

Abstract
Fluorescent indicators are widely used to visualize calcium dynamics downstream of membrane depolarization or G protein-coupled receptor activation, but are poorly suited for non-invasive imaging in mammals. Here, we report a bright calcium-modulated bioluminescent indicator named Orange CaMBI. Orange CaMBI reports calcium dynamics in single cells and, in the context of a transgenic mouse, reveals calcium oscillations in whole organs in an entirely noninvasive manner.
Cytosolic calcium is dynamically regulated as a second messenger for a variety of extracellular and intracellular signals1. Fluorescent genetically encoded calcium indicators (GECIs) are routinely used for calcium imaging in living cells2, and enable high temporal resolution due to their high photon output rates3. However, fluorescence imaging in deep mammalian tissues requires insertion of optical elements to minimize autofluorescence, phototoxicity, and attenuation of excitation light4–6. GECIs that use bioluminescence, in which catalysis of a chemical substrate by a luciferase enzyme produces light7, are potentially well suited for noninvasive whole-animal imaging. As excitation light is not needed, bioluminescence imaging involves simply detecting emitted light. As no autofluorescence is generated and as mammalian tissues lack endogenous bioluminescence, even relatively small numbers of photons can be detected over background with bioluminescence8.
However, bioluminescent imaging of cytosolic calcium from deep locations in mammals has not yet been described. Limiting factors have been the rate of light production and the efficiency of light transmission through tissue, which together must produce enough light to track dynamic calcium transients9. Existing bioluminescent GECIs either exhibit low emission rates or produce blue or green light that is highly attenuated by tissue. For example, Redquorin, a fusion of the natural calcium-dependent luciferase aequorin and the orange fluorescent protein tdTomato, produces orange emission via resonance energy transfer (RET)9, but is hindered by aequorin’s catalytic rate of 1 reaction per 22 min10. BAF-Y and Nanolantern(Ca2+), created from Renilla luciferase 8 (RLuc8), calcium-sensing domains, and yellow fluorescent proteins (YFPs), are more highly catalytic but have negligible emission in the more tissue-penetrating wavelengths above 600 nm11,12.
We set out to engineer a bioluminescent calcium indicator with high emission rates above 600 nm starting from the protein Antares, which produces bright orange-red bioluminescence13. Antares consists of Nano luciferase (NanoLuc) fused to two CyOFP1 fluorescent protein domains. NanoLuc catalyzes 6.6 reactions per s per molecule, 1.7-fold faster than RLuc8 and 8700-fold faster than aequorin14. To make Antares calcium-sensitive, we inserted the CaM-GGS-M13 cassette from the YC-Nano140 fluorescent GECI15 into loops in the NanoLuc domain (Supplementary Fig. 1a). Insertion at position 133 (Fig. 1a) conferred responsivity to calcium while maintaining high maximal brightness (Supplementary Table 1). Linker optimization then further improved responsivity (Supplementary Table 2), yielding a protein with 8-fold responsivity to calcium and 44% of the brightness of Antares in the calcium-saturated state (Fig. 1b, Supplementary Fig. 1b). This protein, named Orange Calcium-Modulated Bioluminescent Indicator (Orange CaMBI), preserves the original linkages between NanoLuc and CyOFP1 domains of Antares (Supplementary Fig. 1c). The calcium-independence of the spectral shape (Fig. 1b) indicates that calcium modulates the catalytic rate of NanoLuc rather than its luminescence quantum yield (QY). The spectral shape is also unchanged from that of Antares, indicating that the luminescence QY of the NanoLuc domain is not affected by the CaM-GGS-M13 insertion (Fig. 1b). We were thus able to create a Blue CaMBI with similar calcium responsivity by removal of the CyOFP1 domains (Supplementary Fig. 2a), and then a Green CaMBI by attachment of EGFP to Blue CaMBI (Supplementary Fig. 2b).
Figure 1. CaMBI characteristics and Imaging calcium activity with Orange CaMBI reporters.
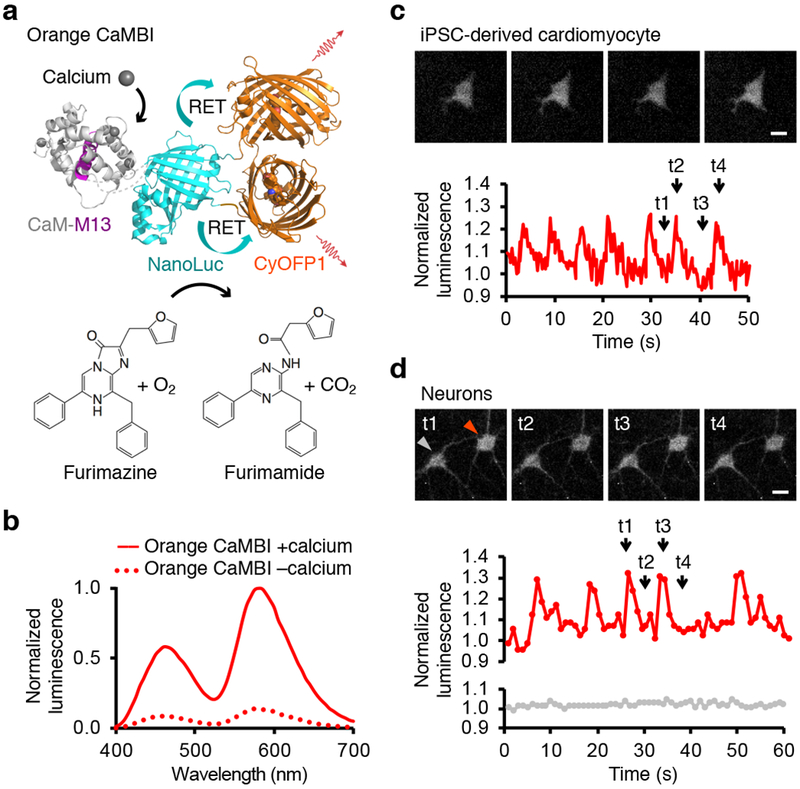
(a) Model of Orange CaMBI in the calcium-bound state. CaM and M13 domains are inserted after Leu133 of NanoLuc, which is fused at its amino and carboxyl termini to two copies of CyOFP1. Binding of calcium (gray spheres) to CaM induces binding of CaM to M13, enhancing NanoLuc activity. RET to CyOFP1 shifts emission to orange and red wavelengths and increases quantum yield. Model based on PDB files 2BBM for CaM-M13, 5IBO for NanoLuc, and 5BQ1 for CyOFP1. (b) Emission spectrum and calcium responsivity of Orange CaMBI. Spectra were acquired from bacterial lysates diluted into buffers with 0 or 39 μM free calcium, then normalized to peak emission in the calcium-bound state. (c) Imaging spontaneous calcium transients in cardiomyocytes differentiate from human induced pluripotent stem cells (iPSCs) with Orange CaMBI 110. Images were collected at 4 fps. Top, luminescence images at time points t1 through t4. Bottom, chart of integrated luminescence signal from one cell normalized to beginning value. Normalization to a monoexponential decay function fit to an Antares signal acquired under identical conditions was performed to correct for signal decay. (d) Imaging spontaneous calcium transients in a neuron with Orange CaMBI 110. Images were collected at 1 fps. Top, luminescence images at time points t1 through t4, with arrow indicating analyzed cell. Bottom, integrated luminescence signal from one cell body normalized to beginning value. Baseline correction was not performed as luminescence decay was negligible in these conditions. Scale bars, 10 μm.
RET from NanoLuc to fluorescent protein acceptors could increase photon output per reaction if the acceptor fluorescence QY exceeds the NanoLuc luminescence QY. We calculated the total luminescence QY of each CaMBI from measured RET efficiencies (Supplementary Fig. 2c,d) and previously measured QYs for NanoLuc (0.37) and fluorescent protein acceptors, yielding 0.47 for Orange CaMBI and 0.40 for green CaMBI (Supplementary Fig. 2e, Supplementary Table 3). Above 600 nm, Orange CaMBI has a QY of 0.16, 16-fold higher than Green CaMBI and 34-fold higher than Blue CaMBI. Comparing measured brightness to QYs revealed that the attachment of fluorescent protein domains in Orange CaMBI and Green CaMBI did not impede activity, with Orange CaMBI actually exhibiting a small increase relative to Blue CaMBI (Supplementary Fig. 2f).
The original Orange CaMBI exhibited a dissociation constant (KD) for calcium of 110 nM and a Hill coefficient of 2.5 (Supplementary Fig. 3a, Supplementary Table 4), and was thus designated Orange CaMBI 110. Orange CaMBI 110 should respond to small changes in cytosolic calcium from typical resting concentrations of ~100 nM with large absolute changes in luminescence. We also shortened the linker between CaM and M13 to decrease affinity12,15, resulting in variants with KD values of 200, 270, and 300 nM (Supplementary Fig. 3a, Supplementary Table 4). Brightness of calcium-saturated Orange CaMBI 110 and Orange CaMBI 300 increased with pH, with brightness at pH 7.0 reaching half that of pH 8.5 (Supplementary Fig. 3b). Coincidentally, this is similar to the GCaMP6 series of fluorescent GECIs16, although the pH sensitivity of Orange CaMBI likely derives from the CaM-M13-NanoLuc component and not from CyOFP1, as pH changes from 7.0 to 8.5 have only a minor effect on CyOFP1 brightness13.
We next tested whether CaMBIs could detect calcium influx in living cells. Green and Orange CaMBIs reported calcium oscillations in HeLa cells induced by histamine stimulation (Supplementary Fig. 4a,b, Supplementary Videos 1–2). Orange CaMBI 110 also detected periodic calcium transients in spontaneously beating cardiomyocytes derived from human induced pluripotent stem cells (Fig. 1c, Supplementary Video 3). In these experiments, Antares served as a negative control and to reveal any bioluminescence decay from substrate depletion (Supplementary Fig. 4c,d). Finally, both Orange CaMBI 110 and Orange CaMBI 300 detected spontaneous calcium fluctuations in cultured rat hippocampal neurons (Fig. 1d, Supplementary Fig. 4e, Supplementary Videos 4–5). As these calcium events are transient, fill entire neurons, and irregularly spaced, they most likely represent action potentials resulting from network activity.
We next compared Orange CaMBIs in living cells to other bioluminescent GECIs, including CalfluxVTN17 and GeNL(Ca2+)14, which were developed while this work was in progress. In CalfluxVTN, NanoLuc is fused to troponin C (TnC) and a YFP (Supplementary Fig. 5a) so that calcium induces a 4-fold increase in YFP emission. In GeNL(Ca2+), NanoLuc is fused to mNeonGreen to improve luminescence QY (Supplementary Fig. 5b), and CaM and M13 domains are inserted into a different site in NanoLuc to produce 5-fold maximum responses to calcium. As in Orange CaMBI 110, the presence of the fluorescent protein acceptor in these reporters has the effect of increasing the luminescence QY of the calcium-bound state (Supplementary Table 3). We characterized the responses of CalfluxVTN, GeNL(Ca2+), Orange CaMBI 110, and Orange CaMBI 300 to calcium entry upon KCl-mediated depolarization in rat hippocampal neurons, using the preferred substrate for each indicator (Supplementary Fig. 6a). As the blue emission of NanoLuc would be well absorbed by thick tissue, we collected only the acceptor channel for each indicator. Responses of the Orange CaMBI variants after depolarization reached maximum values in less time than CalfluxVTN or GeNL(Ca2+) (Supplementary Fig. 6b,c), and exhibited a transient peak similar to those observed with calcium dyes upon KCl-induced depolarization of neurons18,19. GeNL(Ca2+) and Orange CaMBI 300 showed a trend to larger responses than the other indicators, although this was not statistically significant (Supplementary Fig. 6b,c).
We also tested the aequorin-based Redquorin responses in neurons, as photons above 600 nm are preferentially detected through mammalian tissues13, and Redquorin is the only other bioluminescent GECI with a large emission fraction above 600 nm. However, Redquorin is predicted to produce photons at a ~1000-fold slower rate than Orange CaMBI 110 (Supplementary Table 3). Consistent with this, Redquorin bioluminescence from individual neurons was not detectable above camera noise, although it was expressed to similar levels as Orange CaMBI 110 based on direct fluorescence imaging of the tdTomato component (Supplementary Fig. 6a,b), and although its luminescence could be detected in mouse liver following hydrodynamic transfection (Supplementary Fig. 6d). As Redquorin has been used to report calcium in individual cells9, our inability to detect responses is likely due to differences in experimental setup; we imaged neurons with a standard charge-coupled device (CCD)-based camera with 1-s exposures, whereas previous work used an electron-multipled CCD with 3-s exposures9.
The bright red output and high calcium responsivity of Orange CaMBI suggests that it may be useful for non-invasive calcium imaging in mice. We tested whether Orange CaMBI 110 could report calcium dynamics in the liver without performing endoscopy or surgery. Calcium oscillations have been extensively characterized in cultured hepatocytes in response to G-protein coupled receptor (GPCR) activation20. These oscillations spread between hepatocytes in excised liver preparations, an effect believed to be mediated by IP3 flow though gap junctions21. However, calcium oscillations have yet to be observed in the liver in a non-invasive manner, and thus the spatial extent of calcium synchronization has not been known. We generated transgenic mice expressing Orange CaMBI 110 in a Cre-dependent manner (Supplementary Fig. 7a) and delivered Cre to the liver using adeno-associated virus, then imaged Orange CaMBI 110 at baseline or following a therapeutic dose of vasopressin. A patchy pattern of bioluminescence corresponded with distinct lobes of the liver (Fig. 2a). After furimazine injection alone, bioluminescence rose and fell gradually without oscillations (Supplementary Fig. 7b). In contrast, furimazine and vasopressin co-injection elicited striking calcium oscillations (Fig. 2b, Supplementary Fig. 7c, Supplementary Video 6). These oscillations exhibited periods of ~20 s and involved entire lobes, but were not phase-locked between lobes. We also created mice transgenic for both Cre-dependent Orange CaMBI 110 and a gene in which the mouse albumin promoter drives Cre recombinase expression. In these mice, luminescence was brighter and evenly distributed throughout the liver, making individual lobes more difficult to delineate, but we could still observe asynchronous oscillations in different regions (Supplementary Fig. 8, Supplementary Video 7). These results demonstrate that Orange CaMBI 110 allows observation of calcium dynamics in a living mouse in a truly non-invasive manner, and reveal that calcium oscillations in the liver can be synchronized throughout entire lobes.
Figure 2. Non-invasive calcium imaging in the liver of a living mice with Orange CaMBI.
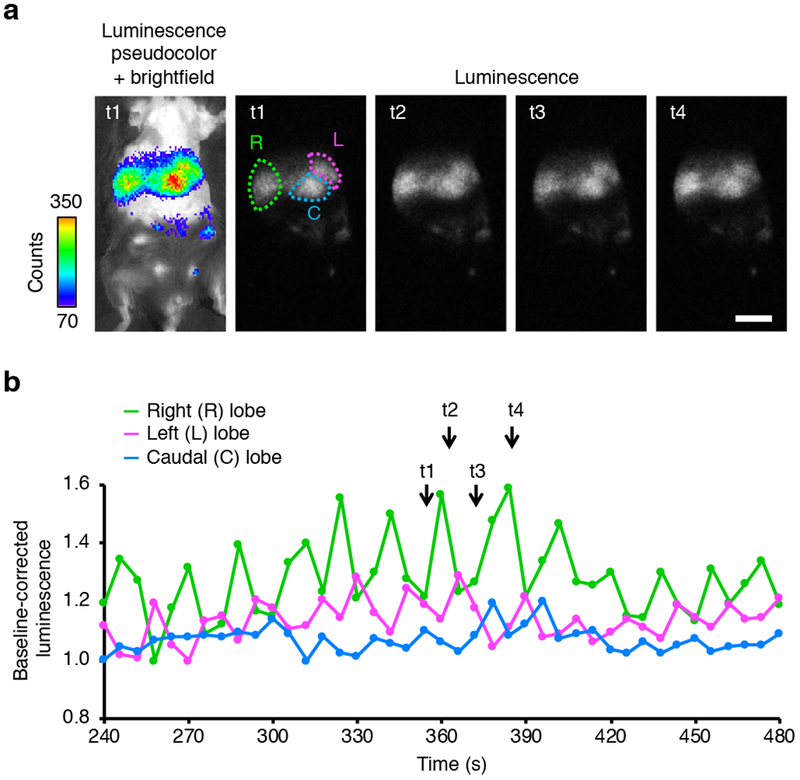
(a) A mouse transgenic for a floxed-stop Orange CaMBI 110 gene was injected with adeno-associated virus expressing Cre recombinase to activate gene expression in the liver. Raw bioluminescence images and overlay of pseudocolored bioluminescence signal over a bright-field image at the indicated time points t1 through t4. Color bar, linear lookup table relating pseudocolor to raw counts per pixel. Scale bar, 1 cm. (b) Time-course of bioluminescence of Orange CaMBI 110 from different lobes of mouse liver. Interspersed 2-s exposures of luminescence and bright field were acquired every 6 s. Integrated luminescence signal from each lobe normalized to its value at 150 s after substrate injection, when baseline luminescence stabilized, is shown. Normalization to a fitted second-order polynomial function was performed to correct for signal decay.
In summary, we have created a series of bright orange-emitting calcium-modulated bioluminescent indicators named Orange CaMBIs that respond to calcium with ~8-fold changes in photon production. Orange CaMBIs tracked calcium fluctuations in single cells and in whole animals using only a standard light microscope and a simple bioluminescence imaging cabinet, in both cases using inexpensive cooled CCD detectors. We estimate that Orange CaMBI can detect synchronized calcium transients in several thousand cells in deep locations (~6 mm) with 1-s temporal resolution and a signal-to-noise ratios of 3 (see Methods). Orange CaMBIs is thus well suited for tracking calcium dynamics in genetically defined cell types when they are located deeply, single-cell resolution is not required, and noninvasive imaging is preferred.
Finally, the Cre-dependent Orange CaMBI 110 transgenic mouse we have produced may be useful for researchers who wish to image calcium dynamics in organ systems in vivo. For example, various classes of GPCRs, such as those for vasopressin, endothelin, angiotensin, epinephrine, acetylcholine, histamine, lysophosphatidic acid, prostaglandins, and leukotrienes, regulate intracellular calcium to modulate vascular and airway smooth muscle contraction22,23. As calcium oscillations are an easily identifiable output of GPCR signaling, the Orange CaMBI 110 transgenic mouse could be used to test the effects of various GPCR agonists or antagonists on vascular or pulmonary physiology. For this purpose, they offer flexibility and physiological information not attainable with bioluminescent indicators derived from a specific GPCR, which report activation of only that GPCR subtype, do not report downstream events, and would be inappropriately expressed unless native regulatory elements are used24,25. In contrast, Orange CaMBI reporters could provide a direct readout of how activation or inhibition of multiple endogenous GPCRs influence the physiologically relevant outcome of calcium release in cells.
METHODS
Methods and any associated references are available in the online version of the paper.
ONLINE METHODS
Calcium indicator engineering.
The atomic structure of NanoLuc was predicted using the Robetta and I-TASSER internet servers26,27, with similar results. A CaM-M13 cassette with a E104Q mutation in CaM and a GGS linker between CaM and M13 was inserted at various sites within the NanoLuc domain of Antares by overlap PCR. Constructs were ligated into the pBAD bacterial expression vector (Thermo Fisher Life Technologies) and transformed into competent XL10-Gold cells (Agilent). Colonies were grown on LB-agar plates containing ampicillin at 37°C for 12 to 20 hours. Bacteria from single colonies were grown in LB broth at 37° C for another 12 to 20 hours, then plasmids were purified and sequenced. Bacteria containing correct constructs were grown in LB broth with 0.1% arabinose at 30° C for one day, then incubated at 4° C for another day in preparation for protein purification and bioluminescence screening.
Protein production and bioluminescence characterization.
Bacteria expressing indicators were pelleted, then lysed in B-PER II (Pierce) with cOmplete EDTA-free Protease Inhibitor Cocktail (Sigma) and 1U DNase (Life Technologies). After pelleting of cell debris, soluble fractions were diluted 50-fold into MOPS buffer (100 mM KCl, 30 mM MOPS, pH 7.2), then 5 μL of each sample was further diluted 20-fold into MOPS buffer with 0 μM free calcium (10 mM EGTA, 100 mM KCl, 30 mM MOPS, pH 7.2) or MOPS buffer with 39 μM free calcium (10 mM CaEGTA, 100 mM KCl, 30 mM MOPS, pH 7.2) as previously described28. 50 μL was placed in a 96-well plate with opaque white walls and equilibrated at room temperature for 10 min. 50 μL of 40 μM furimazine, prepared by diluting 5 mM furimazine stock solution (NanoGlo Luciferase Assay Substrate, Promega) into MOPS buffer with 0 or 39 μM free calcium, was then added to the well and mixed. Bioluminescence emission spectra were then immediately acquired in an Infinite M1000 Pro microplate reader (TECAN) from 400 to 700 nm at 10-nm intervals, with further processing in Excel (Microsoft). Measurements shown are mean of three biological replicates.
For protein purification, indicators were produced with amino-terminal His tags from pBAD expression plasmids in XL10-Gold bacteria (Agilent), lysed in B-PER II containing protease inhibitors and DNAse as above, then purified using HisPur Cobalt Resin (Pierce) and exchanged into PBS using PD-10 desalting columns (Bio-Rad). Protein concentration was assessed by absorbance at 280 nm, with the extinction coefficient of the protein at 280 nm determined by the program ProtParam29. The purified proteins were diluted to 2 nM in MOPS buffer with 0.1% BSA, then analyzed as above. For measurements of dissociation constants and Hill coefficients, bioluminescent emission from 2 nM purified protein was collected in MOPS buffers with a series of free calcium concentrations prepared from the Calcium Calibration Buffer Kit (Thermo Fisher Life Technologies). To determine the KD and Hill coefficient for calcium, emission intensities were normalized to maximum and minimum values, then fit to the Hill equation using the program OriginPro 8 (Origin Labs) with further processing in Excel. Measurements shown are mean of three biological replicates.
For pH-sensitivity measurements, bacteria expressing Orange CaMBI 110 or Orange CaMBI 300 were pelleted, then lysed in B-PER II (Pierce) with cOmplete EDTA-free Protease Inhibitor Cocktail (Sigma) and 1U DNase (Thermo Fisher Life Technologies). After pelleting of cell debris, soluble fractions were diluted 1000-fold into buffer solutions of 130 mM KCl, 50 μM CaCl2 and 20 mM of one of the following: MOPS pH 6.5, MOPS pH 7.0, MOPS pH 7.5, Tris pH 7.5, Tris pH 8.0, or Tris pH 8.5. 50 μL was placed in a 96-well plate with opaque white walls and allowed to equilibrate at room temperature for 10 min. 50 μL of 40-μM furimazine substrate solution, prepared by diluting 5 mM furimazine stock solution (NanoGlo Luciferase Assay Substrate, Promega) into the same buffer solution, was then added to the well and mixed. Bioluminescence emission spectra were then immediately acquired in an Infinite M1000 Pro microplate reader (TECAN) from 400 to 700 nm at 10-nm intervals. Measurements were performed in duplicate rows. Peak luminescence was normalized to the average value at pH 8.5, then values were fit to a logistic function using the solver function of Excel.
For measurements of brightness per protein molecule, emission spectra were obtained from lysates of bacteria expressing Orange, Blue, and Green CaMBIs as above, and the relative amounts of CaMBI protein detected in the same lysates was determined by immunoblotting with a rabbit polyclonal anti-calmodulin primary antibody (One World Lab, AP8549) followed by an IRDye800-conjugated goat anti-rabbit secondary antibody (LI-COR Biosciences). Immunoblot images were acquired with a Odyssey CLx imaging system (LI-COR Biosciences) and analyzed in ImageJ 2.0, Fiji version (NIH)30. The emission spectrum for each indicator was then normalized by its abundance relative to Blue CaMBI using the program Excel 14.0 for Macintosh (Microsoft). Measurements shown are mean of three biological replicates.
Calculation of total quantum yields and catalytic rates of bioluminescent calcium indicators.
The RET efficiency (E) in Orange CaMBI was calculated by fitting the emission spectrum to modelled emission spectra for various E values. For each value of E, an expected emission spectra was generated as follows. NanoLuc emission intensity was normalized so that the Riemann sum of intensities over all wavelengths with 1-nm intervals was equal to 0.28, the NanoLuc luminescence quantum yield (NLQY)14. Likewise CyOFP1 emission intensity was normalized so that the Riemann sum of intensities over all wavelengths with 1-nm intervals was equal to 0.76, the CyOFP1 fluorescence quantum yield (FQY)13. The emission spectrum of the RET system for a given E was then modelled as the sum of the normalized NanoLuc emission spectrum scaled by 1 – E and the normalized CyOFP1 emission spectrum scaled by E, as previously described31,32. The value of E that produced the observed ratio of RET acceptor and donor peaks for Orange CaMBI was determined to be 0.39. The total QY of the RET system was then calculated as (1 – E) • NLQY + E • FQY = 0.61 • 0.28 + 0.39 • 0.76 = 0.47. A similar procedure was performed for Green CaMBI using the emission spectrum and FQY (0.60) of EGFP, and for CalFluxVTN using the calcium-saturated emission spectrum and FQY (0.56) of Venus. For GeNL(Ca), the total QY of the RET system was assumed to be the same as the published value for GeNL (0.45), given their identical emission spectra shapes.
To calculate the kcat of Antares, emission intensities at 450 nm of Antares and of NanoLuc (I450,Antares and I450,NanoLuc) were measured with equimolar amounts of purified proteins and a saturating concentration (20 μM) furimazine. If kcat is unchanged between NanoLuc and Antares, I450,Antares would be expected to equal 0.61 • I450,NanoLuc (where 0.61 = 1 – E, where E is the RET efficiency in Antares). The actual kcat of Antares was thus calculated as the kcat value of NanoLuc alone14 scaled by I450,Antares / (0.61 • I450,NanoLuc). The kcat of calcium-saturated Orange CaMBI was then calculated as the kcat of Antares multiplied by 0.44, the relative molar brightness of calcium-saturated Orange CaMBI compared to Antares. The kcat for GeNL(Ca) was calculated as the kcat of GeNL scaled by 0.32, the relative brightness of GeNL(Ca) in the calcium-saturated state compared to GeNL. The kcat for CalFluxVTN was calculated as the value that would produce the observed relative brightness of CalFluxVTN above 505 nm in depolarized neurons compared to GeNL(Ca), adjusted for their total quantum yields above 505 nm.
Differentiation of human cardiomyocytes from induced pluripotent stem cells.
Beating cardiomyocytes were differentiated from iPSCs as previously described33. Briefly, iPSCs were seeded in Matrigel (Corning, 356231) pre-coated 6-well plates, and were maintained with Essential 8 Medium (Life Technologies). When iPSCs reached 80% confluence, cells were treated with 6 µM CHIR99021 (Selleck) in insulin-free media, comprising RPMI 1640 medium with insulin-free B-27 supplement (Life Technologies), for 48 hr. Cells were then cultured in insulin-free media alone for 24 hr. Cells were then treated with 5 µM IWR-1 (Sigma) in insulin-free media for 2 days, then cultured in insulin-free medium alone for 2 days, and finally in RPMI 1640 medium with insulin-containing B-27 supplement (Life Technologies) for 2 days. Afterwards, culture medium was refreshed every another day. After beating cells were observed, iPSC-derived cardiomyocytes were purified by passaging through glucose-free medium, comprising RPMI 1640 medium without D-glucose (Life Technologies), 213 μg/mL L-ascorbic acid 2-phosphate (Sigma-Aldrich), 500 μg/mL O. sativa-derived recombinant human albumin (Sigma Aldrich), and 5 mM sodium DL-lactate (Sigma-Aldrich), for 7–10 days.
Bioluminescence calcium imaging in HeLa cells and cardiomyocytes.
HeLa cells (obtained from ATCC and tested for mycoplasma) were used to test detection of cellular calcium transients by Orange CaMBI as HeLa cells were previously characterized as demonstrating calcium oscillations in response to histamine stimulation17. HeLa cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% fetal bovine serum (FBS, Hyclone) and transfected with pcDNA3.1 (Life Technologies) mammalian expression plasmids expressing CaMBI variants using Fugene HD (Roche). After 24–36 hr, luminescence images were acquired in culture media with 0.04 mM furimazine (purchased as NanoGlo, Promega) on an Axiovert 200M inverted fluorescence microscope with a 40×/1.2-numerical aperture objective (Zeiss) and an Orca-ER camera (Hamamatsu) using 8×8 binning, exposure time of 200 ms, and frame rate of 4 Hz. Histamine was added to a final concentration of 20 μM in the middle of continuous time-lapse imaging. This experiment was repeated five times and a representative result shown. For bioluminescence imaging of spontaneously beating cardiomyocytes, differentiated beating iPSC-derived cardiomyocytes were dissociated by Accutase (Innovative Cell Technologies) and replated in coverglass imaging chambers (Nunc) pre-coated with Matrigel (Corning). Cells were cultured for 3–4 days after seeding, then transfected with pcDNA3.1 expression plasmids expressing CaMBI variants using Lipofectamine 3000 (Life Technologies) and imaged as above. This experiment was repeated five times and a representative result reported.
Bioluminescence calcium imaging in neurons.
Embryonic day 18 primary rat neurons were cultured on a poly-L-ornithine-coated 4-chamber glass-bottom 35-mm dishes in Neurobasal with B27 serum-free supplement and glutamax (Life Technologies) and 1% FBS (Hyclone), then transfected at 8 days in vitro (DIV) with Lipofectamine 2000 (Life Technologies). On 11 DIV, growth media was replaced with 0.5 mL of a lab-made luminescence imaging solution (2 mM CaCl2, 0.49 mM MgCl2, 0.4 mM MgSO4, 5.3 mM KCl, 0.44 mM KH2PO4, 4.2 mM NaHCO3, 138 mM NaCl, 0.34 mM Na2HPO4, 10 mM Hepes pH 7.2, 15 mM D-glucose, 0.1 mM sodium pyruvate, 0.02 mM furimazine), and cells were imaged on the Axiovert 200M microscope with the 40×/1.2-numerical aperture and the Orca-ER camera. A fluorescence image was first acquired in the acceptor channel, then a luminescence time course was acquired using 8×8 binning, exposure time of 1 s, and frame rate of 1 Hz. After 4–8 min, 0.25 mL of a 3× KCl stimulation solution (2 mM CaCl2, 0.49 mM MgCl2, 0.4 mM MgSO4, 143.2 mM KCl, 0.44 mM KH2PO4, 4.2 mM NaHCO3, 0.34 mM Na2HPO4, 10 mM Hepes pH 7.2, 15 mM D-glucose, 0.1 mM sodium pyruvate, 0.02 mM furimazine) was added using a perfusion pump at a rate of 0.125 mL/s.
Generation of Cre-dependent CaMBI transgenic mice.
The Orange CaMBI coding sequence was cloned into the plasmid pBT378-LSL between the floxed stop cassette and the polyadenylation signal. The resulting plasmid contained a CAGG promoter, the floxed stop cassette, Orange CaMBI, and the polyadenylation signal all flanked by attB sites. This plasmid was then used by the Stanford Transgenic, Knockout, and Tumor Model Center to perform recombinase-mediated integration of the above transcription unit in C57BL/6J ES cells containing an attP site at the H11 locus as previously described34. Mice derived from these ES cells were verified by contain the Orange CaMBI insert at H11 by PCR.
Bioluminescence imaging in liver of living mice.
For the experiment in Fig. 2 and Supplementary Fig. 7, a heterozygous floxed-stop Orange CaMBI transgenic female C57Bl/6 mouse of 16 weeks age was administered 1.1×1012 genome copies of AAV-GFP-cre in 100 μL by tail vein injection. On day 12 after infection, the mouse was administered isoflurane inhalation and placed supine. Fur overlaying the abdomen was removed with Nair depilatory cream followed by rinsing with PBS and cleaning with 70% ethanol on a piece of sterile gauze. Bioluminescence imaging was performed in the IVIS Spectrum imaging system by intraperitoneal injection of 400 nmol of furimazine in 150 uL of 8% glycerol, 12% ethanol, 10% hydroxy-propyl-cyclodextrin, and 35% polyethylene glycol 300 in water, without or with 120 uL of 12 ng vasopressin in PBS. Bioluminescence images were acquired in sequential luminescence scan mode with open emission filter, 2-s exposure time each 6 s (the minimum interval allowed by the system), 8×8 binning, f/1 aperture, and a 13-cm field of view. Image analysis was performed in the program Fiji with further quantitation in the program Excel. Liver lobes were identified by referring to published tomographic images35. Integrated signal at each timepoint within regions of interest were baseline-corrected by normalization to a fitted second-order polynomial (Fig. 2), or plotted without baseline correction (Supplementary Fig. 7). For the experiment in Supplementary Fig. 8, a double heterozygous albumin-cre and floxed-stop Orange CaMBI male C5Bl/6 mouse was imaged at 8 weeks of age. Imaging was performed as above except 50 nmol of furimazine was injected and images were acquired with 1-s exposure time each 5 s (the minimum interval allowed by the system). Animal procedures were performed in compliance with USDA and NIH ethical regulations and approved by the Stanford Institutional Animal Care and Use Committee.
Statistics.
For the comparison of times to peak of various bioluminescent calcium indicators, an effect size observed in s preliminary experiment and the observed variance was used to determine the minimum sample number required to determine statistical confidence to an alpha level of 0.05 using the program G*Power36. Experiments were then repeated until this sample number was obtained for all indicators. Normality was verified by the Shapiro Wilk test and differences were analyzed by one-way ANOVA followed by Tukey’s post-hoc test in the program Prism 6 (GraphPad). Experimenters could not be blinded during data collection as appropriate microscope filters had to be selected for each construct, but analyses were performed blinded by a separate experimenter. All findings in this study were replicatable across multiple preparations.
Estimation of detection limits.
Fluorescent probes require excitation light, and light is attenuated over distance by absorption and scattering with half-attenuation distances of hundreds of microns37,38. Thus excitation of structures deeper than several hundred microns is inefficient, as well as nonuniform due to tissue heterogeneity. Emitted signals are similarly attenuated before collection, reducing signal over noise. Multi-photon excitation optimizes contrast, as excitation is confined to the focal plane, allowing collection of all emitted light. Beyond 1 mm, however, multi-photon imaging becomes limited by heating38,39. Time-lapse imaging beyond 1 mm can be performed in one-photon mode, but this generates autofluorescence all along the light path. Deep one-photon fluorescence imaging is possible under limited conditions, e.g. > 2500 cells colocalized together to create a large accumulation of fluorophores and placed relatively superficially in a subcutaneous position, using fluorophores excitable with light beyond 600 nm5,40. However, if the fluorescence source is less dense, signal will be overcome by noise from autofluorescence generated throughout the illuminated volume. By eliminating the need for illumination, bioluminescence overcomes these limitations. Because signal can be integrated over long time periods without integrating autofluorescence, bioluminescence reporters can allow the detection of smaller numbers of cells. Based on intensities recorded in a typical bioluminescence imaging setup with a 35mm-radius camera lens placed ~300 mm from the mouse, 100 photons/s above background were detected per firefly luciferase (FLuc)-expressing cell implanted in a mouse abdomen, integrated across the whole abdomen41. This is consistent with other reports that 10000 photons can be produced by FLuc per cell40,42, as only 1% of photons will enter the camera lens with the above geometry. Calcium-saturated Orange CaMBI can be expected to exhibit similar photon flux (because Antares generates 2.6-fold more signal than firefly luciferase from deep locations3, and calcium-saturated Orange CaMBI reaches 44% of the brightness of Antares). If Orange CaMBI 110 is used, baseline brightness from one cell would be approximately 50 photons/s, and maximum induced signal would be 50 photons/s.
The signal from a single cell in the abdomen (or any other body cavity) would also be expected to emerge from a wide area of the overlying skin due to internal scattering, although an approximate location in the xy plane can be discerned from the point of maximal signal emergence. This area can be magnified and then projected to fill a 64×64-pixel photodetector array to allow some spatial resolution, useful for verifying the localized nature of a signal, while collecting more signal per pixel than possible if projected to a larger array. The 64×64-pixel image could be obtained by projecting the body region onto a chip with a native 64×64-pixel arrangement, by projecting onto a larger number of pixels binned to 64×64 superpixels, or projecting onto a 64×64-pixel region of a larger chip (as was done in Fig. 3). In any case, 50 photons/s of signal and 50 photons/s of background would be collected in ~4100 effective bins, resulting in an mean signal (S) of 0.012 photons/s above a background (B) of 0.012 photons/s. In a typical imaging system such as the IVIS Spectrum, camera dark current (D) adds another 0.013 counts/s per 8×8 superpixel and read noise (R) is 3 counts/frame per superpixel, so total noise is predicted by the formula (t•S + t•B + t•D + R2)1/2, where t is integration time. To obtain signal from a single cell equivalent to noise, we can solve the equation t•S = (t•S + t•B + t•D + R2)1/2, obtaining 410 s for the required exposure time. However, a 410-s exposure is obviously not useful for revealing calcium transients of biological importance, which have durations in the time frame of seconds. To determine the number of cells (n) that would be required to produce a signal/noise (S/N) ratio of x in a 1-s exposure, we can solve the equation 0.012•n = x•(0.012•n + 0.012•n + 0.013 + 9)1/2 photons. Calculations indicate 350, 1100, and 1800 cells would produce S/N ratios of 1.0, 2.0, or 3.0.
The minimum number of detectable cells per unit time can also be estimated from our empirical observations with luminescence imaging in vitro. Using a Hamamatsu Orca-ER CCD camera, Orange CaMBI reported calcium oscillations in single HeLa cells and cardiomyocytes with SNR of 2 when signals were acquired with exposure times of 1 s and quantified throughout a 64×64-pixel region centered on the cell. The camera used here featured read noise of 6 counts per frame and negligible dark current at 0.1 counts per s, and so has slightly worse performance characteristics as the camera in the IVIS Spectrum used for whole-animal bioluminescence imaging. About half of red photons are lost for each mm of tissue traversed37,38, so we expect retrieval of 64 times fewer photons for a structure located 6 mm deep, sufficient to reach most areas of the brain and liver. In addition, using the standard bioluminescence imaging cabinet captures 31 times fewer photons compared to a 1.1-NA water-immersion lens (1% v. 31% of photons emitted, based on solid angle calculation). Thus we predict ~2000 fewer photons will be collected per unit time from deep locations with typical bioluminescence imaging equipment than from a cultured cell with a microscope. Thus to maintain a SNR of 2 with 1-s exposures and a 64×64-pixel array, we would need to image 2000 times more cells. From calculating the photon flux that produces this SNR on the Orca-ER, and calculating the noise that would be produced on the IVIS Spectrum, we predict that the signal from 2000 cells would produce a S/N ratio of 2.6. This is in good agreement with the first estimation method.
The above calculations assume all cells are producing maximal CaMBI responses with durations > 1 s in a synchronized manner. The actual number of Orange CaMBI-expressing cells needed to detect signals of interest will increase if calcium transients do not saturate the indicator, the signal durations are shorter than 1 s, the signals are not synchronized, or larger SNRs are desired. Nevertheless these calculations provide an estimate of the number of cells that would be required to noninvasively visualize calcium transients in deep locations using widely available CCD cameras.
Data availability.
Nucleotide sequences are available at GenBank for Orange CaMBI 110 (MK558047), Orange CaMBI 300 (MK558048), Blue CaMBI (MK558049), and Green CaMBI (MK558050). Mammalian expression plasmids are available at Addgene for Orange CaMBI 110 (124094), Orange CaMBI 300 (124095), Blue CaMBI (124096), and Green CaMBI (124097). All other data from this study are available from the corresponding author upon request.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Lin laboratory for assistance with experiments and the Stanford Transgenic, Knockout, and Tumor Model Center for generating the Orange 110 CaMBI transgenic mice. This work was supported by an AHA Postdoctoral Fellowship (N.K.), an AHA Innovation Grant 15IRG23290018 (M.Z.L.), a Stanford Discovery Innovation Award (M.Z.L. and Y.P.), NIH grants R01HL133272 (J.C.W.), R01HL128170 (J.C.W.), U01HL099776 (J.C.W.), and U01NS090600 (M.Z.L.); NIH fellowship F32HL119059 (N.K.P.); and NIH Pioneer Award 5DP1GM111003 (M.Z.L.).
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
REFERENCES
- 1.Clapham DE Calcium signaling. Cell 131, 1047–1058 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Russell JT Imaging calcium signals in vivo: a powerful tool in physiology and pharmacology. Br. J. Pharmacol. 163, 1605–1625 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsien RY, Ernst L & Waggoner A in Handbook Of Biological Confocal Microscopy (ed Pawley J) 338–352 (Springer, Boston, 2006). [Google Scholar]
- 4.Chu J et al. Non-invasive intravital imaging of cellular differentiation with a bright red-excitable fluorescent protein. Nat Methods 11, 572–578 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin MZ et al. Autofluorescent proteins with excitation in the optical window for intravital imaging in mammals. Chem. Biol. 16, 1169–1179 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao H et al. Emission spectra of bioluminescent reporters and interaction with mammalian tissue determine the sensitivity of detection in vivo. J Biomed Opt 10, 41210 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Wang A, Feng J, Li Y & Zou P Beyond Fluorescent Proteins: Hybrid and Bioluminescent Indicators for Imaging Neural Activities. ACS Chem Neurosci 9, 639–650 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Martin JR In vivo brain imaging: fluorescence or bioluminescence, which to choose. J Neurogenet 22, 285–307 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Bakayan A, Domingo B, Miyawaki A & Llopis J Imaging Ca(2+) activity in mammalian cells and zebrafish with a novel red-emitting aequorin variant. Pflugers Arch 467, 2031–2042 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shimomura O, Kishi Y & Inouye S The relative rate of aequorin regeneration from apoaequorin and coelenterazine analogues. Biochem. J. 296, 549–551 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoshino H, Nakajima Y & Ohmiya Y Luciferase-YFP fusion tag with enhanced emission for single-cell luminescence imaging. Nat Methods 4, 637–639 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Saito K et al. Luminescent proteins for high-speed single-cell and whole-body imaging. Nat Commun 3, 1262 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chu J et al. A bright cyan-excitable orange fluorescent protein facilitates dual-emission microscopy and enhances bioluminescence imaging in vivo. Nat Biotechnol 34, 760–767 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suzuki K et al. Five colour variants of bright luminescent protein for real-time multicolour bioimaging. Nat Commun 7, 13718 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horikawa K et al. Spontaneous network activity visualized by ultrasensitive Ca(2+) indicators, yellow Cameleon-Nano. Nat Methods 7, 729–732 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Chen TW et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang J et al. Coupling optogenetic stimulation with NanoLuc-based luminescence (BRET) Ca(++) sensing. Nat Commun 7, 13268 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reddy R et al. Voltage-sensitive adenylyl cyclase activity in cultured neurons. A calcium-independent phenomenon. J. Biol. Chem. 270, 14340–14346 (1995). [DOI] [PubMed] [Google Scholar]
- 19.Stanika RI, Villanueva I, Kazanina G, Andrews SB & Pivovarova NB Comparative impact of voltage-gated calcium channels and NMDA receptors on mitochondria-mediated neuronal injury. J. Neurosci. 32, 6642–6650 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gaspers LD, Pierobon N & Thomas AP in Signaling Pathways in Liver Diseases (eds Dufour JF, Clavien PA, Trautwein C & Graf R) 211–221 (Springer, Berlin, Heidelberg, 2005). [Google Scholar]
- 21.Dupont G, Combettes L & Leybaert L Calcium dynamics: spatio-temporal organization from the subcellular to the organ level. Int Rev Cytol 261, 193–245 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Kuo IY & Ehrlich BE Signaling in muscle contraction. Cold Spring Harb Perspect Biol 7, a006023 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Penn RB & Benovic JL Regulation of heterotrimeric G protein signaling in airway smooth muscle. Proc Am Thorac Soc 5, 47–57 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kono M et al. Bioluminescence imaging of G protein-coupled receptor activation in living mice. Nat Commun 8, 1163 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takakura H, Hattori M, Takeuchi M & Ozawa T Visualization and quantitative analysis of G protein-coupled receptor-β-arrestin interaction in single cells and specific organs of living mice using split luciferase complementation. ACS Chem Biol 7, 901–910 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Kim DE, Chivian D & Baker D Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 32, W526–31 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsien R & Pozzan T Measurement of cytosolic free Ca2+ with quin2. Methods Enzymol. 172, 230–262 (1989). [DOI] [PubMed] [Google Scholar]
- 29.Wilkins MR et al. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol 112, 531–552 (1999). [DOI] [PubMed] [Google Scholar]
- 30.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bajar BT et al. Improving brightness and photostability of green and red fluorescent proteins for live cell imaging and FRET reporting. Sci Rep 6, 20889 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lam AJ et al. Improving FRET dynamic range with bright green and red fluorescent proteins. Nat Methods 9, 1005–1012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burridge PW et al. Chemically defined generation of human cardiomyocytes. Nat Methods 11, 855–860 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tasic B et al. Site-specific integrase-mediated transgenesis in mice via pronuclear injection. Proc. Natl. Acad. Sci. U. S. A. 108, 7902–7907 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fiebig T et al. Three-dimensional in vivo imaging of the murine liver: a micro-computed tomography-based anatomical study. PLoS One 7, e31179 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Faul F, Erdfelder E, Lang AG & Buchner A G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods 39, 175–191 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Chuong AS et al. Noninvasive optical inhibition with a red-shifted microbial rhodopsin. Nat Neurosci 17, 1123–1129 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Horton NG et al. In vivo three-photon microscopy of subcortical structures within an intact mouse brain. Nat Photonics 7, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Podgorski K & Ranganathan G Brain heating induced by near-infrared lasers during multiphoton microscopy. J. Neurophysiol. 116, 1012–1023 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levin RA et al. An optimized triple modality reporter for quantitative in vivo tumor imaging and therapy evaluation. PLoS One 9, e97415 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sarkar S, Malekshah OM, Nomani A, Patel N & Hatefi A A novel chemotherapeutic protocol for peritoneal metastasis and inhibition of relapse in drug resistant ovarian cancer. Cancer Med 7, 3630–3641 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baklaushev VP et al. Luciferase Expression Allows Bioluminescence Imaging But Imposes Limitations on the Orthotopic Mouse (4T1) Model of Breast Cancer. Sci Rep 7, 7715 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Nucleotide sequences are available at GenBank for Orange CaMBI 110 (MK558047), Orange CaMBI 300 (MK558048), Blue CaMBI (MK558049), and Green CaMBI (MK558050). Mammalian expression plasmids are available at Addgene for Orange CaMBI 110 (124094), Orange CaMBI 300 (124095), Blue CaMBI (124096), and Green CaMBI (124097). All other data from this study are available from the corresponding author upon request.