

Abstract
Twenty-five years ago, Filiano and Kinney (1994) proposed that a critical period of postnatal development constitutes one of the three risk factors for sudden infant death syndrome (SIDS). The underlying mechanism was poorly understood. In the last 17 years, much has been uncovered on this period in the rat. Against several expected trends of development, abrupt neurochemical, metabolic, ventilatory, and electrophysiological changes occur in the respiratory system at P12-13. This results in a transient synaptic imbalance with suppressed excitation and enhanced inhibition, and the response to acute hypoxia is the weakest at this time, both at the cellular and system’s levels. The basis for the synaptic imbalance is likely to be contributed by a reduced expression of brain-derived neurotrophic factor (BDNF) and its TrkB receptors in multiple brain stem respiratory-related nuclei during the critical period. Exogenous BDNF or a TrkB agonist partially reverses the synaptic imbalance, whereas a TrkB antagonist accentuates the imbalance. A transient down-regulation of pituitary adenylate cyclase-activating polypeptide (PACAP) at P12 in respiratory-related nuclei also contributes to the vulnerability of this period. Carotid body denervation during this time or perinatal hyperoxia merely delays and sometimes prolongs, but not eliminate the critical period. The rationale for the necessity of the critical period in postnatal development is discussed.
Keywords: BDNF, hypoxia, KCC2, NKCC1, PACAP, synaptic imbalance
1. Introduction
The central nervous system in most mammals is not mature at birth. A considerable amount of growth and development takes place postnatally. Even for vital functions such as respiration, its control system undergoes both structural and functional elaboration and molding after birth. In the rat, such growth includes alveolar development and septation in the lung, strengthening of laryngeal and pharyngeal structures, maturation of diaphragm and intercostal muscles (Kelly and Zacks, 1969; Thurlbeck, 1975; Sahebjami and Domino, 1989; Fratacci et al., 1996), as well as the establishment and refinement of the brain stem respiratory network. This review will focus on the last item, and specifically on the critical period of respiratory development, mainly in the rat.
What is a critical period? In the CNS, it was first noted by the classical studies of Hubel and Wiesel (1970; Hubel et al., 1977) as a vulnerable period, when monocular deprivation leads to structural and functional changes in the visual cortex. Perturbations before or after that period did not cause any significant change. In the respiratory system, the critical period has been proposed by Filiano and Kinney (1994) as one of the three risk factors for Sudden Infant Death Syndrome (SIDS) (the other two are “a vulnerable infant” and “external stressors”). The period has also been defined by Carroll (2003) as a time window during development that is “devoted to structural and/or functional shaping of the neural system subserving respiratory control”. Clearly, there is a developmental aspect that is linked to vulnerability, and a plasticity aspect that is associated with a more persistent adaptive or maladaptive alteration of the nervous system. This review will be centered on the developmental aspect and especially on the basic neurochemical, metabolic, electrophysiological and ventilatory mechanisms underlying the vulnerable tendencies during this period. It will focus on acute responses to environmental perturbations, but will not cover the topic of adaptive “plasticity”, which has been elegantly reviewed recently by Bavis and MacFarlane (2017).
2. The critical period is a time of synaptic imbalance
2.1. Neurochemical evidence
What makes a “critical period” so critical for both rodents and humans? The underlying mechanism was poorly understood. The exact timing of sensitivity varied among reports because typically only a few postnatal days were investigated. Earlier studies in rats indicated that postnatal days (P) 7-8 might be the most vulnerable, as the mortality rate after carotid body denervation was the highest (Serra et al., 2001) and chemosensitivity to hypercapnia was the lowest at that time (Stunden et al., 2001; Wickström et al., 2002). The initial study by Liu and Wong-Riley (2001) also focused on the first postnatal week, and skipped from P7 to P14, and from P14 to P21. Cytochrome oxidase (CO) was used as a metabolic marker for neuronal activity (Wong-Riley, 1989), and a gradual increase in CO expression with age was noted, which was not surprising. However, when the metabolic activity was monitored every day for the first 3 postnatal weeks in rats, a surprising pattern emerged in multiple brain stem respiratory-related nuclei: i.e., against a gradual increase in CO activity with age, there was a sudden fall at P12, before partially recovering at P13 (Liu and Wong-Riley, 2002, 2003). This suggests a transient, reduced need for energy.
As the bulk of energy consumption by neurons is for repolarization after excitatory depolarization (Wong-Riley, 1989), a fall in CO activity indicates a decrease in excitatory synaptic activity. Indeed, the expressions of glutamate and NMDA receptor subunit 1 were abruptly reduced at P12, but, concomitantly, the expressions of GABA, GABAB receptors, and glycine receptors were significantly increased (Liu and Wong-Riley, 2002, 2005). This resulted in a brief period of synaptic imbalance, with enhanced inhibition and suppressed excitation among nuclei of the respiratory network in the brain stem (Wong-Riley and Liu, 2005). These included the pre-Botzinger complex (PBC; the proposed center of respiratory rhythmogenesis; Smith et al., 1991), the nucleus tractus solitarius ventral lateral subnucleus (NTSVL; which receives direct projections from carotid chemoafferent fibers and is a part of the dorsal respiratory group known to project to the ventral respiratory group; Smith et al., 1989; Holtman et al., 1990; Finley and Katz, 1992; Bonham, 1995), the nucleus ambiguus (Amb; which innervates airway muscles of the pharynx and larynx and receives input from the central respiratory network; Jordan, 2001), the hypoglossal nucleus (XII; whose motoneurons control tongue extruder muscles for upper airway patency), and the dorsal motor nucleus of the vagus (DMNX; which innervates the airways and the lungs). Changes were not observed in the non-respiratory somatosensory relay, the cuneate nucleus (CN), indicating a developmental adjustment primarily, if not exclusively, in the respiratory system at that time (Liu and Wong-Riley, 2002, 2003).
2.2. Electrophysiological evidence
Is there any synaptic correlate to these neurochemical changes? By means of whole-cell patch-clamp recordings of hypoglossal motoneurons innervating the tongue muscles, Gao et al. (2011) found that the amplitudes and frequencies of both miniature (m) and spontaneous (s) excitatory postsynaptic currents (EPSCs) were significantly reduced at P12-13, whereas those of the inhibitory postsynaptic currents (IPSCs) were markedly enhanced. The kinetics (rise time and decay time) of mEPSCs and mIPSCs accelerated with age, although there was a distinct fall in the rise time of mEPSCs at P12. Both GABA and glycine contributed to mIPSCs, but the dominance switched from GABA to glycine with development. The combination of enhanced inhibition and reduced excitation during the critical period was also evident electrophysiologically in a key respiratory chemosensor, the NTSVL (Gao et al., 2015).
Thus, there appears to be a transient period of synaptic imbalance, with heightened inhibition and suppressed excitation, demonstrable both neurochemically and electrophysiologically, in the brain stem respiratory network toward the end of the second postnatal week in the rat.
3. Other neurochemical changes
3.1. Switching of dominance in neurotransmitter receptor subunits and of chloride transporters during the critical period
The synaptic imbalance that marks the critical period of the respiratory system in rats is accentuated by switches in dominance of several neurochemicals. A switch from the neonatal GABAA receptor subunit α3 to the mature α1 and a switch from the neonatal glycine receptor subunits α2/α3 to the adult α1 (Liu and Wong-Riley, 2004, 2006, 2013c) strengthen inhibition at that time. More importantly, a switch in dominance of the Cl− transporters from the Cl− importer Na+-K+-2Cl− co-transporter 1 (NKCC1) to the Cl− exporter K+-Cl− co-transporter 2 (KCC2) occurs around P12 in multiple brain stem respiratory-related nuclei (Liu and Wong-Riley, 2012) (Fig. 1). As the predominance of NKCC1 favors depolarizing GABAergic and glycinergic transmission, whereas that of KCC2 facilitates hyperpolarizing currents (Ben-Ari, 2002; Löhrke et al., 2005; Blaesse et al., 2009; Viemari et al., 2011).), a switch in dominance to KCC2 reverses the direction of the Cl− gradient and enhances inhibitory neurotransmission, thereby heightening the synaptic imbalance during the critical period.
Fig. 1.
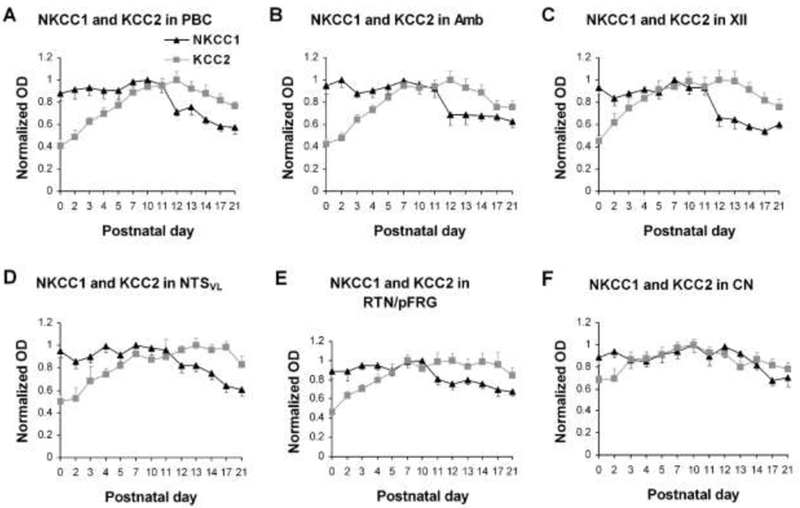
Developmental trends of age-dependent decrease in NKCC1 and age-dependent increase in KCC2 immunoreactivity during the first 3 postnatal weeks in the PBC (A), Amb (B), XII (C), NTSVL (D), RTN/pFRG (E), and CN (F). The highest optical densitometric (OD) value of each marker for each nucleus was set at 1 for comparison. The two trends intersected around P11 for the first five nuclei (A – E). (Modified from Liu and Wong-Riley, 2012).
Very little, if any, is known about the NKCC1/KCC2 switch or about GABAAR subunit expressions in brain stems of humans during postnatal development. In the cortex, the ratio of NKCC1 to KCC2 expression is very high in the youngest infant (1.2 months), decreases rapidly until around 2 years of age, and then remains low into adulthood (Jansen et al., 2010). The exact time point of switch in dominance between the two is not known. As for GABAAR, the expression of the α1 subunit increases logarithmically over the first 5-6 years of age, and then plateaus through adolescence. On the other hand, the expression of the α4 subunit decreases over the first several years before leveling off (Jansen et al., 2010). Again, the point at which the dominance reverses is not known. The expressions of α2 and α3 subunits were not examined.
3.2. Development of serotonergic neurochemicals
Serotonin is involved in the modulation of respiratory rhythmogenesis, respiratory motoneuron excitability, phrenic long-term facilitation, upper airway reflexes, and central chemosensitivity (Bonham, 1995; Hilaire and Duron, 1999; Fuller et al., 2001; Haxhiu et al., 1998; Richerson, 2004). The development of the serotonergic system is of special interest, as reduced binding of serotonergic receptors (5-HT1A-D and 2) and serotonin transporter (SERT) has been reported in the medulla of SIDS infants (Panigrahy et al., 2000; Paterson et al., 2006; Kinney, 2009). Unexpectedly, however, the expressions of the serotonin-synthesizing enzyme, tryptophan hydroxylase (TPH), SERT, as well as several 5-HT receptors (1A, 1B, and 2A) are all significantly reduced at P12 in multiple brain stem respiratory-related nuclei of normal rats duriing development (Liu and Wong-Riley, 2008; 2010a,b). This implies a weakening of the serotonergic system during the critical period and raises the intriguing question of whether reduced serotonergic neurochemicals in SIDS victims reflects a normal phase of development or if additional abnormalities were present.
4. Response to environmental perturbations during the critical period
The critical period is marked by increased vulnerability to environmental perturbations. Challenges that have been tested for acute responses include hypoxia, hyperoxia, hypercapnia, carotid body denervation, infection and inflammation.
4.1. Hypoxia
4.1.1. Ventilatory studies on acute hypoxia
Mammals are born with low sensitivity to hypoxia, with increasing sensitivity over the subsequent days or weeks (Carroll and Kim, 2005). The ventilatory response to acute hypoxia (HVR) is biphasic, with an initial increase in ventilation followed by a roll-off known as the hypoxic ventilatory depression (Eden and Hanson, 1987; Mortola, 1999; Moss, 2002). In analyzing HVR to 5 min of hypoxia (10% O2 + 90% N2) in rats daily from P0 to P21, Liu et al. (2006) found that the biphasic response commenced from P3 onward. However, the HVR underwent developmental changes. The ratio of minute ventilation (VE) in hypoxia to that in normoxia was generally close to or greater than 1 during development. However, it started to fall at P12 and was significantly below 1 at P13 before returning almost to 1 at P14. This indicates that HVR is the weakest during the critical period. The initial phase of HVR is mediated by NMDA receptors, whereas the later roll-off phase is mediated primarily by GABA (Simakajornboon and Kuptanon, 2005). The reduced expression of NMDAR and the elevated expression of GABA during the critical period caused a more depressed response to hypoxia at that time. The weakened reaction was also contributed, to a large extent, by a significant decrease in the metabolic rate (rate of O2 consumption and CO2 production) when challenged by hypoxia at that time (Liu et al., 2009).
4.1.2. Electrophysiological studies on acute hypoxia
How did single neurons respond to acute hypoxia? By means of whole-cell patch-clamp recordings, Gao et al. (2015) tested NTSVL neurons in brain slices at P11, P13, and P15 before and every 30 sec after exposure to 95% N2 and 5% CO2 for 5 min. They found that both the amplitude and frequency of sEPSCs were markedly increased during the first 0.5 to 1 min followed by a fall to a much lower level for the remainder of the 5 min exposure at both P11 and P15 (Fig. 2A, B). This is reminiscent of the biphasic response observed in whole-animals (see above). However, hypoxia did not induce any initial rise in sEPSCs during the critical period (P13), when the values remained a plateau and was the lowest throughout the 5 min of hypoxia.
Fig. 2.
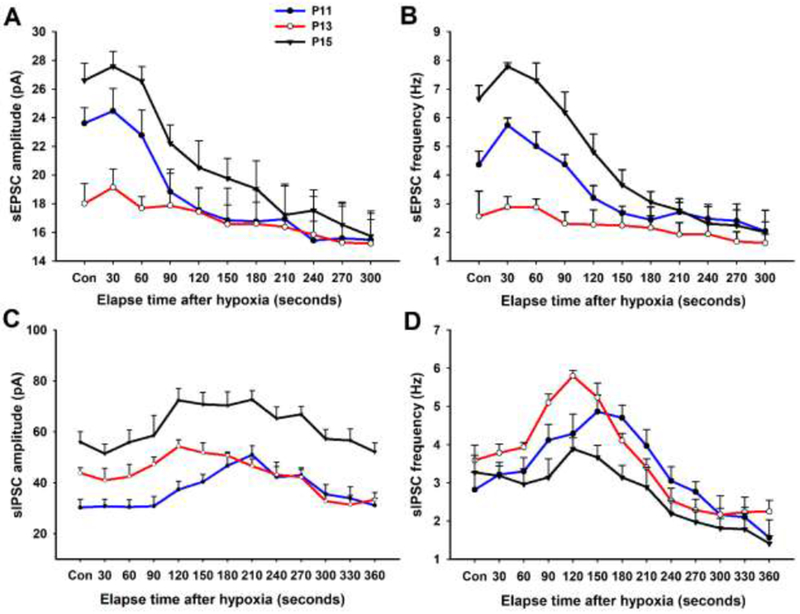
Effect of acute hypoxia on sEPSCs (A and B) and sIPSCs (C and D) of NTSVL neurons at P11 (before the critical period), P13 (during the critical period), and P15 (after the critical period). Note that the amplitude and frequency of sEPSCs were the lowest at P13 and that hypoxia induced a biphasic response at P11 and P15, but not at P13. On the other hand, the amplitude of sIPSCs at P13 was between those of the other two time-points. Hypoxia induced a rise in the frequency of sIPSCs above control levels after 1.5-3 min of exposure that was greater at P13 than at the other two time-points. (Modified from Gao et al., 2015).
On the other hand, the amplitude of sIPSCs at baseline (control) level during the critical period was in between those of the other two time-points, whereas the frequency at P13 was comparable to the other ones (Gao et al., 2015). Acute hypoxia induced at all three ages a dip in the amplitude of sIPSCs during the initial 60 sec followed by a rise and a plateau for the next 3 min (Fig. 2C, D), a pattern that was exactly opposite that of sEPSCs. Remarkably, however, hypoxia induced a significant rise in the frequency of sIPSCs during the second minute of exposure that was much greater at P13 than at the other two time-points.
Thus, a combination of synaptic imbalance, enhanced inhibition, reduced metabolic rate, and weakened ventilatory as well as synaptic responses to hypoxia all contribute to heightened vulnerability during the critical period (Wong-Riley and Liu, 2008; Wong-Riley et al., 2013).
4.1.3. Sustained hypoxia
When rats were exposed to 5 days of sustained hypoxia (11% O2), those treated from P11 to P15 showed absent HVR and reduced response to hypercapnia as well as a high incidence of unexplained mortality, whereas such effects were not seen in the younger (P1-5) or older (P21-25) age groups (Mayer et al., 2014). MacFarlane et al. (2016) also found increased microglial cell number and reduced 5-HT immunoreactivity in the NTS and DMNX of rats exposed to sustained hypoxia, and both effects were prevented by an inhibitor of activated microglia, minocycline, indicating that microglia could modulate serotonergic system at that time. An increase in extracellular matrix protein aggrecan expression was also found in the NTS and DMNX with sustained hypoxia induced between P11 and P15, and the effect was abrogated with intracisternal microinjection of chondroitinase ABC, suggesting that the matrix proteins may be associated with increased vulnerability to hypoxia during the critical period (Stryker et al., 2018).
4.2. Hyperoxia
Hyperoxia at birth is known to impair carotid body chemosensitivity, blunt hypoxic phrenic response, and inhibit lung development (Bavis et al., 2002; Kim et al., 2012; Teng et al., 2017). Hyperoxia during the critical period induces both transient and permanent respiratory defects linked primarily to developmental abnormality of the carotid body (Bavis et al., 2013). Exposure of rat pups to 90% oxygen from P0 to P10 disrupted neurochemical development in multiple brain stem respiratory-related nuclei, but the critical period was simply delayed for 2 days (i.e., from P12 to P14) and not eliminated (Mu et al., 2018). This implies that the critical period is innately determined and is part of the developmental program not easily eradicated.
4.3. Hypercapnia
Newborn rats reportedly have lower ventilatory response to hypercapnia at P2 than at P8 (Matsuoka and Mortola, 1995). They respond with a sustained hyperventilation, which involves increased tidal volume with minimal increase in frequency and is determined mainly by an increase in central inspiratory activity (Saetta and Mortola, 1985). In both brain slices and in culture, the degree and incidence of chemosensitivity of rat medullary raphé neurons were found to increase with age, being greater after P12 than before (Wang and Richerson, 1999). Stunden et al. (2001) found that locus coeruleus neurons in rats had high sensitivity to CO2 at P1 followed by a reduction throughout the first postnatal week to a lowest level at around P8, before rising to a level at P10 that was sustained through adulthood. Similar findings in rats were reported by Wickström et al. (2002), and P7 was considered the lowest point for ventilatory hypercapnic response. However, they examined only four time-points: P1, 3, 7, and 10, and responses at later ages were not investigated. Davis et al. (2006) carried out a longer study and concluded that CO2 sensitivity did not appear in rats until around P15 and reached a peak at P19-21. At the cellular level, Stunden et al. (2001) noted that locus coeruleus neurons responded to increased CO2 with increased firing rate (by ~ 44%) at all ages examined. Clearly, there is not yet a straight-forward correlation between in vivo and in vitro studies, and variations exist among animals and among studies. Maturation of central chemosensitive mechanism appears to be needed to complete the process, but the exact critical period for hypercapnic response in rats has not yet been definitively determined.
4.4. Carotid body denervation
Carotid bodies are the major peripheral chemoreceptors in mammals. Denervating these structures at P2-3 or P7-8 led to lower pulmonary ventilation during eupnea and even lower in acute hypoxia as compared to sham-operated controls (Serra et al., 2001). Denervated animals showed a significant loss in body weight (Liu et al., 2003). When carotid body denervation was performed at P11-13, it simply delayed and prolonged the critical period without eliminating it (Liu et a., 2003).
4.5. Infection and inflammation
Histopathological indications of inflammatory response to infection have been documented in organs of many SIDS victims, but there is no single infectious agent that has been definitively associated with SIDS (reviewed in Blackwell et al., 2015; Blood-Siegfried, 2015). Using a combination of a nonlethal strain of influenza A virus followed by a sublethal dose of endotoxin in rats, Blood-Siegfried and colleagues (2002) found that mortality only occurred when the timing between infectious insults and developmental age were met, which, in their study, was on P12. In a recent study, the administration of the endotoxin lipopolysaccharide attenuated the early and late phases of the acute HVR in only P10 but not P5 or P20 rats (Rourke et al., 2016). Moreover, the authors found that the P10 age group had the largest increase in brain stem TNFα and iNOS mRNA and the highest mortality rate following LPS than the other two age groups. The timing of these periods of heightened vulnerability to infection and inflammatory response corresponds temporally with the critical period described by Liu and Wong-Riley in the rat (see above) and emphasizes the catastrophic outcome of an abnormal or weakened response to an infectious attack at this time.
4.6. In utero effects
It may be argued that the first risk factor proposed by Filiano and Kinney (1994) and others, that of a vulnerable infant, encompasses a variety of conditions, most of which are likely to be present in utero. These include nonlethal abnormal brain development, maternal cigarette smoking (nicotine), injury causing astrogliosis, reduced serotonergic expressions, maternal anemia, and premature birth (reviewed in Guntheroth and Spiers, 2002). However, less is known as to why these conditions should render the infant more prone to death during the critical period rather than in the perinatal stage. In humans, the peak period of SIDS (2nd to 4th postnatal months) also coincides with a loss of maternal antibodies, a drop in night-time core body temperature and cortisol levels, and a male-specific, distinct fall in testosterone level (reviewed in Blackwell et al., 2015). These confounding factors, in addition to changes discussed above, may render the infant less able to respond to external stressors at this time.
5. Role of brain-derived neurotrophic factor (BDNF)
What could be the major underlying mechanism for the synaptic imbalance and increased vulnerability during the critical period? A foremost candidate deserving of consideration is a known regulator of neuronal development and plasticity, i.e., brain-derived neurotrophic factor (BDNF) and its high-affinity tyrosine protein kinase B (TrkB) receptors (Barde et al., 1982; Klein et al., 1993). BDNF plays an important role in neuronal differentiation, cell survival, migration, neurite outgrowth, synapse formation, stabilization, and plasticity (Hofer and Barde, 1988; Wardle and Poo, 2003; Yoshii and Constantine-Paton, 2010). BDNF-TrkB signaling is essential for the development of brain stem respiratory system, and their disruption contributes to severe respiratory dysfunction such as in Rett’s syndrome (Erickson et al., 1996; Ogier et al., 2007; Katz et al., 2009). BDNF knockout animals die within weeks after birth, most likely from respiratory complications (Erickson et al., 1996). Exogenous BDNF or TrkB agonist can ameliorate breathing disorder induced by BDNF knockout or deficit (Kline et al., 2010; Schmid et al., 2012). As the normal function of BDNF is to enhance glutamatergic neurotransmission and attenuate GABAergic and glycinergic ones (Wardle and Poo, 2003; Bardoni et al., 2007), a reduced expression of BDNF-TrkB during the critical period would contribute, at least in part, to suppressed excitation and heightened inhibition at that time.
5.1. Immunohistochemical evidence
The developmental expressions of BDNF and TrkB were analyzed with immunohistochemistry and single neuron optical densitometry by Liu and Wong-Riley (2013a) in multiple brain stem respiratory-related nuclei (including PBC, Amb, XII, NTSVL, the commissural subnucleus of the solitary tract nucleus [NTSCOM], and the retrotrapezoid nucleus/parafacial respiratory group [RTN/pFRG]) during the first three postnatal weeks. They found that the expression of BDNF was relatively high in all of the nuclei from P0 to P11, but a significant fall occurred at P12 that often continued to P13 before returning to P11 levels by P14, followed by a gradual decline until P21 (Fig. 3A–F). Such a fall at P12-13 was not observed in the non-respiratory nucleus, CN (Fig. 3G). The developmental expression of TrkB lagged slightly behind that of BDNF during the first postnatal week, when it exhibited a gradual increase before peaking at P10-11. However, it too had a significant fall at P12 that often continued into P13 before rising again at P14 and plateaued thereafter (Fig. 4A–F). Again, CN did not show a reduction at P12-13 (Fig. 4G). Thus, the abrupt down-regulation of BDNF and TrkB occurred only in respiratory-related nuclei during the critical period. The developmental trends of BDNF and TrkB transcripts were also examined in XII, and they were similar to those of the respective proteins, with a significantly reduced expression at P12-13 (Figs. 3H and 4H).
Fig. 3.
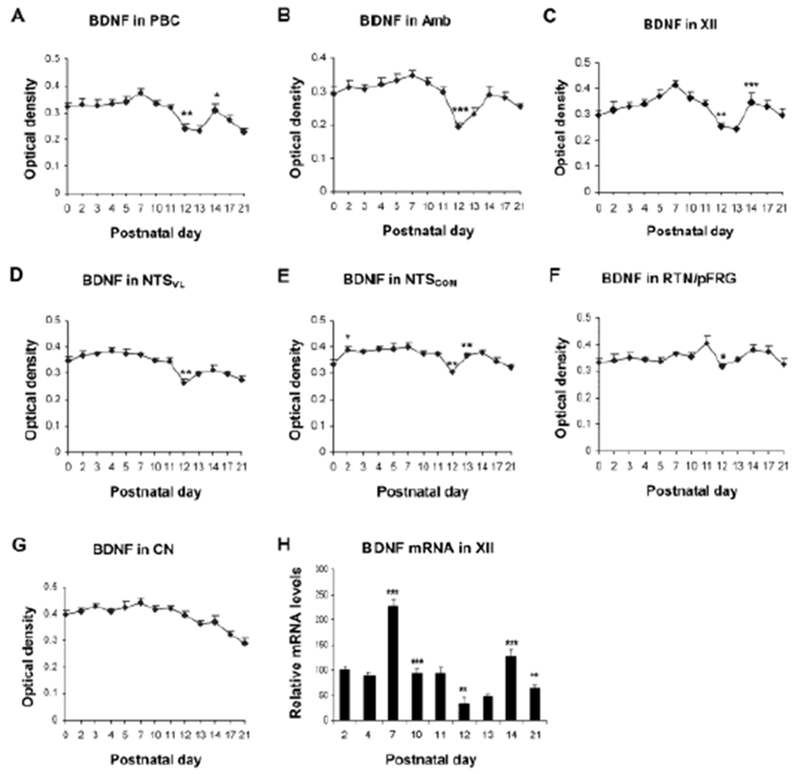
Developmental trends of BDNF immunoreactivity during the first 3 postnatal weeks in the PBC (A), Amb (B), XII (C), NTSVL (D), NTSCOM (E), RTN/pFRG (F), and CN (G). A significant reduction occurred at P12 in all nuclei except for CN. (H) Developmental trend of BDNF mRNA in XII analyzed with RT-qPCR, showing the lowest value at P12. Tukey’s Studentized test comparing one age group with its immediately adjacent younger age group. *P < 0.05; **P < 0.01; ***P < 0.001. (Modified from Liu and Wong-Riley, 2013 for all except C and H, which are modified from Gao et al., 2014).
Fig. 4.
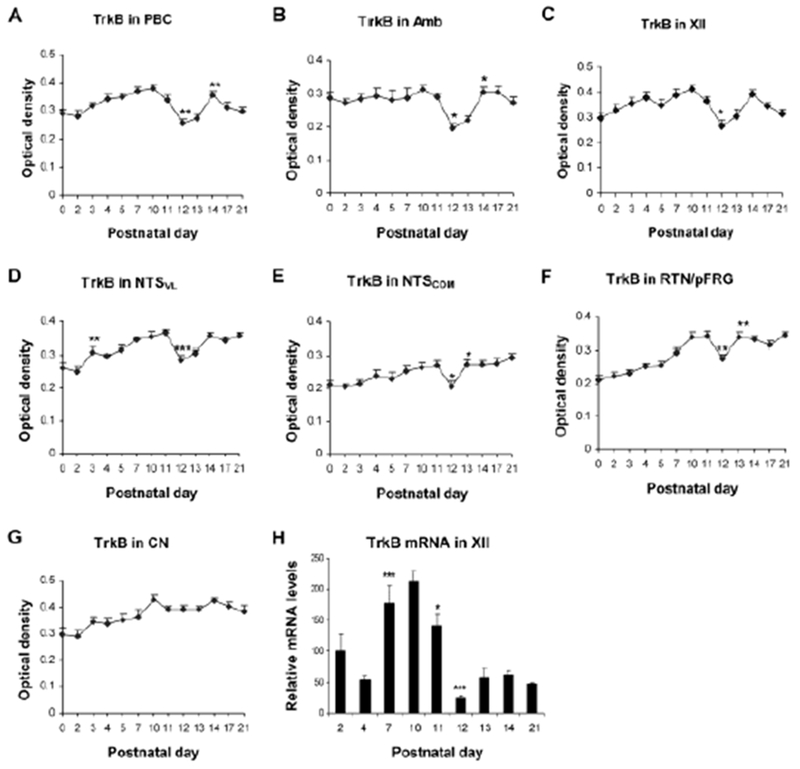
Developmental trends of TrkB immunoreactivity during the first 3 postnatal weeks in the PBC (A), Amb (B), XII (C), NTSVL (D), NTSCOM (E), RTN/pFRG (F), and CN (G). A significant reduction occurred at P12 in all nuclei except for CN. (H) Developmental trend of TrkB mRNA in XII analyzed with RT-qPCR, showing the lowest value at P12. Tukey’s Studentized test comparing one age group with its immediately adjacent younger age group. *P < 0.05; **P < 0.01; ***P < 0.001. (Modified from Liu and Wong-Riley, 2013 for all except C and H, which are modified from Gao et al., 2014).
Interestingly, the down-regulation of BDNF and TrkB occurred 2 days earlier (at P10) in the lateral paragigantocellular nucleus and the parapyramidal region (Liu and Wong-Riley, 2013a). These nuclei have extensive projections to the ventral and dorsal respiratory groups (Van Bockstaele et al., 1989; Ribas-Salgueiro et al., 2005) and may exert an inductive role on the other respiratory-related nuclei in the brain stem.
5.2. Electrophysiological evidence
What are the electrophysiological correlates of neurochemical changes? Would exogenous BDNF have any effect on synaptic imbalance during the critical period? These questions were addressed with whole-cell patch-clamp recordings of hypoglossal motoneurons in brain stem slices, daily, from P0 to P16 (Gao et al., 2014). Exogenous BDNF significantly increased the normally lowered frequency of sEPSCs but decreased the normally heightened amplitude and frequency of sIPSCs during the critical period (Fig. 5). Exogenous BDNF also decreased the normally elevated frequency of mIPSCs at P12-13. These effects were partially blocked by a TrkB receptor antagonist, K252a. However, exogenous BDNF did not affect the kinetics (rise time and decay time) of synaptic responses during development.
Fig. 5.
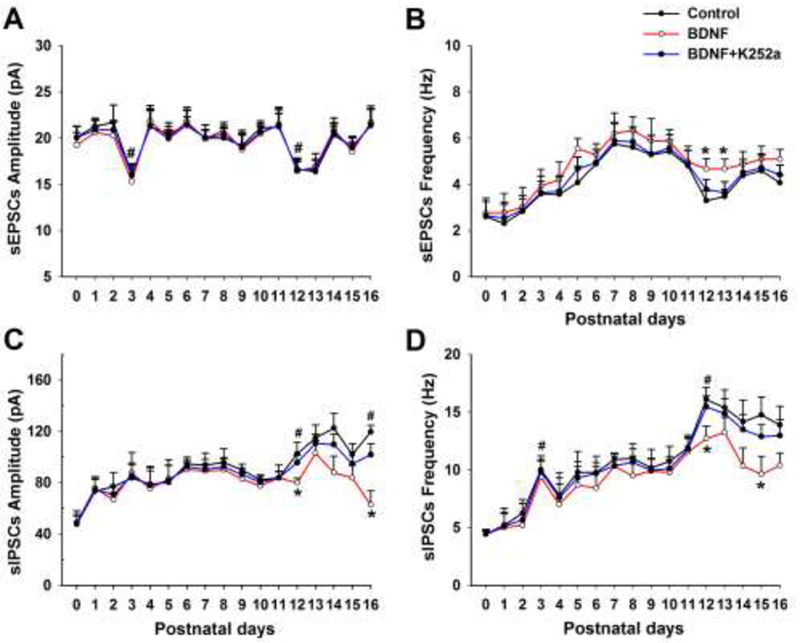
Mean amplitude and frequency of sEPSCs (A and B, respectively) and sIPSCs (C and D, respectively) from P0 to P16 before (control) and after BDNF or BDNF + K252a application in hypoglossal motoneurons. BDNF induced a significant rise in the frequency of sEPSCs at P12-13 (B) but a significant fall in the amplitude of sIPSCs at P12 and P16 (C) as well as the frequency of sIPSCs at P12 and P15 (D) when compared to controls (*P < 0.05). These effects were blocked by K252a. (modified from Gao et al., 2014).
To further substantiate the role of BDNF in contributing to the synaptic imbalance during the critical period, Gao et al. (2015) administered a TrkB agonist, 7,8-DHF, in vivo (5 mg/kg intraperitoneally, once a day for 2 days) and found it to increase the amplitude of sEPSCs by 46.67% and the frequency by 113.01% above controls (P < 0.01) during the critical period (Fig. 6A, B). However, it significantly reduced the normally high amplitude of sIPSCs during the critical period by 63.77% (P < 0.01) (Fig. 6C, D). 7,8-DHF also increased the frequency of sEPSCs and decreased the amplitude of sIPSCs after the critical period (at P14-16). However, it had no effect on responses before the critical period (P10-11), presumably because the level of endogenous BDNF was already high at that time (Liu and Wong-Riley, 2013a).
Fig. 6.
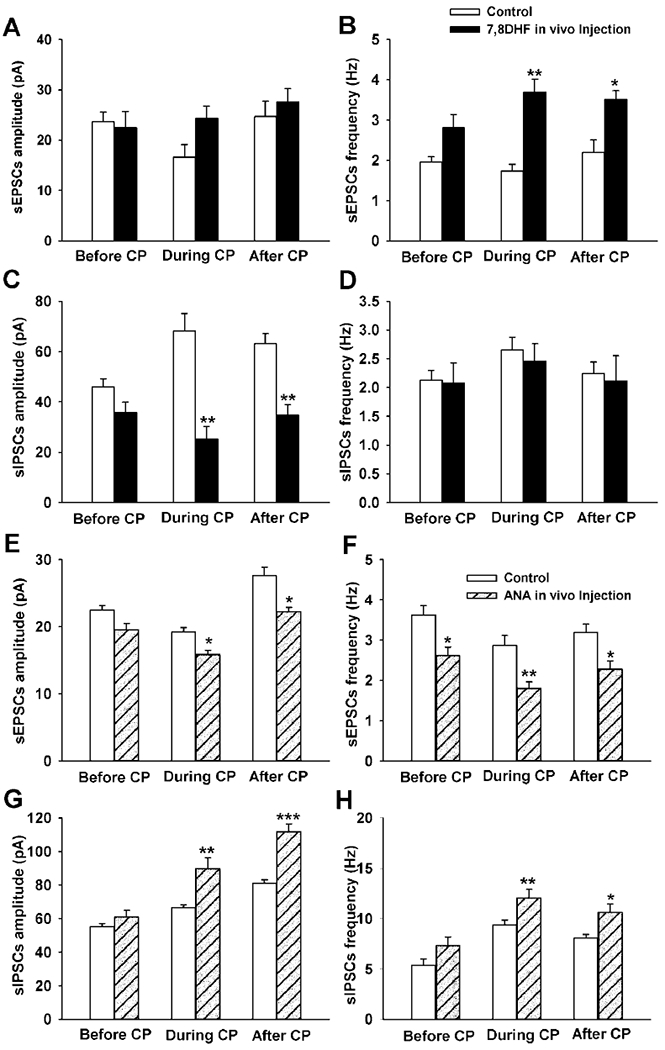
Effect of in vivo application of 7,8-DHF (A-D) and ANA-12 (E-H) on amplitude and frequency of sEPSCs (A, B, E and F) and sIPSCs (C, D, G and H) in NTSVL neurons before (P10-11), during (P12-13), and after (P14-15) the critical period. Note that 7,8-DHF significantly increased the frequency of sEPSCs (B) but decreased the amplitude and frequency of sIPSCs (C) during and after the critical period. The converse is true for ANA-12, which significantly reduced the amplitude and frequency of sEPSCs during and after the critical period (E and F) but increased the amplitude and frequency of sIPSCs (G and H) at those times. ANA-12 also suppressed the amplitude of sEPSCs before the critical period (F). All comparisons were made between treated and respective controls. *P < 0.05; **P < 0.01; ***p < 0.001. (Modified from Gao et al., 2015).
In vivo administration of a TrkB antagonist, ANA-12, had an effect opposite to that of 7,8-DHF (Gao et al., 2015). As little as a single intraperitoneal injection (2.46 mmol/kg) was sufficient to significantly reduce both the amplitude and frequency of sEPSCs, but enhanced those of sIPSCs, during and after the critical period (Fig. 6E–G).
Thus, both in vitro and in vivo studies point to BDNF and TrkB as playing a significant role in contributing to a synaptic imbalance during the critical period of respiratory development in the rat.
5.3. Interaction between BDNF, excitation, inhibition, serotonergic system, and KCC2
How strong is the case of BDNF playing a pivotal role in neurochemical and physiological adjustments during the critical period? Is it merely a coincidence that its level is reduced together with changes in the other neurochemicals? Several lines of evidence point to the relevance of BDNF. First, BDNF is known to enhance excitation by increasing presynaptic glutamate release and increasing the amplitude and frequency of EPSCs as well as the spontaneous firing rate of neurons (Levine et al., 1995; Schinder et al., 2000). At the same time, BDNF significantly reduces both evoked and spontaneous IPSCs and attenuates GABAA receptor-mediated responses to exogenous GABA (Tanaka et al., 1997). A marked decrease in both the message and protein levels of BDNF (and of TrkB receptors) during the critical period would decrease excitation and facilitate inhibition. Second, BDNF reportedly down-regulates KCC2 protein and mRNA expressions, thereby suppressing fast GABAergic inhibition (Rivera et al., 2002; Wardle and Poo, 2003). When BDNF itself is down-regulated during the critical period, it would enable increased expression of KCC2 and enhance inhibition. Third, BDNF has a reciprocal relationship with serotonin in that it stimulates serotonin synthesis, but its own synthesis can be serotonin-dependent (Eaton et al., 1995; Siuciak et al., 1998; Baker-Herman et al., 2004; Homberg et al., 2014). A reduction of BDNF during the critical period would down-regulate serotonin and that, in turn, would inhibit BDNF expression. Fourth, exogenous BDNF or TrkB agonist should at least partially reverse, whereas a TrkB antagonist should accentuate synaptic imbalance during the critical period. Indeed, data obtained thus far (see above) are consistent with all of the above scenarios. These converging lines of evidence do not support a mere coincidence of events, but rather, point to a significant role that reduced BDNF plays in contributing to the suppressed excitation and heightened inhibition during the vulnerable period of respiratory development, at least in the rat.
6. Role of pituitary adenylate cyclase-activating polypeptide (PACAP)
Does BDNF function alone or could there be an upstream signaling protein? A plausible candidate is pituitary adenylate cyclase-activating polypeptide (PACAP), which is known to stimulate BDNF expression by potentiating glutamatergic action (Pelligri et al., 1998). PACAP is a neuropeptide isolated originally from ovine hypothalamus and is a potent stimulator of adenylate cyclase in the anterior pituitary (Miyata et al., 1989). It is widely distributed in the brain and peripheral organs, and is implicated in neural development, neurotransmission, neuromodulation, neuroprotection, metabolic homeostasis, as well as in transcription with central and peripheral targets (Morio et al., 1996; Vaudry et al., 2000, 2009). PACAP powerfully stimulates breathing by activating arterial chemoreceptors (Runcie et al., 1995), and PACAP-null mice die suddenly, mainly during the second postnatal week, due to reduced respiratory chemoresponse and apnea (Gray et al., 2001; Cummings et al., 2004).
The timing of the sudden death in PACAP−/− mice is reminiscent of the critical period in rats. To probe this relationship further, Liu and Wong-Riley (2019) analyzed the developmental expression of PACAP38, a major contributor to the total PACAP immunoreactivity (Masuo et al., 1993), in the PBC, Amb, XII, and NTSVL during the first three postnatal weeks. They found that PACAP immunoreactivity was significantly reduced at P12 in all of these nuclei, but not in the control, non-respiratory cuneate nucleus CN (Fig. 7). These results imply that PACAP down-regulation during normal postnatal development may contribute to the critical period of vulnerability.
Fig. 7.
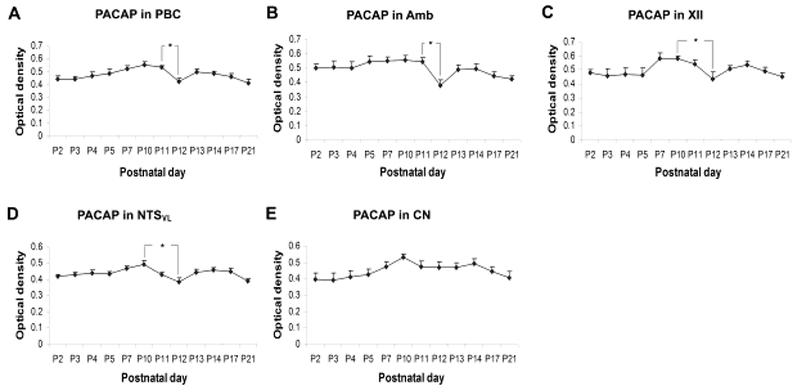
Developmental trends of PACAP immunoreactivity during the first 3 postnatal weeks in the PBC (A), Amb (B), XII (C), NTSVL (D), and CN (E). Tukey’s Studentized tests indicated a significant fall in immunoreactivity from P11 to P12 in the PBC and Amb, and from P10 to P12 in the XII and NTSVL. However, no significant changes were observed in the CN. *P < 0.05. (Modified from Liu and Wong-Riley, 2019).
7. Critical period: gender consideration
Does the critical period exist in one or both genders? This question is of interest, as the prevalence of SIDS is reportedly higher in male infants (Mage and Donner, 2009). Holley et al. (2012) stated that “P10-15 includes a critical developmental period in male but not female rats”. However, an extensive analysis of ventilatory and metabolic responses in hundreds of male versus female rats in both normoxia and acute hypoxia, daily, throughout the first three postnatal weeks indicated that both genders exhibited a significantly weakened response to acute hypoxia at P13 (Liu and Wong-Riley, 2013b). Thus, the critical period exists in both genders in the rat. Significantly, the peak incidence of SIDS in humans is between 2-4 months postnatally (Moon et al., 2007), which corresponds in brain development to P11-P14 (encompassing the critical period) in rats (Ballanyi, 2004).
8. Critical period: Is it necessary?
Is the critical period necessary in postnatal respiratory system development? It is present in both genders and appears to be experienced by all normal rats examined (see above). It is not caused by any intrinsic defect, prematurity, genetic abnormality, or environmental perturbations. It is innately determined and can be delayed and/or prolonged, but generally not eliminated. Yet, it is a period of synaptic imbalance, of major neurochemical and physiological alterations, and it renders the animal vulnerable to external stressors. So, why does it persist and is it necessary for normal development?
A strong rationale for the existence of the critical period is that excitatory synapses in the respiratory system develop and mature earlier than inhibitory ones (Gao et al., 2011). BDNF and PACAP most likely play important roles in nurturing this process for the establishment of the respiratory network. However, the refinement of this system requires the growth and maturation of inhibitory synapses, which necessitates a transient down-regulation of BDNF, PACAP, and excitatory neurotransmission. Thus, this period represents a normal progression of development, but it needs to be relatively brief in the respiratory system to minimize serious threat to a vital function for survival. A similar period exists during postnatal development of the visual cortex in rats (Zhang et al., 2018). However, the timing is later and lasts much longer than that in the respiratory system. Nevertheless, the basic mechanisms remain virtually the same, indicating that the critical period is an obligatory process during normal brain development. Once the critical period is passed, the system will attain a more stabilized state of balanced excitation and inhibition for optimal neuronal functioning.
9. Future studies
Much has been learned about the neurochemical, metabolic, ventilatory, and electrophysiological bases of the critical period of normal respiratory development in the rat. Detailed verifications (or rejection) of such mechanisms in other species would be helpful. The ultimate goal, obviously, is to unravel similar and/or other mechanisms in humans. The challenge is to obtain true control (i.e., normal) specimens from normal infants at varying ages. As closely timed studies in the rat have revealed that postnatal development does not follow a strict continuum, so widely-spaced time points should not be assumed to be connected by a straight line. Finally, global hormonal changes, such as thyroid hormone, that may induce metabolic and neurochemical adjustments, deserve to be monitored during the critical period.
Highlights.
Basic mechanisms underlying a critical period of respiratory development are discussed.
Abrupt neurochemical, ventilatory and electrophysiological changes occur at P12-13 in rats.
BDNF and TrkB play important roles in synaptic imbalance during the critical period.
A reduction in PACAP also contributes to vulnerability during the critical period.
Acknowledgements
Supported by the Children’s Hospital and Health System Foundation, Milwaukee, WI and by NIH Grant R01 HD048954.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baker-Herman TL, Fuller DD, Bavis RW, Zabka AG, Golder FJ, Doperalski NJ, Johnson RA, Watters JJ, Mitchell GS, 2004. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nat. Neurosci 7, 48–55. [DOI] [PubMed] [Google Scholar]
- Ballanyi K, 2004. Neuromodulation of the perinatal respiratory network. Curr. Neuropharm 2, 221–243. [DOI] [PubMed] [Google Scholar]
- Barde YA, Edgar D, Thoenen H, 1982. Purification of a new neurotrophic factor from mammalian brain. Embo J 1, 549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardoni R, Ghirri A, Salio C, Prandini M, Merighi A, 2007. BDNF-mediated modulation of GABA and glycine release in dorsal horn lamina II from postnatal rats. Dev. Neurobiol 67, 960–975. [DOI] [PubMed] [Google Scholar]
- Bavis RW, Fallon SC, Dmitrieff EF, 2013. Chronic hyperoxia and the development of the carotid body. Respir. Physiol. Neurobiol 185, 94–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavis RW, MacFarlane PM, 2017. Developmental plasticity in the neural control of breathing. Exp. Neurol 287, 176–191. [DOI] [PubMed] [Google Scholar]
- Bavis RW, Olson EB Jr., Mitchell GS, 2002. Critical developmental period for hyperoxia-induced blunting of hypoxic phrenic responses in rats. J. Appl. Physiol 92, 1013–1018. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, 2002. Excitatory actions of GABA during development: the nature of the nurture. Nat. Rev. Neurosci 3, 728–739. [DOI] [PubMed] [Google Scholar]
- Blackwell C, Moscovis S, Hall S, Burns C, Scott RJ, 2015. Exploring the risk factors for sudden infant deaths and their role in inflammatory responses to infection. Front. Immunol 6, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaesse P, Airaksinen MS, Rivera C, Kaila K, 2009. Cation-chloride cotransporters and neuronal function. Neuron 61, 820–838. [DOI] [PubMed] [Google Scholar]
- Blood-Siegfried J, 2015. Animal models for assessment of infection and inflammation: contributions to elucidating the pathophysiology of sudden infant death syndrome. Front. Immuno 6, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blood-Siegfried J, Nyska A, Lieder H, Joe M, Vega L, Patterson R, Germolec D, 2002. Synergistic effect of influenza a virus on endotoxin-induced mortality in rat pups: a potential model for sudden infant death syndrome. Pediatr. Res 52, 481–490. [DOI] [PubMed] [Google Scholar]
- Bonham AC, 1995. Neurotransmitters in the CNS control of breathing. Respir. Physiol 101, 219–230. [DOI] [PubMed] [Google Scholar]
- Carroll JL, 2003. Developmental plasticity in respiratory control. J. Appl. Physiol 94, 375–389. [DOI] [PubMed] [Google Scholar]
- Carroll JL, Kim I, 2005. Postnatal development of carotid body glomus cell O2 sensitivity. Respir. Physiol. Neurobiol 149, 201–215. [DOI] [PubMed] [Google Scholar]
- Cummings KJ, Pendlebury JD, Sherwood NM, Wilson RJ, 2004. Sudden neonatal death in PACAP-deficient mice is associated with reduced respiratory chemoresponse and susceptibility to apnoea. J. Physiol. (Lond.) 555, 15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis SE, Solhied G, Castillo M, Dwinell M, Brozoski D, Forster HV, 2006. Postnatal developmental changes in CO2 sensitivity in rats. J. Appl. Physiol 101, 1097–1103. [DOI] [PubMed] [Google Scholar]
- Eaton MJ, Staley JK, Globus MY, Whittemore SR, 1995. Developmental regulation of early serotonergic neuronal differentiation: the role of brain-derived neurotrophic factor and membrane depolarization. Dev. Biol 170, 169–182. [DOI] [PubMed] [Google Scholar]
- Eden GJ, Hanson MA, 1987. Maturation of the respiratory responses to acute hypoxia in the newborn rat. J. Physiol 392, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JT, Conover JC, Borday V, Champagnat J, Barbacid M, Yancopoulos G, Katz DM, 1996. Mice lacking brain-derived neurotrophic factor exhibit visceral sensory neuron losses distinct from mice lacking NT4 and display a severe developmental deficit in control of breathing. J. Neurosci 16, 5361–5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filiano JJ, Kinney HC, 1994. A perspective on neuropathologic findings in victims of the sudden infant death syndrome: the triple-risk model. Biol. Neonate 65, 194–197. [DOI] [PubMed] [Google Scholar]
- Finley JC, Katz DM, 1992. The central organization of carotid body afferent projections to the brainstem of the rat. Brain Res 572, 108–116. [DOI] [PubMed] [Google Scholar]
- Fratacci MD, Levame M, Rauss A, Bousbaa H, Atlan G 1996. Rat diaphragm during postnatal development. I. Changes in distribution of muscle fibre type and in oxidative potential. Reprod. Fertil. Dev 8, 391–398. [DOI] [PubMed] [Google Scholar]
- Fuller DD, Zabka AG, Baker TL, Mitchell GS, 2001. Phrenic long-term facilitation requires 5-HT receptor activation during but not following episodic hypoxia. J. Appl. Physiol 90, 2001–2006. [DOI] [PubMed] [Google Scholar]
- Gao XP, Liu QS, Liu Q, Wong-Riley MTT, 2011. Excitatory-inhibitory imbalance in hypoglossal neurons during the critical period of postnatal development in the rat. J. Physiol. (Lond.) 589, 1991–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao XP, Liu Q, Nair B, Wong-Riley MTT, 2014. Reduced levels of brain-derived neurotrophic factor contribute to synaptic imbalance during the critical period of respiratory development in rats. Eur. J. Neurosci 40, 2183–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao XP, Zhang H, Wong-Riley MTT, 2015. Role of brain-derived neurotrophic factor in the excitatory-inhibitory imbalance during the critical period of postnatal respiratory development in the rat. Physiol. Rep 3, e12631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray SL, Cummings KJ, Jirik FR, Sherwood NM, 2001. Targeted disruption of the pituitary adenylate cyclase-activating polypeptide gene results in early postnatal death associated with dysfunction of lipid and carbohydrate metabolism. Mol. Endocrinol 15, 1739–1747. [DOI] [PubMed] [Google Scholar]
- Guntheroth WG, Spiers PS, 2002. The triple risk hypothesis in sudden infant death syndrome. Pediatrics 110, e64. [DOI] [PubMed] [Google Scholar]
- Haxhiu MA, Erokwu B, Bhardwaj V, Dreshaj IA, 1998. The role of the medullary raphe nuclei in regulation of cholinergic outflow to the airways. J. Auton. Nerv. Syst 69, 64–71. [DOI] [PubMed] [Google Scholar]
- Hilaire G, Duron B, 1999. Maturation of the mammalian respiratory system. Physiol. Rev 79, 325–360. [DOI] [PubMed] [Google Scholar]
- Hofer MM, Barde YA, 1988. Brain-derived neurotrophic factor prevents neuronal death in vivo. Nature 331, 261–262. [DOI] [PubMed] [Google Scholar]
- Holley HS, Behan M, Wenninger JM, 2012. Age and sex differences in the ventilatory response to hypoxia and hypercapnia in awake neonatal, prepubertal and young adult rats. Respir. Physiol. Neurobiol 180, 79–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtman JR Jr., Marion LJ, Speck DF, 1990. Origin of serotonin-containing projections to the ventral respiratory group in the rat. Neurosci 37, 541–552. [DOI] [PubMed] [Google Scholar]
- Homberg JR, Molteni R, Calabrese F, Riva MA, 2014. The serotonin-BDNF duo: developmental implications for the vulnerability to psychopathology. Neurosci. Biobehav. Rev 43, 35–47. [DOI] [PubMed] [Google Scholar]
- Hubel DH, Wiesel TN, 1970. The period of susceptibility to the physiological effects of unilateral eye closure in kittens. J. Physiol. (Lond.) 206, 419–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubel DH, Wiesel TN, LeVay S, 1977. Plasticity of ocular dominance columns in monkey striate cortex. Philos. Trans. R. Soc. Lond. B Biol. Sci 278, 377–409. [DOI] [PubMed] [Google Scholar]
- Jansen LA, Peugh LD, Roden WH, Ojemann JG, 2010. Impaired maturation of cortical GABAa receptor expression in pediatric epilepsy. Epilepsia 51, 1456–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan D, 2001. Central nervous pathways and control of the airways. Respir. Physiol 125, 67–81. [DOI] [PubMed] [Google Scholar]
- Katz DM, Dutschmann M, Ramirez JM, Hilaire G, 2009. Breathing disorders in Rett syndrome: progressive neurochemical dysfunction in the respiratory network after birth. Respir. Physiol. Neurobiol 168, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly AM, Zacks SI 1969. The histogenesis of rat intercostal muscle. J. Cell Biol 42, 135–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Donnelly DF, Carroll JL, 2012. Postnatal hyperoxia impairs acute oxygen sensing of rat glomus cells by reduced membrane depolarization. Adv. Exp. Med. Biol 758, 49–54. [DOI] [PubMed] [Google Scholar]
- Kinney HC, 2009. Brainstem mechanisms underlying sudden infant death syndrome: evidence from human pathologic studies. Dev. Psychobiol 51, 223–233. [DOI] [PubMed] [Google Scholar]
- Klein R, Smeyne RJ, Wurst W, Long LK, Auerbach BA, Joyner AL, Barbacid M, 1993. Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell 75, 113–122. [PubMed] [Google Scholar]
- Kline DD, Ogier M, Kunze DL, Katz DM, 2010. Exogenous brain-derived neurotrophic factor rescues synaptic dysfunction in Mecp2-null mice. J. Neurosci 30, 5303–5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ES, Dreyfus CF, Black IB, Plummer MR, 1995. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc. Natl. Acad. Sci. USA 92, 8074–8077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Fehring C, Lowry TF, Wong-Riley MTT, 2009. Postnatal development of metabolic rate during normoxia and acute hypoxia in rats: implication for a sensitive period. J. Appl. Physiol 106, 1212–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Kim J, Cinotte J, Homolka P, Wong-Riley MTT, 2003. Carotid body denervation effect on cytochrome oxidase activity in pre-Botzinger complex of developing rats. J. Appl. Physiol 94, 1115–1121. [DOI] [PubMed] [Google Scholar]
- Liu Q, Lowry TF, Wong-Riley MTT, 2006. Postnatal changes in ventilation during normoxia and acute hypoxia in the rat: implication for a sensitive period. J. Physiol. (Lond.) 577, 957–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Wong-Riley MTT, 2002. Postnatal expression of neurotransmitters, receptors, and cytochrome oxidase in the rat pre-Botzinger complex. J. Appl. Physiol 92, 923–934. [DOI] [PubMed] [Google Scholar]
- Liu Q, Wong-Riley MTT, 2003. Postnatal changes in cytochrome oxidase expressions in brain stem nuclei of rats: implications for sensitive periods. J. Appl. Physiol 95, 2285–2291. [DOI] [PubMed] [Google Scholar]
- Liu Q, Wong-Riley MTT, 2004. Developmental changes in the expression of GABAA receptor subunits alpha1, alpha2, and alpha3 in the rat pre-Botzinger complex. J. Appl. Physiol 96, 1825–1831. [DOI] [PubMed] [Google Scholar]
- Liu Q, Wong-Riley MTT, 2005. Postnatal developmental expressions of neurotransmitters and receptors in various brain stem nuclei of rats. J. Appl. Physiol 98, 1442–1457. [DOI] [PubMed] [Google Scholar]
- Liu Q, Wong-Riley MTT, 2006. Developmental changes in the expression of GABA(A) receptor subunits alpha1, alpha2, and alpha3 in brain stem nuclei of rats. Brain Res 1098, 129–138. [DOI] [PubMed] [Google Scholar]
- Liu Q, Wong-Riley MTT, 2008. Postnatal changes in the expression of serotonin 2A receptors in various brain stem nuclei of the rat. J. Appl. Physiol 104, 1801–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Wong-Riley MTT, 2010a. Postnatal changes in the expressions of serotonin 1A, 1B, and 2A receptors in ten brain stem nuclei of the rat: implication for a sensitive period. Neurosci 165, 61–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Wong-Riley MTT, 2010b. Postnatal changes in tryptophan hydroxylase and serotonin transporter immunoreactivity in multiple brainstem nuclei of the rat: implications for a sensitive period. J. Comp. Neurol 518, 1082–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Wong-Riley MTT, 2012. Postnatal development of Na+-K+-2Cl− co-transporter 1 and K+-Cl− co-transporter 2 immunoreactivity in multiple brain stem respiratory nuclei of the rat. Neurosci 210, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Wong-Riley MTT, 2013a. Postnatal development of brain-derived neurotrophic factor (BDNF) and tyrosine protein kinase B (TrkB) receptor immunoreactivity in multiple brain stem respiratory-related nuclei of the rat. J. Comp. Neurol 521, 109–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Wong-Riley MTT, 2013b. Gender consideration in ventilatory and metabolic development in rats: special emphasis on the critical period. Resp. Physiol. Neurobiol 188, 200–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Wong-Riley MTT, 2013c. Postnatal development of glycine receptor subunits al, a2, a3, and β immunoreactivity in multiple brain stem respiratory-related nuclei of the rat. Brain Res 1538, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Wong-Riley MTT, 2019. Pituitary adenylate cyclase-activating polypeptide: postnatal development in multiple brainstem respiratory-related nuclei in the rat. Respir. Physiol. Neurobiol 259, 149–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YY, Wong-Riley MT, 2001. Developmental study of cytochrome oxidase activity in the brain stem respiratory nuclei of postnatal rats. J. Appl. Physiol 90, 685–694. [DOI] [PubMed] [Google Scholar]
- Löhrke S, Srinivasan G, Oberhofer M, Doncheva E, Friauf E, 2005. Shift from depolarizing to hyperpolarizing glycine action occurs at different perinatal ages in superior olivary complex nuclei. Eur. J. Neurosci 22, 2708–2722. [DOI] [PubMed] [Google Scholar]
- MacFarlane PM, Mayer CA, Litvin DG, 2016. Microglia modulate brainstem serotonergic expression following neonatal sustained hypoxia exposure: implications for sudden infant death syndrome. J. Physiol. (Lond.) 594, 3079–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mage DT, Donner M, 2009. A unifying theory for SIDS. Int. J. Pediatr 2009, 368270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuo Y, Suzuki N, Matsumoto H, Tokito F, Matsumoto Y, Tsuda M, Fujino M, 1993. Regional distribution of pituitary adenylate cyclase activating polypeptide (PACAP) in the rat central nervous system as determined by sandwich-enzyme immunoassay. Brain Res 602, 57–63. [DOI] [PubMed] [Google Scholar]
- Matsuoka T, Mortola JP, 1995. Effects of hypoxia and hypercapnia on the Hering-Breuer reflex of the conscious newborn rat. J. Appl. Physiol 78, 5–11. [DOI] [PubMed] [Google Scholar]
- Mayer CA, Di Fiore JM, Martin RJ, MacFarlane PM, 2014. Vulnerability of neonatal respiratory neural control to sustained hypoxia during a uniquely sensitive window of development. J. Appl. Physiol 116, 514–521. [DOI] [PubMed] [Google Scholar]
- Miyata A, Arimura A, Dahl RR, Minamino N, Uehara A, Jiang L, Culler MD, Coy DH, 1989. Isolation of a novel 38 residue-hypothalamic polypeptide which stimulates adenylate cyclase in pituitary cells. Biochem. Biophys. Res. Comm 164, 567–574. [DOI] [PubMed] [Google Scholar]
- Moon RY, Horne RS, Hauck FR, 2007. Sudden infant death syndrome. Lancet 370, 1578–1587. [DOI] [PubMed] [Google Scholar]
- Morio H, Tatsuno I, Hirai A, Tamura Y, Saito Y, 1996. Pituitary adenylate cyclaseactivating polypeptide protects rat-cultured cortical neurons from glutamate-induced cytotoxicity. Brain Res 741, 82–88. [DOI] [PubMed] [Google Scholar]
- Mortola JP, 1999. How newborn mammals cope with hypoxia. Respir. Physiol 116, 95–103. [DOI] [PubMed] [Google Scholar]
- Moss IR, 2002. Maturation of respiratory control in the behaving mammal. Respir. Physiol. Neurobiol 132, 131–144. [DOI] [PubMed] [Google Scholar]
- Mu L, Xia D, Michalkiewicz T, Hodges M, Mouradian G, Konduri GG, Wong-Riley MTT, 2018. Effects of neonatal hyperoxia on the critical period of postnatal development of neurochemical expression in brain stem respiratory-related nuclei in the rat. Physiol. Rep 6, e13627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogier M, Wang H, Hong E, Wang Q, Greenberg ME, Katz DM, 2007. Brain-derived neurotrophic factor expression and respiratory function improve after ampakine treatment in a mouse model of Rett syndrome. J. Neurosci 27, 10912–10917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahy A, Filiano J, Sleeper LA, Mandell F, Valdes-Dapena M, Krous HF, Rava LA, Foley E, White WF, Kinney HC, 2000. Decreased serotonergic receptor binding in rhymbic lip-derived regions of the medulla oblongata in the sudden infant death syndrome. J. Neuropathol. Exp. Neurol 59, 377–384. [DOI] [PubMed] [Google Scholar]
- Paterson DS, Trachtenberg FL, Thompson EG, Belliveau RA, Beggs AH, Darnall R, Chadwick AE, Krous HF, Kinney HC, 2006. Multiple serotonergic brainstem abnormalities in sudden infant death syndrome. JAMA 296, 2124–2132. [DOI] [PubMed] [Google Scholar]
- Pelligri G, Magistretti PJ, Martin JL, 1998. VIP and PACAP potentiate the action of glutamate on BDNF expression in mouse cortical neurons. Eur. J. Neurosci, 10, 272–280. [DOI] [PubMed] [Google Scholar]
- Ribas-Salgueiro JL, Gaytan SP, Ribas J, Pasaro R, 2005. Characterization of efferent projections of chemosensitive neurons in the caudal parapyramidal area of the rat brain. Brain Res. Bull 66, 235–248. [DOI] [PubMed] [Google Scholar]
- Richerson GB, 2004. Serotonergic neurons as carbon dioxide sensors that maintain pH homeostasis. Nat. Rev. Neurosci 5, 449–461. [DOI] [PubMed] [Google Scholar]
- Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Vitanen T, Nanobashvili A, Kokaia Z, Airaksinen MS, Voipio J, Kaila K, Saarma M, 2002. BDNF-induced TrkB activation down-regulates the K+-Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. J. Cell Biol 159, 747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rourke K, Mayer CA, MacFarlane PM, 2016. A critical postnatal period of heightened vulnerability to lipopolysaccharide. Resp. Physiol. Neurobiol 232, 26–34. [DOI] [PubMed] [Google Scholar]
- Runcie MJ, Ulman LG, Potter EK, 1995. Effects of pituitary adenylate cyclase-activating polypeptide on cardiovascular and respiratory responses in anesthetized dogs. Regul. Pept 60, 193–200. [DOI] [PubMed] [Google Scholar]
- Saetta M, Mortola JP, 1985. Breathing pattern and CO2 response in newborn rats before and during anesthesia. J. Appl. Physiol 58, 1988–1996. [DOI] [PubMed] [Google Scholar]
- Sahebjami H, Domino M, 1989. Effects of postnatal dexamethasone treatment on development of alveoli in adult rats. Exp. Lung Res 15, 961–973. [DOI] [PubMed] [Google Scholar]
- Schinder AF, Berninger B, Poo M, 2000. Postsynaptic target specificity of neurotrophin-induced presynaptic potentiation. Neuron 25, 151–163. [DOI] [PubMed] [Google Scholar]
- Schmid DA, Yang T, Ogier M, Adams I, Mirakhur Y, Wang O, Massa SM, Longo FM, Katz DM, 2012. A TrkB small molecule partial agonist rescues TrkB phosphorylation deficits and improves respiratory function in a mouse model of Rett syndrome. J. Neurosci 32, 1803–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra A, Brozoski D, Hedin N, Franciosi R, Forster HV, 2001. Mortality after carotid body denervation in rats. J. Appl. Physiol 91, 1298–1306. [DOI] [PubMed] [Google Scholar]
- Simakajornboon N, Kuptanon T, 2005. Maturational changes in neuromodulation of central pathways underlying hypoxic ventilatory response. Respir. Physiol. Neurobiol 149, 273–286. [DOI] [PubMed] [Google Scholar]
- Siuciak JA, Clark MS, Rind HB, Whittemore SR, Russo AF, 1998. BDNF induction of tryptophan hydroxylase mRNA levels in the rat brain. J. Neurosci. Res 52, 149–158. [DOI] [PubMed] [Google Scholar]
- Smith JC, Morrison DE, Ellenberger HH, Otto MR, Feldman JL, 1989. Brainstem projections to the major respiratory neuron populations in the medulla of the cat. J. Comp. Neurol 281, 69–96. [DOI] [PubMed] [Google Scholar]
- Smith JC, Ellenberger HH, Ballanyi K, Richter DW, Feldman JL, 1991. Pre-Botzinger complex: a brainstem region that may generate respiratory rhythm in mammals. Science. 254, 726–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stryker C, Camperchioli DW, Mayer CA, Alilain WJ, Martin RJ, MacFarlane PM, 2018. Respiratory dysfunction following neonatal sustained hypoxia exposure during a critical window of brain stem extracellular matrix formation. Am. J. Physiol. Regul. Integr. Comp. Physiol 314, R216–R227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stunden CE, Filosa JA, Garcia AJ, Dean JB, Putnam RW, 2001. Development of in vivo ventilatory and single chemosensitive neuron responses to hypercapnia in rats. Respir. Physiol 127, 135–155. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Saito H, Matsuki N, 1997. Inhibition of GABAA synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. J. Neurosci 17, 2959–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng RJ, Jing X, Michalkiewicz T, Afolayan AJ, Wu TJ, Konduri GG., 2017, Attenuation of endoplasmic reticulum stress by caffeine ameliorates hyperoxia-induced lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol 312, L586–L598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurlbeck WM, 1975. Postnatal growth and development of the lung. Am. Rev. Respir. Dis 111, 803–844. [DOI] [PubMed] [Google Scholar]
- Van Bockstaele EJ, Pieribone VA, Aston-Jones G, 1989. Diverse afferents converge on the nucleus paragigantocellularis in the rat ventrolateral medulla: retrograde and anterograde tracing studies. J. Comp. Neurol 290, 561–584. [DOI] [PubMed] [Google Scholar]
- Vaudry D, Gonzalez BJ, Basille M, Yon L, Fournier A, Vaudry H, 2000. Pituitary adenylate cyclase-activating polypeptide and its receptors: from structure to function. Pharmacol. Rev 52, 269–324. [PubMed] [Google Scholar]
- Vaudry D, Falluel-Morel A, Bourgault S, Basille M, Burel D, Wurtz O, Fournier A, Chow BK, Hashimoto H, Galas L, Vaudry FL, 2009. Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after the discovery. Pharmcol. Rev 61, 283–357. [DOI] [PubMed] [Google Scholar]
- Viemari JC, Bos R, Boulenguez P, Brocard C, Brocard F, Bras FL, Coulon P, Liabeuf S, Pearlstein E, Sadlaoud K, Stil A, Tazerati S, Vinay L, 2011. Importance of chloride homeostasis in the operation of rhythmic motor networks. Prog. Brain Res 188, 3–14. [DOI] [PubMed] [Google Scholar]
- Wang W, Richerson GB, 1999. Development of chemosensitivity of rat medullary raphe neurons. Neurosci 90, 1001–1011. [DOI] [PubMed] [Google Scholar]
- Wardle RA, Poo MM, 2003. Brain-derived neurotrophic factor modulation of GABAergic synapses by postsynaptic regulation of chloride transport. J. Neurosci 23, 8722–8732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickström R, Hökfelt T, Lagercrantz H, 2002. Development of CO(2) response in the early newborn period in rat. Respir. Physiol. Neurobiol 132, 145–158. [DOI] [PubMed] [Google Scholar]
- Wong-Riley MTT, 1989. Cytochrome oxidase: an endogenous metabolic marker for neuronal activity. Trends Neurosci 12, 94–101. [DOI] [PubMed] [Google Scholar]
- Wong-Riley MTT, Liu Q, 2005. Neurochemical development of brain stem nuclei involved in the control of respiration. In Special Issue on “Development of Respiratory Control”. Bavis R and Carroll J (eds.). Respir. Physiol. Neurobiol 149, 83–98. [DOI] [PubMed] [Google Scholar]
- Wong-Riley MTT, Liu Q, 2008. Neurochemical and physiological correlates of a critical period of respiratory development in the rat. In Special Issue on “Neurochemistry of Respiratory Control”. McCrimmon D, Mitchell G, and Alheid G (eds.) Respir. Physiol. Neurobiol 164, 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong-Riley MTT, Liu Q, Gao X-P., 2013. Peripheral-central chemoreceptor interaction and the significance of a critical period in the development of respiratory control. Invited review. In Special Issue on “Development of the Carotid Body”. Carroll JL, Donnelly D, and Bairm A (eds.) Respir. Physiol. Neurobiol 185, 156–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii A, Constantine-Paton M, 2010. Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev. Neurobiol 70, 304–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Mu L, Wang D, Xia D, Salmon A, Liu Q, Wong-Riley MTT, 2018. Uncovering a critical period of synaptic imbalance during postnatal development of the rat visual cortex: role of brain-derived neurotrophic factor. J. Physiol. (Lond.) 59618, 4511–4536. [DOI] [PMC free article] [PubMed] [Google Scholar]