

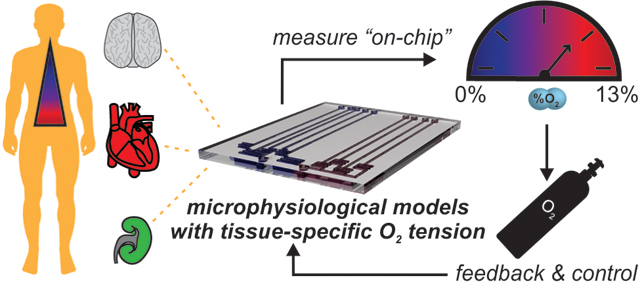
Abstract
As microfabrication techniques and tissue engineering methods improve, microphysiological systems (MPS) are being engineered that recapitulate complex physiological and pathophysiological states to supplement and challenge traditional animal models. Although MPS provide unique microenvironments that transcend common 2D cell culture, without proper regulation of oxygen content, MPS often fail to provide the biomimetic environment necessary to activate and investigate fundamental pathways of cellular metabolism and sub-cellular level. Oxygen exists in the human body in various concentrations and partial pressures; moreover, it fluctuates dramatically depending on fasting, exercise, and sleep patterns. Regulating oxygen content inside MPS necessitates a sensitive biological sensor to quantify oxygen content in real-time. Measuring oxygen in a microdevice is a non-trivial requirement for studies focused on understanding how oxygen impacts cellular processes, including angiogenesis and tumorigenesis. Quantifying oxygen inside a microdevice can be achieved via an array of technologies, with each method having benefits and limitations in terms of sensitivity, limits of detection, and invasiveness that must be considered and optimized. This article will review oxygen physiology in organ systems and offer comparisons of organ-specific MPS that do and do not consider oxygen microenvironments. Materials used in microphysiological models will also be analyzed in terms of their ability to control oxygen. Finally, oxygen sensor technologies are critically compared and evaluated for use in MPS.
Graphical Abstract
Quantifying and regulating oxygen in a microphysiological models can be achieved via an array of technologies, and is an essential component of recapitulating tissue-specific microenvironments.
1. Introduction
The terminology for describing oxygen content in human tissue, or even cell cultures, requires a fairly complex description and has been thoroughly described previously.1 Briefly, the partial pressure of oxygen, i.e. oxygen tension, is how much pressure oxygen alone would exert if it occupied the volume. In a liquid, such cell culture media, dissolved oxygen is proportional to the partial pressure of oxygen in equilibrium with the liquid at the same temperature and pressure. However, in blood, hemoglobin binds and transports oxygen throughout the body, and the quantity of bound oxygen greatly exceeds the dissolved oxygen content. Both in vivo and in vitro, cells, tissues, and organs respond to changes in the oxygen tension, i.e. proportional to dissolved gases, and total oxygen availability, i.e. the combination of dissolved gases and chemically bound gases, in their local microenvironment. For brevity and clarity, we will discuss oxygen content as it relates to the total percentage of oxygen present (<21% O2) in a local microenvironment for the extent of this review.
In the human body, regions of low oxygen (<5% O2), such as the brain and muscle, exist in proximity to regions of much higher oxygen (11% O2), such as the arterial blood and kidneys.1 While atmospheric oxygen levels typically are near 21% O2, the oxygen level in inhaled air decreases to approximately 14.5% O2 by the time it reaches the alveoli, i.e. the primary gas exchange site of the lungs.1, 2 To traverse the alveoli, oxygen binds to hemoglobin in a higher concentration than dissolved O2, and it is subsequently delivered through the body via red blood cells. From the alveoli to capillary beds, oxygen-rich blood moves across into tissues and disseminates through the body along a network of arterioles and arteries. During aerobic cellular respiration, oxygen molecules are consumed during the production of the necessary energy to perform physiological functions.3 Many physiological and pathophysiological pathways are initiated by changes in these fundamental processes due to localized deficiency of oxygen, termed “hypoxia,” including angiogenesis and tumor metastasis.4, 5 During severe or chronic hypoxia, most cells of the human body, apart from gut anaerobes, will experience severe stress and become apoptotic and/or necrotic.
Although there can be no quantitative oxygen level standardized to describe either hypoxic or hyperoxic environments in tissues, as they differ widely across tissues, normoxia can be used to describe the physiologic environment of any healthy tissue. The intricate delineations of normoxic oxygen levels and localized oxygen gradients across tissues impact cell proliferation, angiogenesis, migration, and apoptosis as studied across in vivo and in vitro models.3, 5–12
While the normobaric (1 atm) composition of gases in dry air can be estimated at approximately 21% O2 and 79% N2, studies of cellular physiology using in vitro models are most often performed inside a cell culture incubator under non-ambient conditions. This level of oxygen is much higher than any present in tissue. In a typical cell culture incubator, the controlled supplementation of 5% carbon dioxide (CO2) and maintained relative humidity of 100% results in a normobaric composition of gases at 37°C, as follows: 70.2% nitrogen gas (N2), 6.2% water vapor (H2Og), 5.0% CO2, and 18.6% O2.13 To achieve reduced oxygen levels, a common practice is to use expensive, bulky hypoxic incubators that only supply one specific level of oxygen by either regulating gas composition by using a pre-defined gas mixture or introducing more N2 to reduce the partial pressure of O2. The latter systems are called Tri-Gas incubators, and neither result in the control of microscale oxygen gradients in cell culture systems during the studies. These all or nothing oxygen environments cannot recapitulate any physiological tissue and represent a gap in past model systems.
Microsystems providing precise control of environmental parameters such as oxygen can help bridge the gap between in vivo tissue conditions and in vitro models. To alleviate the burden of animal testing and better inform clinical testing, researchers are using microphysiological systems (MPS) that aim to replicate specific micro-anatomies or physiological function by integrating engineered human tissue constructs and microfluidic devices. By replicating specific physiological function at the micro-scale with human cells and tissues, precise questions concerning cell-cell and cell-environment interactions are investigated in a more physiologically-relevant manner.14, 15
The exquisite control of both fluid transport and subsequent experimental conditions in MPS provides a novel capability to control oxygen levels across the engineered tissue; nevertheless, measurement and control of oxygen levels in microfluidic devices and MPS has not become a standard practice. MPS are typically gas-permeable microdevices kept inside an incubator operating at hyperoxic oxygen levels (19% O2), as endogenous oxygen levels experienced by cells within tissues are much less (see Table 1).1 If a MPS requires an oxygen concentration different from that of an incubator or ambient air, this necessitates a closed off environment and constant reduction or addition of oxygen via perfusion of fluid.
Table 1.
Physiologic oxygen concentrations in selected normal tissues1
| Organ |
pO2 (mmHg) |
% O2 |
|---|---|---|
| Alveoli | 110 | 14.5 |
| Lung | 43 | 5.6 |
| Arterial blood | 100 | 13.2 |
| Kidney | 72 | 9.5 |
| Liver | 41 | 5.4 |
| Skin (superficial region) | 9 | 1.3 |
| Skin (sub-papillary plexus) | 38 | 4.6 |
| Brain | 35 | 4.4 |
| Intestinal tissue | 58 | 7.6 |
Engineering MPS that provide a means to control oxygen content while maintaining viable cell culture requires consideration of materials science, cellular physiology, and mechanical engineering. One scheme to limit oxygen heterogeneity in a microdevice involves using only materials that have minimal oxygen diffusivity, and oxygen is only introduced into the microdevice through oxygenated media or gas.16 Another method involves using oxygen permeable materials for the microdevice with isolation inside a hypoxic chamber.17, 18 Specifics of diffusion-controlled methods for regulating oxygen levels in microfluidics have been described in prior reviews.19–21
Herein, we review and assess current methods to measure and regulate oxygen levels in MPS. Specifically, we summarize the physiologically relevant oxygen levels for different tissues and organ systems, and we address the implications of hypoxic and hyperoxic conditions on function and performance of the MPS. With this information in hand, we compare and contrast the available methods for measuring and regulating oxygen levels in microfluidic devices, in the framework of engineering better MPS. This tutorial review will first discuss microdevices and systems used for physiologic studies of oxygen in organs. Next, we explore materials used to build microdevices and in silico modeling used to design MPS and predict experimental outcomes. We end with a critical assessment of current methods of measuring oxygen in microdevices. This review is intended to present a general description of design considerations and parameters for measuring and controlling oxygen levels in microphysiological modes; it is not intended to serve as a comprehensive review of this rapidly evolving area of research.
2. Microdevice models of physiological organ systems and phenomena
Microdevice models are rapidly transitioning to function as biological analogues for cell studies in vitro. MPS accommodate 3D architecture, mechanical stressors, and other appropriate biochemical cues that are present in vivo and necessary to study physiological processes.22–27 Despite advancements in modeling cell mechanics, microdevices often fail to recapitulate the air-liquid interfaces known to influence cells in vivo. One of the most important air-liquid interfaces is between oxygen and tissue, as 100 to 200 micrometers (μm) is the upper limit of oxygen diffusion through parenchymal tissue to reach cells before reduced oxygen availability inflicts cellular damage.5 Although 200 μm is the common marker of physiologic oxygen diffusion, the true quantity of oxygen available to individual cells depends more on the microenvironment provided by the organ that encompasses them and the density and proximity of surrounding vasculature.
Translating in vitro cellular responses into accurate predictions of in vivo outcomes requires mimicking the same physiologic oxygen levels found in the native organ. Recent advancements in MPS, both in material fabrication and real-time monitoring, show promise in measuring and controlling oxygen diffusion in cell culture. With each organ presenting unique oxygen environments, it is important to first define the organ-specific normoxic condition for the desired MPS. The normoxic oxygen conditions and relevant cellular composition for the most popular engineered tissue models are illustrated in Figure 1.
Fig.1.
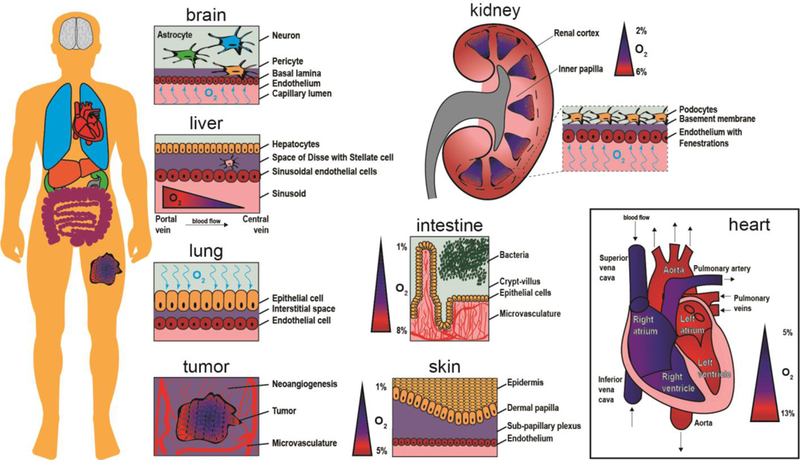
Schematic representation of oxygen content inside organs. The human body is composed of numerous oxygen gradients and organ-specific microenvironments that require microphysiological systems to study oxygen exchange in a tunable and controllable manner. The brain contains neurons, astrocytes, and pericytes situated above and within the basal lamina. Oxygen crosses the endothelium to reach brain cells via the capillary lumen. A tumor creates new vasculature as it grows (neoangiogenesis), while the core of the tumor continues to become more hypoxic. The lung receives oxygen in air, which then binds to hemoglobin molecules after crossing through the epithelium and interstitial space. For the liver, oxygen rich blood travels from the portal vein toward the central vein, providing nutrients to sinusoidal endothelial cells, stellate cells in the perisinusoidal space, and hepatocytes. The kidney contains oxygen gradients within each renal medulla and from the inner papilla to the renal cortex. The intestine has a steep oxygen gradient from the microvasculature underneath the crypt-villus axis to the bacteria populated lumen. The skin is an oxygen barrier with the epidermis at very low oxygen. The heart contains numerous levels of oxygen as oxygen-poor blood flows into the vena cava and oxygen-rich blood exits via the aorta.
2.1. Lung
Oxygen first enters the body via inhalation. During respiration, oxygen interacts with multiple layers of body tissue, from mucosal membranes in nasal passages to alveolar ducts in the lung, resulting in gas exchange with the circulatory system. The unique alveolar-capillary interface of the lung is composed of a symbiotic sandwich, composed of epithelial and endothelial cells separated by an interstitial space at which inspired oxygen-rich air enters the body while expired air composed of carbon dioxide exits. Following pathological foreign invasion, endothelial cells facilitate extravasation of inflammatory cells to the upper layer of the epithelium, where inflammatory cells phagocytose foreign invaders before further damage ensues.28 Diseases that damage the alveolar-capillary interface, such as pulmonary edema, have been studied in MPS of lung tissue.23 Despite the novelty of studying the alveolar-capillary interface with a co-culture model system, cell damage in the lung can be caused by factors dependent on gas exchange, including reduced or excess oxygen.29, 30 Considering the influence of oxygen on the lung, a MPS that includes an air-liquid interface and the appropriate cell types will enable more informative in vitro studies.
In 2010, the first MPS was reported as an alveolar-capillary lung mimic that recapitulated organ-level function.31 Huh et al. created this MPS with an alveolar-capillary interface using human pulmonary epithelial and human pulmonary microvascular endothelial cells to study the impact of breathing motions on lung pathophysiology.23 The lung MPS showed that the cyclic mechanical strain induced to mimic breathing motions could cause increased vascular leakage and was associated with progression of pulmonary edema.23 More recently, the same co-culture MPS of human lung tissue, with an additional non-small cell lung cancer cell component integrated into the upper epithelium, was used to study drug resistance to cancer therapies and found that non-small cell lung cancer cells respond to tyrosine kinase inhibitor treatment and cyclic mechanical strain (breathing motions) by limiting proliferation and spreading.32 These findings associated with pulmonary diseases and cancer could be further explored in relation to oxygen availability in a more advanced lung MPS.
A critical component lacking in this lung MPS is controlled oxygen regulation, as oxygen deficiency can be caused by pulmonary diseases, and hypoxia is an important development during cancer progression. Although this lung MPS was seminal in creating a proof-of-concept for using microdevices to study organ-level functions, no iteration of this MPS included oxygen control or measurements of oxygen transports between the alveolar and endothelial cell layers.
Simultaneously while Dr. Huh and other members of the Wyss Institute worked on integrating multiple cell types into a lung MPS, other researcher teams built lung MPS focused on oxygen regulation. To study how oxygen influences lung epithelial cells, Chen et al. seeded carcinomic human alveolar basal epithelial (A549) cells in a microdevice with varied oxygen gradients.22 Chemical reaction channels on either side of a central cell culture channel provided oxygen scavenging and oxygen generating gas reactions to facilitate the development of an oxygen gradient across the cell culture channel, isolated from the chemical reaction channels by polydimethylsiloxane (PDMS) walls (Figure 2c). The oxygen scavenging channel was created using pyrogallol, a powerful organic reducing agent that absorbs oxygen in an alkaline solution, in combination with sodium hydroxide. The oxygen-generating channel was filled with hydrogen peroxide, which decomposed into water and oxygen gas with the addition of sodium hypochlorite. After seeding and overnight attachment, Chen et al. exposed A549 cells to hypoxia (≈ 0.5% O2) for 48 hours and found that a relatively low oxygen tension did not affect A549 cell growth. In a second set of experiments, the authors found inducing a hyperoxic environment (70% O2) for 6 hours caused A549 cells to peel off the substrate and induced cell death. One major drawback of the microdevice created by Chen et al. was the use of potentially cytotoxic chemicals. Furthermore, Chen et al. revealed how oxygen influences only one cell type found in lung tissue. Oxygen as it relates to more dynamic cell systems should be explored with a MPS that incorporates an endothelial component, if not more cell types. Furthermore, oxygen permeation through each material in the microdevice could potentially impact cellular response to any drug treatment and should be considered when building a lung MPS.
Fig.2.
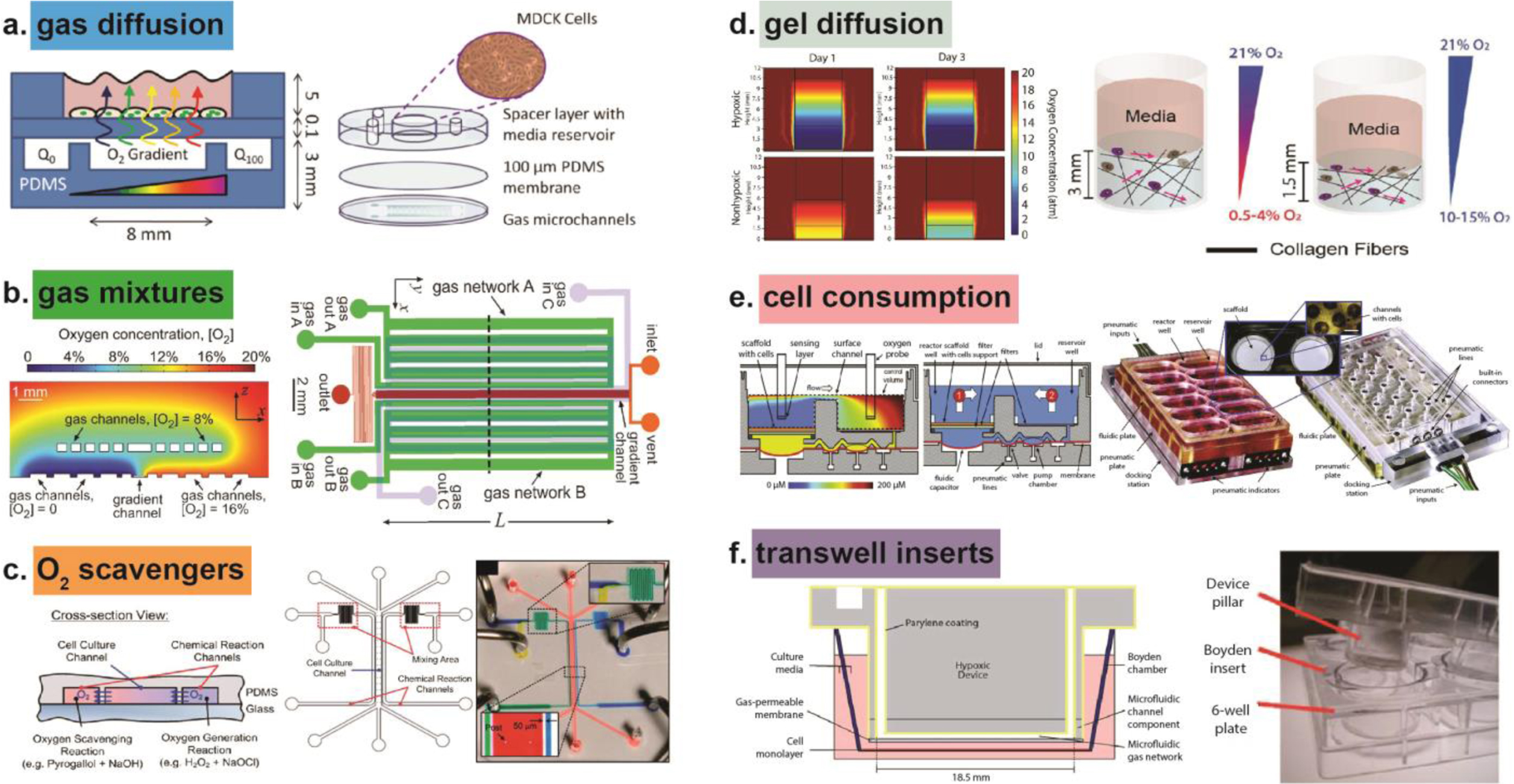
Methods for controlling oxygen in microphysiological systems (MPS) to study cellular responses. (a) Gas diffusion used to generate an oxygen gradient across a layer of Madin-Darby Canine Kidney (MDCK) cells. Reproduced from Ref. 27 with permission from The Royal Society of Chemistry, copyright 2010. (b) Gas filled channels create an oxygen gradient from 0% to 20% oxygen. Reproduced from Ref. 134 with permission from The Royal Society of Chemistry, copyright 2012. (c) Oxygen scavengers in leftmost chemical reaction channel remove oxygen, while oxygen generators in the rightmost channel create a gradient of oxygen between the chemical reaction channels, inside the cell culture channel. Reproduced from Ref. 22 with permission from The Royal Society of Chemistry, copyright 2011. (d) Oxygen diffused through collagen hydrogels of various height and fiber density to create oxygen gradients. Reproduced from Ref. 85 with permission from The Royal Society of Chemistry, copyright 2017. (e) Cells consume oxygen inside the liver MPS to create oxygen-limited environment. Reproduced from Ref. 49 with permission from The Royal Society of Chemistry, copyright 2010. (f) Transwell insert contains Parylene coating to prevent environmental oxygen from contaminating inner hypoxic device. Reproduced from Ref. 115 with permission from The Royal Society of Chemistry, copyright 2010.
2.2. Kidney
The kidneys filter blood to regulate blood pressure, pH and to maintain a balance of water, electrolytes, and hormones. The kidneys generate energy for tubular reabsorption and secretion by consuming oxygen to create ATP. The partial pressure of oxygen in the kidney is low, ranging from 10–15 mmHg (approximately 2% oxygen) in the inner medulla and papilla to 40–45 mmHg (6% oxygen) in the renal cortex (Figure 1).33–36 Due to the higher oxygen levels in the renal cortex, cortical cells are the most sensitive kidney cell type to fluctuations in oxygen. In contrast, the inner medullary and papillae cells demonstrate a reduced sensitivity to hypoxic conditions because they generate ATP from glucose via anaerobic glycolysis.37 Oxygen diffusion through the kidney is tightly regulated to ensure proper pH and molecular composition of the blood. A dense network of arteries channel oxygen-rich blood to the kidneys, where blood flows at the highest rate per gram of tissue in the entire body.38 Reactive oxygen species (ROS) produced during periods of hypoxia have been shown to inhibit kidney function and cause renal damage.39 Studies of kidney function and ROS have thus far been limited to in vivo models, but by developing kidney MPS that include oxygen gradients, the mechanisms of oxygen inhibition can be better understood, especially in oxygen sensitive cortical cells.
ROS are signalling molecules that correlate directly with the local oxygen environment and are released during both hyperoxic and hypoxic events. Kidney cells at the organ level are sensitive to increased ROS production during oxidative stress.40 To study the modulation of intracellular ROS levels in Madin-Darby canine kidney (MDCK) cells, Lo et al. created oxygen gradients in an open-well culture model by diffusing oxygen through PDMS.27 The open-well culture model was fabricated with a 100 μm-thick PDMS diffusion barrier between a 3 mm PDMS bottom with two gas microchannels and a 5 mm PDMS top with a 1 cm diameter reservoir space for cells and media. By flowing 0% and 100% oxygen through either microchannel, a linear oxygen profile was generated that diffused across the barrier to reach MDCK cells in the reservoir space (Figure 2a). MDCK cells exposed to the highest oxygen concentration in a microdevice produced ROS levels 1.5 times more than the initial ROS reading at time zero. MDCK cells exposed to the lowest oxygen concentration in a microdevice produced ROS levels 1.4 times more than the initial ROS reading at time zero. The 1.5 and 1.4-fold increases in ROS production at hyperoxic and hypoxic extremes, respectively, show that MDCK cells are influenced by oxygen and, furthermore, that a diffusion-based MPS can be used to probe oxygen as a controlled and dose-dependent variable.
An important functional unit of the kidney is a capillary network called the glomerulus. Here, circulating blood is filtered into urine via filtration across the capillary wall supported by differentiated epithelial cells called podocytes.41 A recent publication by Musah et al. has defined culture conditions necessary to direct the differentiation of human induced pluripotent stem cells (hiPS) into podocytes.42 Using the hiPS-cell-derived podocytes and human glomerular endothelial cells seeded on opposite sides of a porous flexible PDMS membrane inside a microfluidic device, the authors of this study showed for the first time that a glomerulus-on-a-chip could functionally mimic differential clearance of large and small molecules, including albumin and inulin, respectively, and recapitulate drug-induced podocyte injury in vitro. While the glomerulus-on-a-chip offers a novel platform to study kidney glomerular function, the glomerulus structure is highly influenced by oxygen, as hypoxia-inducible factor (HIF)-1 and HIF-2 regulate podocyte development.43, 44 Incorporating oxygen regulation into such a platform of the glomerulus could reveal mechanistic insight into factors that influence kidney diseases.
2.3. Liver
In the liver, oxygen gradients modulate cellular functions over a median physiological range from 5 to 6% O2.1 The drop in oxygen tension from the periportal (11–13%) to centrilobular (4–5%) regions of the liver is considered a primary regulator of liver zonation.45, 46 Liver zonation, or variant cell function from the portal vein to the central vein, facilitates carbohydrate metabolism and detoxification. Hepatocytes exposed to venous and arterial oxygen levels have shown different cellular responses, including binding activity and gene expression.45
To study liver oxygen gradients, Allen et al. created a parallel-plate perfusion bioreactor system from a polycarbonate block milled with inlet and outlet ports and two microscope slides. Following perfusion of media equilibrated with either 10% or 21% O2, subsequent diffusion gradients of oxygen were generated in the bioreactor comprised of a monolayer of primary rat hepatocytes. Controlled oxygen gradients influenced a heterogeneous distribution of protein levels in hepatocytes, including phosphoenolpyruvate carboxykinase (PEPCK, predominately localized upstream) and cytochrome P450 2B (CYP2B, predominately localized downstream).26 The protein levels correlate with in vivo distribution, as PEPCK is predominately located in the higher oxygen periportal region and CYP2B is found in the lower oxygen pericentral region.47 Despite validation of liver zonal induction and oxygen gradient generation (with miniature Clark-type electrodes), the authors of this study found that the bioreactor was limited to short-term experimentation (3 to 4 days), as primary hepatocytes in their monolayer culture quickly lost their differentiation function.
To better mimic the oxygen gradient in the native liver microenvironment, Bavli et al. expanded upon the work of Allen et al. to create a liver-on-chip that could maintain 3D aggregates of immortalized human hepatocellular carcinoma cells (HepG2/C3A) for 28 days. In the previous parallel-plate perfusion bioreactor system, cells were organized in a single 2D monolayer. In contrast, the microdevice created by Bavli et al. included 9 individual microwells for the 3D HepG2/C3A aggregates. The 3D HepG2/C3A aggregates cultured as spheroids inside microwells introduced complexity to this system, as oxygen was depleted in the center of the spheroid. By incorporating tissue-embedded phosphorescent microprobes and a computer-controlled microfluidic switchboard, oxygen consumption and glucose and lactate levels were monitored in real time.48 This platform also included continuous controlled perfusion of cell culture medium. The sensor-integrated platform tracked in real-time the minute shifts from oxidative phosphorylation to glycolysis or glutaminolysis, signalling mitochondrial dysfunction and chemical toxicity before any effects on cell or tissue viability could be observed.
Another example of a real-time MPS that established an oxygen gradient was created by Domansky et al.49 The authors designed a perfused multiwell with isolated bioreactors, each containing a reactor well, a reservoir well, and an integrated micropump. The reactor well had a collagen-coated scaffold with a diameter of 14.9 mm for cells to self-assemble into an array of ~0.2 mm 3D tissue structures, while the reservoir well contained continuously circulating culture medium. With a co-culture of half murine hepatocytes and half murine liver sinusoidal endothelial cells seeded onto the scaffold in the reactor well, cells consuming oxygen created an oxygen gradient (Figure 2e). The authors found that a flow rate of 0.25 mL min−1 created a tissue outlet oxygen concentration of approximately 50 μM (~5% O2). An oxygen concentration of approximately 50 μM provided a reasonable approximation of physiological oxygen gradients, as reported human in vivo perivenous (sinusoidal exit) region dissolved free oxygen concentrations range from 42 to 49 μM.50 The oxygen consumption rates of hepatocytes in the bioreactor were comparable to rates for perfused rat livers (2.6 versus 1.8 μmol·hour−1·mg protein−1).
The microreactor system used in the study by Domansky et al. has been further advanced with addition of primary human liver cells and commercialized by CN Bio Innovations (Hertfordshire, UK), with multiple researchers using the LiverChip to analyze drug metabolism and drug-drug interactions.51–53 The LiverChip platform includes a pneumatically-driven on-board microfluidic pumping system, distributing positive and negative air pressure to individual valves and pump chambers, to perfuse each scaffold with culture medium and control oxygenation.54 Each reactor well and reservoir well contains a filter with 5 μm pores and a filter support to prevent cells from entering the valves and pumps beneath them. The open well format provides for quick cell seeding and media exchange with pipettes. Overall, the LiverChip is a promising example of a commercialized MPS that considers oxygen regulation for drug studies. The LiverChip platform, containing both primary rat hepatocytes and primary rat sinusoidal endothelial cells, could also be used to investigate the impact of oxygen on liver zonation. Incorporation of additional cells, including Kupffer cells that line the sinusoids and are responsible for phagocytizing bacteria and foreign invaders and stellate cells that are activated during liver injury, would enable for more complex studies of liver function. These additional cells have recently been incorporated in a liver MPS, but this platform fails to include primary human cells or oxygen regulation.55
2.4. Vasculature and Skin
Integrated throughout all tissues is a vascular network critical for supplying nutrients and removing waste, including oxygen and carbon dioxide transport. Oxygen exchange is facilitated largely by blood flow from the macroscale aortic valve of the heart down to the microscale capillary beds within tissue. Many vasculature-like structures have been created using microdevice technologies. In most examples, microscale vasculature is formed using hydrogels seeded with stem cells or mature endothelial cells and growth factors to encourage angiogenesis and induce vascularization.56–59 With a random and spontaneous formation of capillary networks, perfusion of media is impossible and the low density of mature capillary networks limits diffusion of oxygen. Another option for creating vasculature inside a microdevice involves creating a synthetic vessel system composed of a combination of biological and synthetic materials.60–62 The synthetic vessel system provides for perfusion and acts as a nutrient supply pathway.
Although all living tissues require blood supply via a vascular network, most MPS of various organs were first developed without consideration for a vasculature, as the focus was to ensure the key cells relevant to a specific organ were recapitulating in vivo functionality. One organ in which vasculature was not overlooked initially in MPS fabrication was the skin, as oxygen concentration differs significantly across specific layers of the epithelium. Oxygen increases significantly from 1% O2 at the outer epidermis layer of human skin to 3% O2 at the dermal papillae and 5% O2 oxygen at the sub-papillary plexus.1 Many MPS have been made to include a vascular component (i.e. endothelial cells) in a skin equivalent, but few have realized perfusable networks.63–66
Recently, Mori et al. built a skin-on-chip with a perfusable network of channels coated with human umbilical vein endothelial cells (HUVECs).62 The channels were created inside a culture device with 0.52 mm diameter nylon wires strung across connectors of the device. Collagen solution containing normal human dermal fibroblasts was polymerized inside the culture device and then removal of the nylon wires left behind hollow channels, where HUVECs were seeded to form vascular channels. A dermal skin layer seeded with normal human epidermal keratinocytes (NHEKs) was added on top of the collagen hydrogel to complete the fabrication of the skin-equivalent with vascular channels. Characterizing the skin MPS, the authors found perfused vascular channels remained open while non-perfused channels nearly closed. HUVECs and NHEKs were also influenced by nutrient supply, as the perfused skin-equivalent had 3-fold higher average cell density than the non-perfused skin-equivalent. Interestingly, cell density was highest closest to the nutrient supply, a depth range of 0 to 100 μm from the perfused skin-equivalent, and cell density was maintained on the order of 108 cells·mL−1 around the vascular channels. These results are promising support for using perfusion systems to fabricate thick tissues with proper nutrient and oxygen supply. Although levels of oxygen were not measured in this skin-on-chip, measuring and controlling oxygen inside the perfusion system would enable more interesting findings in skin and vascular tissue engineering research.
2.5. Heart
The heart is the only organ relegated to dealing with the burden of handling deoxygenated blood from the body, pumping it to the lungs for oxygen restoration, and delivering oxygenated blood to the rest of the body. Blood flow begins when deoxygenated blood enters the vena cava and fills the right atrium, causing the right atrium to contract. Contraction leads blood to the right ventricle, causing the ventricle to contract, the tricuspid valve to close, and blood to flow through the pulmonary artery to the lungs. The deoxygenated blood becomes oxygenated as it flows through the alveoli and out through the pulmonary vein (Figure 1). From the pulmonary vein, oxygenated blood enters the left atrium. The left atrium contracts, as the mitral valve closes, and blood fills the left ventricle. After filling the left ventricle, the oxygenated blood enters the opened aortic valve and flows through the aorta to the rest of the body. The muscles of the heart also require oxygen to function; therefore, coronary arteries are necessary to pump oxygen-rich blood throughout the heart itself. The meticulous path of oxygenated blood flow throughout the body illuminates why the heart is a vital component to consider when investigating oxygen physiology. If there are any problems with the heart, such as arrhythmia, palpitations, or angina, the rest of the body can immediately be subject to oxygen deficiency. To study oxygen in the heart, cardiac muscle cells or cardiomyocytes have been cultured in various oxygen environments in 2D and 3D.24, 67–70
The impact of periods of low or no blood flow and subsequent low oxygen availability (ischemia) on the heart have been studied using primary porcine cardiomyocytes in a low-shear MPS of ischemia-reperfusion injury.24 Using PDMS bonded to a glass slide to create 2 channels, one for culturing primary porcine cardiomyocytes and one for an oxygen-sensitive dye, a microdevice was created. The cell culture channel was coated with Matrigel, loaded with cardiomyocytes, and placed inside an incubator for cells to attach before inducing an ischemic event. After 2 hours, the microdevice was placed inside a polycarbonate box on a microscope stage. The box had an opening on the bottom for cells to be imaged and inlet and outlet ports on the top for nitrogen flow to displace oxygen and induce ischemia within 30 minutes. Reperfusion was induced via rapid restoration of oxygen to the microdevice. The authors of this study found that during a 3-hour period cardiomyocytes exhibited 0% apoptosis while in “normoxic” conditions. By contrast, the number of cardiomyocytes that underwent apoptosis increased from 3% to 22% O2 from hour 2 to hour 3 in the ischemic condition. Further evidence of ischemic injury came from morphological assessment of cardiomyocytes. After 1 hour of ischemia, cardiomyocytes retracted from the Matrigel-coated surface and displayed spheroid morphologies, indicative of cell stress. Overall, this microdevice with controlled oxygen levels proved useful for revealing cellular responses to controlled ischemic events that would not have been possible in typical cell culture systems. With the perfusable polycarbonate box, oxygen content on the microdevice could be rapidly and precisely controlled, to simulate ischemic events of varying durations, from acute to long-term, to be further investigated using cardiomyocytes harvested from a single animal. The microdevice could be translated to clinical outcomes of ischemia/reperfusion injury by incorporating cardiomyocytes derived from patient-specific human induced pluripotent stem cells (hiPSCs).
Another MPS created to recapitulate myocardial function was designed to include a co-culture of hiPSC derived cardiomyocytes and hiPSC-derived endothelial cells.71 Photolithography and microfabrication techniques were used to form a PDMS microdevice with 2 outer channels for endothelial cells and a central channel for cardiomyocytes encapsulated in a photocrosslinkable hydrogel of gelatin methacrylamide (GelMA). The outer channels served as 1.1 mm wide microvasculature channels separated from the central channel by PDMS posts and supplied nutrients and oxygen to the central channel through media flow. The entire device was fabricated with PDMS and possessed a shallow depth of 160 μm, so diffusion through the top and side walls could deliver oxygen to the cell channels. Using this myocardium-on-chip, Ellis et al. revealed that cardiomyocytes and endothelial cells derived from hiPSCs from a single individual could be functionalized inside a microfluidic device for 7 days without losing phenotypic lineage commitments. Endothelial cells formed tube-like networks that infiltrated into the cardiac muscle channel, closely mimicking the microvasculature of human myocardium. Future studies that include longer culture and additional growth factors could reveal functional networks that transport cytokines from endothelial channels to cardiomyocytes. The myocardium-on-chip could also be enhanced to measure and regulate oxygen diffusion, enabling more in-depth studies of ischemia on cardiac and endothelial cells before and after drug treatment.
Both cardiac MPS described above aim to recapitulate cardiac functions through control of fluid flow and cellular response. To achieve complete cardiac function on the microscale, human cardiomyocytes and endothelial cells must be organized in relevant microarchitecture, form capillary-like networks for fluid exchange, exhibit synchronized beating frequency, and proper stimulation response. Studies of oxygen exchange in a cardiac MPS are only possible after such characterization is achieved in a reproducible, long-term manner. With realized capillary flow and proper diffusion of gas, oxygen can become a single experimental parameter that can be easily manipulated in a cardiac MPS to reveal new relationships and responses on cell-specific functions in the heart.
2.6. Brain
With the highest metabolic activity per unit weight of any organ, the brain consumes 20–25% of the oxygen in the entire body.72, 73 Decreased oxygen concentration in brain tissue can occur with increased depth, but normal brain tissue functions with an oxygen concentration of approximately 4.6% O2.1 Although the brain demands high oxygen, its inability to store metabolic products makes it critically dependent on oxygen delivery. Any slight decrease in oxygen can lead to severe brain damage in minutes. Cell-specific responses to post-ischemic brain damage include degradation of the extracellular matrix and astrocyte and endothelial detachment from basal lamina, causing increased brain microvascular permeability and glial and endothelial apoptosis.74, 75 A variety of brain MPS have been created to investigate cell-specific responses, but controlled delivery of oxygen in these models is still in its infancy.
One important example of controlled delivery of oxygen in a microdevice was created to study hypoxia in 350 μm thick murine hippocampal brain slices.76 Mauleon et al. altered a standard open bath perfusion chamber by adding a gas inlet and outlet and added this chamber to the top of a 100 μm thick PDMS membrane and 200 μm thick PDMS microfluidic channel atop a glass slide. The permeable PDMS membrane allowed oxygen diffusion from the gas channel to brain slice. Overall, by introducing fluid flow above and below the brain slice, the microdevice achieved more stable and uniform oxygen levels throughout the brain slice than had been previously reported with a perfusion chamber alone. The authors could induce physiologically relevant hypoxic insults at controllable rates and at defined locations on the brain slice. This technology was used to study calcium indicator dyes in brain slices from multiple species, including naked mole rats and mice, and multiple age groups, including postnatal (12 to 30 days old) and adult mice.77, 78
A more advanced brain MPS developed by Johnson et al. involved 3D printing complex geometry into microfluidic channels and incorporating multiple cells types, including primary rat embryonic sensory and hippocampal neurons.79 The platform was manufactured using micro-extrusion 3D printing strategies with cell suspensions fed into extruder tips to create 350 μm wide microchannels and 3 adjacent 6 mm chambers directly on protein-coated petri dishes. The 3 adjacent chambers created a compartmentalized neural system with primary rat neurons and axons in the first chamber, self-assembled Schwann cells (S16, CRL-2941, ATCC) in the second chamber, and porcine kidney epithelial cells in the third chamber. The highly functional neural system provided a route for axon termini from the first chamber to interact with both cell types in the adjacent chambers to create aligned axonal networks. Recreating the glial cell-axon interface inside a biomimetic system could be further expanded upon by introducing relevant fluid flow and oxygen exchange in 3D microchannel networks, as considered in the previous Mauleon et al. brain slice platform. Further examination of neural activity in an in vitro model of the brain requires more control of relevant brain physiology, including oxygen distribution.
2.7. Tumor
Oxygen also plays a significant role in tumor development and metastasis. As a tumor grows, the distance from the tumor center to the surrounding vasculature increases, leading to the generation of a hypoxic core. Tumor cells farther away from native blood vessels receive less oxygen and have limited mass transfer. The tumor stroma develops an altered extracellular matrix with more fibroblasts that synthesize growth factors, chemokines, and adhesion molecules.80 Tumor cells respond to hypoxia by secreting chemokines that induce neoangiogenesis, or new vascular formation. As the tumor becomes denser and cells form close contacts, therapeutic drugs meant to deliver lethal concentrations of a cytotoxic agent are limited to outer boundaries and heterogonous distribution, thereby targeting only a fraction of tumor cells. Thus, a hypoxic environment can lead to drug resistance and tumor progression.4, 81 Understanding how hypoxia-induced changes create tumor masses that are more resistant toward chemotherapy and radiotherapy could significantly improve cancer therapies.
Microdevices with controlled oxygen mechanisms provide better experimental conditions for studies of hypoxia-induced changes in cancer cells and the tumor microenvironment.4, 82–85 One method for creating a hypoxic environment is to place a microdevice in a small metal hypoxic chamber and displace oxygen via nitrogen flow in the chamber.4 Khanal et al. showed that even with constant flow of oxygenated medium, a PDMS microdevice inside a hypoxic chamber could be deoxygenated to approximately 1% oxygen within five minutes. The PDMS microdevice then served as an oxygen sink, quickly deoxygenating media perfused through the microdevice. By displacing oxygen rapidly to establish a hypoxic environment, shorter pre-conditioning of cells was necessary to evaluate drug response. Preconditioning human prostate cancer cells to hypoxia lead to significantly different drug responses, including resistance to the cancer drug staurosporine and decreased apoptosis. By quickly preconditioning cells to hypoxic environments using microdevices, more research into drug resistance mechanisms could lead to improved anti-cancer compounds that are more effective against hypoxic tumor cells.
Recently, methods for generating intratumoral hypoxic microenvironments have focused on regulating oxygen diffusion through hydrogels and adjusting cellular oxygen consumption by altering cell-seeding density.85, 86 In one example, an oxygen gradient was made in a fibrous collagen hydrogel by changing the thickness of the hydrogel. The fibrous collagen hydrogel created had an oxygen gradient increasing from approximately 1% oxygen at the bottom to 21% oxygen at the top.85 Collagen was chosen as the hydrogel matrix because it has a fibrous structure that can be adjusted and it has a relatively low oxygen diffusion coefficient.87 By adjusting the pre-incubation period of collagen gel solution (at 4C), but keeping the collagen concentration constant, collagen fibers aggregated without cross-linking to form either high fiber density (2-hour pre-incubation) or low fiber density (30 minute pre-incubation) collagen hydrogels. After fabricating low and high fiber density gels of heights 3 mm tall (gradient from 1 to 15% O2) and 1.5 mm tall (gradient from 10 to 15% O2), murine undifferentiated pleomorphic sarcoma cells encapsulated in high fiber density hypoxic gels were reported to migrated faster and degraded the matrix more rapidly than the low fiber density hypoxic gels (Figure 2d). The increased speed of sarcoma cell motility suggests that fiber density, hydrogel size, and stress relaxation of hydrogels cause different cellular responses under hypoxic gradients. These findings are relevant to tumor progression in vivo as the tumor microenvironment changes from a lower to a higher density as tumorigenesis progresses. Using oxygen concentration to define the density of a tumor could help determine potential treatment options as increased fiber density may require higher doses of chemotherapy delivered to specific regions of the tumor.
2.8. Intestine
Hypoxic and near anoxic environments are also found in normal physiologic regions. One example of a normal or physiologic hypoxic microenvironment in vivo is the intestinal lumen, where intestinal epithelial cells and anaerobic microbes co-exist within a steep oxygen gradient. The steep oxygen gradient is formed through layers of tissue, from the oxygenated, highly vascularized lamina propria and submucosa (8% O2) to the tip of the villi (1.5% O2).88 At the tip of the lumen, facultative and anaerobic microbes flourish in a near anoxic environment (<1% O2).89 Interestingly, the low oxygen environment necessary to sustain a diverse and enormous population of facultative and anaerobic bacteria, approximately 1014 bacteria strong, fluctuates dramatically following digestion and metabolic demand. Within seconds of epithelial contact with nutrients, intestinal blood flow and oxygen extraction locally increase and then, following absorptive hyperemia (accumulation of blood in the underlying vessels), oxygen tension within the villus decreases from approximately 2% to <1% O2 as the microbial zone expands to break down nutrients.90 In the rest of the gastrointestinal tract, hypoxia is associated with poor function, as patients with active inflammatory diseases are often plagued by loss of epithelial barrier function and development of lesions.91 Any damage to the epithelial barrier increases the possibility of bacteria escaping the lumen and infiltrating the body. This devastating phenotype of a complicated inflammatory pathway requires inflammatory disease studies that include controlled oxygen environments. Typically, in vitro cultures of intestinal epithelial cells only include constant oxygen concentrations, in contrast to the cyclic and gradated intestinal oxygen levels.
To study intestinal oxygen, MPS have been designed where precise manipulation of flow and diffusion support co-culture of anaerobic bacteria and mammalian cells.92–95 One version of a co-culture intestinal MPS was created by Kim et al. using a PDMS microdevice with top and bottom cell culture microchannels separated by a porous PDMS membrane.93 On the left and right sides of the cell culture microchannels, hollow vacuum chambers facilitated peristalsis-like cyclic mechanical strain to promote epithelial cell differentiation and villus formation. The microdevice included human epithelial colorectal cells (Caco-2 cell line) that spontaneously formed intestinal villi on the top microchannel after approximately 100 hours of medium flow and cyclic mechanical stretching. On the bottom microchannel, human capillary endothelial cells were cultured to form a monolayer on the opposite side of the porous PDMS membrane. Probiotic gut microbes Lactobacillus rhamnosus GG (LGG) were added to the top cell culture of Caco-2 cells and grew in direct contact with the epithelium for more than two weeks. Kim et al. found that microbial cells predominately colonized the intervillus spaces, despite uniform seeding, and that confined intervillus spaces within the densely packed microchannel potentially caused facultative microbes to rapidly consume oxygen, thereby producing a local anoxic microenvironment to support anaerobic microbial growth. Increased microbial cell colonization in the intervillus spaces could be confirmed by measurements of oxygen in the gut MPS. Further investigation into the oxygen consumption facilitated within the cell culture microchannels would support the use of oxygen controllable systems to study host-microbe interactions in the gut.
Another example of a colon MPS with a co-culture of Caco-2 cells and Lactobacillus rhamnosus GG (LGG) was fabricated using a polycarbonate (PC) outer enclosure with sandwich silicone rubber gaskets and semi-permeable PC membranes.94 The device included three microchambers, the bottom for medium perfusion, the middle for the Caco-2 cell chamber, and the top chamber for LGG microbial cells. Separating each chamber were either micro-or nanoporous membranes to permit exchange of biomolecular and oxygen gradients. The microporous membrane (pore diameter, 1 μm) separated the perfusion chamber from the epithelial cell chamber to allow for perfusion-dominant exchange, while the nanoporous membrane (pore diameter, 50 nm) separated the epithelial cell chamber from the microbial chamber to prevent microorganism infiltration. Both normoxic and anoxic culture media were perfused to the respective microchamber, with the concentration of oxygen in the anoxic culture media maintained at 0.1% by bubbling with nitrogen gas. Shah et al. used this microdevice system to show that when co-cultured with LGG, Caco-2 cells display transcriptional responses in agreement with host-microbe in vivo interactions.
Despite the novelty of co-culture systems, the intestinal MPS described above are limited by the tumorigenic cell line used to generate Caco-2 cells because Caco-2 cells differ from normal human enterocytes in regards to contact inhibition, as they possess tighter cellular junctions than normal epithelial cells.96 A co-culture model of human-microbial crosstalk with oxygen control could be further enhanced with more representative cell types found in the human small intestine or colon, such as primary intestinal epithelial cells and multiple anaerobic and aerobic bacteria that occupy the native human lumen.
3. Design considerations for engineering physiologically-relevant oxygen gradients in MPS
3.1. Materials
To engineer a MPS with an oxygen gradient or hypoxic region, a material impermeable to oxygen must be used and any oxygen introduced into the MPS must be removed quickly. Cellular respiration, deoxygenation and reoxygenation of media, and removal of waste will alter oxygen levels, and the material must allow for rapid adjustment and reestablishment of the desired concentration of oxygen. To measure and control oxygen, the first parameter to optimize is the potential oxygen exchange through each material. Some key materials used for cell culture and microdevice fabrication include polydimethylsiloxane (PDMS), polystyrene (PS), polycarbonate (PC), poly(chloro-p-xylyene) (Parylene-C) and polymethyl methacrylate (PMMA). PDMS and PMMA are chosen for easy and repeatable fabrication that can provide microscale architecture needed for cell attachment and proliferation. Due to limited oxygen diffusivity, PC and Parylene-C provide microenvironments with controlled gas exchange and limited oxygen contamination from the surrounding environment. Collecting data from a MPS should be considered when choosing a material, as studies focused only on endpoint readouts cannot readily benefit from the real-time monitoring offered by optically transparent materials. Deciding on a material to fabricate a MPS involves consideration of critical characteristics, including oxygen diffusivity (see Table 2) and workability, as explored for each material below.
Table 2.
Oxygen diffusivity of common plastics
| Material |
O2 diffusivity (cm2·s−1) |
Ref. |
|---|---|---|
| Polydimethylsiloxane (PDMS) | 3.4 ± 1 × 10−5 | 97 |
| Polyetheretherketone (PEEK) | 1.38 × 10−6 | 98 |
| Polytetrafluoroethylene (PTFE) | 2.83 × 10−7 | 99 |
| Polypropylene (PP) | 2.33 ± 0.4 × 10−7 | 100 |
| Polystyrene (PS) | 2.3± 0.2 × 10−7 | 101 |
| Polyurethane | 1.16 × 10−7 | 102 |
| Polycarbonate (PC) | 8.0 × 10−8 | 103 |
| Polyethylene terephthalate (PET) | 2.89 × 10−8 | 104 |
| Polymethyl methacrylate (PMMA) | 2.7 ± 0.2 × 10−8 | 105 |
3.1.1. Polydimethylsiloxane (PDMS).
Many MPS are constructed from the elastomer, PDMS, in part due to ease of fabrication and optical transparency, but also due to its high gas permeability, which is often desired to transport oxygen from the incubator into the cell culture device.106 PDMS allows for microscale features to be designed and molded for prototyping via soft lithography.107 With control of part thickness, semi-permeable membranes of PDMS can facilitate transport of gases through walls or layers. Despite its ubiquity in microfabrication, PDMS may not be the best material for microdevice applications focused on controlling oxygen because oxygen possesses a high diffusivity (3.4 × 10−5 cm2) and solubility in PDMS compared to other common materials and thick PDMS is required for mechanical strength (Table 2).97 PDMS can also negatively impact cell studies because it can absorb small hydrophobic molecules, such as drugs, and leaches un-crosslinked oligomers into solution.107–109 The absorption of hydrophobic molecules is especially an issue for MPS that intend to test drugs or rely on perfusion of media; therefore, the use of PDMS may contribute confounding variables in such studies. PDMS is also expensive and could be replaced with polymers more resistant to small molecule absorption, such as polyurethane or polycarbonate.110 PDMS can be chemically modified using oxygen plasma treatment to enhance hydrophilicity and allow for better cell attachment and viability.111 PDMS microdevices can also be coated with ECM-components or integrated with hydrogels, but these additions may negatively impact the optical transparency of the microdevice surface, sacrificing visibility needed for monitoring cell morphology via real-time microscopy.
3.1.2. Polystyrene (PS).
Polystyrene (PS) is the most popular cell culture material used to manufacture cell culture flasks and plates. Its optical clarity, ease of molding, and stability during sterilization via irradiation make it a gold-standard material for mass production of cell culture platforms. PS has a relatively low oxygen diffusion coefficient (2.3 × 10−7 cm2 ) compared to other plastics and polymers, therefore, it can be used for microdevices which aim to control oxygen permeation (Table 2).101 PS can create 3D oxygen gradients, as it is a semi-impermeable barrier on the sides and bottom of any culture dish and allows for oxygen to only traverse the space between, which can be filled with hydrogel for a cell system.85 One disadvantage of PS for cell culture is its hydrophobic surface, which limits cell adhesion. Most PS plates are treated using either gas-plasma under vacuum or corona discharge under atmospheric conditions to be more hydrophilic.112, 113 If an MPS is to be fabricated with PS walls or layers and requires cell attachment, then the surface will require chemical modification. PS is also more difficult and expensive to prototype on the microscale, as it requires expertise in micromilling and thermal bonding, as compared to the soft-lithography techniques used for PDMS prototypes.114
3.1.3. Polycarbonate (PC).
Polycarbonate (PC) can be added to microdevices to control oxygen.24, 26, 94 PC has a very low oxygen diffusion coefficient (8.0 × 10−8 cm2·s−1) compared to other microdevice materials and PC films have been used to surround PDMS devices and prevent oxygen diffusion from an incubator into a microdevice.83, 103 PC is also easy to machine and can be sterilized via autoclave. In one study, a PDMS device was created with a PC film layered above microchannels composed of a central gel region with media channels and gas channels flanking each side (Figure 2).83 Normoxia was created using a humidified gas mixture of 21% O2, 5% CO2, and 74% N2, each supplied to the microdevice channels at a flow rate of 18 ml min−1, corresponding to fluid flow dominated by advection. A 10 wt.% sodium sulfite (Na2SO3) solution humidified a 0% O2 gas mixture to remove dissolved oxygen and generate uniform hypoxia in the microdevice. Next, one gas channel was switched from 0% to 21% O2 to generate an oxygen gradient. The authors of this study found that with the PC film approximately 2% O2 could be achieved within a microdevice, whereas without the PC film the lowest oxygen tension achieved was 6.5% O2. Once validated, the microdevice was used in studies comparing how normoxic and hypoxic environments affect human breast cancer cell migration. Using 3D confocal imaging in real-time, the authors reported that hypoxic conditions promoted directional cancer cell migration and increased migration speed as compared to normoxic conditions.
3.1.4. Poly(chloro-p-xylyene) (Parylene-C).
Parylene-C has been used to coat PDMS microdevices and prevent oxygen contamination from ambient air. Using a film as thin as 2.5 μm, a Parylene-C coating has been shown to inhibit oxygen diffusion through a PDMS membrane.62, 106, 115, 116 In experiments designed to compare the diffusion of oxygen through Parylene-C, PDMS microchannels were fabricated and bonded to PDMS membranes with and without Parylene coatings.115 Using fluorescent oxygen sensors placed directly below the membranes, researchers found that an infusion of 0% oxygen gas (5% carbon dioxide and balanced nitrogen) through the PDMS gas channel caused an uncoated PDMS membrane to equilibrate to 0% O2 within a minute. In contrast, a Parylene-coated PDMS membrane sustained an oxygen concentration of approximately 21% O2, remaining at the same concentration of oxygen as present in the surrounding environment despite the flow of anoxic gas on the opposite side of the coated membrane.115 Similarly, when challenged with an infusion of pure O2, the uncoated PDMS membrane reached 100% O2 within a minute, whereas the Parylene-coated PDMS membrane remained at approximately 21% O2. These results demonstrated that Parylene-C is a useful coating to add on to any microdevice material to prevent oxygen contamination from ambient air when coated on the outer walls of the microdevice or between different layers when coated on partitioned membranes.
One major disadvantage to coating any surface with Parylene-C is its hydrophobicity, which can lead to extremely low cell adhesion. O2 plasma treatment can be used to alter a Parylene-coated surface to make it more amenable to cell culture as the treatment alters the surface from hydrophobic to hydrophilic.117 O2 plasma treatment has been shown to increase the adhesion of collagen and cells on Parylene by permanent addition of hydrophilic groups, such as hydroxyl and carboxyl groups that increase adsorption of adhering molecules of the culturing medium.118, 119 Comparing a plasma-treated Parylene-C surface to a non-treated Parylene-C surface, Hoshino et al. found that plasma-treatment increased neural cell adhesiveness more than twenty times.118 These findings significantly support the use of O2 plasma treatment to increase cell attachment, because neuronal cells typically have worse cell adhesiveness than any previously tested cells. Another option when using Parylene-C is to only coat the walls and outside of a microdevice, thereby allowing for cells to adhere to an uncoated surface or membrane inside the device and still limiting outside environmental oxygen contamination.115 Coating a microdevice with Parylene-C can be performed using a commercial chemical deposition system to complete vacuum vapor deposition polymerization.
3.1.5. Polymethyl methacrylate (PMMA).
Polymethyl methacrylate (PMMA) has a very low oxygen permeability (2.7 × 10−8 cm2·s−1) compared to other common microdevice materials.105 PMMA, also known as acrylic, is biocompatible and amenable to micromachining, therefore it has been used to create replicate microdevices.30, 48, 120–123 PMMA is often used as a support layer on the top and/or bottom of microdevices to prevent environmental oxygen contamination. PMMA is mechanically and thermally robust enough to support multiple microfabrication techniques including, laser cutting/engraving, milling, hot embossing, and injection molding, while maintaining high-resolution microscale geometries.124–127 PMMA is transparent to visible and near infrared (NIR) light, allowing for in situ imaging and fluorescence-based characterization.128 PMMA can be fabricated into porous microfluidic films to transform the material from possessing a very low permeability to one that possess a high permeability comparable to PDMS.129 Gas diffusion is possible across the PMMA porous film, which can be used to oxygenate media on the opposing side of the film without disturbing cell aggregates or monolayers beneath the film. With PMMA porous films, multiple layers in a microdevice can be easily stacked, allowing for different functions and morphology across a single microdevice. For MPS, a PMMA porous film could facilitate oxygen diffusion through specific layers and offers an alternative material to bypass the potential leaching of oligomers into solution observed in PDMS.
3.2. Methods of controlling oxygen content and distribution in MPS
3.2.1. Diffusion through hydrogel
Hydrogels form 3D extracellular matrix (ECM)-like environments for cells to align within, on top of, and alongside which are much more like native tissue in the body than standard flat tissue culture plates. Hydrogels can also be modifiable, as cells secrete their own ECM proteins and remake a hydrogel matrix. Cellular hydrogels can be made of many biological materials such as agarose, collagen, gelatin, and Matrigel and supplemented with additional ECM-like components such as RGD-binding motifs.83, 86, 87, 130, 131 One example of an oxygen gradient in a microdevice created with a hydrogel was made with agarose gel and PDMS.131 The microdevice was made with two perfused microchannels, the bottom channel filled with 0% O2 gas (in balanced nitrogen and 5% CO2) and the top channel filled with either 5% or 21% O2 gas. With continuous perfusion of oxygen, a steady-state oxygen gradient was formed in an agarose hydrogel sandwiched between the two microchannels. A platinum-based fluorescent oxygen sensor was used to confirm the presence of an oxygen gradient across the agarose gel and will be described in more detail below. Generating an oxygen gradient across a hydrogel can be easily realized inside MPS designed with inlet and outlet ports for gas flow. One consideration when deoxygenated via gas diffusion is that the defined oxygen levels at each step across the oxygen boundaries cannot be measured without multiple oxygen sensors measuring in parallel along the length of the hydrogel.
3.2.2. Transwell
Transwells can be used for hypoxic studies with an additional insert. A transwell, or Boyden chamber, is a porous membrane-based platform polarized for migration and invasion cell studies. A transwell is composed of a chamber insert with a Polyethylene terephthalate (PET) porous membrane placed inside a PS tissue culture well plate well. Typically, two different cell types are cultured inside the transwell on opposing sides of the porous membrane, facilitating cellular interaction. Using microdevice fabrication techniques, a hypoxic chamber has been made inside a transwell with a Parylene-coated PDMS microfluidic network for gas infusion, as shown in Figure 2f.115 By enclosing the entire well within the Parylene-coated insert, a hypoxic environment in the device was achieved, equilibrating to 1% O2 in just 20 minutes.115
Although transwell inserts offer a simple method to studying cell-cell interactions, generating a controlled oxygen environment in a transwell by adding more components is more difficult than creating a controlled oxygen environment with a microdevice alone. Transwells are limited to studies of only two cell types, whereas microdevices have been designed to include as many as four cell types thus far.55 Microdevices offer higher sensitivity and control over diffusion of soluble factors than transwell derivatives because microdevices can be fabricated with channels and compartments to provide greater spatial organization.132 Some initial co-culture studies of oxygen can be performed using modified transwell inserts, but the number of cells and medium required will remain limiting factors in Transwells that are typically in volumes of milliliters, whereas microdevices offer an alternative approach to investigate oxygen in cell culture systems while minimizing resources.
3.2.3. Diffusion through liquid medium
Microdevices can rely on rapid diffusion of small molecules to generate an oxygen gradient.26, 48, 85, 86, 94 Diffusion through liquid medium can be estimated inside a microdevice, assuming steady-state transport in a uniform flow field. With lateral diffusion in a given direction, oxygen concentration at any location is dependent on the channel height and length, as well as oxygen diffusivity and the Peclet number. The Peclet number is the ratio of advection to diffusion, calculated using the average flow velocity in a channel, the width of the channel, and coefficient of diffusion.26 A Peclet number of 100 characterizes transport of fluid flow dominated by advection. As flow velocity decreases, the Peclet number decreases and diffusive transport dominates convective transport. Dominately diffusive transport at lower flow rates allows for oxygen gradients to form along the length of a microdevice, as shown by Allen et al. in 2003.26 By perfusing media across the length of a 28 mm x 55 mm x 100 μm channel at a rate of 0.35 mL·min−1 for 24 hours, Allen et al. created a MPS of the liver with appropriate zonation of oxygen, from 21% O2 at the inlet to <1.5% O2 at the oulet. Developing an oxygen gradient across a channel with simple Cartesian geometry provides a uniform flow field, but deleterious fluid shear and cell seeding conditions can lead to non-uniformity in the flow field and must be considered in any model predictions.
3.2.4. Gas mixtures (Tri-Gas Systems)
Oxygen gradients in microdevices have been generated with O2-N2 mixtures.4, 24, 121, 133–135 Using an off-chip multichannel gas mixer, Alder et al. fabricated oxygen concentration gradients with linear, exponential, and non-monotonic profiles ranging from 0 to 21% O2 in a single device. By optimizing the thickness of PDMS between the flow and gas channels (120 μm), a discrete series of oxygen concentrations in the gas channels translated to smooth gradients of oxygen in the flow channels.133 Oxygen gradients created using O2-N2 mixtures are limited by the number of gas channels, the period of the gas channel array, and the thickness of PDMS between the gas and flow channels. Polinkovsky et al. fabricated microdevices that included on-chip gas mixing networks to generate a series of nine different gas mixtures using only two gas inlets, each fed with pure N2 and O2.121 With serpentine channels and three stages of mixing, one microdevice exhibited a linear dependence of oxygen concentration on the channel number. The oxygen concentration increased from 0% to 100% O2 across the array of microchannels in the device. Another microdevice created by Alder, Polinkovsky and colleagues with serpentine gas-mixing networks but with gas inlets of pure N2 and 20.9% O2, produced an exponential dependence of oxygen concentration on channel number, shifting from 0% O2 in channel one to 20.9% O2 in channel nine (Figure 2b). A feasibility study looking at how an exponential change in oxygen affected Escherichia coli (E. coli) growth found that cells grew faster at a higher oxygen concentration but colony growth rates displayed the largest increase at near 0% O2.134 Although gas mixtures can achieve well-defined oxygen gradients, the disadvantages include intricate system design and setup and potential issues drying out the chamber when gas flow is used.
3.2.5. Oxygen scavenging agents
Liquid oxygen scavengers can remove oxygen chemically to achieve a desired oxygen concentration and bypass some potentially complicated microdevice designs. Chemicals that removed oxygen from solutions, or oxygen scavenging agents, have been used in PDMS microdevices to consume oxygen in culture and create oxygen gradients.22, 122, 136, 137 One potential drawback to using an oxygen scavenging agent is its potential cytotoxicity at high chemical concentrations, which can lead to unwarranted cell stress and apoptosis.138 Separating the oxygen scavenging liquid from cultured cells is paramount to limit chemical toxicity, and with microdevice fabrication techniques, separate channels can be manufactured to prevent unwarranted chemical interactions. In the lung model created by Chen et al., pyrogallol, a powerful organic reducing agent, was combined with sodium hydroxide to create an oxygen scavenging reaction, while hydrogen peroxide and sodium hypochlorite were combined in another chemical channel to generate oxygen.22 The chemical channels created an oxygen gradient across a microdevice without interfering with the cell culture channel in between them, because each chemical channel was separated by 50 μm thick PDMS walls (Figure 2c). The thickness of the walls was significant as it allowed for oxygen to diffuse readily but limited toxic chemical by-products from harming cells.
Using oxygen scavenging liquids with a gas permeable membrane to control oxygen gradients was first proposed by Skolimowski et al.122 The novelty of Skolimowski et al. comes from a microfluidic device design that separates channels containing oxygen-scavenging liquid from chambers with media. The complete device includes two PMMA bottom layers for inlets and outlets and the oxygen scavenging liquid is composed of 10 mM sodium citrate. Next, there is a 60 μm thick PDMS oxygen permeable membrane. Lastly, there is a 150 μm thick cell culture chamber and a glass slide covered with oxygen sensing dye. The device was used to culture bacteria Pseudomonas aeruginosa (P. aeruginosa), which is a pathogenic, recurrent cause of lung infections in patients with cystic fibrosis. During metabolic challenges associated with varying oxygen availability within the mucus layers of the CF respiratory system, P. aeruginosa sustains colonization in the human airways by switching from aerobic to anaerobic respiration. Studies of P. aeruginosa in different dissolved oxygen environments required a device with controlled oxygen concentrations. The authors of this study reported that P. aeruginosa was negatively influenced by reduced oxygen, as a decrease in oxygen availability caused a reduction in the number of attached cells to the surface.
3.3. Computational modeling
When designing MPS, in silico modelling is an invaluable tool for simulating physical parameters. Computational modelling not only saves resources, but also it can inform expected results or potential limitations of a MPS design. In silico models provide a theoretical data set to compare to in vitro results, a necessary engineering process that can simultaneously characterize and validate a model. Important categories to distinguish in an in silico model include steady-state versus dynamic, stochastic versus deterministic, continuous versus discrete and local versus distributed. Finite element analysis (FEA) is a form of discrete simulation described by local and deterministic calculations of ordinary or partial differential equations that represent the physical phenomenon at steady-state or dynamically. Other methods include lumped element modelling and Monte Carlo simulation, which can both evaluate and refine the underlying theory of models. The main physical phenomena simulated in in silico modelling include fluid dynamics, mass transfer, and solid mechanics, and, to a lesser extent, electronics, acoustics, and magnetics.
FEA is perhaps the most popular method for simulating fluid dynamics and mass transfer. The typical workflow of running a simulation involves describing the physics, building the model, establishing the boundary conditions, discretizing the model into elements, applying the relevant equations to the elements and boundaries, calculating the numerical solutions to the equations and, finally, visualizing the simulated model solutions. With known material properties and reasonable assumptions, thorough mathematical models have been used to predict experimental outcomes for various MPS.49, 86, 122, 133, 139, 140
Inside MPS, dynamic fluid flow is finely tuned and controlled in ways not possible on the macroscale due to inertial forces. Fluid dynamics of MPS are described by the Navier-Stokes equations and the fluid continuity equation, which maintain local conservation of momentum and mass, respectively. Cell media is considered incompressible, Newtonian, and fully developed, with no-slip boundary conditions. This simplifies the Navier-Stokes equations and continuity equations, which can be further simplified if the model is simulated at steady-state and the fluid is in the laminar flow regime. In microchannels, fluid flows are generally considered laminar, because viscous forces far exceed inertial forces resulting in laminar flow.14, 141
Steady-state versus time-dependent is a parameter that can be changed based on the model and the experiment. At steady state, an oxygen transport model uses fluid dynamics equations to calculate flow profiles and mass transfer equations to calculate the oxygen concentration.49 Navier-Stokes and fluid continuity are equations of fluid dynamics, while convection-diffusion, Brownian motion, Fick’s Laws, and the mass continuity equation, are equations of mass transfer. Fluid dynamics equations are used to calculate the velocities and momentum of liquid and gaseous fluids, while mass transfer equations are used to calculate chemical concentrations and diffusion in those fluids. In order to accurately model oxygen levels in microfluidic devices both fluid dynamics and mass transport should be coupled. Typically, the solution to the fluid dynamics simulation is calculated and used as a variable in the mass transfer equations to solve for the concentration profile of dissolved oxygen due to convection and diffusion. On the microscale, there is no turbulent flow to enhance mixing, but laminar flow can be significant when dealing with gradients, therefore the convection-diffusion equation is used to define the concentration of oxygen species throughout a microchannel. The change in concentration over time can be solved for by calculating the flux of oxygen due to diffusion and convection then adjusting for any flux sources or sinks. Flux is the amount of a chemical species, oxygen, moving through an area over time. Cellular consumption rate, a value dependent on both cell type and cell density, is an example of an oxygen sink. Concentration profiles are generated across the geometric space. With known values for parameters including density, viscosity, and diffusivity of oxygen in media, gas, and microdevice materials, the optimal oxygen tension for a respective cell culture can be simulated before experimental validation. Varying degrees of shear stress in a channel can be predicted with a simulation, but, experimentally, shear stress should be minimized to ensure long-term cell survival in any MPS.
Experimental cellular respiration values are important to consider for models that are meant to study cell mechanics in vitro compared to in vivo.49, 86 Many different gases are generated as by-products of cellular respiration and metabolism; therefore, anaerobic microdevices must be able to evacuate gaseous products to sustain low oxygen levels. Oxygen consumption values for bacteria and mammalian cells are often reported in units of millimoles (mmol) per hour per number of cells and must be converted to oxygen flux for use in most computational models. Using Henry’s Law and a known cellular respiration, the oxygen flux necessary to sustain a tightly packed monolayer of cells can be calculated. Henry’s Law states that the concentration of a dissolved gas is equal to the partial pressure times the solubility coefficient of that gas.142 At a constant temperature, the oxygen flux of cells in a microdevice can be calculated by multiplying a known concentration of oxygen by Henry’s constant. The oxygen flux can represent a flux discontinuity at the individual cell boundary or entire monolayer surface. The cell density can then be altered to achieve a limited change in oxygen concentration across a material.121 Analytical solutions to computational models of cellular respiration typically overestimate oxygen consumption in low oxygen environments because constant oxygen uptake is assumed, although many cell types report oxygen uptake decline following isolation.139, 143, 144 An explicit approximation of Michaelis-Menten oxygen consumption kinetics offers numerical solutions that more closely correlate with measured values.
An in silico model of a physiological phenomenon, such as oxygen exchange in cells, can have its shortcomings, including assumptions such as constant influx of nutrients or constant cellular uptake. Such shortcomings can be bypassed with in-depth cellular experiments beforehand and knowledge of the differences between a given cell type in vivo versus in vitro culture. To better predict and control oxygen in a MPS, in silico models should account for fluctuations in oxygen levels due to cellular respiration and proliferation and be experimentally validated. Vertical oxygen gradients generated by highly active cells, such as hepatocytes that deplete oxygen at the media-cell surface interface faster than it is replenished by media oxygen diffusion, must be considered when comparing computations to experimental measurements.145 Computational modelling of physiologic systems using the appropriate equations and assumptions can offer insight into complicated processes experienced by cells at the microscale level.
2D numerical simulations of oxygen profiles in microdevices have been created to simplify geometry and allow for quick computations.133 Some negligible differences in peak oxygen concentration have been observed when comparing experimental results to 2D numerical simulations of non-monotonic oxygen concentrations in a microdevice with 9 gas channels containing various oxygen-nitrogen gas mixtures.133 3D numerical simulations of oxygen transport offer more defined oxygen profiles at the expense of computation time and simulation complexity, as an extra dimension necessitates more input parameters, detailed geometry, and boundary conditions to generate an accurate simulation that can be experimentally validated. One example of a 3D computational model of oxygen profiles in a liver MPS was created by Domansky et al.49 The liver model, described above, included a reactor well and a reservoir well, fitted together inside a larger well where cell culture medium circulates between an open channel. Oxygen concentration varied with position and oxygen was replenished via the air-liquid interface on top. The control volume region within the larger well extended from the air-liquid interface down to the scaffold in the reactor well and across the filter in the reservoir well. Fluid moved from the reservoir well to the reactor well with cells. It was assumed that fluid was completely mixed from the underlying pump and fluid entering the lower scaffold where cells were seeded had a uniform concentration. Fluid exiting the scaffold was assumed constant as no significant differences were observed experimentally at different regions of the scaffold due to uniform cell distributions. As fluid moved upward from the scaffold, oxygen concentration profiles were altered by the boundary conditions at the walls and convection and diffusion from the gas-liquid interface. Fluid moved back across an open channel into the reservoir well, creating a non-uniform oxygen concentration profile in the reservoir well. The control volume region within the larger well extended from the air-liquid interface down to the scaffold in the reactor well and across the filter in the reservoir well. Oxygen probes placed inside the reactor and reservoir wells confirmed the concentration of oxygen with an estimated error of less than 5%.
4. Measuring oxygen in microdevices
Although simulations of gas and fluid exchange offer insight into design parameters, real-time oxygen measurements are necessary to adjust and maintain desired oxygen levels in a MPS, as oxygen fluctuates continuously during cellular respiration and metabolism.88 Measuring oxygen in MPS can be challenging due to the microscale features, complex flow profiles, need for sterility, and need for minimal oxygen consumption such that the oxygen concentration in the microenvironment does not change significantly.
Fortunately, there is a considerable body of literature on oxygen sensing. Much attention has been paid to gas-phase oxygen sensing146, 147; nevertheless, there are multiple reports options for dissolved oxygen sensing in MPS. Oxygen can be measured in MPS by integrating either electrochemical or optical sensors, which are the most commonly reported technologies for dissolved oxygen measurement.148–158 For further detail on the specific operation and performance of recent oxygen sensors, we suggest reading the focused review on microscale oxygen sensors which has been published by Oomen et al.21 Herein, we will briefly define the most common oxygen sensors, which have been adapted to in vitro models—(1) amperometric oxygen sensors and (2) fluorescence or phosphorescence quenching oxygen sensors.
4.1. Electrochemical oxygen measurement
Electrochemical oxygen sensors can be categorized by their principle of operation: amperometric or potentiometric. Amperometric sensors are the most reported electrochemical technology for quantification of gaseous and dissolved oxygen.159, 160 While potentiometric measurement of gaseous oxygen, potentiometric measrument of dissolved oxygen is emerging as a potential alternative.161, 162
For this review, we will focus on the more common amperometric measurement techniques, but we will briefly discuss the principles and emerging benefits of potentiometric measurement of dissolved oxygen. Equilibrium potential measurements enable oxygen measurement via the Nernst Equation (Equation 1), where E (Volts) is the potential difference of the electrochemical cell, E0, is the potential of the reduction or oxidation reaction or the standard potential, R is the gas constant (mol−1·K−1), F is Faraday’s constant (C·mol−1), T is temperature (K), n is the number of electrons active in the reaction, and [O2] is the dissolved oxygen concentration (mol·L−1). The sensor output varies logarithmically with oxygen concentration, and if the solid electrolyte is an ionic conductor, the sensitivity of the equilibrium potentiometric sensors is given by RT/nF and depends only on temperature, as the number of active electrons is fixed for the specific reaction at the electrode.
| (1) |
Potentiometric measurement have long been employed for the measurement of oxygen in the gas phase.153, 163, 164 There have been few demonstrations of potentiometric dissolved oxygen sensors, using working electrodes that range from heavy metal alloys and ceramics to polymer films.165–168 Potentiometric sensors for dissolved oxygen at physiologically-relevant oxygen concentrations have been most recently demonstrated by Zimmermann et al., but they do not demonstrate any operation in a cell culture system.162 It should be noted, oxygen concentrations were measured from 0.1–228 μM, but their measurements significantly deviated from the classical Nernstian logarithmic transfer function (Eq. 1); therefore, potentiometry measurements in liquid phase systems may have to consider alternative models during operation. Ultimately, this demonstration may pave the way for future application of potentiometric sensors in MPS.
A vast majority of reported and prospective electrochemical oxygen sensors integrated into MPS operate via amperometry. Amperometric measurement of correlates the concentration of dissolved oxygen to the electrical current produced by an electrochemical reaction. A concentration gradient of oxygen, caused by its depletion at the electrode surface during the electrochemical reaction, leads to mass transport by diffusion, and the flux of oxygen is directly proportional to the reduction current according to Faraday’s Law (Equation 2), where ireduction is the electrical current, F is Faraday’s constant (C·mol−1), n is the number of electrons active in the reaction, A is the area of the electrode (m2), and JO2 is the flux of oxygen (mol·m−2·s−1). Accordingly, by measuring the reduction current, the flux of oxygen can be related to the moles of oxygen per unit volume, i.e. oxygen concentration, via Fick’s Law. This conversion requires that the diffusion constant of oxygen in the selected medium is known and assumes the concentration of oxygen at the electrode surface is zero.
| (2) |
In general, for amperometric systems the sensor performance (e.g., magnitude of current, sensitivity, response time) depends on the electrode material (e.g., gold or platinum)169, surface area of the electrode (i.e., improved sensitivity and response time result with larger electrode areas)170, 171, temperature171, 172, solubility of the oxygen in electrolyte172, and oxygen diffusion rate through the electrolyte and any incorporated gas-permeable membrane.171, 172 Due to these parameters, it is most common for electrochemical oxygen sensors to be operated via comparison to a calibration standard, rather than a Coulometric manner.
The most established of the amperometric oxygen sensors was developed by Clark et al.173 The Clark-type (CT) electrode consists of a silver-silver chloride anode, a platinum cathode, an electrolyte, and a gas-permeable membrane (e.g., cellophane or Teflon®).
The gas-permeable membrane covers all electrodes in the system, as illustrated in yellow in Figure 3a. Oxygen diffuses through the selective, gas-permeable membrane to the internal electrolyte covering the electrodes. The addition of a selective, gas-permeable membrane separates the medium under test from the electrolyte solution at the surface of the electrodes (typically potassium chloride). In addition, the gas-permeable membrane creates a diffusion profile where the response of the sensor is controlled by the oxygen diffusion instead of reaction kinetics on the electrode surface, i.e. the steady-state current is proportional to the concentration of dissolved oxygen.
Fig.3.
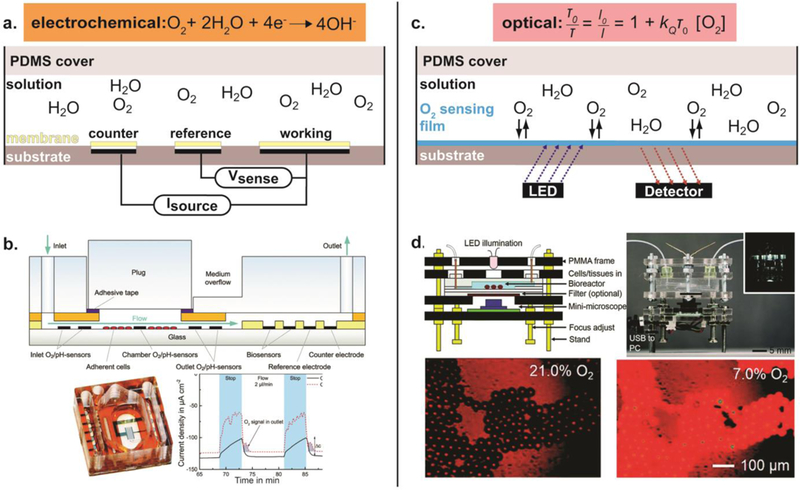
Measuring oxygen in microdevices. (a) Schematic of non-Clark type amperometric oxygen sensing. An optional membrane or coating can be applied over the electrodes to limit fouling or cell-electrode interactions. An oxygen-selective membrane can also be integrated to achieve a Clark-type system, i.e. using only an anode and cathode. (b) An example of amperometric oxygen sensing. (Top) Schematic of the fabricated microfluidic device. (Bottom, left) Fabricated microfluidic device with integrated electrochemical O2 sensors. (Bottom, right) Amperometric continuous oxygen measurement. Reproduced from Ref. 123 with permission from The Royal Society of Chemistry, copyright 2014. (c) Schematic of optical oxygen sensing. (d) An example of optical oxygen sensing. (Top, left) Schematic of the fabricated microfluidic device. (Top, right) Fabricated microfluidic device with integrated optical O2 sensors. (Bottom) Fluorescent images of the microbeads doped with two dyes, oxygen sensitive ruthenium dye and oxygen-irresponsive Nile blue, to 21% and 7% oxygen concentrations. Reproduced from Ref. 243 with permission from The Royal Society of Chemistry, copyright 2015.
During operation, oxygen is reduced to water at the cathode, and metallic silver is oxidized to silver chloride at the anode. Current is generated by reduction of oxygen at the cathode surface according to the reaction presented in Equation 3.174 The current generated at the necessary overpotential for the electrochemical cell is proportional to the concentration of dissolved oxygen in the system. CT electrodes in microfluidic systems have been shown to operate in arrange of −0.6 V to −0.8 V.173, 175 The applied potential is set such that all oxygen molecules are consumed at the cathode and the generated current is directly proportional to the oxygen diffusion from medium, across the membrane, and to the cathode.176
| (3) |
It should be noted that the consumption of oxygen in CT electrodes is not trivial, and stable signals can be difficult to generate in small, microscale volumes. To overcome this challenge, there have been efforts for miniaturization of CT electrodes177–181, but there are limited examples of CT electrodes being miniaturized and integrated into microfluidic devices.182 Early attempts exhibited operational stability problems due to dissolution of the reference electrode material when operating in two-electrode configuration (i.e. no counter electrode). This deleterious effect could be limited by using three-electrode configurations, which drive most of the current through a counter electrode and little current flows through the reference electrode.183 Park et al. created a miniaturized Clark electrode with a three electrode configuration for measuring cellular oxygen consumption, and it was reported that membrane type and working electrode area were the two dominant factors in determining both the stability and sensitivity of the oxygen sensor.184
As an alternative to CT-electrodes, direct amperometric oxygen sensors have been reported, including two electrode (working and reference/counter)170, 174, 185 or three electrode (working, reference, and counter)172, 186 configurations. Figure 3b illustrates an example MPS containing bare, thin-film platinum electrodes used as oxygen sensors. In this example, Weltin et al. incorporated amperometric oxygen sensors with small-molecule biosensors to achieve dynamic monitoring of human glioblastoma multiform T98G cells (Figure 3b).123 In direct amperometric oxygen sensors, oxygen is reduced at the working electrode without a selective membrane. The system is designed for diffusion-limited operation, such that the oxygen concentration is linearly proportional to the current. These direct amperometric oxygen sensors are typically operated as chronoamperometric measurements, in which a potential step is applied to the working electrode with respect to the reference electrode and the resulting current response is measured. The generated current density is linearly proportional to the concentration of dissolved oxygen.
As demonstrated in many examples, amperometric detection is advantageous for fast and sensitive detection of oxygen in the microchannels of MPS and many other bioreactors.174, 187–190 Challenges to facile adoption and integration of oxygen sensing electrodes into MPS include: electrode fouling due to biological-electrode interactions, resultant signal drift, local oxygen consumption and depletion of internal electrolyte, material instability and dissolution, susceptibility to electrical interference, and signal fluctuation under flow.106, 135
While many of these challenges can be overcome by utilizing alternatives to CT electrodes or employing alternative electrochemical modalities, like potentiometry, there are also material and engineering design considerations that can improve the operation of integrated oxygen sensors.
Selective, gas-permeable and electrolyte-permeable membranes may also be considered for application as protective barriers in non-CT electrochemical oxygen sensors. Selective membranes or coatings of the sensor electrodes can be employed to reduce fouling and biological-electrode interactions. Bare metal electrodes are vulnerable to contamination from cell adhesion, protein adsorption and interaction with other electroactive species.191 Many oxygen selective membranes are made from oxygen permeable polymers such as silicone170, 192, polyethylene193, polypropylene171, and PDMS.171, 175 However, the slower diffusional characteristic of such materials, as previously described (Table 2), may limit the dynamic sensing of oxygen in a rapidly evolving system. It should also be noted, that these gas-permeable coatings do not permit electrolyte transmission, and therefore are only suitable in solid-state gas oxygen sensors.194–196 Select membrane materials, such as Nafion™, polyelectrolytes, and hydrogels, may better serve as electrode protectants, while enabling rapid diffusion of oxygen to and from the electrode surface and also behave as electrolyte and ionic conductors in aqueous media.197–200
The method of sensor fabrication and integration may also play a critical design role for instrumented MPS. Planar amperometric oxygen sensing electrodes on glass170, 185 and flexible Kapton® 201 have been integrated in-line with microfluidic channels and are the most common. Bare metal electrodes can be fabricated via myriad vapor deposition processes. Rodrigues et al. designed planar amperometric oxygen and glucose sensors at the inlet and outlet of a PDMS chamber for cell culture analysis of hepatocytes; this approach provided real-time monitoring of the composition and consumption of nutrients in the cell culture medium before and after its contact with cells.202 Weltin et al. also utilize a distributed sensing design in which the system included two oxygen sensors in the inlet, one electrode in the cell culture area, and two electrodes in the outlet channel to investigate the oxygen concentration inside and outside of the cell culture and provide some spatial resolution of oxygen concentration. More complex microelectrode architectures have also been explored for oxygen sensing “on-chip.” Lee et al. created amperometric gold oxygen sensing microneedles for evaluation of dissolved oxygen microprofiles. The fabricated microneedles were used to penetrate into the microfluidic channel for vertical measurement of dissolved oxygen levels in the microchannel, and as a pre-conditioning step, the electrodes were polarized before calibration.174
Lastly, preconditioning of electrodes is carried out when a dry sensor is first introduced to the solution for measurement. In bare metal electrodes, near-zero oxygen levels at the electrode surface can be achieved in less than one second; whereas, the introduction of high-surface area electrodes, hydrogel coatings, solid polyelectrolytes, or other membranes, may require increased preconditioning to achieve a stable baseline and accurate measurement of oxygen. Numerous strategies have been employed to reduce preconditioning times for planar amperometric oxygen sensors203, and various materials have been used as a gas permeable membrane to control the time to and stability of the internal electrolyte for fabricating planar type oxygen sensors.204–207
4.2. Fluorescence/phosphorescence-based oxygen measurements
Luminescence is the cold-body radiation of light. Photoluminescence, which is luminescence due to the absorption of photons, is a property exploited for the quantification of oxygen concentration in the local microenvironment of the selected light-emitting substances, i.e. luminophores. Specifically, fluorescence and phosphorescence quenching are the predominant forms of luminescence-based optical sensors that can be integrated into both in vitro tissue cultures and microfluidic devices for oxygen quantification. Fluorescence and phosphorescence are forms of photoluminescence that result from singlet-singlet electronic relaxation with nanosecond lifetimes and triplet-singlet electronic relaxation with microsecond or greater lifetimes, respectively.
Popular fluorescence and phosphorescence-based oxygen sensors operate by quenching of the excited states of luminophores by molecular oxygen.208 Fluorescent and phosphorescent radiative emissions are generated when the excited molecule returns to the ground state from its singlet and triplet electronic states, respectively. If oxygen is present in the environment, it acts as an energy acceptor and non-radiatively quenches the excited singlet or triplet states of the molecule prior to relaxation and emission of photons. This energy transfer, i.e. quenching, leads to a decrease of the intensity and lifetime of the fluorescence and phosphorescence of the luminophore. The degree of quenching is determined by the probability of an encounter between an oxygen molecule and an indicator dye in its excited state.209 Therefore, the change in fluorescence and phosphoresce intensity or lifetime is proportional to the molecular oxygen concentration. The quenching of the indicator dye can be modelled according to Stern-Volmer equation (Equation 4):210
| (4) |
where and are excited-state lifetime and luminescence intensity in the absence of O2, is the oxygen concentration, and are the lifetime and intensity, respectively, at a given oxygen pressure, and is the quenching rate constant. According to the Stern-Volmer equation, the quenching of luminescence intensity by oxygen is non-linear. In practice, the quenching of luminescence intensity is non-linear due to inhomogeneity of the system’s optical properties. Compared to amperometric sensors, oxygen sensing by luminescence quenching is well suited for small fluid volumes and offers fast and stable detection of oxygen with minimal signal drift.135 Optical sensing via luminescence quenching enables oxygen sensing without disturbing the microfluidic setup and provides spatiotemporal imaging of cell cultures during long-term studies.211
Luminophores for quantification of oxygen in microfluidic devices are affected by various factors such as efficiency of the quenching process, lifetime of the indicator dye, stability of the luminophore (i.e., photobleaching and leaching), absorption and emission of spectra of the indicator dye, availability of optical components (i.e., excitation and detection devices), and background interference (i.e., autofluorescence from plasma and cells).156
There are two main groups of oxygen-sensitive indicators: ruthenium-based complexes and metalloporphyrin complexes (Figure 4, Table 3). Tris(4,7-diphenyl-1,10-phenanthroline) ruthenium(II) dichloride ([Ru(dpp)3]Cl2) 24, 212–216 and ruthenium tris(2,2′-dipyridyl) dichloride ([Ru(bpy)3]Cl2) 22, 106, 133, 211, 217, 218 are commonly used ruthenium-based compounds for microfluidic devices. [Ru(dpp)3]Cl2 has longer excited state lifetimes (5.34 μs vs. 0.6 μs) and higher quantum yield of luminescence (ΦL: 0.3 vs. 0.0042) than [Ru(bpy)3]Cl2.219 Ruthenium-based dyes are photo-stable and have short excited state lifetimes (<10 μs) as compared to the excited state lifetimes of metalloporphyrin dyes.219, 220 The ability of oxygen to quench a luminophore increases if the luminescent probe has long lived excited states (i.e., natural lifetime of the excited state,, in the absence of oxygen), which in turn results in more sensitive oxygen sensors. Platinum (II)-5,10,15,20-tetrakis-(2,3,4,5,6-pentafluorphenyl)-porphyrin (PtTFPP) 221–224, palladium(II) or platinum(II) meso-tetra(4-fluorophenyl) tetrabenzoporphyrin (PdTPTBPF and PtTPTBPF) 225, palladium-meso-tetra (4-carboxyphenyl) porphyrin (Pd-TCPP) 226, platinum octaethylporphyrin (PtOEP) 137, platinum octaethylporphine-ketone (Pt-OEPK) 135, 227, and Pt(II) meso-di(pentafuorophenyl)diphenyl porphyrin 228 are some of the commonly used metal porphyrin based oxygen sensors in microfluidic devices. Therefore, the oxygen sensors made by metal porphyrins are more sensitive to oxygen than ruthenium-based sensors popularized in earlier studies.
Fig. 4.
Common oxygen sensing via luminescent optical dyes. (a-d) Chemical structures of Ru(II) complexes and metalloporphyrins. M stands for Pt(II) or Pd(II) metal ions.
Table 3.
Microfluidic devices with Ruthenium or Metalloporphyrin-based oxygen sensors
| Probe | Detection Scheme | Culture or reaction | O2 Range | Reference |
|---|---|---|---|---|
| Oxygen Sensitive Foil | Fluorescence (Intensity) |
hepatocytes; endothelial cells; Escherichia coli | <20% | 237, 241 |
| [Ru(bpy)3]Cl2 | Fluorescence (Intensity) |
HepG2 cells, C2C12 cells (mouse myoblasts); MC3T3- E1 cells (mouse preosteoblast); human dermal microvascular endothelial cells; human lung adenocarcinoma (A549); porcine cardiomyocytes; b- TC-6 (transgenic insulin-secreting murine cells) |
1–6%; 0–15%; 0–11 mg·L−1 | 22, 24, 133, 212, 213, 218, 217 |
| Fluorescence (Lifetime) |
C2C12 cells; L2 and H4IIE cells (lung and liver) | 1–8 mg·L−1 | 106, 211, 242 | |
| Ruthenium + Secondary Compound |
Fluorescence (Ratiometric Intensity) |
pancreatic islets; Neuronal tissue; HepG2 human hepatocellular carcinoma cells; NIH/3T3 fibroblasts; neonatal rat cardiomyocytes; Corynebacterium glutamicum |
0–21% | 243, 76, 229, 244 |
| Pt or Pd porphyrins | Phosphorescence (Intensity) |
oxidation of sulphite to sulphate human lung adenocarcinoma (A549); human cervical carcinoma (HeLa); Escherichia coli |
<20%; 0–100%; 0–40 mg·L−1; 0.03–0.2 atm |
137, 223, 227, 245 |
| Phosphorescence (Lifetime) |
enzymatic reactions; hepatocytes | 0–21 kPa; 0–150 mmHg; < ~125 hPa; 0–42.5 ppm |
135, 225, 226, 228 | |
| PtTFPP + MFY | Phosphorescence (Ratiometric Intensity) |
human cervical carcinoma (HeLa); human dermal fibroblasts |
0–21%; 0–200 hPa | 222, 224 |
Indicator dyes are usually immobilized in polymeric or sol-gel matrices to protect the dyes from degradation and to reduce dye leaching. Several polymers are commonly used for immobilization of dyes in microfluidic devices. Indicator dyes such as PtTFPP and Pt-OEPK were embedded in polystyrene.135, 221, 227 PtOEP, PdTCPP, PtTFPP and [Ru(dpp)3]Cl2 were incorporated into PDMS. 137, 214, 223, 226 Other matrices such as silica gels 212, 229, poly(sodium styrenesulfonate) (PSS) 217, and polyfluorenes 228 were also utilized as immobilization medium.
Properties of the luminescent dyes (i.e., sensitivity, response time and Stern-Volmer calibration graph) are strongly dependent on the type of polymer used as it controls the oxygen diffusion through the polymer matrix.230 Permeation of oxygen through a polymeric matrix is dependent on diffusion constant and solubility of oxygen within that matrix (, where P: permeation rate, D: diffusion constant (cm2 s−1), S: solubility of oxygen (cm3 (STP) cm−3 (cmHg)−1)). According to Amao et al., PDMS has higher permeation rate than organic glassy polymers such as polystyrene (PS), poly(methyl methacrylate), and poly(vinly chloride) (PVC). Fluoropolymers exhibit high permeability to oxygen due to the high electronegativity of fluorine.231 Andrew et al. studied the sensitivity, response and recovery times of Pt-OEP and Pd-OEP immobilized in cellulose acetate butyrate (CAB) and PMMA.219 Pt-OEP and Pd-OEP embedded in CAB polymer had higher oxygen sensitivities and smaller response and recovery times than similar luminophores embedded in PMMA polymer, which was attributed to higher permeability of oxygen in CAB. It was reported that incorporation of plasticizers, such as tributyl phosphate, increased the oxygen sensitivity of the sensors due to localization of the luminophores in plasticizer rich areas within the polymer. Moreover, substitution of Pt-OEP with Pd-OEP in CAB or PMMA film produced much more sensitive films because of a long lifetime of Pd-OEP than Pt-OEP (0.99 vs 0.0091 ms). Hartmann et al. investigated sensitivity of Pt-OEPK and Pd-OEPK in PVC and PS films and reported that Pd-OEPK had the highest oxygen sensitivity among other combinations of the Pt and Pd metal complexes in PVC and PS matrices.
There have been various methods for integration of luminescent oxygen sensors into microfluidic devices. Regardless of the integration technique, several criteria should be met for successful oxygen sensor integration. First, the oxygen sensitive dyes should not detach from the surface of the microchannel. Second, the indicator dye must withstand repeated cleaning steps (i.e., solvent stability), and should not present any toxicological effects to cell cultures.156 Lastly, the distribution of the indicator dye in the matrix and within the microfluidic channel should be homogenous as it changes the light intensity and spatial resolution of the intensity-based readout techniques.232 Measuring oxygen inside microfluidic devices has been done using a commercial handheld oxygen probe (e.g., Neofox, Ocean Optics™), which includes oxygen sensitive dyes at the tip of the probe.76, 233–236 This method of measurement lacks the integration of oxygen sensors with microfluidics.
An alternative method for measuring oxygen inside a microchannel involves perfusing oxygen sensitive dye with media, where the fluorescent luminophore (e.g., [Ru(bpy)3]Cl2) is injected into channels for oxygen monitoring.22, 211, 218 This method may require replenishment of the medium and does not provide an integrated sensing approach. An integrated oxygen sensing approach was reported by Ochs et al., where a microfluidic chip was fabricated by spin coating PDMS polymer on a commercially available oxygen sensor foil. The PDMS surface was then treated with an oxygen plasma for bonding it to a complementary pre-molded PDMS piece for oxygen level measurement during cell culturing.237 Sato et al. mixed an oxygen sensitive phosphorescent Pd-TCPP dye with PDMS polymer to create a thin oxygen sensing membrane by spin coating on a glass slide. Afterwards, the membrane was attached to the sealed O2 and N2 PDMS microchannels for detection of oxygen gradients in a microfluidic device.226, 227 Wang et al. and Thomas et al. used a similar spin coating technique to spin coat phosphorescent PtOEP and PtTFPP embedded PDMS polymers on glass slides, respectively.137, 223 Subsequently, the pre-molded PDMS pieces were combined with the spin coated O2 sensing layers via plasma bonding to measure oxygen gradients in the microchannels. This way of luminescent dye integration into polymeric matrices yields more mechanically durable sensing layers as compared to physical adhesion. Lasave et al. and Chang-Yen et al. studied physical adsorption of the luminophore on glass substrates.22, 228 Lasave et al. filled the microfluidic channel with oxygen sensitive conjugated polymer nanoparticles. Due to the positively charged quaternary ammonium groups, the oxygen sensitive sensor was adhered to the negatively charged glass surface. Similarly, Chang-Yen et al. used a layer-by-layer deposition technique for physical adsorption of the indicator dye on a glass substrate. The glass slides were dipped into a mixture of interpolyelectrolyte complex ([Ru(bpy)3]Cl2 with PSS polymer) to form a negatively charged surface. The slides were subsequently dipped into a poly(diallyl dimethylammonium) chloride (PDDA) solution to form a positively charged surface. Repeated dipping formed alternating layers of a desired thickness. Other integration techniques such as airbrush spraying of indicator dyes 225, wet etching of glass substrate and subsequent luminophore pipetting 135, knife-coating of luminophore solution onto glass slides 224, and recess-filling with an indicator dye, where the recesses were created via reactive ion etching of a silicon wafer or micro molding of a PDMS polymer have been reported.213, 214
Quenching of a luminophore dye results in a decrease in luminescence intensity and excited-state lifetime. Therefore, two main readout techniques (e.g., intensity and lifetime) have been developed for measurement of oxygen concentration. The first readout technique, intensity-based oxygen sensing, is based on detection of luminescence intensity, which can be easily implemented compared to the lifetime-based readout techniques. In this technique, the luminophore is excited with an excitation light source and the emitted light intensity is measured with a detector. Intensity-based oxygen sensing has been implemented for quantification of oxygen in microfluidic devices.22, 24, 133, 212, 213, 217 However, intensity-based oxygen measurement is dependent on concentration of luminophore, photobleaching, optoelectronic properties of excitation source and detector.238 Therefore, ratiometric sensing has been proposed to overcome the disadvantages of the intensity-based measurement. In this technique, an oxygen-sensitive dye and an oxygen-insensitive dye were incorporated into the sensing film. The emission spectra of the dyes differ from each other and only the oxygen-sensitive dye is quenched by oxygen. The quantification of oxygen is made by taking the ratio of the luminescence intensity of the oxygen-sensitive dye to that of the oxygen-insensitive dye (Figure 3d). This method reduces the effects of factors that are common to both dyes, such as sample inhomogeneity and variations in excitation light and detector sensitivity.158 John et al. used ratiometric sensing based on fluorescent [Ru(dpp)3]Cl2 and safranin dyes for measurement of oxygen concentration in microtiter plates. The ratio of the luminescence intensities of both dyes in the presence and absence of oxygen was related to the oxygen concentration by using the Stern-Volmer equation.229 In another study, Ungerbock et al. used red emitting phosphorescent PtTFPP and green emitting Macrolex Fluorescent Yellow (MFY) dyes for measurement of the respiratory activity of HeLa carcinoma cells and human dermal fibroblasts via ratiometric sensing. MFY dye acts as an antenna dye for collection of blue light and transfers part of its energy to the Q-bands of the oxygen sensitive PtTFPP indicator dye, thus, eliminating the need for two different excitation sources.224
The second type of readout technique, lifetime-based oxygen sensing, measures the lifetime of the luminophore. The lifetime is defined as the average amount of time a fluorophore remains in the excited state following excitation.239 Frequency 135, 225, 228 and time-domain 226, 238 lifetime measurements have been studied to measure oxygen concentration in microfluidics. Lasave et al. used phase fluorometer for measurement of enzyme activity of glucose oxidase.228 The frequency domain technique applied a modulating excitation light to the luminophore with an LED and measured the emission of the light with a photodiode. The phase lag between the excitation light and the emitted light was measured using a reference LED for phase correction. The lifetime of the luminophore was calculated from the luminescent phase shifts according to Equation 6.
| (5) |
where f is the modulation frequency and is the phase shift. The frequency-domain lifetime measurement is advantageous over the time-domain method for measurement of lifetimes of dyes with close emission wavelengths. In contrast to the frequency-domain lifetime measurement, the time-domain technique is based on “pulse and gate” method (i.e., rapid lifetime detection). The measurement starts with switching on the modulated excitation source (i.e., square wave) for a short period of time. Then, the light source is turned off and the detector is turned on to allow measurement of the luminescent light at two different times (t1 and t2). The lifetime of the luminophore is calculated from the emission intensity decay curve according to Equation 6.240
| (5) |
where A1 and A2 are the photon counts at two different times. Sato et al. designed a microdevice for measurement of oxygen consumption rate of hepatocytes. The device used a pulsed laser at 532 nm and a photomultiplier for irradiation of the sensor and detection of the emitted phosphorescence, respectively.226 Similarly, Sud et al. designed a microfluidic bioreactor for continuous O2 monitoring of mouse myoblasts using fluorescence lifetime imaging microscopy (FLIM). The FLIM system included an excitation source (337 nm to 960 nm) and a gated CCD camera for image recording.211 Overall, lifetime-based measurement is advantageous over intensity-based measurements because it is independent of concentration of luminophore, photobleaching, drifts in source intensity and detector sensitivity.155
5. Conclusion
Measuring and regulating oxygen levels in microdevices is paramount to develop the next generation of in vitro tools for studying human physiology and pathophysiology. Often microdevices used to study cell biology, tissue or organ function, or drug response studies are criticized for their simplicity. By using primary human cells and accurately recapitulating the in vivo environment inside a microdevice, basic scientific questions can be answered, and the design of more complex experiments can be better informed. Before designing a MPS, a desired oxygen gradient or constant concentration must be defined using previous knowledge attained from in vivo measurements of oxygen in a human organ or hypothesized oxygen concentrations extrapolated from reported animal models. Materials should be chosen to generate cells-specific microenvironments and in silico models can help determine appropriate seeding densities and expected oxygen concentrations at material interfaces. Overall, the most important factors determining the success of a MPS are the ability to measure oxygen in real-time and adjust oxygen concentration easily. Microdevice architecture should inform the best oxygen sensor integration.
With more knowledge of the intricate and critical role of oxygen in physiologic and pathophysiologic phenomenon, microdevices can be fabricated that are more representative of true human organ and cell-level responses to damage or repair and lead to better predictions of drugs and cell therapies. It is important to consider how organ systems respond to oxygen fluctuations based on function. MPS should incorporate fluctuating oxygen, but this will only be realized with continuous, real-time oxygen sensing.
Supplementary Material
Acknowledegements
K.R.R. is supported by the National Institutes of Health under award number F31DK118859–02.
Footnotes
Conflicts of interest
There are no conflicts to declare.
References
- 1.Carreau A, El Hafny-Rahbi B, Matejuk A, Grillon C and Kieda C, J Cell Mol Med, 2011, 15, 1239–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kump LR, Nature, 2008, 451, 277–278. [DOI] [PubMed] [Google Scholar]
- 3.Hatefi Y, Annu Rev Biochem, 1985, 54, 1015–1069. [DOI] [PubMed] [Google Scholar]
- 4.Khanal G, Hiemstra S and Pappas D, Analyst, 2014, 139, 3274–3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Secomb TW, Hsu R, Dewhirst MW, Klitzman B and Gross JF, Int J Radiat Oncol Biol Phys, 1993, 25, 481–489. [DOI] [PubMed] [Google Scholar]
- 6.Akimoto T, Liapis H and Hammerman MR, Am J Physiol Regul Integr Comp Physiol, 2002, 283, R487–495. [DOI] [PubMed] [Google Scholar]
- 7.Fleury C, Mignotte B and Vayssiere JL, Biochimie, 2002, 84, 131–141. [DOI] [PubMed] [Google Scholar]
- 8.Fong GH, J Mol Med (Berl), 2009, 87, 549–560. [DOI] [PubMed] [Google Scholar]
- 9.Helmlinger G, Endo M, Ferrara N, Hlatky L and Jain RK, Nature, 2000, 405, 139–141. [DOI] [PubMed] [Google Scholar]
- 10.Mizukami Y, Li J, Zhang X, Zimmer MA, Iliopoulos O and Chung DC, Cancer Res, 2004, 64, 1765–1772. [DOI] [PubMed] [Google Scholar]
- 11.Noda T, Iwakiri R, Fujimoto K, Matsuo S and Aw TY, Am J Physiol, 1998, 274, G270–276. [DOI] [PubMed] [Google Scholar]
- 12.Uchida T, Rossignol F, Matthay MA, Mounier R, Couette S, Clottes E and Clerici C, J Biol Chem, 2004, 279, 14871–14878. [DOI] [PubMed] [Google Scholar]
- 13.Wenger RH, Kurtcuoglu V, Scholz CC, Marti HH and Hoogewijs D, Hypoxia (Auckl), 2015, 3, 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whitesides GM, Nature, 2006, 442, 368–373. [DOI] [PubMed] [Google Scholar]
- 15.Pampaloni F, Reynaud EG and Stelzer EH, Nat Rev Mol Cell Biol, 2007, 8, 839–845. [DOI] [PubMed] [Google Scholar]
- 16.Abaci HE, Devendra R, Soman R, Drazer G and Gerecht S, Biotechnol Appl Biochem, 2012, 59, 97–105. [DOI] [PubMed] [Google Scholar]
- 17.Bhaskara VK, Mohanam I, Rao JS and Mohanam S, Plos One, 2012, 7, e30905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han M, Wang Y, Liu M, Bi X, Bao J, Zeng N, Zhu Z, Mo Z, Wu C and Chen X, Cancer Sci, 2012, 103, 1058–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brennan MD, Rexius-Hall ML, Elgass LJ and Eddington DT, Lab Chip, 2014, 14, 4305–4318. [DOI] [PubMed] [Google Scholar]
- 20.Kieninger J, Weltin A, Flamm H and Urban GA, Lab Chip, 2018, 18, 1274–1291. [DOI] [PubMed] [Google Scholar]
- 21.Oomen PE, Skolimowski MD and Verpoorte E, Lab Chip, 2016, 16, 3394–3414. [DOI] [PubMed] [Google Scholar]
- 22.Chen YA, King AD, Shih HC, Peng CC, Wu CY, Liao WH and Tung YC, Lab on a Chip, 2011, 11, 3626–3633. [DOI] [PubMed] [Google Scholar]
- 23.Huh D, Leslie DC, Matthews BD, Fraser JP, Jurek S, Hamilton GA, Thorneloe KS, McAlexander MA and Ingber DE, Sci Transl Med, 2012, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khanal G, Chung K, Solis-Wever X, Johnson B and Pappas D, Analyst, 2011, 136, 3519–3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xia C and Fang NX, Biomed Microdevices, 2009, 11, 1309–1315. [DOI] [PubMed] [Google Scholar]
- 26.Allen JW and Bhatia SN, Biotechnol Bioeng, 2003, 82, 253–262. [DOI] [PubMed] [Google Scholar]
- 27.Lo JF, Sinkala E and Eddington DT, Lab Chip, 2010, 10, 2394–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bastarache JA, Wang L, Geiser T, Wang ZM, Albertine KH, Matthay MA and Ware LB, Thorax, 2007, 62, 608–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fridovich I, Science, 1978, 201, 875–880. [DOI] [PubMed] [Google Scholar]
- 30.Burgess KA, Hu HH, Wagner WR and Federspiel WJ, Biomed Microdevices, 2009, 11, 117–127. [DOI] [PubMed] [Google Scholar]
- 31.Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Hsin HY and Ingber DE, Science, 2010, 328, 1662–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hassell BA, Goyal G, Lee E, Sontheimer-Phelps A, Levy O, Chen CS and Ingber DE, Cell Rep, 2017, 21, 508–516. [DOI] [PubMed] [Google Scholar]
- 33.Schurek HJ, Klin Wochenschr, 1988, 66, 828–835. [DOI] [PubMed] [Google Scholar]
- 34.Schurek HJ, Jost U, Baumgartl H, Bertram H and Heckmann U, Am J Physiol, 1990, 259, F910–915. [DOI] [PubMed] [Google Scholar]
- 35.Welch WJ, Baumgartl H, Lubbers D and Wilcox CS, Kidney Int, 2001, 59, 230–237. [DOI] [PubMed] [Google Scholar]
- 36.Brezis M, Heyman SN, Dinour D, Epstein FH and Rosen S, J Clin Invest, 1991, 88, 390–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kalogeris T, Baines CP, Krenz M and Korthuis RJ, Int Rev Cell Mol Biol, 2012, 298, 229–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blantz RC and Weir MR, Curr Opin Nephrol Hy, 2004, 13, 67–71. [DOI] [PubMed] [Google Scholar]
- 39.Koivisto A, Pittner J, Froelich M and Persson AE, Kidney Int, 1999, 55, 2368–2375. [DOI] [PubMed] [Google Scholar]
- 40.Vaziri ND, Dicus M, Ho ND, Boroujerdi-Rad L and Sindhu RK, Kidney Int, 2003, 63, 179–185. [DOI] [PubMed] [Google Scholar]
- 41.Greka A and Mundel P, Annu Rev Physiol, 2012, 74, 299–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Musah S, Mammoto A, Ferrante TC, Jeanty SSF, Hirano-Kobayashi M, Mammoto T, Roberts K, Chung S, Novak R, Ingram M, Fatanat-Didar T, Koshy S, Weaver JC, Church GM and Ingber DE, Nat Biomed Eng, 2017, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brukamp K, Jim B, Moeller MJ and Haase VH, Am J Physiol Renal Physiol, 2007, 293, F1397–1407. [DOI] [PubMed] [Google Scholar]
- 44.Freeburg PB, Robert B, St John PL and Abrahamson DR, J Am Soc Nephrol, 2003, 14, 927–938. [DOI] [PubMed] [Google Scholar]
- 45.Kietzmann T and Jungermann K, Cell Biol Toxicol, 1997, 13, 243–255. [DOI] [PubMed] [Google Scholar]
- 46.Godoy P, Hewitt NJ, Albrecht U, Andersen ME, Ansari N, Bhattacharya S, Bode JG, Bolleyn J, Borner C, Bottger J, Braeuning A, Budinsky RA, Burkhardt B, Cameron NR, Camussi G, Cho CS, Choi YJ, Craig Rowlands J., Dahmen U, Damm G, Dirsch O, Donato MT, Dong J, Dooley S, Drasdo D, Eakins R, Ferreira KS, Fonsato V, Fraczek J, Gebhardt R, Gibson A, Glanemann M, Goldring CE, Gomez-Lechon MJ, Groothuis GM, Gustavsson L, Guyot C, Hallifax D, Hammad S, Hayward A, Haussinger D, Hellerbrand C, Hewitt P, Hoehme S, Holzhutter HG, Houston JB, Hrach J, Ito K, Jaeschke H, Keitel V, Kelm JM, Kevin Park B., Kordes C, Kullak-Ublick GA, LeCluyse EL, Lu P, Luebke-Wheeler J, Lutz A, Maltman DJ, Matz-Soja M, McMullen P, Merfort I, Messner S, Meyer C, Mwinyi J, Naisbitt DJ, Nussler AK, Olinga P, Pampaloni F, Pi J, Pluta L, Przyborski SA, Ramachandran A, Rogiers V, Rowe C, Schelcher C, Schmich K, Schwarz M, Singh B, Stelzer EH, Stieger B, Stober R, Sugiyama Y, Tetta C, Thasler WE, Vanhaecke T, Vinken M, Weiss TS, Widera A, Woods CG, Xu JJ, Yarborough KM and Hengstler JG, Arch Toxicol, 2013, 87, 1315–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Giffin BF, Drake RL, Morris RE and Cardell RR, J Histochem Cytochem, 1993, 41, 1849–1862. [DOI] [PubMed] [Google Scholar]
- 48.Bavli D, Prill S, Ezra E, Levy G, Cohen M, Vinken M, Vanfleteren J, Jaeger M and Nahmias Y, P Natl Acad Sci USA, 2016, 113, E2231–E2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Domansky K, Inman W, Serdy J, Dash A, Lim MHM and Griffith LG, Lab on a Chip, 2010, 10, 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jungermann K and Kietzmann T, Hepatology, 2000, 31, 255–260. [DOI] [PubMed] [Google Scholar]
- 51.Vivares A, Salle-Lefort S, Arabeyre-Fabre C, Ngo R, Penarier G, Bremond M, Moliner P, Gallas JF, Fabre G and Klieber S, Xenobiotica, 2015, 45, 29–44. [DOI] [PubMed] [Google Scholar]
- 52.Sarkar U, Rivera-Burgos D, Large EM, Hughes DJ, Ravindra KC, Dyer RL, Ebrahimkhani MR, Wishnok JS, Griffith LG and Tannenbaum SR, Drug Metab Dispos, 2015, 43, 1091–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsamandouras N, Kostrzewski T, Stokes CL, Griffith LG, Hughes DJ and Cirit M, J Pharmacol Exp Ther, 2017, 360, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dash A, Inman W, Hoffmaster K, Sevidal S, Kelly J, Obach RS, Griffith LG and Tannenbaum SR, Expert Opin Drug Metab Toxicol, 2009, 5, 1159–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Du Y, Li N, Yang H, Luo CH, Gong YX, Tong CF, Gao YX, Lu SQ and Long M, Lab on a Chip, 2017, 17, 782–794. [DOI] [PubMed] [Google Scholar]
- 56.Black AF, Berthod F, L’Heureux N, Germain L and Auger FA, Faseb J, 1998, 12, 1331–1340. [DOI] [PubMed] [Google Scholar]
- 57.Klar AS, Guven S, Biedermann T, Luginbuhl J, Bottcher-Haberzeth S, Meuli-Simmen C, Meuli M, Martin I, Scherberich A and Reichmann E, Biomaterials, 2014, 35, 5065–5078. [DOI] [PubMed] [Google Scholar]
- 58.Marino D, Luginbuhl J, Scola S, Meuli M and Reichmann E, Sci Transl Med, 2014, 6, 221ra214. [DOI] [PubMed] [Google Scholar]
- 59.Tremblay PL, Berthod F, Germain L and Auger FA, J Pharmacol Exp Ther, 2005, 315, 510–516. [DOI] [PubMed] [Google Scholar]
- 60.DiVito KA, Daniele MA, Roberts SA, Ligler FS and Adams AA, Biomaterials, 2017, 138, 142–152. [DOI] [PubMed] [Google Scholar]
- 61.Truskey GA and Fernandez CE, Expert Opin Drug Metab Toxicol, 2015, 11, 1021–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mori N, Morimoto Y and Takeuchi S, Biomaterials, 2017, 116, 48–56. [DOI] [PubMed] [Google Scholar]
- 63.Atac B, Wagner I, Horland R, Lauster R, Marx U, Tonevitsky AG, Azar RP and Lindner G, Lab Chip, 2013, 13, 3555–3561. [DOI] [PubMed] [Google Scholar]
- 64.Shepherd BR, Enis DR, Wang F, Suarez Y, Pober JS and Schechner JS, Faseb J, 2006, 20, 1739–1741. [DOI] [PubMed] [Google Scholar]
- 65.Supp DM, Wilson-Landy K and Boyce ST, Faseb J, 2002, 16, 797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wufuer M, Lee G, Hur W, Jeon B, Kim BJ, Choi TH and Lee S, Sci Rep, 2016, 6, 37471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gordon LI, Burke MA, Singh AT, Prachand S, Lieberman ED, Sun L, Naik TJ, Prasad SV and Ardehali H, J Biol Chem, 2009, 284, 2080–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ganitkevich V, Reil S, Schwethelm B, Schroeter T and Benndorf K, Circ Res, 2006, 99, 165–171. [DOI] [PubMed] [Google Scholar]
- 69.McDougal AD and Dewey CF Jr., J Biol Chem, 2017, 292, 11760–11776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Unterberg C, Buchwald AB, Mindel L and Kreuzer H, Basic Res Cardiol, 1992, 87, 148–160. [DOI] [PubMed] [Google Scholar]
- 71.Ellis BW, Acun A, Can UI and Zorlutuna P, Biomicrofluidics, 2017, 11, 024105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee JM, Grabb MC, Zipfel GJ and Choi DW, J Clin Invest, 2000, 106, 723–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kristian T, Cell Calcium, 2004, 36, 221–233. [DOI] [PubMed] [Google Scholar]
- 74.Dirnagl U, Iadecola C and Moskowitz MA, Trends Neurosci, 1999, 22, 391–397. [DOI] [PubMed] [Google Scholar]
- 75.Winquist RJ and Kerr S, Neurology, 1997, 49, S23–26. [DOI] [PubMed] [Google Scholar]
- 76.Mauleon G, Fall CP and Eddington DT, Plos One, 2012, 7, e43309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Peterson BL, Larson J, Buffenstein R, Park TJ and Fall CP, Plos One, 2012, 7, e31568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mauleon G, Lo JF, Peterson BL, Fall CP and Eddington DT, J Neurosci Methods, 2013, 216, 110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Johnson BN, Lancaster KZ, Hogue IB, Meng F, Kong YL, Enquist LW and McAlpine MC, Lab Chip, 2016, 16, 1393–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aznavoorian S, Stracke ML, Krutzsch H, Schiffmann E and Liotta LA, J Cell Biol, 1990, 110, 1427–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tredan O, Galmarini CM, Patel K and Tannock IF, J Natl Cancer Inst, 2007, 99, 1441–1454. [DOI] [PubMed] [Google Scholar]
- 82.Acosta MA, Jiang X, Huang PK, Cutler KB, Grant CS, Walker GM and Gamcsik MP, Biomicrofluidics, 2014, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Funamoto K, Zervantonakis IK, Liu Y, Ochs CJ, Kim C and Kamm RD, Lab Chip, 2012, 12, 4855–4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lewis DM, Park KM, Eisinger-Mathason TSK, Simon C and Gerecht S, Cancer Res, 2017, 77. [Google Scholar]
- 85.D. M. T. Lewis V; Jain N; Isser A; Xia Z; Gerecht S, ACS Biomaterials Science and Engineering, 2017, DOI: 10.1021/acsbiomaterials.7b00056, 10. [DOI] [PubMed] [Google Scholar]
- 86.Lewis DM, Blatchley MR, Park KM and Gerecht S, Nat Protoc, 2017, 12, 1620–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Abaci HE, Truitt R, Tan S and Gerecht S, Am J Physiol Cell Physiol, 2011, 301, C431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Espey MG, Free Radic Biol Med, 2013, 55, 130–140. [DOI] [PubMed] [Google Scholar]
- 89.Colgan SP and Taylor CT, Nat Rev Gastroenterol Hepatol, 2010, 7, 281–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bohlen HG, Am J Physiol, 1980, 238, H164–171. [DOI] [PubMed] [Google Scholar]
- 91.Turner JR, Nat Rev Immunol, 2009, 9, 799–809. [DOI] [PubMed] [Google Scholar]
- 92.Kim HJ, Huh D, Hamilton G and Ingber DE, Lab Chip, 2012, 12, 2165–2174. [DOI] [PubMed] [Google Scholar]
- 93.Kim HJ, Lee J, Choi JH, Bahinski A and Ingber DE, J Vis Exp, 2016, DOI: 10.3791/54344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shah P, Fritz JV, Glaab E, Desai MS, Greenhalgh K, Frachet A, Niegowska M, Estes M, Jager C, Seguin-Devaux C, Zenhausern F and Wilmes P, Nat Commun, 2016, 7, 11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim HJ, Li H, Collins JJ and Ingber DE, Proc Natl Acad Sci U S A, 2016, 113, E7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Artursson P, Palm K and Luthman K, Adv Drug Deliv Rev, 2001, 46, 27–43. [DOI] [PubMed] [Google Scholar]
- 97.Merkel TC, Bondar VI, Nagai K, Freeman BD and Pinnau I, J Polym Sci Pol Phys, 2000, 38, 415–434. [Google Scholar]
- 98.J. X. W. D. Leong WR; Ghasemi M; Ahmad A; Ismail M; Liew KB, International Journal of Hydrogen Energy, 2015, 40, 11604–11614. [Google Scholar]
- 99.W. J. W. Koros J; Felder RM, J Appl Polym Sci, 1981, 26, 2805–2809. [Google Scholar]
- 100.A. K. Mirzadeh M, Eur Polym J, 2007, 43, 3757–3765. [Google Scholar]
- 101.Gao YP, Baca AM, Wang BJ and Ogilby PR, Macromolecules, 1994, 27, 7041–7048. [Google Scholar]
- 102.Chen SAJ, H.L., J Appl Polym Sci, 1980, 25, 1105–1116. [Google Scholar]
- 103.Barbari TA, Koros WJ and Paul DR, J Polym Sci Pol Phys, 1988, 26, 709–727. [Google Scholar]
- 104.G. A. J. S. Orchard P; Ward IM, J Polym Sci Pol Phys, 1990, 28, 603–621. [Google Scholar]
- 105.Klinger M, Tolbod LP, Gothelf KV and Ogilby PR, Acs Appl Mater Inter, 2009, 1, 661–667. [DOI] [PubMed] [Google Scholar]
- 106.Mehta G, Mehta K, Sud D, Song JW, Bersano-Begey T, Futai N, Heo YS, Mycek MA, Linderman JJ and Takayama S, Biomed Microdevices, 2007, 9, 123–134. [DOI] [PubMed] [Google Scholar]
- 107.Duffy DC, McDonald JC, Schueller OJ and Whitesides GM, Anal Chem, 1998, 70, 4974–4984. [DOI] [PubMed] [Google Scholar]
- 108.Toepke MW and Beebe DJ, Lab Chip, 2006, 6, 1484–1486. [DOI] [PubMed] [Google Scholar]
- 109.Regehr KJ, Domenech M, Koepsel JT, Carver KC, Ellison-Zelski SJ, Murphy WL, Schuler LA, Alarid ET and Beebe DJ, Lab Chip, 2009, 9, 2132–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Domansky K, Leslie DC, McKinney J, Fraser JP, Sliz JD, Hamkins-Indik T, Hamilton GA, Bahinski A and Ingber DE, Lab Chip, 2013, 13, 3956–3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Olah A, Hillborg H and Vancso GJ, Appl Surf Sci, 2005, 239, 410–423. [Google Scholar]
- 112.Ramsey WS, Hertl W, Nowlan ED and Binkowski NJ, In Vitro, 1984, 20, 802–808. [DOI] [PubMed] [Google Scholar]
- 113.Amstein CF and Hartman PA, J Clin Microbiol, 1975, 2, 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.K. T. L. C. Trinh NM; Lee NY, Anal. Methods, 2018, DOI: 10.1039/C8AY01707G. [DOI] [Google Scholar]
- 115.Oppegard SC, Blake AJ, Williams JC and Eddington DT, Lab Chip, 2010, 10, 2366–2373. [DOI] [PubMed] [Google Scholar]
- 116.Rowe L, Almasri M, Lee K, Fogleman N, Brewer GJ, Nam Y, Wheeler BC, Vukasinovic J, Glezer A and Frazier AB, Lab Chip, 2007, 7, 475–482. [DOI] [PubMed] [Google Scholar]
- 117.Chang TY, Yadav VG, De Leo S, Mohedas A, Rajalingam B, Chen CL, Selvarasah S, Dokmeci MR and Khademhosseini A, Langmuir, 2007, 23, 11718–11725. [DOI] [PubMed] [Google Scholar]
- 118.Hoshino T, Saito I, Kometani R, Samejima K, Matsui S, Suzuki T, Mabuchi K and Kato YX, J Biosci Bioeng, 2012, 113, 395–398. [DOI] [PubMed] [Google Scholar]
- 119.Trantidou T, Rao C, Barrett H, Camelliti P, Pinto K, Yacoub MH, Athanasiou T, Toumazou C, Terracciano CM and Prodromakis T, Biofabrication, 2014, 6, 025004. [DOI] [PubMed] [Google Scholar]
- 120.Abaci HE, Devendra R, Smith Q, Gerecht S and Drazer G, Biomed Microdevices, 2012, 14, 145–152. [DOI] [PubMed] [Google Scholar]
- 121.Polinkovsky M, Gutierrez E, Levchenko A and Groisman A, Lab Chip, 2009, 9, 1073–1084. [DOI] [PubMed] [Google Scholar]
- 122.Skolimowski M, Nielsen MW, Emneus J, Molin S, Taboryski R, Sternberg C, Dufva M and Geschke O, Lab on a Chip, 2010, 10, 2162–2169. [DOI] [PubMed] [Google Scholar]
- 123.Weltin A, Slotwinski K, Kieninger J, Moser I, Jobst G, Wego M, Ehret R and Urban GA, Lab Chip, 2014, 14, 138–146. [DOI] [PubMed] [Google Scholar]
- 124.Day D and Gu M, Opt Express, 2005, 13, 5939–5946. [DOI] [PubMed] [Google Scholar]
- 125.Frazer RQ, Byron RT, Osborne PB and West KP, J Long Term Eff Med Implants, 2005, 15, 629–639. [DOI] [PubMed] [Google Scholar]
- 126.Klank H, Kutter JP and Geschke O, Lab Chip, 2002, 2, 242–246. [DOI] [PubMed] [Google Scholar]
- 127.Yao L, Liu B, Chen T, Liu S and Zuo T, Biomed Microdevices, 2005, 7, 253–257. [DOI] [PubMed] [Google Scholar]
- 128.Rivera KR, Pozdin VA, Young AT, Erb PD, Wisniewski NA, Magness ST and Daniele MA, Biosens Bioelectron, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.de Jong J, Ankone B, Lammertink RG and Wessling M, Lab Chip, 2005, 5, 1240–1247. [DOI] [PubMed] [Google Scholar]
- 130.Park KM and Gerecht S, Nat Commun, 2014, 5, 4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Oppegard SC and Eddington DT, Biomed Microdevices, 2013, 15, 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Domenech M, Yu H, Warrick J, Badders NM, Meyvantsson I, Alexander CM and Beebe DJ, Integr Biol (Camb), 2009, 1, 267–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Adler M, Polinkovsky M, Gutierrez E and Groisman A, Lab Chip, 2010, 10, 388–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Adler M, Erickstad M, Gutierrez E and Groisman A, Lab Chip, 2012, 12, 4835–4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Vollmer AP, Probstein RF, Gilbert Rand Thorsen T, Lab on a Chip, 2005, 5, 1059–1066. [DOI] [PubMed] [Google Scholar]
- 136.Aitken CE, Marshall RA and Puglisi JD, Biophys J, 2008, 94, 1826–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wang L, Liu W, Wang Y, Wang JC, Tu Q, Liu R and Wang J, Lab Chip, 2013, 13, 695–705. [DOI] [PubMed] [Google Scholar]
- 138.Reist M, Marshall KA, Jenner P and Halliwell B, J Neurochem, 1998, 71, 2431–2438. [DOI] [PubMed] [Google Scholar]
- 139.Mathur A, Loskill P, Shao K, Huebsch N, Hong S, Marcus SG, Marks N, Mandegar M, Conklin BR, Lee LP and Healy KE, Sci Rep, 2015, 5, 8883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Allen JW, Khetani SR and Bhatia SN, Toxicol Sci, 2005, 84, 110–119. [DOI] [PubMed] [Google Scholar]
- 141.Kamholz AE and Yager P, Biophys J, 2001, 80, 155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Pera-Titus M, El-Chahal R, Rakotovao V, Daniel C, Miachon S and Dalmon JA, Chemphyschem, 2009, 10, 2082–2089. [DOI] [PubMed] [Google Scholar]
- 143.Foy BD, Rotem A, Toner M, Tompkins RG and Yarmush ML, Cell Transplant, 1994, 3, 515–527. [DOI] [PubMed] [Google Scholar]
- 144.Gracz AD, Williamson IA, Roche KC, Johnston MJ, Wang F, Wang Y, Attayek PJ, Balowski J, Liu XF, Laurenza RJ, Gaynor LT, Sims CE, Galanko JA, Li L, Allbritton NL and Magness ST, Nat Cell Biol, 2015, 17, 340–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lee-Montiel FT, George SM, Gough AH, Sharma AD, Wu J, DeBiasio R, Vernetti LA and Taylor DL, Exp Biol Med (Maywood), 2017, 242, 1617–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Shuk P and Jantz R, 2015 9th International Conference on Sensing Technology (Icst), 2015, 12–17. [Google Scholar]
- 147.Eguchi T, Electrochemistry, 1999, 67, 819–819. [Google Scholar]
- 148.Hauser CJ, Locke RR, Kao HW, Patterson J and Zipser RD, J Lab Clin Med, 1988, 112, 68–71. [PubMed] [Google Scholar]
- 149.Sheridan WG, Lowndes RH and Young HL, Am J Surg, 1990, 159, 314–319. [DOI] [PubMed] [Google Scholar]
- 150.Cooper GJ, Sherry KM and Thorpe JA, Eur J Cardiothorac Surg, 1995, 9, 158–160; discussion 160. [DOI] [PubMed] [Google Scholar]
- 151.He G, Shankar RA, Chzhan M, Samouilov A, Kuppusamy P and Zweier JL, Proc Natl Acad Sci U S A, 1999, 96, 4586–4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Albenberg L, Esipova TV, Judge CP, Bittinger K, Chen J, Laughlin A, Grunberg S, Baldassano RN, Lewis JD, Li H, Thom SR, Bushman FD, Vinogradov SA and Wu GD, Gastroenterology, 2014, 147, 1055–1063 e1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ramamoorthy R, Dutta PK and Akbar SA, J Mater Sci, 2003, 38, 4271–4282. [Google Scholar]
- 154.Yi C, Zhang Q, Li CW, Yang J, Zhao J and Yang M, Anal Bioanal Chem, 2006, 384, 1259–1268. [DOI] [PubMed] [Google Scholar]
- 155.Wang XD and Wolfbeis OS, Chemical Society Reviews, 2014, 43, 3666–3761. [DOI] [PubMed] [Google Scholar]
- 156.Grist SM, Chrostowski L and Cheung KC, Sensors-Basel, 2010, 10, 9286–9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Helm I, Karina G, Jalukse L, Pagano T and Leito I, Environ Monit Assess, 2018, 190. [DOI] [PubMed] [Google Scholar]
- 158.Feng Y, Cheng JH, Zhou L, Zhou XG and Xiang HF, Analyst, 2012, 137, 4885–4901. [DOI] [PubMed] [Google Scholar]
- 159.Akmal N and Lauer J, Acs Sym Ser, 1998, 690, 149–160. [Google Scholar]
- 160.Pham AQ and Glass RS, J Electrochem Soc, 1997, 144, 3929–3934. [Google Scholar]
- 161.Martinez-Manez R, Camino JS, Garcia-Breijo E, Sanchez LG, Civera JI and Morant EG, Sensors and Actuators B: Chemical, 2004, 101, 295–301. [Google Scholar]
- 162.Zimmermann P, Weltin A, Urban GA and Kieninger J, Sensors, 2018, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ramamoorthy R, Akbar SA and Dutta PK, Sensor Actuat B-Chem, 2006, 113, 162–168. [Google Scholar]
- 164.Maskell WC and Steele BCH, J Appl Electrochem, 1986, 16, 475–489. [Google Scholar]
- 165.Hahn PB, Wechter MA, Johnson DC and Voigt AF, Anal Chem, 1973, 45, 1016–1021. [Google Scholar]
- 166.Yim HS and Meyerhoff ME, Anal Chem, 1992, 64, 1777–1784. [DOI] [PubMed] [Google Scholar]
- 167.Meruva RK and Meyerhoff ME, Biosens Bioelectron, 1998, 13, 201–212. [DOI] [PubMed] [Google Scholar]
- 168.Li M, Ornelas IM, Liu W, Niu Y, Correia JP, Viana AS and Jin G, Electroanalysis, 2015, 27, 1427–1435. [Google Scholar]
- 169.Linsenmeier RA and Yancey CM, J Appl Physiol, 1987, 63, 2554–2557. [DOI] [PubMed] [Google Scholar]
- 170.Gao C, Choi JW, Dutta M, Chilukuru S, Nevin JH, Lee JY, Bissell MG and Ahn CH, Eng Med Biol Soc Ann, 2002, DOI: Doi 10.1109/Mmb.2002.1002318, 223–226. [DOI] [Google Scholar]
- 171.Park J, Pak YK and Pak JJ, Sensor Actuat B-Chem, 2010, 147, 263–269. [Google Scholar]
- 172.Mitrovski SM and Nuzzo RG, Lab on a Chip, 2005, 5, 634–645. [DOI] [PubMed] [Google Scholar]
- 173.Clark LC, Wolf R, Granger D and Taylor Z, J Appl Physiol, 1953, 6, 189–193. [DOI] [PubMed] [Google Scholar]
- 174.Lee JH, Lim TS, Seo Y, Bishop PL and Papautsky I, Sensor Actuat B-Chem, 2007, 128, 179–185. [Google Scholar]
- 175.Wu CC, Luk HN, Lin YTT and Yuan CY, Talanta, 2010, 81, 228–234. [DOI] [PubMed] [Google Scholar]
- 176.Khine M, Thompson JK, Bierer SM, Altamirano LJ, Freeman RD and Lee LP, I Ieee Embs C Neur E, 2003, DOI: Doi 10.1109/Cne.2003.1196793, 204–207. [DOI] [Google Scholar]
- 177.Luo J, Dziubla T and Eitel R, Sensor Actuat B-Chem, 2017, 240, 392–397. [Google Scholar]
- 178.Suzuki H, Kojima N, Sugama A and Takei F, Sensor Actuat B-Chem, 1990, 2, 185–191. [Google Scholar]
- 179.Suzuki H, Sugama A and Kojima N, Anal Chim Acta, 1990, 233, 275–280. [Google Scholar]
- 180.Suzuki H, Sugama A, Kojima N, Takei F and Ikegami K, Biosens Bioelectron, 1991, 6, 395–400. [DOI] [PubMed] [Google Scholar]
- 181.Wu CC, Yasukawa T, Shiku H and Matsue T, Sensor Actuat B-Chem, 2005, 110, 342–349. [Google Scholar]
- 182.Yamagishi A, Tanabe K, Yokokawa M, Morimoto Y, Kinoshita M and Suzuki H, Anal Chim Acta, 2017, 985, 1–6. [DOI] [PubMed] [Google Scholar]
- 183.H. S. Suzuki A; Kojima N., Sensor Actuat B-Chem, 1990, 2, 297–303. [Google Scholar]
- 184.Park J, Youngmi KP, James JP, Sensors and Actuators B: Chemical, 2010, 147, 263–269. [Google Scholar]
- 185.Date Y, Takano S, Shiku H, Ino K, Ito-Sasaki T, Yokoo M, Abe H and Matsue T, Biosens Bioelectron, 2011, 30, 100–106. [DOI] [PubMed] [Google Scholar]
- 186.Amatore C, Arbault S, Chen Y, Crozatier C and Tapsoba I, Lab on a Chip, 2007, 7, 233–238. [DOI] [PubMed] [Google Scholar]
- 187.Kieninger J, Aravindalochanan K, Sandvik JA, Pettersen EO and Urban GA, Cell Proliferat, 2014, 47, 180–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kieninger J, Dannenberg A, Aravindalochanan K, Jobst G, Pettersen EO and Urban GA, Transducers ‘07 & Eurosensors Xxi, Digest of Technical Papers, Vols 1 and 2, 2007. [Google Scholar]
- 189.Park JH, Song YS, Ha JG, Kim YK, Lee SK and Bai SJ, Sensor Actuat B-Chem, 2013, 188, 1300–1305. [Google Scholar]
- 190.Demuth C, Varonier J, Jossen V, Eibl R and Eibl D, Appl Microbiol Biot, 2016, 100, 3853–3863. [DOI] [PubMed] [Google Scholar]
- 191.Brischwein M, Motrescu ER, Cabala E, Otto AM, Grothe H and Wolf B, Lab on a Chip, 2003, 3, 234–240. [DOI] [PubMed] [Google Scholar]
- 192.Ahn CH, Choi JW, Beaucage G, Nevin JH, Lee JB, Puntambekar A and Lee JY, P Ieee, 2004, 92, 154–173. [Google Scholar]
- 193.Clark LC and Clark EW, Int Anesthesiol Clin, 1987, 25, 1–29. [DOI] [PubMed] [Google Scholar]
- 194.Azad AM, Akbar SA, Mhaisalkar SG, Birkefeld LD and Goto KS, J Electrochem Soc, 1992, 139, 3690–3704. [Google Scholar]
- 195.Fratoddi I, Venditti I, Cametti C and Russo MV, Sensor Actuat B-Chem, 2015, 220, 534–548. [Google Scholar]
- 196.Llobet E, Sensor Actuat B-Chem, 2013, 179, 32–45. [Google Scholar]
- 197.Stetter JR and Li J, Chem Rev, 2008, 108, 352–366. [DOI] [PubMed] [Google Scholar]
- 198.Glasspool W and Atkinson J, Sensor Actuat B-Chem, 1998, 48, 308–317. [Google Scholar]
- 199.Wang P, Liu Y, Abruna HD, Spector JA and Olbricht WL, Sensor Actuat B-Chem, 2011, 153, 145–151. [Google Scholar]
- 200.Zhang H, Liu JYY, Liu Q, Chen RR, Zhang HS, Yu J, Song DL, Jing XY, Zhang ML and Wang J, J Electrochem Soc, 2018, 165, B779–B786. [Google Scholar]
- 201.Moya A, Illa X, Prats-Alfonso E, Zine N, Gabriel G, Errachid A and Villa R, 2014 Ieee Sensors, 2014. [Google Scholar]
- 202.Rodrigues NP, Sakai Y and Fujii T, Sensor Actuat B-Chem, 2008, 132, 608–613. [Google Scholar]
- 203.Choi SH, Ha J, Shin JH, Choi YS, Han SH, Nam H and Cha GS, Anal Chim Acta, 2001, 431, 261–267. [Google Scholar]
- 204.Severinghaus JW and Bradley AF, J Appl Physiol, 1958, 13, 515–520. [DOI] [PubMed] [Google Scholar]
- 205.Arquint P, Vandenberg A, Vanderschoot BH, Derooij NF, Buhler H, Morf WE and Durselen LFJ, Sensor Actuat B-Chem, 1993, 13, 340–344. [Google Scholar]
- 206.Ratner BD and Miller IF, J Biomed Mater Res, 1973, 7, 353–367. [DOI] [PubMed] [Google Scholar]
- 207.Naylor PFD and Evans NTS, Nature, 1963, 199, 911-&. [DOI] [PubMed] [Google Scholar]
- 208.Rumsey WL, Vanderkooi JM and Wilson DF, Science, 1988, 241, 1649–1651. [DOI] [PubMed] [Google Scholar]
- 209.Bergman I, Nature, 1968, 218, 396-&. [Google Scholar]
- 210.Wolfbeis OS, Bioessays, 2015, 37, 921–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Sud D, Mehta G, Mehta K, Linderman J, Takayama S and Mycek MA, J Biomed Opt, 2006, 11. [DOI] [PubMed] [Google Scholar]
- 212.Deepa N and Ganesh AB, Measurement, 2015, 59, 337–343. [Google Scholar]
- 213.Lin Z, Cherng-Wen T, Roy P and Trau D, Lab on a Chip, 2009, 9, 257–262. [DOI] [PubMed] [Google Scholar]
- 214.Sin A, Chin KC, Jamil MF, Kostov Y, Rao G and Shuler ML, Biotechnol Progr, 2004, 20, 338–345. [DOI] [PubMed] [Google Scholar]
- 215.Acosta MA, Ymele-Leki P, Kostov YV and Leach JB, Biomaterials, 2009, 30, 3068–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Lambrechts D, Roeffaers M, Kerckhofs G, Roberts SJ, Hofkens J, Van de Putte T, Van Oosterwyck H and Schrooten J, Biomaterials, 2013, 34, 922–929. [DOI] [PubMed] [Google Scholar]
- 217.Chang-Yen DA, Lvov Y, McShane MJ and Gale BK, Sensor Actuat B-Chem, 2002, 87, 336–345. [Google Scholar]
- 218.Mehta G, Lee J, Cha W, Tung YC, Linderman JJ and Takayama S, Anal Chem, 2009, 81, 3714–3722. [DOI] [PubMed] [Google Scholar]
- 219.Mills A and Lepre A, Anal Chem, 1997, 69, 4653–4659. [Google Scholar]
- 220.Papkovsky DB and O’Riordan TC, J Fluoresc, 2005, 15, 569–584. [DOI] [PubMed] [Google Scholar]
- 221.Gitlin L, Hoera C, Meier RJ, Nagl S and Belder D, Lab on a Chip, 2013, 13, 4134–4141. [DOI] [PubMed] [Google Scholar]
- 222.Grist SM, Schmok JC, Liu MC, Chrostowski L and Cheung KC, Sensors, 2015, 15, 20030–20052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Thomas PC, Raghavan SR and Forry SP, Anal Chem, 2011, 83, 8821–8824. [DOI] [PubMed] [Google Scholar]
- 224.Ungerbock B, Charwat V, Ertl P and Mayr T, Lab on a Chip, 2013, 13, 1593–1601. [DOI] [PubMed] [Google Scholar]
- 225.Ehgartner J, Sulzer P, Burger T, Kasjanow A, Bouwes D, Kruhne U, Klimant I and Mayr T, Sensor Actuat B-Chem, 2016, 228, 748–757. [Google Scholar]
- 226.Sato A, Kadokura K, Uchida H and Tsukada K, Biochem Bioph Res Co, 2014, 453, 767–771. [DOI] [PubMed] [Google Scholar]
- 227.Lee HLT, Boccazzi P, Ram RJ and Sinskey AJ, Lab on a Chip, 2006, 6, 1229–1235. [DOI] [PubMed] [Google Scholar]
- 228.Lasave LC, Borisov SM, Ehgartner J and Mayr T, Rsc Adv, 2015, 5, 70808–70816. [Google Scholar]
- 229.John GT, Klimant I, Wittmann C and Heinzle E, Biotechnol Bioeng, 2003, 81, 829–836. [DOI] [PubMed] [Google Scholar]
- 230.Hartmann P and Trettnak W, Anal Chem, 1996, 68, 2615–2620. [DOI] [PubMed] [Google Scholar]
- 231.Amao Y, Microchim Acta, 2003, 143, 1–12. [Google Scholar]
- 232.Pfeiffer SA and Nagl S, Methods Appl Fluores, 2015, 3. [DOI] [PubMed] [Google Scholar]
- 233.Nourmohammadzadeh M, Lo JF, Bochenek M, Mendoza-Elias JE, Wang Q, Li Z, Zeng LY, Qi MG, Eddington DT, Oberholzer J and Wang Y, Anal Chem, 2013, 85, 11240–11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Carter WO, Narayanan PK and Robinson JP, J Leukoc Biol, 1994, 55, 253–258. [DOI] [PubMed] [Google Scholar]
- 235.Oppegard SC and Eddington DT, Conf Proc IEEE Eng Med Biol Soc, 2009, 2009, 2097–2100. [DOI] [PubMed] [Google Scholar]
- 236.Oppegard SC, Nam KH, Carr JR, Skaalure SC and Eddington DT, Plos One, 2009, 4, e6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Ochs CJ, Kasuya J, Pavesi A and Kamm RD, Lab on a Chip, 2014, 14, 459–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Hartmann P, Ziegler W, Holst G and Lubbers DW, Sensor Actuat B-Chem, 1997, 38, 110–115. [Google Scholar]
- 239.Lakowicz JR, in Principle of fluorescence spectroscopy, Springer US, 3 edn., 2006, DOI: 10.1007/978-0-387-46312-4, p. 99. [DOI] [Google Scholar]
- 240.Liebsch G, Klimant I, Frank B, Holst G and Wolfbeis OS, Appl Spectrosc, 2000, 54, 548–559. [Google Scholar]
- 241.Zanzotto A, Szita N, Boccazzi P, Lessard P, Sinskey AJ and Jensen KF, Biotechnol Bioeng, 2004, 87, 243–254. [DOI] [PubMed] [Google Scholar]
- 242.Sin A, Chin KC, Jamil MF, Kostov Y, Rao G and Shuler ML, Biotechnol Prog, 2004, 20, 338–345. [DOI] [PubMed] [Google Scholar]
- 243.Zhang YS, Ribas J, Nadhman A, Aleman J, Selimovic S, Lesher-Perez SC, Wang T, Manoharan V, Shin SR, Damilano A, Annabi N, Dokmeci MR, Takayama S and Khademhosseini A, Lab Chip, 2015, 15, 3661–3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Nourmohammadzadeh M, Lo JF, Bochenek M, Mendoza-Elias JE, Wang Q, Li Z, Zeng L, Qi M, Eddington DT, Oberholzer J and Wang Y, Anal Chem, 2013, 85, 11240–11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Gitlin L, Hoera C, Meier RJ, Nagl S and Belder D, Lab Chip, 2013, 13, 4134–4141. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.