneration. Further studies are necessary to determine the role of MMP9 in the pathogenesis of the neurologic catastrophe of the brain edema of DKA. Inhibition of MMP9 expression might be helpful in preserving neuronal function and BBB integrity during DKA.
Keywords: matrix metalloproteinase 9, diabetic ketoacidosis, C5b-9, RGC-32, tissue inhibitor of metalloproteinases 1
Graphical abstract
Introduction
Varying degrees of metabolic dysregulation and the associated oxidative stress (Lee et al., 2002) initiate and perpetuate an inflammatory response that can progress from a chronic low-grade inflammatory milieu (Heier et al., 2015) to the metabolic and immunologic crisis of DKA (Hoffman et al., 2003a). Similarly, acute and more severe dysregulation results in perturbation with an increased morbidity (Peeters et al., 2017). Evidence for the early pathogenesis of the dysregulated metabolic stress of DKA comes from in-vitro studies of ketone bodies and hyperglycemia activating both rodent (Isales et al., 1999) and human cerebral capillary endothelial cells (Hoffman et al., 2002), resulting in an increased expression of proinflammatory cytokines and chemokines such as CXCL1 and CXCL8 and promoting neutrophil adhesiveness (Omatsu et al, 2014). Among the numerous perturbations in systemic metabolic/immunological variables that have been suggested to increase the stress of type 1 diabetes mellitus/DKA include: 1) deoxy carbonyls (Hoffman et al., 2003a; Turk et al, 2006); 2) advanced glycation end products (AGE)/RAGE axis (Skrha et al., 2012; Peeters et al., 2018); 3) AGE and oxidized low density lipoproteins (LDL) (Lopes-Virella et al., 2012); 4) inflammatory cytokines (Hoffman et al., 2003b; Karavanaki et al., 2011; Close et al., 2013); 5) complement activation (Jerath et al., 2005; Speidl et al., 2011) and MMP9 (Woo et al., 2016a, 2016b; Garro et al., 2017).
Matrix metalloproteinases (MMPs) are potent zinc-dependent endopeptidases produced by numerous cell types including leukocytes, microglia, astrocytes and neurons (Fedorova et al., 2018). MMP9 is one of the few MMPs enzymes that are constitutively expressed. It has a diverse spectrum of activity: 1) degrades tight junction proteins and all components of the extracellular matrix proteins including laminin, collagen and fibronectin (Feng et al., 2011); 2) facilitates leukocyte migration and invasion (Lavini-Ramos et al., 2017); 3) integration of immunoregulatory pathways (Lavini-Ramos et al., 2017); 4) maintenance of tissue barriers by processing a range of non-matrix proteins, including cytokines and chemokines (Van Lint and Libert, 2007; Nissinen and Kahari, 2014; Smigiel and Parks, 2017); and 5) staging serum and tissue in metastatic conditions (Wieczorek et al., 2015). They are also involved in the pathogenesis of acute complications such as acute coronary syndromes (Suzuki et al., 2006). Although MMPs have broad substrate specificity, they are tightly regulated in inflammation and play a significant role in biological processes of autoimmune diseases (Ram et al., 2006).
MMP9, MMP2 and TIMP1 expression were reported in T1D (Maxwell et al., 2001; Jacqueminet et al., 2006; Symeonidis et al., 2013). Woo et al. (2016a) reported an increase of MMP9 in DKA and after its treatment along with other leukocyte azurophilic enzymes, such as proteinase 3 (PR-3), whose level correlated with DKA severity and was shown to be involved in tight junction protein disruption in vitro. Recent publications by Woo et al. (2016b) and by Garro et al. (2017) reported a positive correlation between DKA severity and the prevalence of MMP8 and MMP9, and hypothesized that these enzymes are playing a role in the disruption of the blood-brain barrier by extracellular matrix cleavage. Activation of MMP9 before DKA treatment likely involves: hyperglycemia, ketones, methylglyoxal and endothelin 1 (ET-1) (Isales et al., 1999; Kim et al., 2012; Koyama et al., 2017); hypercortisolemia and free fatty acids (Boden and Song, 2008), in addition to the bidirectional modulation by inflammatory cytokines and chemokines (Strazielle et al., 2003), whose cleavage by MMP9 can result in a gain of function of some substrates (Van Lint and Libert, 2007). Also, of potential importance is that insulin has been reported to activate MMP9 (Fedorova et al., 2018). This possibility suggests a similar pattern of activation for inflammatory cytokines, like in systemic inflammatory response (SIR) and for complement activation during DKA treatment (Hoffman et al., 2003b; Jerath et al., 2005).
We investigated the intracerebral expression of MMP9 and TIMP1, along with the previously reported membrane attack complex (MAC) or C5b-9 (Hoffman et al., 2006) using immunohistochemistry in five brain regions (basal ganglia, choroid plexus, hippocampus, frontal cortex and cerebellum) of two adolescent cases with fatal DKA/brain edema (Hoffman et al., 2006, 2007). Our results show that the neuronal expression of MMP9 in the hippocampal areas of both BE/DKA cases was associated with decreased expression of TIMP1. In addition, significant expression of MMP9 was found on the cells present in brain intravascular areas. The presence of MMP9 in intravascular cells indicates a possible role in BBB tight junction protein disruption, possibly leading to neurological complications including BE.
MATERIALS AND METHODS
Case 1:
An adolescent female had a four-year history of poorly controlled T1D, which resulted in recurrent hospitalizations for DKA. The admission was preceded by a 12- hour history of abdominal pain and several episodes of emesis. There was no history of fever or enteritis. On physical examination, the patient was orientated but drowsy. There was no evidence of infection. The Tanner stage of puberty was B3 and P3. Admission laboratory tests consisted of a pH 7.10; pCO2 15 mmHg; pO2 106 mmHg; glucose 810 mg/dL; Na 132 mEq/L; K 5.7 mEq/L; Cl 93 mEq/L; HCO3 5 mEq/L; and BUN 30 mg/dL. The patient was treated in a Pediatric Intensive Care Unit lead for the correction of hyperglycemia and metabolic acidosis. Twelve hours following treatment, the child developed a mild headache. Seven hours later, she developed sudden onset of labored respirations and within 20 minutes had a cardio-respiratory arrest. An emergency CT scan of the head showed sulcal effacement and cerebral and pontine edema with evidence of herniation. Efforts at resuscitation were unsuccessful and she was pronounced dead after the cardio-respiratory arrest.
Case 2:
An adolescent female had an eight-year history of poorly controlled T1D, which had resulted in recurrent hospitalizations for DKA. The admission was preceded by an 18-hour history of capillary blood glucoses of over 300 mg/dl; ketonuria; a four-hour history of headache; and several episodes of emesis. There was no history of fever or enteritis. On physical examination, the patient was slightly confused and lethargic. The Tanner stage of puberty was B5 and P4. There was no evidence of infection. Admission laboratory tests consisted of: pH 7.16; pCO2 17 mm Hg; pO2 100 mmHg; blood glucose 581 mg/dL; Na 130 mEq/L; K 4.8 mEq/L; Cl 89 mEq/L; HCO3 6 mEq/L; and BUN 28 mg/dL. Treatment was in a Pediatric Intensive Care Unit and correction of the hyperglycemia metabolic acidosis was performed. Ten hours following initiation of treatment, she became unresponsive and was treated with mannitol and hyperventilation and placed on mechanical ventilation. An emergency CT scan of the head showed diffuse cerebral edema. She was pronounced dead approximately ten hours after the cardiorespiratory arrest.
Control normal brain tissue arrays were obtained from Pro Sci Incorporated (Poway, CA) and Chemicon (Temecula, CA).
Immunohistochemistry
The paraffin sections were processed as described previously (Hoffman et al., 2006). After xylene deparaffinization and epitope retrieval using a Target Retrieval Solution (DAKO, Carpinteria, CA), sections were treated with 3% H2O2 to remove endogenous peroxidase. Sections were preincubated for 30 min with normal goat serum then with a rabbit monoclonal antibody (mAb) against human MMP9 (CST, Danvers, MA) or a rabbit polyclonal antibody against human TIMP1 (Millipore Sigma, Burlington, MA) over-night at 4°C in a humid chamber. MMP9 (D6O3H) XP® Rabbit mAb recognizes the full-length, proenzyme (92 kDa) and the cleaved, active enzyme (84 kDa) of MMP9. In addition, we were unable to perform additional analysis by Western blot due to the lack of tissue to further confirm the specificity of the MMP9 antibody. Similar steps were performed for RGC-32 using a rabbit IgG anti-RGC-32 generated by us (Fosbrink et al., 2005) and for C5b-9 neoantigens with the monoclonal antibody against C5b-9 (Quidel, San Diego, CA) as described (Hoffman et al., 2006). Then sections were washed 3 times for 3 min at room temperature with Tris buffer saline pH 7.4 (TBS), and then incubated for 1hr at room temperature with HRP-conjugated goat anti-mouse IgG or HRP-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA). After washing, the specific deposits were developed using NovaRed (Vector Laboratories, Burlingame, CA). The nuclei were counterstained with Mayer’s hematoxylin (Sigma Chemical Co, St. Louis, MO). Controls for the specificity of each immunohistochemical reaction were performed by replacing the primary antibody with PBS, mouse, or rabbit IgG. Immunostaining was independently evaluated by two investigators in a blinded fashion.
Statistical analysis
Comparisons between groups were performed using unpaired two-tailed Student’s t-test. p values <0.05 were considered significant. Statistical analysis was performed using Graph Pad software, version 7.
Results
Immunohistochemical localization of MMP9 in DKA and control brains
Hippocampal neurons exhibit extensive MMP9 specific deposits (Figure 1A and B). We did not found similar MMP9 deposits in midbrain, substantia nigra or Purkinje neurons (Table 1). MMP9 deposits were also present on choroid plexus (Figure 1C). Controls of the immunoperoxidase reaction were negative (Figure 1D). MMP9 deposits were absent on neurons or choroid plexus of the control normal brains (Table 1). The number of neurons with MMP9 deposits in DKA patients and control brains was quantified by two independent observers. For each brain, the number of positive neurons was determined by counting 200 cells. We found a statistically significant increase of MMP9 expression in neurons in DKA brains as compared to control brains (p=0.002) (Figure 2A). In addition, we found that many brain blood vessels had positive intravascular cells for MMP9 (Figure 3A–D). The presence of MMP9 in intravascular cells and that of MMP9+ cells seen passing the BBB (Figure 3C–D, arrows) indicates a possible role in tight junction protein disruption of the BBB. Some MMP9+ cells are attached to the blood vessel wall (Figure 3D, arrowhead). Some of the intravascular cells had morphology compatible with neutrophils and lymphocytes (Figure 3B). Since the anti-MMP9 antibody used recognized both the latent and active form, we were unable to delineate the functional role of the molecule. Controls of immunoperoxidase reaction were negative (Figure 3E). There were no positive MMP9 intravascular cells in control normal brains (Table 1). These data suggest an increased and selective neuronal vulnerability of hippocampus in DKA/BE.
Figure 1. Immunohistochemical localization of MMP9 in DKA brains.

MMP9 was localized by indirect immunoperoxidase in DKA brains as described in Material and Methods section. We found that hippocampal neurons exhibit extensive MMP9 intracytoplasmatic staining (A, B). In addition, immunostaining for MMP9 was found in choroid plexus epithelial cells (C), while controls of immunoperoxidase reaction were negative (D). Original magnification: x400 (in A–C) and x200 (D).
Table 1. Expression of MMP9 and TIMP1 in DKA and normal brains.
−, negative; +, slightly positive; ++, positive; +++, highly positive; ND, not determined.
| Region | Intravascular | Neurons | |||
|---|---|---|---|---|---|
| MMP9 | TIMP1 | MMP9 | TIMP1 | ||
| DKA brain #1 | Basal ganglia | − | + | − | + |
| Hippocampus | ++ | + | ++ | + | |
| Choroid plexus | − | + | − | − | |
| Frontal cortex | + | + | − | + | |
| Cerebellum | − | + | − | ++ | |
| DKA brain #2 | Basal ganglia | ++ | + | − | ++ |
| Hippocampus | +++ | ++ | +++ | ++ | |
| Choroid plexus | + | ++ | − | − | |
| Frontal cortex | ++ | + | − | ++ | |
| Cerebellum | ++ | ++ | − | ++ | |
| Normal brain | Frontal cortex | − | ++ | − | ++ |
| Occipital cortex | − | ND | − | ND | |
| Parietal cortex | − | ++ | − | ++ | |
| Temporal cortex | − | ND | − | ND | |
| Cerebellum | − | ++ | − | ++ | |
| Pons | − | ND | − | ND | |
Figure 2. Quantitative analysis of the expression of MMP and TIMP1 in control and DKA brains.

The percentage of MMP9+ (A) and TIMP1+ (B) hippocampal neurons was determined by counting 200 cells from 3 controls and 2 DKA brains. We did not find MMP9+ neurons in control brains (A) and an arbitrary value of 1 was given. There was a statistically significant increase of MMP9+ neurons in DKA brains as compared to control brains (p=0.002). In addition, we found significantly higher levels of TIMP1 expression in neurons in the control brains as compared to DKA brains (p=0.03) (B). Data are shown as mean ± SEM.
Figure 3. Immunohistochemical localization of MMP9 in blood vessels in DKA brains.

MMP9 was localized by indirect immunoperoxidase in DKA brains. MMP9 was found to be localized in cells present in brain blood vessels (A–D). MMP9 positive cells were seen passing the BBB or to be present in the brain parenchyma (C, D, arrows). Some MMP9+ cells are attached to the blood vessel wall (D, arrowhead). The presence of MMP9 in intravascular cells and that of MMP9+ cells seen passing the BBB indicates a possible role in BBB disruption. Controls of immunoperoxidase reaction were negative (E). Original magnification: x400 (A, B, D) and x200 (C, E).
Immunohistochemical localization of TIMP1 in DKA and control brains
The main function of TIMPs is their inhibitory effect on MMPs. TIMPs irreversibly inactivate MMPs by direct binding to their catalytic zinc cofactor and resultant inhibition of proteinase function (Smigiel and Parks, 2017). It has been also shown that pro- MMP9 binds TIMP1 (Smigiel and Parks, 2017). In Case #2, TIMP1 was present intravascularly and in the neurons in all five regions studied (Figure 4A) but not in the choroid plexus (Table 1). Intravascular expression of TIMP1 was present in all five brain regions investigated for Case#1, with low degree of expression (Figure 4B, C). Low expression of TIMP1 was also found in the neurons of all regions studied (Table 1). Some of the intravascular cells which were TIMP1 positive had morphology typical for neutrophils while others for lymphocytes (Figure 4C). Controls of immunoperoxidase reaction were negative (Figure 4D). Concerning expression in hippocampus, the number of positive TIMP1 neurons was determined by counting 200 cells in DKA and control brains. We found significantly higher levels of TIMP1 expression in neurons in controls as compared to DKA brains (p=0.03) (Figure 2B).
Figure 4. Immunohistochemical localization of TIMP1 in DKA brains.

TIMP1 was localized by indirect immunoperoxidase in DKA brains as described in Materials and Methods section. Hippocampal neurons exhibit low TIMP1 staining (A). In addition, reduced staining of TIMP1 was found in cells present in the blood vessels (B, C). Some intravascular TIMP1+ cells had morphology typical for neutrophils (C, arrows), while others for lymphocytes (C, arrowhead). Controls of Immunoperoxidase were negative (D). Magnification: x400 (in A, C and D) and x200 (in B).
Immunohistochemical localization of C5b-9 and RGC-32 in DKA brains
We investigated the presence of C5b-9 and RGC-32 in hippocampal areas of the brains as these areas also had MMP9 expression. As shown in Figure 5A and B, C5b-9 (MAC) deposits were found to be present on hippocampal neurons as previously described (Hoffman et al., 2006). In addition, RGC-32 was found to be expressed in the cytoplasm of hippocampal neurons (Figure 5C, D). Since we previously showed that C5b-9 induces RGC-32 expression (Badea et al., 2002), it is possible that RGC-32 was induced in hippocampal neurons by the C5b-9 assembly.
Figure 5. Immunohistochemical localization of C5b-9 neoantigens and RGC-32 in DKA brains.

C5b-9 and RGC-32 were localized by indirect immunoperoxidase in DKA brains. We found that hippocampal neurons exhibit extensive intracytoplasmatic C5b-9 deposits (A, B); RGC-32 was found to be highly expressed in cytoplasm of hippocampal neurons (C, D). Magnification: x200 (A, C); x400 (B, D).
Discussion
MMP9 expression in the pathogenesis of acute cerebrovascular accidents unrelated to DKA has been well studied. Recently, it has been hypothesized that MMP9 is a candidate for the disruption of BBB tight junction proteins (TJP) in DKA (Woo et al., 2016a, 2016b; Garro et al., 2017). Important contributors to this perturbation include: 1) systemic leukocytosis with neutrophil predominance, as seen in DKA, which can result in increased MMP9 levels (Turner and Sharp, 2016); 2) MMP9 production by astrocytes, important cellular components of the neurovascular unit of the BBB (Hsieh et al., 2013, 2014); 3) activation by increased oxidative stress (Pun et al., 2009), a major product of DKA’s dysfunctional metabolic state that initiates the superoxide-MMP9 cascade; 4) activation by the endothelin (ET) system, along with vascular endothelial growth factor (VEGF) (a perturbator of the BBB) (Koyama and Michinaga, 2012); and 5) modulation of both systemic and local inflammation (Manicone and McGuire, 2008; Nissinen and Kahari, 2014). Unfortunately, the metabolic (Lee et al., 2002; Carl et al., 2002; Hoffman et al., 2003a) and immunologic crisis of DKA (Hoffman et al., 2003b; Jerath et al., 2005; Karavanaki et al., 2011; Close et al., 2013; Hoffman et al., 2013) can occur frequently in young patients, resulting in insults to the brain and other organs and leading to early cognitive deficits (Jessup et al., 2015; Nunley et al., 2015) and progressive chronic subclinical renal complications. Both comorbidities have much a greater frequency (Hursh et al., 2017) than does the tragic acute complication of clinical BE (Tasker and Acerini, 2014; Patel et al., 2016; Peeters et al., 2018).
The hippocampus was the only location in which neuronal expression of MMP9 was found. This hippocampal location of MMP9 expression is in keeping with the expression of the other inflammatory components, particularly C5b-9 and RGC-32, along with the increased neuronal deficits and neuronal loss occurring in this region (Figure 6) (Hoffman et al., 2006, 2008, 2009; Cameron et al., 2014). C5b-9 has been shown to induce MMP9 expression (Wu et al., 2010), which is also dependent on RGC-32 (Xu et al., 2014). It is possible that MMP9 expression in hippocampal neurons during DKA is induced by C5b-9 assembly (Hoffman et al., 2006), with a possible contribution from RGC-32. RGC-32 mRNA and protein levels have been found to be upregulated in the brains of patients with mild cognitive impairment and Alzheimer disease. In addition, high brain levels of RGC-32 protein are associated with poorer antemortem global cognitive performance (Counts and Mufson, 2017). The increased expression of RGC-32 found in hippocampal neurons may reflect neuronal degeneration during DKA. Published reports indicate that neurons are capable of synthesizing MMP9 (Kaplan et al., 2014), and this synthesis may result in an increased stress on vulnerable neurons, which could trigger the degeneration of these cells (Kaplan et al., 2014). Thus, is possible that MMP9 inhibition or modulation can be beneficial for neuronal preservation. The goal of protecting against MMP9’s harmful effects in DKA is in line with therapeutic strategies used in inflammatory diseases and cancer that would block the damaging effects of MMP9 without inhibiting its beneficial actions.
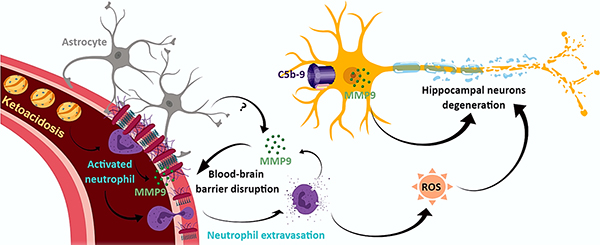
Figure 6. Schematic representation of the hypothetical role of MMP9 in brain dysfunction during DKA.

Our data suggest that during DKA, MMP9 is secreted by inflammatory cells and may be involved in BBB disruption, which leads to neutrophil infiltration into the brain parenchyma. In addition, activation of complement system with the assembly of terminal complement complex C5b-9 occurs both in plasma and in the brain tissue. It is possible that C5b-9 induces the expression of MMP9 particularly in hippocampal neurons, possibly by activating RGC-32. This increase in MMP9 expression may lead to hippocampal neuron dysfunction and degeneration. MMP9 intravascular and neuronal expression may also be facilitated also by the decreased expression of its natural inhibitor, TIMP1.
We have also reported significant neuroinflammation of the choroid plexus (CP) (Hoffman et al., 2007), a site of major importance in view of its role in bioactive organic anion and neuro-immune regulation (Strazielle et al., 2003). Unfortunately, studying the CP was precluded because of limited tissue availability. The previous MMP9/DKA studies (Woo et al., 2016a, 2016b; Garro et al., 2017) did not allow for identifying a peak time for the systemic concentration in relation to the 6- to 12-hour duration of the SIR following the initiation of insulin.
Although ischemia of the cerebrovascular tight junctions and the presence of MMP9 have been extensively researched (Turner and Sharp, 2016), we were unable to identify any histopathologic evidence of hemorrhage to indicate the occurrence of vascular compromise in either of the two brains, suggesting that there are other metabolic and immunologic initiating/activating factors of MMP9 in DKA. The intracerebral presence of MMP9 in DKA extends the previously described inflammatory phenotype and adds another mediator for consideration as a participant in BBB perturbation during DKA (Figure 6). This study is limited by the fact that it relies only on immunohistochemical analysis, and therefore there are no functional data to show a role in alerting BBB or neuronal function. In addition, while we believe that our study extends the previous observations by Woo et al. (Woo et al., 2016a, 2016b) and Garro et al. (Garro et al., 2017), our investigation is limited to brain samples obtained postmortem from two DKA patients, and therefore the results must be interpreted with caution.
The reduction in TIMP1 expression associated with other possible inhibitors may affect MMP9 levels. Other possible MMP9 inhibitors include: 1) heat shock protein (HSP) 70 (Lee et al., 2002), which is increased in DKA, paralleling changes in serum glucose levels (Oglesbee et al., 2005); and 2) IL-10, an anti-inflammatory molecule, which, like HSP70 is elevated prior to DKA treatment; both these anti-inflammatory molecules decrease rapidly with the initiation of insulin treatment (Hoffman et al., 2003a; Oglesbee et al., 2005). In contrast, insulin treatment is reported to increase MMP9 expression (Mostafa et al., 2001), akin to the increase in inflammatory cytokines that occurs during SIR/DKA (Hoffman et al., 2003b; Karavanaki et al., 2011; Close et al., 2013; Hoffman et al., 2013) and complement activation (Jerath et al., 2005) during DKA treatment.
In conclusion, we speculate that MMP9 plays a dual role during DKA/BE by perturbing BBB permeability and possibly contributing to neuronal dysfunction and loss. Further studies are necessary to establish that MMP9 plays a definite role in the pathogenesis of subclinical and clinical BE in DKA.
Highlights:
MMP9 is extensively expressed in intravascular cells from DKA brains.
MMP9 is expressed in hippocampal neurons in DKA brains.
TIMP1 is expressed at lower levels in intravascular cells and hippocampal neurons.
Hippocampal neurons from DKA brains express high levels of RGC-32 and C5b-9.
Further studies are needed to establish MMP9 role in DKA brains.
ACKNOWLEDGEMENTS
This work was supported in part by US Public Health Grants, RO1 NS42011 (to HR).
Abbreviations
- BBB
blood-brain barrier
- BE
brain edema
- CP
choroid plexus
- DKA
diabetic ketoacidosis
- HSP
heat shock protein
- MAC
membrane attack complex
- MMP9
matrix metalloproteinase 9
- RGC-32
Response Gene to Complement-32
- SIR
systemic inflammatory response
- T1D
type 1 diabetes mellitus
- TJP
tight junction proteins
- TIMP1
tissue inhibitor of metalloproteinases 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Badea T, Niculescu F, Soane L, Fosbrink M, Hila H, Rus V, Shin ML, Rus H, 2002. RGC-32 increases p34CDC2 kinase activity and entry of aortic smooth muscle cells into S-phase. J Biol Chem. 277, 502–8. [DOI] [PubMed] [Google Scholar]
- Boden G, Song WW, 2008. Effects of insulin and free fatty acids on matrix metalloproteinases. Curr. Diab. Rep 8, 239–242. [DOI] [PubMed] [Google Scholar]
- Cameron FJ, Scratch SE, Nadebaum C, Northam EA, Koves I, Jennings J, Finney K, Neil JJ, Wellard RM, Mackay M, Inder TE; DKA Brain Injury Study Group, 2014. Neurological consequences of diabetic ketoacidosis at initial presentation of type 1 diabetes in a prospective cohort study of children. Diabetes Care. 37, 1554–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carl GF, Hoffman WH, Blankenship PR, Litaker MS, Hoffman MG, Mabe PA, 2002. Diabetic ketoacidosis depletes plasma tryptophan. Endo. Res 28, 91–102. [DOI] [PubMed] [Google Scholar]
- Close TE, Cepinskas G, Omatsu T, Rose KL, Summers K, Patterson EK, Fraser DD. 2013. Diabetic ketoacidosis elicits systemic inflammation associated with cerebrovascular endothelial dysfunction. Microcirculation. 20, 534–543. [DOI] [PubMed] [Google Scholar]
- Counts SE, Mufson EJ, 2017. Regulator of Cell Cycle (RGCC) Expression during the Progression of Alzheimer’s Disease. Cell Transplant. 26, 693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorova NV, Ksenofontov AL, Serebryakova MV, Stadnichuk VI, Gaponova TV, Baratova LA, Sud’ina GF, Galkina SI, 2018. Neutrophils release metalloproteinases during adhesion in the presence of insulin, but cathepsin G in the presence of glucagon. Mediators Inflamm. 14,1574928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Cen J, Huang Y, Shen H, Yao L, Wang Y, Chen Z, 2011. Matrix metalloproteinase-2 and-9 secreted by leukemic cells increase the permeability of blood-brain barrier by disrupting tight junction proteins. PLoS One. 6, e20599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fosbrink M, Cudrici C, Niculescu F, Badea TC, David S, Shamsuddin A, Shin ML, Rus H, 2005. Overexpression of RGC-32 in colon cancer and other tumors. Exp Mol Pathol. 78, 116–22. [DOI] [PubMed] [Google Scholar]
- Garro A, Chodobski A, Szmydynger-Chodobska J, Shan R, Bialo SR, Bennett J, Quayle K, Rewers A, Schunk JE, Casper TC, Kuppermann N, Glaser N; Pediatric Emergency Care Applied Research Network (PECARN), 2017. Circulating matrix metalloproteinases in children with diabetic ketoacidosids. Pediatr. Diabetes 18, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heier M, Margeirsdottir HD, Brunborg C, Hanssen KF, Dahl-Jørgensen K, Seljeflot I, 2015. Inflammation in childhood Type 1 diabetes; influence of glycemic control. Atherosclerosis. 38, 233–237. [DOI] [PubMed] [Google Scholar]
- Hoffman WH, Cheng C, Passmore GG, Carroll JE, Hess D, 2002. Acetoacetate increases expression of intercellular adhesion molecule-1 (ICAM-1) in human brain microvascular endothelial cells. Neurosci. Lett 334, 71–74. [DOI] [PubMed] [Google Scholar]
- Hoffman WH, Kappler F, Passmore GG, Mehta R, 2003a. Diabetic ketoacidosis and its treatment increase plasma 3-deoxyglucosone. Clin. Biochem 36, 269–273. [DOI] [PubMed] [Google Scholar]
- Hoffman WH, Burek CL, Waller JL, Fisher LE, Khichi M, Mellick LB, 2003b. Cytokine response to diabetic ketoacidosis and its treatment. Clin. Immunol 108, 175–181. [DOI] [PubMed] [Google Scholar]
- Hoffman WH, Cudrici CD, Zafranskaia E, Rus H, 2006. Complement activation in diabetic ketoacidosis brains. Exp. Mol. Pathol 80, 283–288. [DOI] [PubMed] [Google Scholar]
- Hoffman WH, Casanova MF, Cudrici CD, Zakranskaia E, Venugopalan R, Nag S, Oglesbee MJ, Rus H. 2007. Neuroinflammatory response of the choroid plexus epithelium in fatal diabetic ketoacidosis. Exp. Mol. Pathol 83, 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman WH, Artlett CM, Zhang W, Kreipke CW, Passmore GG, Rafols JA, Sima AA, 2008. Receptor for advanced glycation end products and neuronal deficit in fatal brain edema of diabetic ketoacidosis. Brain Res. 1238, 154–162. [DOI] [PubMed] [Google Scholar]
- Hoffman WH, Stamatovic SM, Andjelkovic AV, 2009. Inflammatory mediators and blood brain disruption in fatal brain edema of diabetic ketoacidosis. Brain Res. 1254, 138–148. [DOI] [PubMed] [Google Scholar]
- Hoffman WH, Passmore GG, Hannon DW, Talor MV, Fox P, Brailer C, Haislip D, Keel C, Harris G, Rose NR, Fiordalisi I, Čiháková D, 2013. Increased systemic Th17 cytokine are associated with diastolic dysfunction in children and adolescents with diabetic ketoacidosis. PLoS One. 8, e71905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh HL, Lin CC, Hsiao LD, Yang CM, 2013. High glucose induces reactive oxygen species-dependent matrix metalloproteinase-9 expression and cell migration in brain astrocytes. Mol. Neurobiol 48, 601–614. [DOI] [PubMed] [Google Scholar]
- Hsieh HL, Chi PL, Lin CC, Yang CC, Yang CM, 2014. Up-regulation of ROS- dependent matrix metalloproteinase-9 from high-glucose-challenged astrocytes contributes to the neuronal apoptosis. Mol. Neurobiol 50, 520–533. [DOI] [PubMed] [Google Scholar]
- Hursh BE, Ronsley R, Islam N, Mammen C, Panagiotopoulos C, 2017. Acute kidney injury in children with type 1 diabetes hospitalized for diabetic ketoacidosis. JAMA Pediatr. 171, e170020. [DOI] [PubMed] [Google Scholar]
- Isales CM, Min L, Hoffman WH, 1999. Acetoacetate and beta-hydroxybutyrate differentially regulate endothelin-1 and vascular endothelial growth factor in mouse brain microvascular endothelial cells. J. Diabetes Complications. 13, 91–97. [DOI] [PubMed] [Google Scholar]
- Jacqueminet S, Ben Abdesselam O, Chapman MJ, Nicolay N, Foglietti MJ, Grimaldi A, Beaudeux JL, 2006. Elevated circulating levels of matrix metalloproteinase-9 in type 1 diabetic patients with and without retinopathy. Clin. Chim. Acta 367, 103–107. [DOI] [PubMed] [Google Scholar]
- Jerath RS, Burek CL, Hoffman WH, Passmore GG, 2005. Complement activation in diabetic ketoacidosis and its treatment. Clin. Immunol 116, 11–17. [DOI] [PubMed] [Google Scholar]
- Jessup AB, Grimley MB, Meyer E, Passmore GP, Belger A, Hoffman WH, Çalıkoğlu AS1, 2015. Effects of diabetic ketoacidosis on visual and verbal neurocognitive function in young patients presenting with new-onset type 1 diabetes. J. Clin. Res. Pediatr. Endocrinol 7, 203–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan A, Spiller KJ, Towne C, Kanning KC, Choe GT, Geber A, Akay T, Aebischer P, Henderson CE, 2014. Neuronal matrix metalloproteinase-9 is a determinant of selective neurodegeneration. Neuron. 81, 333–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karavanaki K, Karanika E, Georga S, Bartzeliotou A, Tsouvalas M, Konstantopoulos I, Fotinou A, Papassotiriou I, Karayianni C, 2011. Cytokine response to diabetic ketoacidosis (DKA) in children with type 1 diabetes (T1DM). Endocr. J 58, 1045–1053. [DOI] [PubMed] [Google Scholar]
- Kim J, Kim CS, Lee YM, Jo K, Shin SD, Kim JS, 2012. Methylglyoxal induces hyperpermeability of the blood-retinal barrier via the loss of tight junction proteins and the activation of matrix metalloproteinases. Graefes Arch. Clin. Exp. Ophthalmol 250, 691–697. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Michinaga S, 2012. Regulations of astrocytic functions by endothelins: roles in the pathophysiological responses of damaged brains. J. Pharmacol. Sci 118, 401–407. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Ukita A, Abe K, Iwamae K, Tokuyama S, Tanaka K, Kotake Y, 2017. Dexamethasone downregulates endothelin receptors and reduces endothelin-induced production of matrix metalloproteinases in cultured rat astrocytes. Mol. Pharmacol 92, 57–66. [DOI] [PubMed] [Google Scholar]
- Lavini-Ramos C, Silva HM, Soares-Schanoski A, Monteiro SM, Ferreira LRP, Pacanaro AP, Gomes S, Batista J, Faé K, Kalil J, Coelho V, 2017. MMP integrates multiple immunoregulatory pathways that discriminate high suppressive activity of human mesenchymal cells. Sci. Report 7, 874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes-Virella MF, Baker NL, Hunt KJ, Lyons TJ, Jenkins AJ, Virella G; DCCT/EDIC Study Group, 2012. High concentrations of AGE-LDL and oxidized LDL in circulating immune complexes are associated with progression of retinopathy of type 1 diabetes. Diabetes Care. 35, 1333–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DM, Hoffman WH, Carl GF, Khichi M, Cornwell PE, 2002. Lipid peroxidation and antioxidant vitamins prior to, during, and after correction of diabetic ketoacidosis. J. Diabetes Complications. 16, 294–300. [DOI] [PubMed] [Google Scholar]
- Manicone AM, McGuire JK, 2008. Matrix metalloproteinases as modulators of inflammation. Semin. Cell. Dev. Biol 19, 34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell PR, Timms PM, Chandran S, Gordon D, 2001. Peripheral blood level alterations of TIMP-1, MMP-2 and MMP-9 in patients with type 1 diabetes. Diabet. Med 18, 777–780. [DOI] [PubMed] [Google Scholar]
- Mostafa Mtairag E, Chollet-Martin S, Oudghiri M, Laquay N, Jacob MP, Michel JB, Feldman LJ, 2001. Effects of interleukin-10 on monocyte/endothelial cell adhesion and MMP-9/TIMP-1 secretion. Cardiovasc. Res 49, 882–890. [DOI] [PubMed] [Google Scholar]
- Nissinen L, Kahari VM, 2014. Matrix metalloproteinases in inflammation. Biochim. Biophys. Acta 1840, 2571–2580. [DOI] [PubMed] [Google Scholar]
- Nunley KA, Rosano C, Ryan CM, Jennings JR, Aizenstein HJ, Zgibor JC, Costacou T, Boudreau RM, Miller R, Orchard TJ, Saxton JA, 2015. Clinically Relevant Cognitive Impairment in Middle-Aged Adults with Childhood-Onset Type 1 Diabetes. Diabetes Care. 38, 1768–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oglesbee MJ, Herdman AV, Passmore GG, Hoffman WH, 2005. Diabetic ketoacidosis increases extracellular levels of the major inducible 70-kDa heat shock protein. Clin. Biochem 38, 900–904. [DOI] [PubMed] [Google Scholar]
- Omatsu T, Cepinskas G, Clarson C, Patterson EK, Alharfi IM, Summers K, Couraud PO, Romero IA, Weksler B, Fraser DD; Canadian Critical Care Translational Biology Group, 2014. CXCL1/CXCL8 (GROα/IL-8) in human diabetic ketoacidosis plasma facilitates leukocyte recruitment to cerebrovascular endothelium in vitro. Am J Physiol Endocrinol Metab. 306, E1077–84. [DOI] [PubMed] [Google Scholar]
- Patel A, Singh D, Bhatt P, Thakkar B, Akingbola OA, Srivastav SK, 2016. Incidence, trends, and outcomes of cerebral edema among children with diabetic ketoacidosis in the United States. Clin. Pediatr 55, 943–951. [DOI] [PubMed] [Google Scholar]
- Peeters SA, Engelen L, Buijs J, Jorsal A, Parving HH, Tarnow L, Rossing P, Schalkwijk CG, Stehouwer CDA, 2017. Plasma matrix metalloproteinases are associated with incident cardiovascular disease and all-cause mortality in patients with type 1 diabetes: a 12-year follow-up study. Cardiovasc. Diabetol 16, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters SA, Engelen L, Buijs J, Theilade S, Rossing P, Schalkwijk CG, Stehouwer CDA, 2018. Associations between advanced glycation end-products and matrix metalloproteinases and its inhibitor in individuals with type 1 diabetes. J Diabetes Complications. 32, 325–329. [DOI] [PubMed] [Google Scholar]
- Pun PB, Lu J, Moochhala S, 2009. Involvement of ROS in BBB dysfunction. Free Radic. Res 43, 348–364. [DOI] [PubMed] [Google Scholar]
- Ram M, Sherer Y, Shoenfeld Y, 2006. Matrix metalloproteinase-9 and autoimmune diseases. J. Clin. Immunol 26, 299–307. [DOI] [PubMed] [Google Scholar]
- Skrha J Jr, Kalousová M, Svarcová J, Muravská A, Kvasnička J, Landová L, Zima T, Skrha J, 2012. Relationship of soluble RAGE and RAGE ligands HMGB1 and EN- RAGE to endothelial dysfunction in type 1 and type 2 diabetes mellitus. Exp. Clin. Endocrinol. Diabetes 120, 277–281. [DOI] [PubMed] [Google Scholar]
- Smigiel KS, Parks WC, 2017. Matrix metalloproteinases and leukocyte activation. Prog. Mol. Biol. Transl. Sci 147, 167–195. [DOI] [PubMed] [Google Scholar]
- Speidl WS, Kastl SP, Hutter R, Katsaros KM, Kaun C, Bauriedel G, Maurer G, Huber K, Badimon JJ, Wojta J, 2011. The complement component C5a is present in human coronary lesions in vivo and induces the expression of MMP-1 and MMP-9 in human macrophages in vitro. FASEB J. 25, 35–44. [DOI] [PubMed] [Google Scholar]
- Strazielle N, Khuth ST, Murat A, Chalon A, Giraudon P, Belin MF, Ghersi-Egea JF, 2003. Pro-inflammatory cytokines modulate matrix metalloproteinase secretion and organic anion transport at the blood-cerebrospinal fluid barrier. J. Neuropathol. Exp. Neurol 62, 1254–1264. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Saito M, Nagai T, Saeki H, Kazatani Y, 2006. Systemic versus coronary levels of inflammation in acute coronary syndromes. Angiology. 57, 459–463. [DOI] [PubMed] [Google Scholar]
- Symeonidis C, Papakonstantinou E, Galli A, Tsinopoulos I, Mataftsi A, Batzios S, Dimitrakos SA, 2013. Matrix metalloproteinase (MMP-2,−9) and tissue inhibitor (TIMP-1,−2) activity in tear samples of pediatric type 1 diabetic patients: MMPs in tear samples from type 1 diabetes. Graefes Arch Clin. Exp. Ophthalmol 251, 741–749. [DOI] [PubMed] [Google Scholar]
- Tasker RC, Acerini CL, 2014. Cerebral edema in children with diabetic ketoacidosis: vasogenic rather than cellular? Pediatr. Diabetes 15, 261–270. [DOI] [PubMed] [Google Scholar]
- Turk Z, Nemet I, Varga-Defteardarović L, Car N, 2006. Elevated level of methylglyoxal during diabetic ketoacidosis and its recovery phase. Diabetes Metab. 32, 176–180. [DOI] [PubMed] [Google Scholar]
- Turner RJ, Sharp FR, 2016. Implications of MMP9 for Blood Brain Barrier Disruption and Hemorrhagic Transformation Following Ischemic Stroke. Front Cell Neurosci. 10, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint P, Libert C, 2007. Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration. J. Leukoc. Biol 82, 1375–1381. [DOI] [PubMed] [Google Scholar]
- Wieczorek E, Jablonowski Z, Tomasik B, Konecki T, Jablonska E, Gromadzinska J, Fendler W, Sosnowski M, Wasowicz W, Reszka E, 2015. MMP, VEGF and TIMP as prognostic factors in recurring bladder cancer. Clin. Biochem 48, 1235–1240. [DOI] [PubMed] [Google Scholar]
- Woo MM, Patterson EK, Clarson C, Cepinskas G, Bani-Yaghoub M, Stanimirovic DB, Fraser DD, 2016a. Elevated leukocyte azurophilic enzymes in human diabetic ketoacidosis plasma degrade cerebrovascular endothelial junctional proteins. Crit. Care Med. 44, e846–853. [DOI] [PubMed] [Google Scholar]
- Woo M, Patterson EK, Cepinskas G, Clarson C, Omatsu T, Fraser DD, 2016b. Dynamic regulation of plasma matrix metalloproteinases in human diabetic ketoacidosis. Ped. Res 79, 295–300. [DOI] [PubMed] [Google Scholar]
- Wu G, Chen T, Shahsafaei A, Hu W, Bronson RT, Shi GP, Halperin JA, Aktas H, Qin X. 2010. Complement regulator CD59 protects against angiotensin II-induced abdominal aortic aneurysms in mice. Circulation. 121, 1338–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Shang C, Zhao J, Han Y, Liu J, Chen K, Shi W, 2014. Knockdown of response gene to complement 32 (RGC-32) induces apoptosis and inhibits cell growth, migration, and invasion in human lung cancer cells. Mol Cell Biochem. 394, 109–18. [DOI] [PubMed] [Google Scholar]