

Abstract
Fission yeast Rad3 is a member of a family of phosphoinositide 3-kinase -related kinases required for the maintenance of genomic stability in all eukaryotic cells. In fission yeast, Rad3 regulates the cell cycle arrest and recovery activities associated with the G2/M checkpoint. We have developed an assay that directly measures Rad3 kinase activity in cells expressing physiological levels of the protein. Using the assay, we demonstrate directly that Rad3 kinase activity is stimulated by checkpoint signals. Of the five other G2/M checkpoint proteins (Hus1, Rad1, Rad9, Rad17, and Rad26), only Rad26 was required for Rad3 kinase activity. Because Rad26 has previously been shown to interact constitutively with Rad3, our results demonstrate that Rad26 is a regulatory subunit, and Rad3 is the catalytic subunit, of the Rad3/Rad26 kinase complex. Analysis of Rad26/Rad3 kinase activation in rad26.T12, a mutant that is proficient for cell cycle arrest, but defective in recovery, suggests that these two responses to checkpoint signals require quantitatively different levels of kinase activity from the Rad3/Rad26 complex.
INTRODUCTION
All organisms monitor genomic integrity to ensure proper development and reproduction. When this integrity is compromised in eukaryotes, checkpoint systems prevent passage into the next cell cycle phase (Hartwell and Weinert, 1989). This cell cycle arrest allows time for maintenance activities to occur before the next cell cycle phase begins. Without checkpoint systems, problems such as unreplicated DNA in S phase go uncorrected and lead to unequal chromosome segregation after mitosis. Checkpoint systems also enable cells to sustain viability during S-phase arrest, a poorly understood process known as “recovery” that may direct DNA repair and recombination activities (Enoch et al., 1992; Stewart et al., 1997; Desany et al., 1998; Lindsay et al., 1998).
The G2/M checkpoint of fission yeast, Schizosaccharomyces pombe, delays mitosis in response to unreplicated or damaged DNA. Screens designed to isolate G2/M checkpoint control genes led to the identification of six “checkpoint rad” genes, hus1+, rad1+, rad3+, rad9+, rad17+, and rad26+ (Al-Khodairy and Carr, 1992; Enoch et al., 1992; Rowley et al., 1992; Al-Khodairy et al., 1994), as well as rhp9/crb2+, chk1+, and cds1+ (Walworth and Bernards, 1996; Willson et al., 1997; Lindsay et al., 1998). Although none of these genes is essential, deletion of any one of the six checkpoint rad genes (hus1+, rad1+, rad3+, rad9+, rad17+, and rad26+) abolishes G2/M checkpoint control and allows mitosis to proceed in the presence of either unreplicated or damaged DNA. Recovery activities are also abolished in checkpoint radΔ cells (Enoch et al., 1992; Al-Khodairy et al., 1994). On the other hand, deletion of cds1+ only disrupts aspects of the checkpoint response to unreplicated DNA, whereas deletion of rhp9/crb2+ or chk1+ only disrupts the checkpoint response to damaged DNA. Structurally and functionally related proteins have been found in many other eukaryotes, suggesting that aspects of the cellular response to genotoxic stresses have been highly conserved during evolution (reviewed by O'Connell et al., 2000).
One of the checkpoint Rad proteins, Rad3, is a member of the phosphoinositide 3-kinase-related kinase (PIKK) family (Abraham, 2001). PIKKs have been implicated in the cellular response to genotoxic stresses in many eukaryotes. For example, other family members include human ATM, ATR, and DNA-dependent protein kinase; TEL1 and MEC1 of Saccharomyces cerevisiae; Mei-41 of Drosophila; and UVSB of Aspergillus nidulans (De Souza et al., 1999; Hofmann and Harris, 2000; reviewed by Elledge, 1996; Hoekstra, 1997). All the proteins in this family are large (>200 kDa) and have a domain related to the catalytic domain of phosphoinositide 3-kinases. However, phosphoinositide 3-kinases can phosphorylate lipids, whereas PIKKs are only known to phosphorylate proteins.
It is not known how PIKK family members monitor genomic integrity. It is attractive to speculate that they are activated by particular DNA structures and then phosphorylate downstream substrates involved in genome maintenance activities. In fission yeast, a number of checkpoint proteins are phosphorylated in response to checkpoint signals in a Rad3-dependent manner (Walworth and Bernards, 1996; Kostrub et al., 1998; Lindsay et al., 1998; Edwards et al., 1999; Caspari et al., 2000). Although these observations predict that Rad3 kinase activity changes in response to checkpoint signals, a direct test of this hypothesis has not been presented.
Both Cds1 and Chk1 are protein kinases that are phosphorylated in Rad3-dependent manners in response to checkpoint signals (Walworth and Bernards, 1996; Lindsay et al., 1998). This phosphorylation of Cds1 and Chk1 may activate these kinases, which control the activity of proteins that regulate the cyclin-dependent kinase, Cdc2 (Furnari et al., 1997; O'Connell et al., 1997; reviewed by Weinert 1997; Zeng et al., 1998). Thus, checkpoint signals may cause a change in Rad3 kinase activity that allows Rad3 to phosphorylate and activate the downstream kinases Cds1 and Chk1. Once activated, these kinases then target substrates that regulate the Cdc2 kinase and thereby delay entry into mitosis (reviewed by Walworth, 2001).
The checkpoint response in fission yeast also requires the five other checkpoint Rad proteins: Hus1, Rad1, Rad9, Rad17, and Rad26. Structural predictions of Hus1, Rad1, and Rad9 have led to the proposal that all three fold similarly to the subunits of proliferating cell nuclear antigen (PCNA) (Thelen et al., 1999; Caspari et al., 2000; Venclovas and Thelen, 2000). Consistent with these predictions, PCNA exists as a homotrimer (Krishna et al., 1994), whereas Hus1, Rad1, and Rad9 all interact and possibly form a heterotrimer (Kostrub et al., 1998; Caspari et al., 2000). In addition, Rad17 has sequence similarity and physical interactions with subunits of replication factor C (Griffiths et al., 1995; Shimada et al., 1999). A model follows, whereby loading of the PCNA-like Hus1-Rad1-Rad9 heterotrimer onto DNA by Rad17 may allow the complex to scan the genome for problems. When damage is detected, the complex may recruit Rad3 to the site of damage and activate Rad3 kinase activity. This activation would then allow Rad3 to phosphorylate and activate Cds1 and Chk1 and, in turn, delay entry into mitosis.
This model is not consistent with a recent study that characterized Rad26 phosphorylation in response to DNA damage in fission yeast (Edwards et al., 1999). This group showed that Rad26 constitutively binds to Rad3 and is phosphorylated in a Rad3-dependent manner after DNA damage. Phosphorylation of Hus1, Cds1, and Chk1 also occur in Rad3-dependent manners after DNA damage, and these events require all of the checkpoint Rad proteins (Hus1, Rad1, Rad9, Rad17, and Rad26) (Walworth and Bernards, 1996; Kostrub et al., 1998; Lindsay et al., 1998). In contrast, Rad26 phosphorylation after DNA damage was shown to require only Rad3 and not the other checkpoint Rad proteins (Hus1, Rad1, Rad9, and Rad17) (Edwards et al., 1999). Although the physiological significance of any of these Rad3-dependent phosphorylation events is not understood, these studies suggest that Hus1, Rad1, Rad9, and Rad17 may not be involved in sensing checkpoint signals.
Herein, we describe an in vitro kinase assay that can be used to measure the kinase activity of physiological levels of Rad3. Using this assay, we show that Rad3 has a basal level of kinase activity that increases after cells are treated with a DNA-damaging agent or DNA replication inhibitor. This provides the first direct evidence that the Rad3 kinase is activated by checkpoint signals.
Rad3 kinase activity could be precipitated with antibodies raised against Rad26, and both basal and induced levels of Rad3 kinase activity were dependent on Rad26. These results suggest that the checkpoint response in fission yeast involves activation of a kinase complex consisting of a catalytic subunit (Rad3) and a regulatory subunit (Rad26). Basal and induced levels did not require Hus1, Rad1, Rad9, Rad17, Cds1, or Chk1, suggesting that these proteins are not involved in the recognition of checkpoint signals.
Although we found that both replication arrest and DNA damage similarly activated the Rad3/Rad26 complex, only treatment with a DNA-damaging agent resulted in Rad3-dependent modification of Rad26. This difference shows that the biochemistry of the checkpoint-activated Rad3/Rad26 complex depends on the nature of the checkpoint signal, suggesting that the complex may interact directly with different checkpoint signals. Last, analysis of Rad26/Rad3 kinase activation in a rad26 mutant that is proficient for cell cycle arrest, but defective in recovery, suggests that these two responses to checkpoint signals may require quantitatively different levels of Rad3/Rad26 kinase activity.
MATERIALS AND METHODS
Strains, Growth Conditions, and Physiological Methods
The strains used in this study (Table 1) were grown at 30°C under standard conditions (Moreno et al., 1991). Hydroxyurea (HU; Sigma, St. Louis, MO) was prepared as a 0.2 M stock solution in water and kept at 4°C, and bleomycin (Bleo; Sigma) was prepared as a 3 U/ml stock solution in water and kept at −20°C.
Table 1.
Fission yeast strains and plasmids
| Strain | Genotype | Origin of strain |
|---|---|---|
| TE235 | leul-32 h | Chapman et al. (1999) |
| TE253 | rad26.T12 ura4-D18 leu1-32 ade6-704 h+ | Al-Khodairy et al. (1994) |
| TE257 | rad26∷ura4+ ade6-704 leu1-32 ura4-D18 h− | Al-Khodairy et al. (1994) |
| TE459 | rad1∷ura4+ leu1-32 his− | Russell and Nurse et al. (1987) |
| TE484 | hus1∷LEU2 leu1-32 ura4-D18 h- | Kostrub et al. (1997) |
| TE571 | rad3.a (kinase null) ura4-D18 | Gift a A. Carr |
| TE794 | rad9∷ura4+ ade6-704 leu1-32 ura4-D18 h− | Murray et al. (1991) |
| TE864 | rad17∷ura4+ ade6-704 leu1-32 ura4-D18 h− | Griffiths et al. (1995) |
| TE892 | chk1∷ura4+ cds1∷ura4+ ura4-D18 leu1-32 | Gift of C. Kostrub |
| TE1029 | myc-rad3 ura4-D18 h− | Edwards et al. (1999) |
| TE1050 | rad17∷ura4+ myc-rad3 | This study |
| TE1051 | rad1∷ura4+ myc-rad3 | This study |
| TE1053 | cds1∷ura4+ chk1∷ura4+ myc-rad3 | This study |
| TE1055 | rad9∷ura4+ myc-rad3 | This study |
| TE1057 | rad26∷ura4+ myc-rad3 ade6-704 | This study |
| TE1059 | hus1∷LEU2 myc-rad3 ade6-704 ura4-D18 | This study |
| TE1102 | rad26.T12 myc-rad3 | This study |
To collect cell pellets used for making protein extracts, overnight cultures were used to inoculate 400 milliliters of YE5S liquid medium. When the optical density reached 0.5, half of the culture (200 ml/2 × 109 cells) was collected by centrifugation, washed twice with ice-cold Stop Buffer (150 mM NaCl, 50 mM NaF, 10 mM EDTA, 1 mM NaN3), and stored at −80°. To activate the S-phase checkpoint, HU was added to the other half of the culture to a concentration of 10 mM and the culture was allowed to grow for another 3 h before collection. To activate the DNA damage checkpoint, bleomycin was added to the other half of the culture to a concentration of 5 mU/ml and the culture was allowed to grow for another 3 h before collection.
To observe the ability of rad26.T12 cells to undergo checkpoint-induced cell cycle arrest (Figure 5), rad26+ myc-rad3 (wild type [WT], TE1029), rad26Δ myc-rad3 (TE1057), and rad26.T12 myc-rad3 (TE1102) cells were treated with bleomycin for 4 h. Cells were then centrifuged, washed once with water, fixed with 70% cold ethanol, and stored at −20°. To observe phenotypes, the fixed cells were centrifuged, rehydrated in water, and heat-fixed onto glass slides. With the use of fluorescence microscopy, phenotypes were observed after cells were stained with 1 μg/ml 4,6-diamidino-2-phenylindole (to visualize nuclei) and 50 μg/ml calcofluor (to visualize septa).
Figure 5.

Normal kinase activity of the Rad26/Rad3 complex is not required for cell cycle arrest. (A) rad26. T12 strain has low Rad26/Rad3 kinase activity. Anti-Rad26 immunoprecipitations from different cell extracts were split in half. Half of each immunoprecipitation was assayed for kinase activity (top), whereas the other half was Western blotted and probed with anti-Rad26 (middle) and anti-Myc (bottom) antibodies. The extracts were collected from untreated rad26Δ myc-rad3 (lane 1, TE1057) cells, as well as untreated and Bleo-treated rad26+ myc-rad3 (myc-rad3, lanes 2 and 3; TE1029) and rad26. T12 myc-rad3 (rad26. T12, lanes 4 and 5; TE1102) cells. All of the lanes shown are from the same SDS-PAGE Western blot. (B) rad26. T12 strain undergoes cell cycle arrest after Bleo treatment. Samples from cell cul-tures were collect before Bleo treatment (− Bleo) or 4 h after Bleo treatment (+ Bleo). To observe cell phenotypes, these samples were prepared for fluorescence microscopy (see MATERIALS AND METHODS) and stained with 4,6-diamidino-2-phenylindole and calcofluor. Untreated and Bleo-treated cells of rad26+ myc-rad3 (rad26+, panels 1 and 2; TE1029), rad26Δ myc-rad3 (rad26Δ, panels 3 and 4; TE1057), and rad26. T12 myc-rad3 (rad26. T12, panels 5 and 6; TE1102) are shown. Each arrow points to a septum. In panel 4, these septa of Bleo-treated rad26Δ cells have partitioned unequal amounts of DNA. Bar, 6.5 μm.
Constructing Strains
The myc-rad3 allele (kindly provided by A. Carr; Edwards et al., 1999) is not marked, but it can be followed through crosses by probing Western blots with anti-Myc monoclonal antibodies (see “Immunoprecipitation and Kinase Assays”). To place the myc-rad3 allele into the hus1Δ, rad1Δ, rad9Δ, rad17Δ, rad26Δ, and chk1Δcds1Δ backgrounds, TE1029 (myc-rad3) was crossed to the appropriate deletion strains (Table 1; TE484, TE459, TE794, TE864, TE257, and TE892, respectively). Crosses were plated on YE5S and then replica plated to YE5S + HU (10 mM). Protein extracts were prepared (see “Immunoprecipitation and Kinase Assays”) from 10 HU-sensitive segregants of each cross. Strains containing Myc-Rad3 were identified by immunoprecipitating cell extracts with anti-Myc antibodies and probing the Western blots with anti-Myc antibodies. To construct the rad26.T12 myc-rad3 strain (TE1102), the rad26.T12 strain (TE253) was crossed to the rad26Δ myc-rad3 strain (TE1057; the rad26Δ allele is marked with ura+) and ura− segregants were selected. Protein extracts were prepared from 10 ura− segregants and the presence of Rad26.T12 and Myc-Rad3 was examined after immunoprecipitating with anti-Rad26 antibodies and probing Western blots with both anti-Rad26 and anti-Myc antibodies. The genotype of all strains was confirmed using Southern blot analysis (our unpublished data).
Immunoprecipitation and Kinase Assays
To produce Rad26 antibodies, rabbit polyclonal antibodies were raised against a bacterially expressed β-galactosidase-Rad26 fusion protein. Procedures to produce yeast protein extracts from 1 × 109 cells, and perform immunoprecipitations and immunodetections by Western blots are described by Kostrub et al. (1998). Importantly, 1% Triton X-100 (Sigma) was included in the IP-buffer to facilitate visualization of Myc-Rad3 on Western blots (Edwards et al., 1999). We specify below where other changes to these procedures occurred. Kinase assays and Rad3 Western blots are described by Chapman et al. (1999).
To observe Rad26, anti-Rad26 immunoprecipitations were separated by 10% SDS-PAGE and Western blots were probed with anti-Rad26 polyclonal antibodies at a concentration of 1:5000 in 1 × Tris-buffered saline/Tween 20 (TBST) blocking solution (Chapman et al., 1999). Immobilon-P transfer membranes (Millipore, Bedford, MA) were used for Western blotting.
To observe Myc-Rad3 that coimmunoprecipitated with Rad26 (Figures 1B, 4B, and 5A), the Western blots produced from anti-Rad26 immunoprecipitations (as described directly above) were probed with anti-c-Myc monoclonal antibodies (9E10; Roche Molecular Biochemicals, Indianapolis, IN) at a concentration of 10 μg/ml in 1 × TBST blocking solution. Secondary antibody (horseradish peroxidase-conjugated anti-mouse; Amersham Biosciences, Arlington Heights, IL) was added at a 1:5000 dilution in 1 × TBST.
Figure 1.
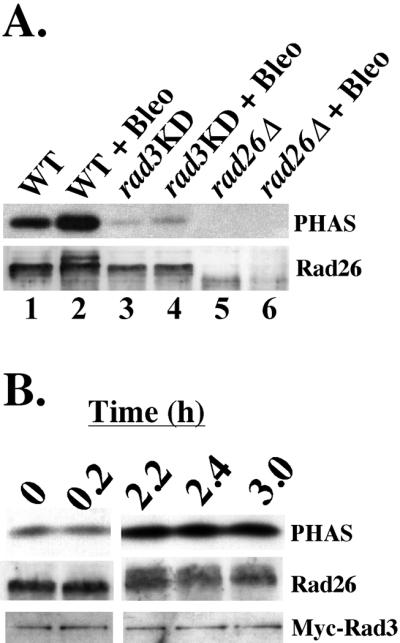
Activation of Rad26-associated Rad3 kinase activity by checkpoint signals. Kinase assays and Western blots of anti-Rad26 immunoprecipitations from fission yeast protein extracts. To induce DNA damage, cells were treated with 5 mU/ml bleomycin for 3 h in liquid YE5S before extracts were collected. (A) Anti-Rad26 immunoprecipitations of extracts from untreated and Bleo-treated cultures of WT (lanes 1 and 2, TE235), rad3KD (lanes 3 and 4, TE571), and rad26Δ (lanes 5 and 6, TE257) cells were split in half. Top, half of each anti-Rad26 immunoprecipitation was assayed for the ability to phosphorylate PHAS-1. Bottom, Western blot of the other half of each anti-Rad26 immunoprecipitation was probed with anti-Rad26 antibody. The phosphorylated form of Rad26 appears after WT cells are treated with bleomycin (lane 2), but not when rad3KD cells are treated with this DNA-damaging agent (lane 4). (B) Rad3 kinase is activated after Bleo treatment. Samples from a single, rad26+ myc-rad3 (WT; TE1029) culture were collected before Bleo treatment (T = 0) and at times after Bleo treatment (T = 0.2, 2.2, 2.4, and 3.0 h). Anti-Rad26 immunoprecipitations of each sample were divided in half and either used to assay for the ability to phosphorylate PHAS (top), or Western blotted and probed with anti-Rad26 (middle) and anti-Myc (bottom) antibodies. The doublet that appears in the Rad26 blot (middle) after 2 h of Bleo treatment represents Rad3-dependent phosphorylation of Rad26 (Edwards et al., 1999).
To immunoprecipitate Myc-Rad3 directly from protein extracts (Figure 3), 10 μl of a 160 μg/ml stock solution of anti-Myc in phosphate-buffered saline was added to protein extracts for 1 h. Next, 25 μl of Protein G-Sepharose beads (Roche Diagnostics, Indianapolis, IN) were washed three times in IP-buffer (Kostrub et al., 1998) before incubating them with the extracts for an hour. The beads were then washed three times in IP-buffer and split in half. One half of each immunoprecipitation was separated using 5% SDS-PAGE, and Western blot analysis and detection were preformed using the method described by Chapman et al. (1998) for visualizing full-length Rad3. The other half was assayed for kinase activity (see below) or run on 10% SDS-PAGE followed by Western blotting to determine whether Rad26 coimmunoprecipitated with Myc-Rad3 in this experiment (Figure 3).
Figure 3.
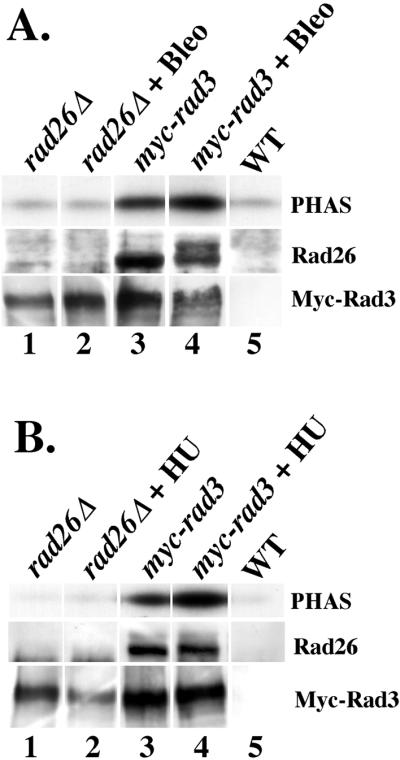
Rad26 is required for basal and induced levels of Rad3 kinase activity after HU and Bleo treatments. (A) Anti-Myc immunoprecipitations of extracts from untreated and Bleo-treated cells were split in half and assayed for the ability to phosphorylate PHAS (top), or Western blotted and probed with anti-Rad26 (middle) and anti-Myc (bottom) antibodies. Each lane shows the amount of kinase activity, Rad26, and Myc-Rad3 that coimmunoprecipitated with anti-Myc antibodies in different extracts. Extracts used are from untreated and Bleo-treated rad26Δ myc-rad3 (rad26Δ, TE1057; lanes 1 and 2) cells, rad26+ myc-rad3 (myc-rad3, TE1029; lanes 3 and 4) cells, and untreated rad26+ rad3+ (WT, TE257; lane 5) cells. The middle panel of lane 4 shows that Rad26 is significantly phosphorylated after Bleo treatment, because two distinct bands representing Rad26 are visible. (B) Anti-Myc immunoprecipitations of extracts from untreated and HU-treated cells were split in half and assayed for the ability to phosphorylate PHAS (top), or Western blotted and probed with anti-Rad26 (middle) and anti-Myc (bottom) antibodies. Each lane shows the amount of kinase activity, Rad26, and Myc-Rad3 that coimmunoprecipitated with anti-Myc antibodies in different extracts. Extracts used are from untreated and HU-treated rad26Δ myc-rad3 (rad26Δ, TE1057; lanes 1 and 2) cells, rad26+ myc-rad3 (myc-rad3, TE1029; lanes 3 and 4) cells, and untreated rad26+ rad3+ (WT, TE257; lane 5) cells. All of the lanes are from the same PAGE/Western blot experiment.
Kinase assays with PHAS were preformed as described by Chapman et al. (1998). When kinase activity was directed against unknown proteins that coimmunoprecipitated with Rad26, as shown in Figure 2, anti-Rad26 immunoprecipitations were treated the same except that PHAS was not added to the kinase reactions. In addition, phosphorylated proteins were resolved on 10% SDS-PAGE, whereas PHAS reactions were resolved on 12% SDS-PAGE before autoradiography.
Figure 2.

Phosphorylation of endogenous proteins by the activated Rad26/Rad3 complex. Anti-Rad26 immunoprecipitations of extracts from untreated and Bleo-treated cultures of rad26Δ (lanes 1 and 2, TE257); from untreated, HU-treated, and Bleo-treated cultures of WT (lanes 3, 4, and 5, TE235); and from HU-treated and Bleo-treated cultures of rad3KD (lanes 6 and 7, TE571) cells were divided in half. Top, half of each anti-Rad26 immunoprecipitation was assayed for kinase activity that was directed against unidentified proteins that coimmunoprecipitated with Rad26. These proteins were found only in anti-Rad26 immunoprecipitations of extracts from WT cells (lanes 3–5). The arrows to the right of this panel point to the bands that represent these unidentified, phosphorylated proteins. Estimated protein molecular weights are shown to the right of each arrow. ∗ designates the band in lane 5 that likely represents a hyperphosphorylated form of Rad26. Bottom, Western blot analysis was preformed using the other half of each anti-Rad26 immunoprecipitation. Rad26 only appears in anti-Rad26 immunoprecipitations from extracts of WT (lanes 3–5, TE235) and rad3KD (lanes 6 and 7, TE571) cells. ∗ shows the reduced mobility of Rad26 that occurs when WT cells have been treated with bleomycin (lane 5). This is caused by a Rad3-dependent phosphorylation event that occurs after cells are treated with DNA-damaging agents (Edwards et al., 1999).
Quantification of Kinase Activity and Proteins
Kinase activity directed against PHAS was quantified using two different methods. Analysis of kinase activity was performed directly from dried kinase gels with the STORM 860 phosphorimaging system (Molecular Dynamics, Sunnyvale, CA). Kinase activity was also analyzed from autoradiograms made from the dried kinase assay gels. Autoradiograms were scanned using the UMAX Vista-S8 scanner (UMAX, www.umax.com) and kinase activity was measured with NIH image analysis by using the public domain NIH Image program (developed at the U.S. National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image/). These two different techniques measured Rad3 kinase activity similarly (our unpublished data).
For Figure 1A, the fold-induction of Rad3 kinase activity was determined by normalizing kinase activity to the amount of Rad26 in each immunoprecipitation. For all other figures, the fold-induction of Rad3 kinase activity was determined by normalizing kinase activity to the amount of Myc-Rad3 protein in each immunoprecipitate. We have found that the fold-induction of Rad3 kinase activity caused by treatment with either HU or bleomycin generally ranges between 1.5- and 7-fold (our unpublished data) and attribute these differences to experimental variability.
RESULTS
Direct Evidence for Activation of Rad3 Kinase in Response to Checkpoint Signals
The C-terminal catalytic domain of Rad3 is required for G2/M checkpoint signaling in fission yeast (Bentley et al., 1996; Chapman et al., 1999). In addition, several checkpoint proteins (Hus1, Rad26, Cds1, and Chk1) are phosphorylated after checkpoint activation, and these events are Rad3 dependent (Walworth and Bernards, 1996; Kostrub et al., 1998; Lindsay et al., 1998; Edwards et al., 1999). Although these observations suggest that checkpoint signals induce Rad3 kinase activity, to date there has been no direct evidence for this. To obtain such evidence, we developed an in vitro kinase assay for Rad3.
In previous studies, kinase activity was detected in immunoprecipitates prepared from cells overexpressing epitope-tagged versions of Rad3 (Bentley et al., 1996; Chapman et al., 1999). The activity was not, however, DNA damage inducible, possibly because the protein was overexpressed and thus not associated with key regulators.
More recently it has been shown that Rad3 associates with Rad26 (Edwards et al., 1999), a 69-kDa protein with little informative sequence homology (Al-Khodairy et al., 1994). Rad26 interacts physically with Rad3 in normally growing cells, and is phosphorylated in a rad3+-dependent manner after DNA damage (Edwards et al., 1999). These results suggest that Rad26 and Rad3 work closely together before and during checkpoint activation. Therefore, we tested whether we could use Rad26 antibodies to immunoprecipitate an enzymatically active Rad26/Rad3 complex from cells expressing physiological levels of the two proteins.
Rad26 immunoprecipitations were performed on extracts prepared from cell cultures that were either untreated, or treated with Bleo, a drug that activates the G2/M checkpoint by creating double-stranded breaks in DNA (Suzuki et al., 1970; Kostrub et al., 1998). The Rad26 immunoprecipitations were divided, and one-half was incubated with [32P]ATP and the exogenous substrate PHAS-1, a eukaryotic initiation factor-4E binding protein. Previous studies have shown that Rad3, ATM, and ATR efficiently phosphorylate this substrate (Banin et al., 1998; Canman et al., 1998; Sarkaria et al., 1998; Chapman et al., 1999), and that PHAS-1 is a physiological substrate of ATM (Yang and Kastan, 2000). The kinase reactions were resolved using SDS-PAGE and autoradiography, whereas the other half of the immunoprecipitate was subjected to SDS-PAGE and Western blot analysis to monitor the amount of Rad26 in each immunoprecipitate.
Figure 1A shows that a Rad26-associated kinase activity that phosphorylated PHAS was immunoprecipitated from wild-type cells (lane 1). Approximately 1.7-fold more kinase activity was coimmunoprecipitated with Rad26 from Bleo-treated cells (Figure 1A, lane 2; see MATERIALS AND METHODS). Western blot analysis revealed that some of the Rad26 in the Bleo-treated sample had reduced mobility (Figure 1A, compare lanes 1 and 2). This is likely due to rad3+-dependent phosphorylation that has previously been detected in cells treated with UV or γ-irradiation (Edwards et al., 1999). Anti-Rad26 antibodies failed to immunoprecipitate kinase activity from untreated or Bleo-treated rad26Δ cells (Figure 1A, lanes 5 and 6), showing that the activity specifically associates with the Rad26 protein. To determine whether the Rad26-associated activity was due to Rad3, we examined kinase activity in the rad3-KD strain that has a mutation (D2230A) in the conserved kinase domain of Rad3 that eliminates cell cycle arrest in response to DNA-damaging agents (Bentley et al., 1996; Chapman et al., 1999). Lanes 3 and 4 show that PHAS phosphorylation was dramatically reduced and Rad26 phosphorylation was undetectable when extracts from untreated or Bleo-treated rad3-KD cells were used. Therefore, Rad3 is most likely the source of this Bleo-induced, Rad26-associated kinase activity.
These experiments demonstrate directly that the kinase activity of the Rad26/Rad3 complex increases in response checkpoint signals. We show below (Figure 4) that this increase is not due to the DNA damage-dependent cell cycle arrest caused by Bleo treatment.
Figure 4.

Activation of the Rad26/Rad3 complex by Bleo and HU treatments does not require many of the checkpoint proteins. (A) Anti-Rad26 immunoprecipitations from different cell extracts were assayed for kinase activity (top) or Western blotted and probed with anti-Rad26 (bottom). The extracts were collected from untreated and Bleo-treated hus1Δ myc-rad3 (lanes 1 and 2, TE1059), rad1Δ myc-rad3 (lanes 3 and 4, TE1051), rad9Δ myc-rad3 (lanes 5 and 6, TE1055), rad17Δ myc-rad3 (lanes 7 and 8, TE1050), chk1Δ cds1Δ myc-rad3 (lanes 9 and 10, TE1053) and myc-rad3 (WT; lanes 11 and 12, TE1029) cells, in addition to Bleo-treated rad26Δ myc-rad3 (lane 13, TE1057) cells. (B) Procedure as outlined in A was used, except cells were treated with HU instead of bleomycin. In addition, the amount of Myc-Rad3 that coimmunoprecipitated with Rad26 is shown in the lower panel. Anti-Rad26 immunoprecipitations collected from untreated and HU-treated rad1Δ myc-rad3 (TE1051), rad9Δ myc-rad3 (TE1055), and rad17Δ myc-rad3 (TE1050) cell extracts (our unpublished data) behaved similarly to those of untreated and HU-treated hus1Δ myc-rad3 (lanes 1 and 2, TE1059) and myc-rad3 (WT; lanes 5 and 6, TE1029) cell extracts, respectively. As described by Edwards et al. (1999), Rad26 was modified after chk1Δ cds1Δ cells were treated with HU (lane 4).
Regulation of Rad26/Rad3 Complex
We next examined whether the increase in kinase activity after DNA damage was due to increased association of Rad3 with Rad26, or an increase in the intrinsic activity of the Rad26/Rad3 complex. To distinguish between these possibilities, we tested whether the amount of Rad3 associated with Rad26 changed after Bleo treatment. To measure Rad3 levels, we used a strain with an integrated, Myc-tagged version of Rad3 controlled by the rad3+ promoter (Edwards et al., 1999). Extracts were prepared from this strain before Bleo treatment and at the indicated times points afterward (Figure 1B). The extracts were immunoprecipitated with anti-Rad26 antibody. Half of the immunoprecipitation was used to assay kinase activity with PHAS as a substrate (Figure 1B, top) and the other half was resolved on SDS-PAGE and blotted with anti-Rad26 and anti-Myc antibodies. Figure 1B shows that the level of Myc-Rad3 kinase activity directed against PHAS during early time points (T = 0 and 0.2) increased sevenfold (see MATERIALS AND METHODS) after 2 h of Bleo treatment (T = 2.2, 2.4, and 3.0 h). Western analyses showed that Rad26 also became phosphorylated after 2 h of treatment. These analyses also showed that the amount of Myc-Rad3 associated with Rad26 remained constant throughout the experiment, demonstrating that rather than stimulating the Rad26–Rad3 association, Bleo causes an increase in the kinase activity of the Rad26/Rad3 complex.
Together, these data indicate that the Rad26/Rad3 complex has kinase activity and provide the first direct evidence that checkpoint signals activate Rad3 kinase activity. Rad3 is the catalytic subunit of the Rad26/Rad3 complex, because kinase activity is abolished by mutations in the conserved kinase domain of Rad3.
Phosphorylation of Coimmunoprecipitating Proteins by Activated Rad26/Rad3 Complex
We next examined whether the in vitro kinase assay we developed could be used to examine phosphorylation of endogenous Rad3 substrates. To activate the checkpoint, cultures were treated with Bleo or HU, a drug that stalls the progression of replication forks during S phase. Rad26 was immunoprecipitated from cell extracts and half of each immunoprecipitate was incubated with [32P]ATP in kinase buffer without the exogenous substrate, PHAS. These kinase reactions were then resolved using SDS-PAGE and autoradiography. Endogenous substrates of the Rad26/Rad3 complex that coimmunoprecipitate with Rad26 become phosphorylated in the kinase assay and evident as bands on the autoradiograms. The other half of the immunoprecipitation was subjected to SDS-PAGE and Western blot analysis to monitor the amount of Rad26 in each immunoprecipitation.
Anti-Rad26 immunoprecipitates from extracts of untreated or Bleo-treated rad26Δ cultures contained a background kinase activity directed against a similar pattern of unidentified proteins (Figure 2, top, lanes 1 and 2). Anti-Rad26 immunoprecipitates from extracts of untreated WT cells contained this background kinase activity, as well as one directed against proteins of ∼220 and ∼70 kDa (Figure 2, top, lane 3). The ∼70-kDa band may represent in vitro phosphorylation of Rad26 (Edwards et al., 1999) by Rad3, whereas the ∼220-kDa band may represent Rad3 autophosphorylation, which has previously been reported (Bentley et al., 1996; Chapman et al., 1999).
Treatment of cells with HU or bleomycin resulted in the phosphorylation of the 70- and 220-kDa proteins as well as three additional bands (Figure 2, bottom, lanes 4 and 5; ∼47, ∼33, and ∼17 kDa). These differences were not due to the amount of Rad26 in each kinase assay because Western analysis showed that Rad26 levels remained relatively constant (Figure 2, bottom, compare lanes 3, 4, and 5). Because these bands were not observed when kinase assays were preformed using extracts from HU- and Bleo-treated rad3KD cells (Figure 2, bottom, lanes 6 and 7), they may represent proteins phosphorylated in vitro by the activated Rad3 kinase. Possibly, checkpoint activation promotes association of Rad26/Rad3 with additional proteins, including the substrates detected herein. In conclusion, the activity of the Rad26/Rad3 complex toward these putative substrates, as well as toward the exogenous substrate PHAS, is stimulated by checkpoint signals.
This experiment also suggests that, depending on the type of checkpoint signal, activation of the Rad26/Rad3 kinase can have different results. A comparison of Figure 2, top (lanes 4 and 5) revealed that both HU (stalled replication forks) and Bleo (DNA damage) stimulated Rad26/Rad3 activity to similar extents. However, Western blot analysis showed that a form of Rad26 with decreased mobility (indicated by arrowhead in the Figure 2, bottom) was only detected in extracts from Bleo-treated cells, and not in extracts from HU-treated cells. This form of Rad26 may also be represented in the kinase assays by the band that migrated slightly slower than 70 kDa (indicated by ∗). This band was observed only after treatment with Bleo, and not after treatment with HU (Figure 2, top, compare lanes 4 and 5). By Western blotting, Edwards et al. (1999) previously observed a phosphorylated form of Rad26 with reduced mobility after DNA damage, but not after HU treatment. This may mean that phosphorylation of Rad26 does not occur after replication forks are stalled, or that HU treatment may lead to phosphorylation on a different site that does not affect the mobility of the protein. In agreement, our studies show that the mobility of Rad26 does not change after treatment with HU. However, we observed a significant increase in kinase activity despite the absence of this Rad26 modification. We suggest, therefore, that the mobility-shifted, phosphorylated form of Rad26 is not required for Rad3 kinase activation. Rather, the status of Rad26 may depend on the nature of the signal that leads to activation of the Rad26/Rad3 complex.
In the kinase assay (Figure 2, top, lanes 3–5), we did observe in vitro incorporation of 32P into the 70-kDa band in untreated, HU-treated, and Bleo-treated cells. We do not know whether this phosphorylation event is physiologically relevant, or is a consequence of the in vitro assay conditions.
Rad3 Kinase Activity Requires Rad26
As shown above, Rad26 constitutively associates with Rad3 kinase activity. To test whether Rad3 requires Rad26 for basal and/or activated levels of kinase activity, we constructed a strain containing the Myc epitope-tagged version of Rad3 (Edwards et al., 1999) in a rad26Δ background. We then used anti-Myc monoclonal antibodies to immunoprecipitate Myc-Rad3 from myc-rad3 rad26+ and myc-rad3 rad26Δ extracts collected from untreated, HU-, and Bleo-treated cell extracts. As shown in Figure 3A, anti-Myc immunoprecipitations of extracts from a wild-type strain lacking an epitope-tagged version of Rad3 (WT, lane 5) did not contain Rad26 or significant levels of kinase activity. Likewise, anti-Myc immunoprecipitations prepared from extracts of untreated or Bleo-treated myc-rad3 rad26Δ cells (Figure 3A, lanes 1 and 2) contained only background levels of kinase activity, although Myc-Rad3 was present in each of these immunoprecipitations. Anti-Myc immunoprecipitations from untreated myc-rad3 rad26+ cell extracts (Figure 3A, lane 3) did contain kinase activity that became elevated (1.7-fold relative to the amount of Myc-Rad3) when cells were treated with bleomycin (Figure 3A, lane 4). These data establish that Rad26 is required for both basal and induced levels of Rad3 kinase activity.
Next, we investigated whether, like Bleo treatment, HU-treatment also caused an increase in Rad3 activity that depended upon Rad26. As shown in Figure 3B, anti-Myc immunoprecipitations of extracts from a wild-type strain lacking an epitope-tagged version of Rad3 (WT, lane 5) did not contain Rad26 or significant levels of kinase activity. Likewise, anti-Myc immunoprecipitations prepared from extracts of untreated or HU-treated myc-rad3 rad26Δ cell extracts (Figure 3B, lanes 1 and 2) contained only background levels of kinase activity, although Myc-Rad3 was present in each of these immunoprecipitations. Anti-Myc immunoprecipitations from untreated myc-rad3 rad26+ cell extracts (Figure 3B, lane 3) did contain kinase activity that became elevated (threefold relative to the amount of Myc-Rad3) when cells were treated with HU (Figure 3B, lane 4). These data establish that Rad26 is also required for HU-activated levels of Rad3 kinase activity.
In summary, Rad26 is required for basal as well as Bleo- and HU-activated levels of Rad3 kinase activity. These results suggest that the checkpoint kinase in fission yeast consists of a catalytic subunit (Rad3), and a regulatory subunit (Rad26).
Activation of Rad3/Rad26 Kinase Does not Require Hus1, Rad1, Rad9, or Rad17
We next investigated the requirement of other checkpoint Rad proteins for the basal and activated forms of the Rad26/Rad3 checkpoint kinase. Like Rad26 and Rad3, these are also required for full checkpoint activity because disruption of hus1+, rad1+, rad9+, or rad17+ abolishes the checkpoint response to damaged DNA and replication arrest (Al-Khodairy and Carr, 1992; Enoch et al., 1992; Rowley et al., 1992; Al-Khodairy et al., 1994). The roles and biochemical properties of these gene products are not known.
For these studies, Rad26 was immunoprecipitated from extracts prepared before and after cells were treated with bleomycin or HU. Half of each anti-Rad26 immunoprecipitation was used to assay Rad3 kinase activity, and the other half was subjected to SDS-PAGE and Western blotting to observe Rad26. As shown in Figure 4A, Rad3 kinase activity was detectable in extracts from untreated hus1Δ, rad1Δ, rad9Δ, rad17Δ, chk1Δ cds1Δ, and WT cells. Induction of kinase activity in response to Bleo treatment was also observed in all of these strains (Figure 4A, lanes 1–12). This demonstrates that activation of the Rad3 kinase in response to DNA damage does not require these checkpoint proteins. In this experiment, the kinase activity isolated from untreated and Bleo-treated extracts of hus1Δ cells was very low (Figure 4A, lanes 1 and 2). This result, however, was not reproducible because further testing has shown that hus1Δ cells have levels of constitutive and induced Rad26/Rad3 kinase activity that are similar to those of WT cells (our unpublished data). As expected, no activity was observed in extracts from Bleo-treated rad26Δ cells (Figure 4A, lane 13). Western analyses confirmed that similar amounts of Rad26 were present in each kinase assay. Western analyses also confirmed that Rad26 phosphorylation after Bleo treatment did not require any of these checkpoint proteins (Edwards et al., 1999), consistent with this observation that normal kinase activity of the Rad26/Rad3 complex does not require these proteins. These blots also show that slight modification of Rad26 occurred in untreated rad9Δ, rad17Δ, and chk1Δ cds1Δ cells (Figure 4A, lanes 5, 7, and 9), suggesting that the absence of these proteins alone can lead to production of a checkpoint signal.
Figure 4B shows that Rad3 kinase activity was also induced in extracts collected from HU-treated hus1Δ myc-rad3+, chk1Δ cds1Δ myc-rad3+, and myc-rad3+ cells (lanes 1–6). Extracts from HU-treated rad1Δ, rad9Δ, and rad17Δ cells also contained inducible kinase activity (our unpublished data). Kinase activity was not detected in anti-Rad26 immunoprecipitations of extracts from HU-treated rad26Δ myc-rad3+ (Figure 4B, lane 7) and HU-treated rad3KD (our unpublished data) cells. Western analyses showed that similar amounts of Rad26 and Myc-Rad3 were collected in each experiment, and that the Rad26/Rad3 association did not change following HU treatment.
These data demonstrate that the Rad26/Rad3 kinase checkpoint response can be initiated in the absence of the Hus1, Rad1, Rad9, Rad17, Chk1, and Cds1 checkpoint proteins. In addition, these experiments demonstrate that Bleo- and HU-induced kinase activation of the Rad26/Rad3 complex is not a consequence of cell cycle arrest, because hus1Δ, rad1Δ, rad9Δ, rad17Δ, chk1Δ cds1 cells are checkpoint-deficient and progress through the cell cycle in the presence of these genotoxic stresses (Kostrub et al., 1998).
Western analyses reconfirmed that Rad26 was not modified after HU treatment of WT cells (Figure 4B, lane 6). However, we observed Rad26 modification in HU-treated cds1Δ chk1Δ (Figure 4B, lane 4) cells. Edwards et al. (1999) also reported this, and suggested that it occurs in response to replication-associated DNA damage that may be generated when S-phase arrest takes place in the absence of Cds1.
Overall, we find that the Hus1, Rad1, Rad9, and Rad17 checkpoint Rad proteins, in addition to Chk1 and Cds1, are not required for basal or elevated levels of Rad3 kinase activity after Bleo and HU treatments. However, genetic and physiological studies have shown that these proteins are required for the checkpoint response in vivo (Al-Khodairy and Carr, 1992; Enoch et al., 1992; Rowley et al., 1992; Al-Khodairy et al., 1994). We conclude that a normal checkpoint response requires more than just activation of the Rad3 kinase. Furthermore, we conclude that of these checkpoint proteins, Rad3 and Rad26 are necessary and sufficient for kinase activation in response to either replication arrest or DNA damage.
Quantitatively Different Levels of Rad3/Rad26 Kinase Activity May be Required for Cell Cycle Arrest and Recovery
Rad3 and Rad26, in cooperation with the other checkpoint Rad proteins, serve at least two functions during checkpoint signaling. To bring about cell cycle arrest, they are required to transduce the checkpoint signal to targets in the cell cycle machinery. To promote recovery, they may regulate components of the DNA repair and/or recombination machinery. The induction of Rad3 kinase activity that we observed after Bleo and HU treatments could be required for cell cycle arrest, recovery, or both. Although null alleles of the checkpoint rads eliminate both activities, a mutant allele of rad26+, rad26.T12, genetically separates these functions. Cells expressing rad26.T12 are proficient for G2 arrest, but lose viability after UV or ionizing-radiation treatments (Al-Khodairy et al., 1994; Lindsay et al., 1998), suggesting that the recovery function is specifically compromised in these cells.
To examine the mechanism by which the rad26.T12 mutation eliminates recovery, we performed kinase assays with rad26.T12 cells before and after Bleo treatment (Figure 5A). For this experiment, we introduced the myc-rad3 gene into a rad26.T12 strain. Anti-Rad26 immunoprecipitations of extracts from untreated and treated cells were split and used for kinase assays and Western blotting. Anti-Rad26 immunoprecipitations from extracts of Bleo-treated myc-rad3+ rad26Δ cells did not contain kinase activity (Figure 5A, lane 1). Immunoprecipitations from extracts of myc-rad3+ rad26+ cells contained activity (Figure 5A, lane 2) that increased after treatment with bleomycin (Figure 5A, lane 3). Immunoprecipitates from myc-rad3+ rad26.T12 cells exhibited kinase activity (Figure 5A, lane 4) that also increased after Bleo treatment (Figure 5A, lane 5). However, basal levels of Rad26.T12/Myc-Rad3 kinase activity (Figure 5A, lane 4) were 10-fold lower than the levels found associated with Rad26/Myc-Rad3 (Figure 5A, lane 2; see MATERIALS AND METHODS), and activated levels of Rad26.T12/Myc-Rad3 kinase activity (Figure 5A, lane 5) were threefold lower than the levels found associated with Rad26/Myc-Rad3 (Figure 5A, lane 3). Western blot analysis revealed that equal amounts of Rad26 and Rad26.T12 were immunoprecipitated throughout the experiment. Western blots also revealed that Rad26.T12 was phosphorylated as efficiently as Rad26 after Bleo treatment (Figure 5A, compare lanes 3 and 5). Together, these kinase and Western blot data suggest that the Rad26.T12 protein does not affect the ability of the complex to be activated by DNA damage.
However, we found that significantly less Myc-Rad3 coimmunoprecipitated with Rad26.T12 than with Rad26 (Figure 5A, compare lanes 2 and 3 with 4 and 5). This suggests that Rad26.T12 associates inefficiently with Myc-Rad3. This reduced association between Rad26.T12 and Myc-Rad3 is likely responsible for the low, absolute levels of kinase activity we found in anti-Rad26 immunoprecipitations of extracts collected from both untreated and Bleo-treated myc-rad3+ rad26.T12 cells.
To examine the ability of rad26.T12 cells to arrest cell cycle progression after checkpoint activation, samples from rad26+ myc-rad3, rad26Δ myc-rad3, and rad26.T12 myc-rad3 strains in liquid cultures were collected before and after Bleo treatment (Figure 5B). Before treatment, the rad26+ myc-rad3 culture contained short, wild-type length cells that were occasionally binucleate and septate (Figure 5B, panel 1). After Bleo treatment, rad26+ myc-rad3 cells were long and mononucleate and did not contain septa, three characteristics of checkpoint-arrested cells (Figure 5B, panel 2) (Enoch et al., 1992). The rad26Δ myc-rad3 cells were short, binucleate, and septate before and after Bleo treatment (Figure 5B, panels 3 and 4). This shows that rad26Δ myc-rad3 cells did not arrest cell cycle progression after Bleo treatment and instead continued to divide, producing both short cells and somewhat longer, septate cells with uneven distributions of DNA (Figure 5B, panel 4). This phenotype is typical of checkpoint-deficient cells (Enoch et al., 1992). Although Rad26/Rad3 kinase activity was very low in Bleo-treated rad26.T12 myc-rad3 cells (Figure 5A, lanes 4 and 5), the cells still underwent normal cell cycle arrest, and appeared as long, mononucleate, unseptate cells after Bleo treatment (Figure 5B, panel 6). This confirms that rad26.T12 cells have an intact cell cycle arrest function (Al-Khodairy et al., 1994; Lindsay et al., 1998).
In conclusion, the rad26.T12 mutation may abolish recovery by destabilizing the Rad26/Rad3 complex, thereby reducing the overall amount of Rad26/Rad3 kinase activity in the cell (Figure 5A). In contrast, the mutation does not affect checkpoint-induced cell cycle arrest (Figure 5B), suggesting that the cells still contain enough Rad26/Rad3 kinase activity to perform this function. Thus, the cell cycle arrest function of the checkpoint seems to require less Rad26/Rad3 activity than recovery.
DISCUSSION
Direct Activation of Rad3 Kinase by Checkpoint Signals
The Rad3 kinase transduces the status of the genome to downstream, checkpoint processes, including cell cycle arrest and recovery. Previous studies have shown that a number of proteins involved in checkpoint control are phosphorylated in a Rad3-dependent manner in response to genotoxic stresses (Walworth and Bernards, 1996; Kostrub et al., 1998; Lindsay et al., 1998; Edwards et al., 1999). These observations predict that Rad3 kinase activity changes in response to checkpoint signals. We have developed an assay that directly measures the kinase activity of physiological levels of Rad3, and used it herein to demonstrate that the intrinsic kinase activity of Rad3 against PHAS (Figure 1) and coimmunoprecipitating proteins (Figure 2) increases in response to checkpoint signals. This activation is not an indirect consequence of cell cycle arrest, because induction of the kinase was also detected in checkpoint arrest-deficient mutants (Figure 4, A and B). This investigation provides the first direct evidence for activation of the Rad3 kinase by checkpoint signals.
We believe that we have been able to observe this regulation because we used cells expressing physiological levels of the Rad3 kinase. In contrast, other studies using strains overexpressing Rad3 did not observe regulation of kinase activity in response to checkpoint signals (Bentley et al., 1996; Chapman et al., 1999). Another study found that HU treatment activated a kinase associated with an overexpressed, tagged version of Rad3; however, the activity was due to Cds1 instead of Rad3 (Moser et al., 2000). Because we observed Bleo- and HU-induced kinase activation in cds1Δ chk1Δ cells, as well as in hus1Δ, rad1Δ, rad9Δ, and rad17Δ cells (Figure 4, A and B) where the Cds1 response to HU treatment is absent (Lindsay et al., 1998), the activity we detected is not due to Cds1. In contrast to our findings, Wakayama et al (2001) did not observe stimulation of Mec1 kinase activity after treatment of cells with methyl methane sulfonate (MMS). While we do not know if Rad3 activity changes after MMS treatment, this result could mean that Mec1 and Rad3 activate the checkpoint response using different mechanisms.
In humans, direct evidence for ATM kinase induction in response to treatment with a DNA-damaging agent has also been obtained (Banin et al., 1998; Canman et al., 1998). Activity of ATM increases after ionizing radiation, and this increase correlates with ATM-dependent phosphorylation of CHK2 (Matsuoka et al., 1998, 2000). In contrast, an increase of ATR kinase activity has not been observed after such treatments. However, relocation of ATR to nuclear foci containing BRCA1 occurs in response to DNA damage and replication blocks, suggesting that downstream signaling by this kinase is regulated by localization (Tibbetts et al., 2000).
Our studies regarding the phosphorylation of Rad26 coimmunoprecipitating substrates (Figure 2) provide preliminary evidence that Rad26/Rad3 kinase activation may also involve association with new proteins. As shown, we detected in vitro phosphorylation of three peptides when extracts from cells treated with HU or bleomycin were used, but not when extracts from untreated cells were used. These peptides could be nonphysiological substrates that were phosphorylated due to general stimulation of Rad26/Rad3 kinase activity. Alternatively, checkpoint activation may induce association of the Rad26/Rad3 complex with other proteins. Such proteins could include the other checkpoint Rad proteins, Chk1, Cds1, or proteins that have not been identified. Genetic and biochemical studies aimed at identifying these proteins are currently underway.
Constitutive Activity of Rad3 Kinase
Although Rad3 kinase activity is stimulated by bleomycin and HU, we also detected significant levels of Rad3 kinase activity in the absence of inducers (Figure 1). Although this result could be an in vitro artifact, it may indicate that low levels of DNA damage or replication intermediates activate the Rad3 kinase during a normal cell cycle. This kinase activity may function in the repair of this spontaneous damage, or possibly in telomere maintenance, an activity that Rad3 has been implicated in Naito et al. (1998). ATM also seems to have constitutive kinase activity (Canman et al., 1998) that may perform similar functions in mammalian cells (reviewed by Shiloh, 1997).
Rad26 Is a Regulatory Subunit of Rad3 Kinase
To investigate whether Rad26 is required for Rad3 kinase activity, we used strains expressing a Myc-epitope tagged version of Rad3 under the control of the rad3+ promoter. These strains allowed us to use antibodies raised against the Myc epitope to directly immunoprecipitate Rad3 from strains expressing physiological levels of the protein. When assayed using PHAS as a substrate (Figure 3), Rad26 is required for constitutive Rad3 kinase activity and induction of this activity in response to HU and bleomycin. Previous studies have shown that phosphorylation of Hus1 and Chk1 in vivo also requires Rad26 (Walworth and Bernards, 1996; Kostrub et al., 1998). Taken together, these observations demonstrate that the combined action of Rad26 and Rad3 is required for Rad3 kinase activity to respond to checkpoint signals under physiological conditions. These findings lead us to propose that the checkpoint kinase consists of two different kinds of subunits. The regulatory subunit is Rad26, which does not have kinase activity of its own (Figure 1A, lanes 3 and 4) but is required for Rad3 activity. The catalytic subunit of the complex is Rad3.
In S. cerevisiae, a protein with weak sequence similarity to Rad26 that physically interacts with Mec1 has been described. This protein has been called LCD1, DDC2, and PIE1 (Paciotti et al., 2000; Rouse and Jackson, 2000; Wakayama et al., 2001). Deletion of the corresponding gene causes phenotypes similar to those caused by deletion of mec1. However, this protein is not required for Mec1 kinase activity (Wakayama et al., 2001), suggesting that Mec1 could be regulated differently than Rad3 in this regard. The predicted product of the uvsD+ gene in Aspergillus nidulans also shows weak similarity to Rad26, and mutations in uvsD+ also abolish the G2/M checkpoint (De Souza et al., 1999). It is not known whether the UVSD protein is required for activity of UVSB, the Rad3-related kinase in this organism (De Souza et al., 1999; Hofmann and Harris, 2000).
Proteins with sequence similarity to Rad26 have not been identified in higher eukaryotes. The low sequence conservation of Rad26 among fungi suggests that if such proteins exist, their relatedness to the fungal proteins may not be obvious. Because these proteins may be tightly associated with the Rad3-related kinases of higher eukaryotes, perhaps they could be identified using biochemical approaches.
Hus1, Rad1, Rad9, and Rad17 Are not Required for Either Constitutive or Activated Rad3 Kinase Activity
The Hus1, Rad1, Rad9, and Rad17 proteins play an essential role in the fission yeast checkpoint response. Edwards et al. (1999) found that these proteins were not required for Rad26 phosphorylation in response to bleomycin, suggesting that they are not required for sensing the DNA damage checkpoint signal. Hus1, Rad1, Rad9, Rad17, and Rad26 are, however, required for the phosphorylation of other Rad3 substrates, such as Chk1 and Cds1. These results raise questions about the roles of Hus1, Rad1, Rad9, and Rad17 in the checkpoint response. They may be required to induce Rad3 kinase activity to a level that allows phosphorylation of Chk1 and Cds1 after the checkpoint signal is recognized. Alternatively, they may not play a role in regulating Rad3 activity, and may act downstream of Rad3 in another manner.
To investigate this issue, we analyzed Rad3 kinase activation in response to DNA damage and replication arrest in a variety of checkpoint-deficient strains (Figure 4). We found that Rad3 kinase activation by either bleomycin or HU did not require Hus1, Rad1, Rad9, or Rad17. Therefore, we suggest that these proteins act after Rad3 activation in the response to both DNA damage and replication arrest. Possibly, Hus1, Rad1, Rad9, and Rad17 are required to direct the kinase activity of the Rad26/Rad3 complex to downstream checkpoint substrates. The DNA damage checkpoint pathway of budding yeast may operate in a similar manner, because Mec1-dependent phosphorylation of the Rad26-like protein DDC2 occurs after DNA damage in the absence of MEC3, DDC1, RAD17, and RAD24 (Paciotti et al., 2000).
Role of Rad26 Phosphorylation in Checkpoint-dependent Kinase Activation
Edwards et al. (1999) have previously reported that Rad26 is phosphorylated in response to DNA damage caused by UV and γ-irradiation. In contrast, Rad26 does not seem to be phosphorylated when replication forks are stalled by treatment with HU. To determine whether Rad26 phosphorylation is required for kinase activation, we compared kinase activity and Rad26 phosphorylation in cells treated with Bleo and HU. Using Rad26 coimmunoprecipitating substrates (Figure 2) or PHAS (Figure 3) to quantitate kinase activity, we found that HU treatment stimulated Rad26/Rad3 kinase activity to roughly the same extent as Bleo treatment. We did not detect phosphorylated Rad26 in HU-treated extracts, although it was readily detected in Bleo-treated extracts (Figures 2–4). These results suggest that Rad26 phosphorylation is not necessary for Rad3 kinase activation.
The phosphorylation of Rad26 in response to bleomycin (Figure 2) and other DNA-damaging agents (Edwards et al., 1999), but not in response to HU, leads us to suggest that the phosphorylation status of Rad26 depends on the nature of the checkpoint signal. This is important, because it is not known how DNA damage and replication arrest activate the same checkpoint response. It has been suggested that a processing pathway converts many different primary checkpoint signals to a common structure, such as single-stranded DNA, which then activates a checkpoint pathway (Lydall and Weinert, 1995; Lee et al., 1998). Because the phosphorylation status of Rad26 depends on the checkpoint signal, we suggest instead that the Rad26/Rad3 complex is directly influenced by the nature of the checkpoint signal. This, together with the fact that none of the other checkpoint proteins are required for activation, is consistent with a model in which the Rad26/Rad3 complex interacts directly with different checkpoint signals.
Cell Cycle Arrest and Recovery May Require Different Levels of Rad3 Kinase Activity
Checkpoint genes were initially identified because of their role in ensuring cell cycle arrest in response to genotoxic insults (Weinert and Hartwell, 1988). Later studies have shown that the same genes are also required for a poorly characterized process that has been termed “recovery” (Enoch et al., 1992; Stewart et al., 1997; Desany et al., 1998; Lindsay et al., 1998). When replication is arrested, this process allows cells to eventually complete S phase, possibly by preventing illegitimate recombination that may occur after the regression of stalled replication forks (Seigneur et al., 1998; Viguera et al., 2000). Cells lacking Rad3 or Rad26 are deficient in both cell cycle arrest and recovery.
A previously identified mutation, rad26.T12, abolishes recovery, but not cell cycle arrest, in response to checkpoint signals (Al-Khodairy et al., 1994; Lindsay et al., 1998). To determine the biochemical basis of this defect, we examined the kinase activity of the Rad3/Rad26.T12 complex. We found that the activity of the Rad3/Rad26.T12 complex was lower than that of the wild-type complex, even although Bleo-treatment induced both (Figure 5A, lanes 4 and 5). Because less Rad3 associated with the mutant Rad26.T12 protein (Figure 5A, compare lanes 2 and 3 with 4 and 5), we suggest that the rad26.T12 mutation may alter the stability of the Rad26/Rad3 complex. Interestingly, although less Rad3 associates with Rad26.T12, the mutant protein is phosphorylated as efficiently as the wild-type protein (Figure 5A, compare lanes 3 and 5). This could mean that the Rad26.T12/Rad3 complex initially forms, but is less stable than the wild-type complex. Alternatively, it may indicate that the complex is stable in vivo, but disrupted by our extraction or assay conditions.
Our results suggest that the rad26.T12 mutation acts by reducing the stability and activity of the checkpoint kinase complex. This may only affect recovery because recovery may require more kinase activity than cell cycle arrest. Possibly, cell cycle arrest requires only a pulse of kinase activity from the Rad26/Rad3 complex. This conclusion is consistent with studies of Martinho et al. (1998). Using a temperature-sensitive allele of rad3, this group showed that Rad3 is required to initiate the Chk1-dependent, cell cycle arrest response, but not at later times to maintain it. Together, these results suggest that quantitative differences in the kinase activity of the Rad3/Rad26 complex may determine whether the cell cycle arrest or recovery pathway is activated in response to checkpoint signals.
ACKNOWLEDGMENTS
We are grateful to Tony Carr for the myc-rad3 strain, to Cory Kostrub and Musetta Leung for raising antibodies against Rad26, and to Cory Kostrub and Sarah Evans for technical help. We are also very thankful to Bob Weiss, Steve Harris, Gary Rathbun, and members of the Enoch lab for many helpful discussions, and to the anonymous MBC reviewers for their very helpful critiques. This work was supported by National Institutes of Health grants (GM-19773-03 to T.W. and GM-50015 to T.E.).
Footnotes
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.01–03-0104. Article and publication date are at www.molbiolcell.org/cgi/10.1091/mbc.01–03-0104.
REFERENCES
- Abraham R. Cell cycle checkpoint signaling through the ATM, and ATR kinases. Genes Dev. 2001;15:2177–2121. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- Al-Khodairy F, Carr AM. DNA repair mutants defining G2 checkpoint pathways in Schizosaccharomyces pombe. EMBO J. 1992;11:1343–1350. doi: 10.1002/j.1460-2075.1992.tb05179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Khodairy F, Fotou E, Sheldrick KS, Griffiths DJF, Lehmann AR, Carr AM. Identification and characterization of new elements involved in checkpoint and feedback controls in fission yeast. Mol Biol Cell. 1994;5:147–160. doi: 10.1091/mbc.5.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banin S, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;11:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- Bentley NJ, Hawkins G, Keegan K, DeMaggio A, Holtzman D, Ford JC, Hoekstra M, Carr AM. The Schizosaccharomyces pombe rad3 checkpoint gene. EMBO J. 1996;15:6641–6651. [PMC free article] [PubMed] [Google Scholar]
- Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- Caspari T, Dahlen M, Kanter-Smoler G, Lindsay H, Hofman K, Papadimitriou K, Sunnerhagen P, Carr A. Characterization of S. pombe Hus1. a PCNA-related protein that associates with Rad1 and Rad9. Mol Cell Biol. 2000;20:1254–1262. doi: 10.1128/mcb.20.4.1254-1262.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman CR, Evans ST, Carr AM, Enoch T. Requirement of sequences outside the conserved kinase domain of fission yeast Rad3p for checkpoint control. Mol Biol Cell. 1999;10:3223–3238. doi: 10.1091/mbc.10.10.3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Souza C, Ye XS, Osmani SA. Checkpoint defects leading to premature mitosis also cause endoreplication of DNA in Aspergillus nidulans. Mol Biol Cell. 1999;10:3661–3674. doi: 10.1091/mbc.10.11.3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desany BA, Alcasabas AA, Bachant JB, Elledge SJ. Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway. Genes Dev. 1998;12:2956–2970. doi: 10.1101/gad.12.18.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards RJ, Bentley NJ, Carr AM. A Rad3-Rad26 complex responds to DNA damage independently of other checkpoint proteins. Nat Cell Biol. 1999;1:393–398. doi: 10.1038/15623. [DOI] [PubMed] [Google Scholar]
- Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science. 1996;274:1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- Enoch T, Carr AM, Nurse P. Fission yeast genes involved in coupling mitosis to completion of DNA replication. Genes Dev. 1992;6:2035–2046. doi: 10.1101/gad.6.11.2035. [DOI] [PubMed] [Google Scholar]
- Furnari B, Rhind N, Russell P. Cdc25 mitotic inducer targeted by Chk1 DNA damage checkpoint kinase. Science. 1997;277:1495–1497. doi: 10.1126/science.277.5331.1495. [DOI] [PubMed] [Google Scholar]
- mcGriffiths DJF, Barbet NC, McCready S, Lehmann AR, Carr AM. Fission yeast rad17: a homologue of budding yeast RAD24 that shares regions of sequence similarity with DNA polymerase accessory proteins. EMBO J. 1995;14:5812–5823. doi: 10.1002/j.1460-2075.1995.tb00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH, Weinert TA. Checkpoints: controls that ensure the order of cell cycle events. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- Hoekstra MF. Responses to DNA damage and regulation of cell cycle checkpoints by the ATM protein kinase family. Curr Opin Genet Dev. 1997;7:170–175. doi: 10.1016/s0959-437x(97)80125-6. [DOI] [PubMed] [Google Scholar]
- Hofmann AF, Harris SD. The Aspergillus nidulans uvsB gene encodes an ATM-related kinase required for multiple facets of the DNA damage response. Genetics. 2000;154:1577–1586. doi: 10.1093/genetics/154.4.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostrub CF, Knudsen K, Subramani S, Enoch T. Hus1p, a conserved fission yeast checkpoint protein, interacts with Rad1p and is phosphorylated in response to DNA damage. EMBO J. 1998;17:2055–2066. doi: 10.1093/emboj/17.7.2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna TSR, Kong XP, Gary S, Burgers P, Kuriyan J. Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell. 1994;79:1233–1243. doi: 10.1016/0092-8674(94)90014-0. [DOI] [PubMed] [Google Scholar]
- Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD, Haber JE. Saccharomyces Ku70, Mre11/Rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell. 1998;94:399–409. doi: 10.1016/s0092-8674(00)81482-8. [DOI] [PubMed] [Google Scholar]
- Lindsay HD, Griffiths DJ, Edwards RJ, Christensen PU, Murray JM, Osman F, Walworth N, Carr AM. S-phase-specific activation of Cds1 kinase defines a subpathway of the checkpoint response in Schizosaccharomyces pombe. Genes Dev. 1998;12:382–395. doi: 10.1101/gad.12.3.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydall D, Weinert T. Yeast checkpoint genes in DNA damage processing: implications for repair and arrest. Science. 1995;270:1488–1491. doi: 10.1126/science.270.5241.1488. [DOI] [PubMed] [Google Scholar]
- Martinho RG, Lindsay HD, Flaggs G, DeMaggio AJ, Hoekstra MF, Carr AM, Bentley NJ. Analysis of Rad3 and Chk1 protein kinases defines different checkpoint responses. EMBO J. 1998;17:7239–7249. doi: 10.1093/emboj/17.24.7239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998;282:1893–1897. doi: 10.1126/science.282.5395.1893. [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge S. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo, and in vitro. Proc Natl Acad Sci USA. 2000;12:10389–10394. doi: 10.1073/pnas.190030497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno SA, Klar A, Nurse P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. In: Guthrie C, Fink GR, editors. Methods in Enzymology: Guise to Yeast Genetics and Molecular Biology. New York: Academic Press; 1991. pp. 795–823. [DOI] [PubMed] [Google Scholar]
- Moser BA, Brondello J, Baber-Furnari B, Russell P. Mechanisms of caffeine-induced checkpoint override in fission yeast. Mol Cell Biol. 2000;20:4288–4294. doi: 10.1128/mcb.20.12.4288-4294.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito T, Matsuura A, Ishikawa F. Circular chromosome formation in a fission yeast mutant defective in two ATM homologues. Nat Genet. 1998;20:203–206. doi: 10.1038/2517. [DOI] [PubMed] [Google Scholar]
- O'Connell M, Walworth N, Carr A. The G2-phase DNA damage checkpoint. Trends Cell Biol. 2000;10:296–303. doi: 10.1016/s0962-8924(00)01773-6. [DOI] [PubMed] [Google Scholar]
- O'Connell MJ, Raleigh JM, Verkade HM, Nurse P. Chk1 is a wee1 kinase in the G2 DNA damage checkpoint inhibiting cdc2 by Y15 phosphorylation. EMBO J. 1997;16:545–554. doi: 10.1093/emboj/16.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paciotti V, Clerici M, Lucchini G, Longhese MP. The checkpoint protein Ddc2, functionally related to S. pombe Rad26, interacts with Mec1, and is regulated by Mec1-dependent phosphorylation in budding yeast. Genes Dev. 2000;14:2046–2059. [PMC free article] [PubMed] [Google Scholar]
- Rouse J, Jackson SP. LCD1. an essential gene involved in checkpoint control and regulation of the MEC1 signaling pathway in Saccharomyces cerevisiae. EMBO J. 2000;19:5801–5812. doi: 10.1093/emboj/19.21.5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley R, Subramani S, Young PG. Checkpoint controls in Schizosaccharomyces pombe: rad1. EMBO J. 1992;11:1335–1342. doi: 10.1002/j.1460-2075.1992.tb05178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkaria JN, Tibbetts RS, Busby EC, Kennedy AP, Hill DE, Abraham RT. Inhibition of phosphoinositol 3-kinase related kinases by the radiosensitizing agent wortmannin. Cancer Res. 1998;58:4375–4382. [PubMed] [Google Scholar]
- Seigneur M, Bidnenko V, Ehrlich SD, Michel B. RuvAB acts at arrested replication forks. Cell. 1998;95:419–430. doi: 10.1016/s0092-8674(00)81772-9. [DOI] [PubMed] [Google Scholar]
- Shiloh Y. Ataxia-telangiectasia and the nijmegen breakage syndrome: related disorders but genes apart. Annu Rev Genet. 1997;31:635–662. doi: 10.1146/annurev.genet.31.1.635. [DOI] [PubMed] [Google Scholar]
- Shimada M, Okuzaki D, Tanaka S, Tougan T, Tamai KK, Shimoda C, Nojima H. Replication factor C3 of S. pombe, a small subunit of replication factor C complex, plays a role in both replication and damage checkpoints. Mol Biol Cell. 1999;10:3991–4003. doi: 10.1091/mbc.10.12.3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart E, Chapman CR, Al-Khodiary F, Carr AM, Enoch T. rqh1+, a fission yeast gene related to the Bloom's and Werner's syndrome genes, is required for reversible S phase arrest. EMBO J. 1997;16:2682–2692. doi: 10.1093/emboj/16.10.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Nagai K, Akutsu E, Yamaki H, Tanaka N. On the mechanism of action of bleomycin. Strand scission of DNA caused by bleomycin and its binding to DNA in vitro. J Antibiot. 1970;23:473–480. [PubMed] [Google Scholar]
- Thelen MP, Venclovas C, Fidelis K. A sliding clamp model for the Rad1 family of cell cycle checkpoint proteins. Cell. 1999;96:769–770. doi: 10.1016/s0092-8674(00)80587-5. [DOI] [PubMed] [Google Scholar]
- Tibbetts RS, Cortez D, Brumbaugh KM, Scully R, Livingston D, Elledge SJ, Abraham RT. Functional interactions between BRAC1, and the checkpoint kinase ATR during genotoxic stress. Genes Dev. 2000;14:2989–3002. doi: 10.1101/gad.851000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venclovas C, Thelen MP. Structure-based predictions of Rad1, Rad9, Hus1, and Rad17 participation in sliding clamp, and clamp-loading complexes. Nucleic Acids Res. 2000;28:2481–2493. doi: 10.1093/nar/28.13.2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viguera E, Hernandez P, Krimer DB, Lurz R, Schvartzman JB. Visualization of plasmid replication intermediates containing reversed forks. Nucleic Acids Res. 2000;28:498–503. doi: 10.1093/nar/28.2.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakayama T, Kondo T, Ando S, Matsumoto K, Sugimoto K. Pie1, a protein interacting with Mec1, controls cell growth, and checkpoint responses in Saccharomyces cerevisiae. Mol Cell Biol. 2001;21:755–764. doi: 10.1128/MCB.21.3.755-764.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walworth NC. DNA damage. Chk1 and Cdc25, more than meets the eye. Curr Opin Genet Dev. 2001;11:78–82. doi: 10.1016/s0959-437x(00)00160-x. [DOI] [PubMed] [Google Scholar]
- Walworth NC, Bernards R. rad-dependent response of the chk1-encoded protein kinase at the DNA damage checkpoint. Science. 1996;271:353–356. doi: 10.1126/science.271.5247.353. [DOI] [PubMed] [Google Scholar]
- Weinert T. A DNA damage checkpoint meets the cell cycle engine. Science. 1997;271:1450–1451. doi: 10.1126/science.277.5331.1450. [DOI] [PubMed] [Google Scholar]
- Weinert TA, Hartwell LH. The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science. 1988;241:317–322. doi: 10.1126/science.3291120. [DOI] [PubMed] [Google Scholar]
- Willson J, Wilson S, Warr N, Watts FZ. Isolation and characterization of the Schizosaccharomyces pombe rhp9 gene: a gene required for the DNA damage checkpoint but not the replication checkpoint. Nucleic Acids Res. 1997;25:2138–2145. doi: 10.1093/nar/25.11.2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Kastan M. Participation of ATM in insulin signaling through phosphorylation of eIF-4E-binding protein 1. Nat Cell Biol. 2000;2:893–898. doi: 10.1038/35046542. [DOI] [PubMed] [Google Scholar]
- Zeng Y, Chrispell Forbes K, Wu Z, Moreno S, Piwnica-Worms H, Enoch T. Replication checkpoint requires phosphorylation of the phosphatase Cdc25 by Cds1 or Chk1. Nature. 1998;395:507–510. doi: 10.1038/26766. [DOI] [PubMed] [Google Scholar]