

Abstract
The targeting of mRNA and local protein synthesis is important for the generation and maintenance of cell polarity. As part of the translational machinery as well as an actin/microtubule-binding protein, elongation factor 1α (EF1α) is a candidate linker between the protein translation apparatus and the cytoskeleton. We demonstrate in this work that EF1α colocalizes with β-actin mRNA and F-actin in protrusions of chicken embryo fibroblasts and binds directly to F-actin and β-actin mRNA simultaneously in vitro in actin cosedimentation and enzyme-linked immunosorbent assays. To investigate the role of EF1α in mRNA targeting, we mapped the two actin-binding sites on EF1α at high resolution and defined one site at the N-terminal 49 residues of domain I and the other at the C-terminal 54 residues of domain III. In vitro actin-binding assays and localization in vivo of recombinant full-length EF1α and its various truncates demonstrated that the C terminus of domain III was the dominant actin-binding site both in vitro and in vivo. We propose that the EF1α–F-actin complex is the scaffold that is important for β-actin mRNA anchoring. Disruption of this complex would lead to delocalization of the mRNA. This hypothesis was tested by using two dominant negative polypeptides: the actin-binding domain III of EF1α and the EF1α-binding site of yeast Bni1p, a protein that inhibits EF1α binding to F-actin and also is required for yeast mRNA localization. We demonstrate that either domain III of EF1α or the EF1α-binding site of Bni1p inhibits EF1α binding to β-actin mRNA in vitro and causes delocalization of β-actin mRNA in chicken embryo fibroblasts. Taken together, these results implicate EF1α in the anchoring of β-actin mRNA to the protrusion in crawling cells.
INTRODUCTION
β-Actin mRNA is localized in the lamellae in chicken embryo fibroblasts (CEFs) and several other cell types (Lawrence and Singer, 1986; Hoock et al., 1991; Hill et al., 1994; Kislauskis et al., 1997). A 54-nucleotide sequence (the “zipcode”) in the 3′-untranslated region (3′-UTR) directs this localization of β-actin mRNA (Kislauskis et al., 1994). Delocalization of β-actin mRNA with antisense oligonucleotides complementary to the zipcode suppresses cell motility (Kislauskis et al., 1997). A recent more rigorous analysis of antisense-treated cells revealed that the delocalization of β-actin mRNA causes the loss of cell polarity and the polarity of actin polymerization (Shestakova et al., 2001).
There could be several reasons for suppression of cell motility and polarity upon delocalization of β-actin mRNA. Both require actin polymerization at the leading edge (Tilney et al., 1981; Wang, 1985; Chan et al., 1998). Cells with delocalized β-actin mRNA may not synthesize sufficient actin to maintain actin filament polymerization over a period of time longer than the duration of the initial protrusion. As a result, the cells would have a slower rate of migration. Alternatively, the site of actin synthesis (and of other functionally related proteins) may affect the location of nucleation of actin polymerization that would define the direction of protrusion and, therefore, polarity of movement. For instance, the translation of the mRNA only while it is at the leading edge of fibroblasts would result in the selective sorting of β-actin protein to this region. β-Actin has been suggested to be an important player in leading edge dynamics by virtue of its isoform-specific interaction with other proteins (such as ezrin and β-Cap73; Shuster et al., 1996).
The sorting of β-actin mRNA requires transport and then selective anchoring of the mRNA on the actin cytoskeleton in the lamella (Sundell and Singer, 1991). We have proposed that the mRNA is translated once it is anchored, to facilitate protein sorting (Kislauskis et al., 1993). For this anchoring component, a protein capable of actin binding as well as participating in protein synthesis would be the ideal candidate for a regulatory molecule(s), coordinating both the location and translation of the mRNA. In this work we present evidence that the protein translation elongation factor 1α (EF1α) has a role in β-actin mRNA anchoring.
The conventional role of EF1α during protein synthesis is to bind and transport aminoacyl-tRNA to the A site of the ribosome in a GTP-dependent mechanism. However, an increasing body of evidence suggests that, in addition to its role in peptide elongation, EF1α may have functions beyond translation (Durso and Cyr, 1994; Condeelis, 1995). One of these proposed unconventional functions for EF1α is its role in the regulation of dynamics of the cytoskeleton (Liu et al., 1996a). In addition to binding to GDP/GTP, aminoacyl-tRNA, EF1β, and the ribosome, EF1α binds and bundles actin and binds microtubules. Because EF1α is an abundant protein in most eukaryotic cells, and it binds to actin filaments with relatively high affinity, it could be a potent regulator of the dynamics of the cytoskeleton. Previous in vitro studies demonstrated that EF1α inhibits the rate of actin polymerization and stabilizes actin filaments (Murray et al., 1996). It has been proposed that, because EF1α cross-links actin filaments via a unique bonding rule that tends to exclude other F-actin cross-linking proteins (Owen et al., 1992), the resulting actin structure could be the scaffold for the transport and/or anchorage of mRNA (Liu et al., 1996a).
It is estimated that 70–80% of mRNA in the cell is associated with, but not bound directly to, microfilaments or microtubules (Lenk et al., 1977; Cervera et al., 1981; van Venrooij et al., 1981; Taneja et al., 1992; Bassell, 1993; Bassell et al., 1994). A linker is therefore required for the association of mRNAs to the cytoskeleton. Minimal qualifications for the linker candidate include 1) ability to bind to both the mRNAs and the cytoskeleton and 2) sufficient abundance for the large number of mRNAs. Several lines of evidence suggest that EF1α is a good linker candidate. EF1α is an abundant actin/microtubule-binding protein, which is in molar excess to the other components of the protein translation machinery (Condeelis, 1995). It is found to colocalize with poly(A) RNA and ribosomes on actin filaments in fibroblasts (Bassell et al., 1994). EF1α is also found in the mRNA particles that are transported on microtubules in oligodendrocytes and neurons (Barbarese et al., 1995; Carson et al., 1997; Bassell et al., 1998). The involvement of EF1α in mRNA localization is also suggested from in vivo studies. For example, Bni1p, a yeast protein that is involved in the asymmetric localization of ASH1 mRNA in the daughter cell (Long et al., 1997), has been identified as an EF1α-binding protein (Umikawa et al., 1998). The EF1α-binding site (EBS) on Bni1p appears to be important for a normal actin cytoskeletal structure and cell polarity (Umikawa et al., 1998).
In this study, we investigated the direct interaction of EF1α with β-actin mRNA in vitro. To elucidate the relationship of binding to F-actin and to mRNA by EF1α, we have mapped the actin-binding sites on EF1α using a series of recombinant truncates of EF1α. We have investigated how these domains of EF1α are involved in F-actin binding and mRNA targeting in vitro and in vivo. Furthermore, we have used the EBS of Bni1p to study the consequences of disruption of the EF1α–F-actin interaction in vitro and in vivo. The results indicate that EF1α is involved in anchoring β-actin mRNA to F-actin in the protrusions of crawling cells.
MATERIALS AND METHODS
Tissue Culture
MTLn3 and MTC are cell lines derived from the same 13762NF rat mammary adenocarcinoma and were cultured according to the method of Segall et al. (1996). CEFs were isolated from 12-d chicken embryos and cultured as described by Kislauskis et al. (1993).
Construction of Expression Vectors for Glutathione S-Transferase (GST) Fusion Proteins
Constructs for expressing GST fusion proteins were made using a vector, pGEX-KG (Guan and Dixon, 1991). Each truncate was obtained by PCR using a pair of primers corresponding to each end of the desired sequence and inserted into the vector. For example, to make a construct to express GST–403–456 fusion protein (containing amino acid residues 403–456 of Dictyostelium EF1α), the fragment was obtained using primers GCG GAA TTC TAC CAA TGT GTG TTG AAT CA and CG CGA AGC TTA TTT CTT CTT TGA TGG AGC AGC and was inserted into the vector at EcoRI and HindIII sites. For GST-408-462 (containing amino acid residues 408–462 of rat EF1α), the fragment was obtained using primers CGG AAT TCA AAT GAA GCC CAT GTG TGT TGA G and CGA AGC TTC ATT TAG CCT TCT GAG CTT T and was inserted into the vector at EcoRI and HindIII sites. This strategy was used to make all of the GST fusion protein expression constructs that were used in this study. All of the expression constructs were validated by DNA sequencing.
Construction of Expression Vectors for EF1α-GFP Fusion Proteins
Constructs for expressing full-length or truncated EF1α as a green fluorescence protein (GFP) fusion protein were made by using an expression vector, pGL-1, originally from Life Technologies (Gaithersburg, MD). To express fusion proteins with GFP fused to the C terminus of EF1α or its truncate, this vector was modified by Dr. Jeffrey Segall (Albert Einstein College of Medicine, New York) by replacing a stop codon with a HindIII site 5′ of the GFP-coding sequence, resulting in pGL-H3. A cDNA sequence encoding rat EF1α was amplified from an expression construct for GST-EF1α fusion protein (a gift from Dr. Richard Stanley, Albert Einstein College of Medicine) by PCR using primers TCA GGA ATT CGA TTC AAA GCA AAA ATG and CTC GTC GAC CTT TAG CCT TCT GAG C for the full-length EF1α and primers TCA GGA ATT CGA TTC AAA GCA AAA ATG and GTT GAG CTC GCT TGC CAG GGA CCA T for truncate rat EF1α containing amino acid residues 1–408. The full-length and truncated rat EF1α sequences were cloned into the pGL-H3 at sites of EcoRI/SalI and EcoRI/SacI, respectively. The rat EF1α domain III (amino acids 330–462) truncate and EBS of yeast Bni1p were obtained similarly by PCR and cloned into the pEGFP-C1 expression vector (CLONTECH, Palo Alto, CA). These constructs were transformed into Escherichia coli cell XL1-Blue (Stratagene, La Jolla, CA) and selected with 100 μg/ml ampicillin or 25 μg/ml kanamycin. Colonies were screened by PCR with the corresponding pair of primers for each construct. Positive colonies were selected and the correct inserts were confirmed by DNA sequencing.
Transfection and Screening for GFP-positive Cells
Transfection of MTLn3 and MTC cells was performed by using LipofectAMINE according to the protocol of Life Technologies. The rat tumor cell lines were plated onto dishes (MaTTek, Ashland, MA) such that each dish contained 100,000 cells 24 h before the transfection (for 50–80% confluence at transfection). The constructs for expression, EF1α-GFP or EF1αΔC-GFP, were cotransfected with vector pSV7 containing the neomycin-resistant gene (kindly provided by Dr. Fishman, Albert Einstein College of Medicine) at a ratio of 10:1 at different doses ranging from 0.125 to 1.0 μg of total DNA per dish. Antibiotic resistance selection was started 72 h after transfection by using 0.8 mg/ml Geneticin (G418). Surviving cells were subsequently screened using fluorescence microscopy. Colonies that emitted green fluorescent light were marked and loop cloned with glass cloning cylinders (Sigma, St. Louis, MO) into Petri dishes, and this procedure was repeated until a stable green fluorescent light-emitting population was obtained. The GFP-EF1α-domain III (domain III) truncate was transiently expressed using LipofectAMINE Plus (Life Technologies) with 10–40% transfection efficiency. The expression of the corresponding GFP fusion proteins were validated by Western blots using antibodies against GFP (CLONTECH) and EF1α (Upstate Biotechnologies, Lake Placid, NY).
Transfection of CEF
Transfection of CEF for transient expression of GFP, GFP-EF1α-domain III, was performed with the Effectene Transfection Reagent kit from Qiagen (Valencia, CA) according to the manufacturer's recommended procedure. Plasmic DNA (1 μg) was used for each coverslip with CEFs in MEM without removing serum.
Immunofluorescence Staining and Fluorescence In Situ Hybridization (FISH)
To study β-actin mRNA localization in CEFs, FISH was performed as described by Kislauskis et al. (1994). For double probing of protein localization and mRNA, cells were first subjected to the normal process of indirect immunofluorescence staining using RNase-free reagents and RNase inhibitor. After secondary antibody binding and washes, the samples were fixed again using 3.7% paraformaldehyde to preserve the antibodies, followed by the normal FISH process. Both signals of FISH and immunostaining are compromised in such double-probed samples compared with either FISH or immunostaining alone. However, the general pattern of distribution of mRNA and protein in double-probed samples is comparable to that for single probing. Nevertheless, samples for either FISH or immunostaining were usually processed along with the double-probed sample to ensure the correct interpretation of signals from both mRNA and protein.
Cell Permeabilization and Fixation
The transfected MTLn3 cells were plated onto MatTek dishes precoated with rat tail collagen I (Collaborative Research, Bedford, MA) and cultured overnight. The cells were serum starved by incubation for 3 h in medium supplemented with 12 mM HEPES, pH 7.0, and 0.35% bovine serum albumin (BSA; MEMH) and then stimulated with 5 nM epidermal growth factor (EGF) to induce the extension of lamellipodia. To assess the actin cytoskeleton localization of EF1α-GFP fusion proteins, these cells were briefly permeabilized after ∼4 min of EGF stimulation to remove soluble cytoplasmic proteins with 100 μl of permeabilization buffer (20 mM 1,4-piperazinediethanesulfonic acid [PIPES], pH 6.5, with 30 mM KOH, 4 mM MgCl, and 10 mM EGTA, pH 6.5 with 20 mM KOH and 0.025% saponin). After 10 s of permeabilization at 37°C, the dishes were gently flooded with 2 ml of fixation buffer containing 3.7% formaldehyde, 5 mM PIPES, 1.1 mM Na2HPO4, 0.4 mM KH2PO4, 137 mM NaCl, 5 mM KCl, 4.0 mM NaHCO3, 2 mM MgCl2, 2 mM EGTA, and 5.5 mM glucose. After 5 min of fixation at 37°C, aldehyde autofluorescence was quenched with 0.1 M glycine for 10 min. The cells were blocked with 1% BSA and 1% fetal calf serum in TBS (20 mM Tris-HCl, pH 8.0, and 150 mM NaCl) and then incubated with rhodamine-conjugated phalloidin (50 nM plus 100 nM nonlabeled phalloidin) for 2 h or overnight. The transfected MTC cells were treated similarly except that they were not EGF stimulated before saponin permeabilization.
Fluorescence Microscopy
All samples were viewed with an Olympus (Tokyo, Japan) microscope equipped with a cooled CCD camera. Optical sections were obtained by deconvolution using IPLab computer software (Scanalytics, Fairfax, VA). Image processing was performed with NIH Image (version 1.6, National Institutes of Health, Bethesda, MD) and Photoshop (version 5.0; Adobe Systems, Mountain View, CA) software.
Actin Cosedimentation Assay
Actin cosedimentation was performed in sedimentation buffer containing 2 mM MgCl2, 1 mM ATP, 1 mM dithiothreitol (DTT), 15 mM KCl, 25 mM PIPES, and 2 mM EGTA (pH to 6.6 with 45 mM KOH). Results of testing indicated that actin isolated from Dictyostelium and rabbit muscle behaves similarly in actin cosedimentation assays (for native and GST fusion of Dictyostelium and rat EF1α, recombinant Dictyostelium EF1α domains I, II, and III). Therefore, rabbit actin was used in all actin-binding assays presented in this report. The tested proteins were mixed without (as control) or with preformed actin filaments and incubated at room temperature for 1 h. The mixtures were then centrifuged in an Airfuge (Beckman Coulter, Fullerton, CA) at 100,000 × g for 20 min. The amount of actin and tested protein in the supernatants and pellets were measured by SDS-PAGE and densitometry using Molecular Dynamics laser scanner and Image Quant software (Amersham Pharmacia Biotech, Piscataway, NJ). Most of the recombinant EF1α truncates are very soluble and showed only trace amounts in the pellet fraction after centrifugation in the absence of F-actin except truncate GST–49–221, which was fairly soluble but showed slightly more in the pellet than other truncates. The amount of the truncates in the pellet fraction in the absence of F-actin was subtracted from that in the presence of F-actin as the net binding. For measurement of β-actin mRNA binding to EF1α/F-actin bundles, EF1α or other actin-binding proteins were allowed to interact with F-actin at room temperature for 1 h. Biotin-labeled β-actin mRNA was then added and incubated for 1 h before being subjected to low-speed centrifugation at 50,000 × g for 2 min. Fractions of the reaction mixture before centrifugation and the supernatant after centrifugation were measured for biotin-labeled mRNA using the modified enzyme-linked immunosorbent assay (ELISA) as described in this report. Under the same conditions, the F-actin–bundling activity of rat liver EF1α, α-actinin (Cytoskeleton, Denver, CO), aprotinin (Sigma), fascin (gift from Dr. Fumio Matsumora), and fimbrin (gift from Dr. Paul Matsudaira) was confirmed. All of these tested proteins showed a single band on SDS-PAGE gel and pelleted with F-actin after low-speed centrifugation.
Protein Purification
Dictyostelium actin and EF1α, rabbit skeletal muscle actin, GST fusion proteins of Dictyostelium and rat EF1α, and corresponding truncations were prepared as described previously (Liu et al., 1996b). All of the GST fusion proteins were clarified by centrifugation at 320,000 × g at 4°C for 30 min and stored in storage buffer (10 mM PIPES, 0.05% NaN3, 0.1 mM EDTA, 1 mM DTT, and 25% glycerol, pH 7.0) on ice for the short term or under liquid nitrogen for the long term.
Affinity Purification of Domain-specific Antibodies
A recombinant fusion protein consisting of a Dictyostelium EF1α C-terminal actin-binding sequence (amino acid residues 403–456), dihydro-folate reductase, and a 6× His-tag was expressed in bacteria using an expression vector, pQE-40, from Qiagen. The fusion protein was purified using a Ni- nitrilotriacetic acid agarose bead affinity column. The fusion protein was injected into rabbits and the antiserum obtained was purified on a GST-EF1α truncate (amino acids 403–456) fusion protein affinity column using a standard procedure described previously (Bresnick et al., 1991).
Immunoprecipitation of Native Dictyostelium EF1α by Anti–C-Terminal Antibody
The affinity-purified C-terminal actin-binding site antibody was first incubated with protein-A beads (Amersham Pharmacia Biotech) in phosphate-buffered saline (PBS) containing 20 mM NaPO4, 300 mM NaCl, 0.1% of Tween 20, and 1 mg/ml BSA, pH7.5. Preimmune serum was used as a control. After 45 min at room temperature, the beads were washed four times with PBS and then were incubated with 0.5 μM Dictyostelium EF1α. After a 1-h incubation at room temperature, the reaction mixture was briefly centrifuged and the supernatant was obtained for SDS-PAGE. The beads were washed three times with PBS containing BSA and two times with PBS in the absence of BSA. The beads were finally boiled with gel sample buffer for SDS-PAGE. Because the heavy chain of rabbit immunoglobulin (Ig) G comigrates with Dictyostelium EF1α on SDS-PAGE gel, the amount of coprecipitated EF1α was detected by Western blotting using a chicken anti-rat EF1α peptide antibody that cross-reacts with Dictyostelium EF1α.
In Vitro Transcription of mRNA
A 1.8-kb full-length cDNA sequence of chicken β-actin was inserted into a pcDNA3 vector (Invitrogen, Carlsbad, CA) at the HindIII and XbaI sites. To linearize this plasmid for in vitro transcription, it was digested with XbaI, BamHI, or HindIII for full-length β-actin mRNA, the antisense mRNA of β-actin 3′-UTR, or the antisense mRNA of β-actin, respectively. The digested DNAs were separated by electrophoresis on a 1% agarose gel followed by purification using the Qiagen DNA extraction kit. These linearized cDNA sequences were then used as templates for in vitro transcription with an SP6 or T7 Maxiscript kit following the manufacturer's recommendations (Ambion, Austin, TX). mRNA transcripts were labeled with either biotin-16-UTP (Boehringer Mannheim, Indianapolis, IN) or [32P]α-CTP (Amersham Pharmacia Biotech). After biotin-labeling, unincorporated nucleotides were removed by using ProbeQuant G50 MicroColumns (Amersham Pharmacia Biotech). 32P-labeled RNA was purified on a 6% denaturing polyacrylamide gel containing 42% (wt/vol) urea. The position of the labeled RNA was identified by brief exposure of the gel to x-ray film. The band containing the labeled RNA was excised, and the RNA was eluted with elution buffer (1% SDS and 1 M ammonium acetate) overnight at 37°C. The RNA was then precipitated and washed with ethanol and redissolved in water.
Gel Mobility Shift Assay
Gel mobility shift was used to detect the RNA-protein interaction. Labeled chicken β-actin mRNA (10,000 cpm of 32P) was incubated with buffer or 5 μg of rabbit muscle actin or rat liver EF1α at room temperature for 20 min in 20 μl of reaction buffer (10 mM Tris-HCl, pH 7.4, 300 mM KCl, 1 mM DTT, 5 mM MgCl2, and 5% glycerol). To minimize nonspecific RNA-protein binding, heparin was added to a final concentration of 5 mg/ml and incubated for a further 10 min at room temperature. Samples were loaded on 3% native gel for electrophoresis. The gel was dried, and the position of the mRNA was identified by autoradiography.
ELISA to Detect the mRNA-EF1α Interaction
Because the ELISA assay can be used to analyze a large number of samples objectively and quickly, we used this assay for most of the characterization of the binding of EF1α to mRNA. The EF1α-mRNA interaction was analyzed by using a modified ELISA assay on a 96-well plate. Briefly, tested proteins were allowed to interact with F-actin to form complexes. The protein samples were then diluted and quickly distributed into the wells of the plate and incubated overnight at 4°C in assay buffer containing 100 mM KCl, 50 mM Tris-HCl, and 10 mM MgCl2, pH 7.4. These proteins were bound tightly and nonspecifically to the surface of the wells through charge and hydrophobic interactions. The wells were then washed five times with assay buffer and blocked with 1% BSA in assay buffer containing 0.05% Tween 20 for 30 min at room temperature. mRNA was added and incubated for 1 h at room temperature. After five washes with assay buffer, horseradish peroxidase-conjugated streptavidin (1:5000) was added and incubated for 30 min followed by 10 washes. Peroxidase substrate TMB (Kirkegaard and Perry Laboratories, Gaithersburg, MD) was added for color development for 10 min before the reaction was stopped by adding stop solution. The samples were read at a wavelength of 450 nm using an MRX Revelation Microplate Reader with Revelation software, version 4.0 (DYNEX Technologies, Chantilly, VA).
Light-scattering Assay
The effect of anti–C-terminal actin-binding site antibody (anti–403–456) on EF1α's cross-linking of actin filaments was analyzed using right angle light scattering as described previously (Liu et al., 1996b). Briefly, rabbit muscle G-actin was allowed to polymerize for 2 h at room temperature before it was used. Native Dictyostelium EF1α was incubated with IgGs for 1 h before being added to the F-actin solution in the cuvette, while the light scattering was being measured at 600 nm.
Electrophoresis and Western Blots
SDS-PAGE was performed according to the method of Laemmli (1970). Western blotting was performed according to the manufacturer's protocol using an ECL kit (Amersham Pharmacia Biotech).
Estimation of Binding Affinity
To estimate the binding affinity of EF1α truncates to F-actin, constant amounts of GST-EF1α recombinant proteins were allowed to interact with various amounts of F-actin. To measure the binding affinity of EF1α for mRNA, a constant amount of EF1α was used to interact with various amounts of biotin labeled β-actin mRNA. To estimate the apparent Kd, the binding titration data were graphed with Origin software (version 4.1, RockWare, Golden, CO) and then curve fitted by nonlinear least squares to a bimolecular binding isotherm according to the following expression (Hulme and Birdsall, 1992):
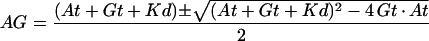 |
where AG is the concentration of bound ligand/receptor, At is the total concentration of ligand, and Gt is the total concentration of the receptor.
In addition, we have also used the GraphPad Prism software package (version 3.0; GraphPad Software, San Diego, CA) to analyze and curve fit the data for one- and two-site binding. The equations for one- and two-site binding are: Y = Bmax × X/(Kd + X) and Y = Bmax1 × X/(Kd1) + Bmax2 × X/(Kd2 + X), where Y is the total concentration of the receptor, X is the bound ligand, and Bmax is the maximal binding of the ligand.
RESULTS
EF1α Is Located in Cell Protrusions where mRNA Is Anchored
In unstimulated cell lines of Dictyostelium and rat carcinoma, EF1α is diffusely distributed (Dharmawardhane et al., 1991; Okazaki and Yumura, 1995; Edmonds et al., 1996). Interestingly, we observed in unstimulated primary CEFs that ∼60% (±12%) of the cells have EF1α in protrusions. Because this is also the region to which β-actin mRNA is localized, we double probed the CEFs for EF1α protein and β-actin mRNA to investigate the relationship of localization of EF1α protein and β-actin mRNA in the same cells. As shown in Figure 1, EF1α protein and β-actin mRNA were found to colocalize in protrusions. Because EF1α is an actin-binding protein and an elongation factor that is capable of binding to RNA (aminoacyl-tRNA), the above observations suggest that EF1α in F-actin–rich protrusions might interact directly with the β-actin mRNA. To ensure that EF1α binds to mRNA, a gel shift assay was used. As shown in Figure 2, EF1α can bind to β-actin mRNA as indicated by the formation of a protein-mRNA complex that has slower migration than the mRNA alone on the native gel.
Figure 1.

Colocalization of EF1α protein and β-actin mRNA in protrusions in unstimulated CEFs. (A–C) A representative cell in which EF1α protein and β-actin mRNA are colocalized at the cell protrusions. (A) EF1α protein; (B) β-actin mRNA; (C) superimposed signals of EF1α protein and β-actin mRNA to show colocalization. Bar, 10 μm.
Figure 2.

EF1α binds to chicken β-actin mRNA in a gel mobility shift assay. EF1α binds to the mRNA and forms a complex that exhibits shifted mobility on a 3% native gel. Lane 1, β-actin mRNA alone; lane 2, β-actin mRNA with rabbit actin; lane 3, β-actin mRNA with rat liver EF1α. Arrow indicates shifted band.
mRNA Binds to EF1α–F-Actin Bundles but Not to Other Types of Bundles Found In Vivo
To investigate whether EF1α, can also bind to β-actin mRNA when bound to F-actin, F-actin bundles formed with either EF1α or other actin-binding proteins were used in actin cosedimentation assays. As shown in Figure 3A, β-actin mRNA is able to bind to EF1α–F-actin bundles but not to fimbrin–F-actin bundles, suggesting that such binding is EF1α specific. A modified ELISA assay was also used to measure binding of mRNA to actin-bundling proteins. As shown in Figure 3, some of these actin-bundling proteins by themselves bind to the mRNA in the ELISA assay. We further tested the binding of mRNA to actin bundles formed using EF1α, α-actinin, aprotinin, fascin, and fimbrin. All of these bundling proteins are known to form F-actin bundles in vivo except aprotinin, which is found in serum. Aprotinin was included as a bundling protein with basic charge as a control to determine whether a basic protein was sufficient to confer mRNA-binding activity. As shown in Figure 3C, EF1α forms a bundle capable of binding to β-actin mRNA significantly above background unlike the other bundling proteins. The binding of the α-actinin/F-actin bundle to β-actin mRNA is barely detectable.
Figure 3.

mRNA binds to EF1α-F-actin but not to other types of bundles. (A) Actin cosedimentation. Either fimbrin or EF1α (1 μM) was incubated with 2 μM F-actin for 1 h at room temperature. Biotin-labeled β-actin mRNA (3 nM) was added and incubated for 1 h before centrifugation as described in MATERIALS AND METHODS. Bound mRNA has been normalized against F-actin in the pellets. Lane 1, F-actin; lane 2, fimbrin/F-actin; lane 3, EF1α/F-actin. Bars, SD (n = 3). (B) Actin-binding proteins interact with mRNA in the absence of actin measured by ELISA. Each tested protein (100 nM) was immobilized in the wells of a microplate and then blocked with BSA. Biotin-labeled β-actin mRNA (3 nM) was added and incubated. After washes, the bound mRNA was quantitated as described in MATERIALS AND METHODS. Lane 1, F-actin; lane 2, EF1α; lane 3, fimbrin; lane 4, fascin. (C) ELISA. Each sample contained 200 nM F-actin on a microplate well, with/without 100 nM tested protein. Biotin-labeled β-actin mRNA (3 nM) was used for each sample. Lane 1, F-actin alone; lane 2, aprotinin-F-actin; lane 3, α-actinin–F-actin; lane 4, fimbrin/F-actin; lane 5, fascin/F-actin; lane 6, EF1α–F-actin. Bars, SD (n = 3).
Binding of mRNA to EF1α Is Specific, Saturable, and of High Affinity
To further address the binding specificity and affinity of EF1α/F-actin bundle to β-actin mRNA, titration assays were performed. As illustrated in Figure 4, the binding of EF1α–F-actin to the mRNA is saturable and shows a dose-dependent relationship. Results of analyses of these data using GraphPad Prism software predict a two-site–binding reaction with an apparent Kd of ∼0.1 nM (±0.1 nM) and ∼3 nM (±3.4 nM) (Figure 4). Alternatively, a less-fit one-site–binding model predicts a Kd ∼ 0.3 nM (±0.05 nM). The binding stoichiometry cannot be determined because the number of EF1α molecules that are accessible in the F-actin bundles is not known. Interestingly, p50, a universal mRNA-binding protein that was also found to bind to F-actin (Ruzanov et al., 1999), was shown to bind to mRNA with a Kd of 25 nM and stoichiometry of one p50 to ∼20 nucleotides (Minich and Ovchinnikov, 1992). To further address the specificity of binding, competition assays were performed. Unlabeled mRNA was able to reduce the binding of labeled mRNA significantly, whereas poly(A) RNA failed to compete with mRNA binding to EF1α (Liu, Grant, Persky, Latham, Singer, and Condeelis, unpublished results).
Figure 4.

Binding of β-actin mRNA to EF1α is saturable and of high affinity. Complexes containing constant amounts of 10 nM EF1α with 20 nM F-actin were titrated with various amounts of biotin-labeled β-actin mRNA. The curve was fitted using Graphpad Prism software (version 3.0). The estimated apparent Kds were ∼0.1 nM (±0.1 nM) and ∼3 nM (±3.4 nM) based on a two-site–binding prediction. Bars, SD (n = 2).
The binding of EF1α–F-actin bundles to β-actin mRNA appears not to depend on the orientation of the mRNA because EF1α binds to β-actin mRNA regardless of whether it is in the sense or antisense orientation. In addition, the binding does not require the 3′-UTR of β-actin mRNA (Liu, Grant, Persky, Latham, Singer, and Condeelis, unpublished results), which has been shown to interact with other proteins required for mRNA targeting (Kislauskis et al., 1993). EF1α binding to β-actin mRNA was not affected by the guanine nucleotide-binding state of EF1α, pH, or salt concentration up to 200 mM KCl (Liu, Grant, Persky, Latham, Singer, and Condeelis, unpublished results).
Domain III Contains the Dominant Actin-binding Site of EF1α
The above data indicate that EF1α is able to bind to F-actin and β-actin mRNA simultaneously. Therefore, it is of interest to know which domains interact with actin and/or mRNA to further define the relationship of EF1α binding to the actin cytoskeleton and the mRNA. Previous studies indicated that there is probably more than one actin-binding site on Dityostelium EF1α (Liu et al., 1996b). EF1α is an evolutionarily conserved protein family that has >80% sequence homology among eukaryotes. We used Dictyostelum EF1α for binding studies related to EF1α of other eukaryotes because it could be readily purified in active form. We quantified the actin-binding affinity of two domains (I and III) that were previously shown to bind to F-actin. The results of the quantitative studies are shown in Figure 5. The titration curve of domain I indicates that the binding of domain I to F-actin is weak, with the curve-fitting software (see MATERIALS AND METHODS) predicting a one-site binding with apparent Kd ∼ 6 μM (±3 μM). In contrast to domain I, domain III binds to F-actin strongly, and the titration curve gives a Kd of 0.15 μM. These results indicate that domain III is the dominant actin-binding domain of EF1α.
Figure 5.

Determination of the F-actin-binding affinity of recombinant Dictyostelium EF1α domains I and III. Each recombinant protein (2 μM) was incubated with various amounts of F-actin at pH 6.6, and the amount of GST-fusion protein bound to F-actin was analyzed using actin cosedimentation assays. These data were graphed and curve fit as described in MATERIALS AND METHODS. The predicted Kds are ∼6 μM (±3 μM) and ∼0.15 μM (±0.04 μM) for domains I and domain III, respectively.
Mapping the Actin-binding Regions in Domains I and III at Higher Resolution
To delineate each of the actin-binding sites in domains I and III at higher resolution, we have expressed and purified a series of recombinant Dictyostelium EF1α truncates from bacteria. Under identical conditions, these truncates were tested for F-actin–binding activities using actin cosedimentation assays. Our mapping focused on domains I and III because these are the two domains that show actin-binding activities. We made a construct to express the N-terminal 49 residues of EF1α by taking advantage of a convenient HindIII restriction site at amino acid residue 49. As shown in Figure 6, this 49-amino acid segment of EF1α exhibited almost the same actin-binding ability as the whole domain I and that of a much bigger truncate EF1αΔC that contained ∼88% of the full-length EF1α but was missing the C-terminal 54 amino acids. Therefore, the 49 amino acids at the N terminus of EF1α represents the F-actin–binding activity of domain I.
Figure 6.

High-resolution mapping of the two actin-binding sites on Dictyostelium EF1α. (A) Representative SDS-PAGE results of actin cosedimentation assays of EF1α truncates. Each truncate (2 μM) was incubated with 4 μM F-actin and analyzed under conditions shown in Figure 4. SDS-PAGE gel images of recombinant full-length EF1α, domains I, II, and III, were previously described (Liu et al., 1996b). Arrowhead, GST-fusion proteins; *, actin; S, supernatant; p, pellet. Numbers under gels indicate the amino acid residues of Dictyostelium EF1α. Molecular mass standards are in kilodaltons. (B) Summary of actin-binding assays of recombinant Dictyostelium EF1α truncates. (n) is the number of each independent experiment and the error bars represent the SD. (C) Illustration of EF1α domains. Gray areas are the predicted actin-binding sites. Black shaded areas are consensus motifs for guanine nucleotide binding.
Interestingly, the actin-binding activity of domain III was localized to the 54 amino acids (403–456) in the extreme C terminus. This region was tested first in domain III because it contained the last two β-sheet structures at the C terminus (see Figure 12 in DISCUSSION) with many positively charged residues (predicted pI = 11.0). Although the actin-binding activity of these 54 residues was somewhat weaker than that of the intact domain III, it exhibited actin-binding activity above background, whereas another truncate representing two-thirds of the length of domain III (314–402) showed no significant actin-binding activity (Figure 6B). The reduced actin-binding activity of the C terminus of domain III (403–456) may be caused by the absence of several amino acid residues upstream of amino acid 403, which are required for maximal actin-binding activity of domain III. Alternatively, the maximal actin-binding activity presented by the whole domain III may require a certain conformation that is lacking in the short truncate of GST–403–456. Furthermore, the EF1αΔC truncate only exhibited an actin-binding activity similar to that of domain I, suggesting that the 54 residues at the C terminus were the essential actin-binding site in domain III.
Figure 12.

Two separated actin-binding sites are predicted to be on the surface of the EF1α molecule. The amino acid residues of the two actin-binding sites were displayed on a three-dimensional model of Dictyostelium EF1α that was constructed previously (Liu et al., 1996b). The three domains of the EF1α are assigned with blue for I, purple for II, and yellow for III. The N-terminal actin-binding site is assigned with red (for only those residues exposed on the surface: amino acids 1–10 and 33–49). The majority of the C-terminal actin-binding sites is on the surface and is in green. Left, a ribbon model and the three domains are displayed in a commonly used face. Right, an atomic model that is turned 180 degrees from the ribbon model on the right to clearly show the positions of the actin-binding sites.
The Actin-binding Sites Are on the Surface of EF1α
Because the proposed actin-binding sites were derived from in vitro data using recombinant truncations of EF1α, it was possible that sequences that were normally buried inside the molecule were exposed and the measured actin-binding activity represented in vitro artifacts. To exclude this possibility, we developed an antibody specific for the C-terminal actin-binding site. Using this antibody, we were able to completely inhibit binding of domain III to F-actin and to specifically immunoprecipitate the native Dictyostelium EF1α protein (Liu, Grant, Persky, Latham, Singer, and Condeelis, unpublished results). Furthermore, this antibody was able to disrupt the cross-linking activity of native Dictyostelium EF1α in a light-scattering assay by >60% (Figure 7). The residual light scattering in the presence of the antibody was likely caused by partial cross-linking of two EF1α molecules bound to actin filaments via domain I. Nonimmune IgG had no effect on cross-linking activity of the EF1α (Figure 7).
Figure 7.

Inhibition of Dictyostelium EF1α binding to F-actin by antibody against amino acids 403–456. Native Dictyostelium EF1α (1 μM) was incubated with or without 1 μM IgG in sedimentation buffer plus 50 μM GTP at pH 6.5 and room temperature for 1 h. The reaction mixture was then added to a F-actin solution (final 2 μM) in the cuvette. EF1α increases the light scattering of F-actin because it cross-links F-actin to form bundles. *, shutter open to add sample. Representative of two experiments.
Deletion of the C-Terminal Actin-binding Site of EF1α Is Correlated with Loss of Actin Binding In Vivo
The highly conservative nature of the EF1α family has made it feasible to predict actin-binding sites on rat EF1α based on the mapping results of actin-binding sites on Dictyostelium EF1α. In addition, we have prepared recombinant full-length domains I and III of rat EF1α and qualitatively confirmed their actin-binding activities in vitro using cosedimentation assays (Liu, Grant, Persky, Latham, Singer, and Condeelis, unpublished results). Based on sequence alignment, amino acid residues 408–462 of rat EF1α make up the corresponding actin-binding site on domain III. To study the importance of this site for actin binding in vivo, we expressed full-length or truncated rat EF1α as GFP fusion proteins in rat adenocarcinoma cells and confirmed expression by Western blotting with an antibody against GFP (Liu, Grant, Persky, Latham, Singer, and Condeelis, unpublished results). To reveal the cytoskeletal localization of these EF1α-GFP fusion proteins in vivo, we briefly permeabilized these cells with saponin to remove most of the soluble cytoplasmic proteins before fixation. Although GFP was seen diffusely distributed in the cytoplasm and in the nucleus in live and unpermeabilized cells, after saponin treatment, there is no cytoskeletal localization of the GFP (Figure 8). The nuclear localization of the GFP is probably a consequence of trapping. In contrast to the GFP localization, in cells expressing the full-length EF1α-GFP fusion protein, the green fluorescence signal was perinuclear (possibly the endoplasmic reticulum compartment) and at the cell edges (Figure 8D), where it was colocalized with F-actin as revealed by rhodamine-phalloidin staining (Figure 8C) indicating that the GFP-fusion protein was bound to cytoskeletal components. This is consistent with the endogenous EF1α localization on actin filaments observed in lightly permeabilized cells detected with anti-EF1α antibodies (Edmonds et al., 1996). It is worth noting that, although there were prominent stress fibers in these cells, there was no prominent colocalization of EF1α-GFP and the stress fibers, suggesting that EF1α-GFP selectively bound to the actin filaments in the cell edges as seen for endogenous EF1α (Figure 8D).
Figure 8.

EF1α-GFP and GFP-domain III, but not EF1αΔC-GFP, colocalize with actin filaments at the cell edge in rat adenocarcinoma cells. The transfected cells were briefly permeabilized with saponin before fixation. (A and B) Control cells expressing GFP; (C and D) Cells expressing full-length rat EF1α-GFP; (E and F) cells expressing EF1αΔC-GFP; (G and H) cells expressing GFP-domain III. (A, C, E, and G) Rhodamine-phalloidin staining for actin filaments. (B, D, F, and H) Light emitted from the GFP chromofluor. Both full-length EF1α and the domain III truncate colocalize with F-actin at the cell edge (arrowhead in C, D, G, and H) but not with stress fibers (arrows in C, D, G, and H). Bars, 10 μm.
To determine the contribution of the C-terminal 54 amino acids of domain III for the binding of EF1α to actin filaments in vivo, EF1α was truncated to remove its C-terminal 54 amino acids and then fused to GFP to create EF1αΔC-GFP. The pattern of intracellular localization of EF1αΔC-GFP was identical to that of GFP alone (in Figure 8, compare F and B). There was no EF1αΔC-GFP localized at the cell edge, and the majority of fluorescence is in the nucleus. Therefore, deletion of the C-terminal dominant actin-binding site on EF1α greatly reduced the ability of the truncated protein to bind to actin filaments in vivo. Furthermore, consistent with the in vitro actin-binding studies, the truncate domain III-GFP fusion protein exhibited localization with F-actin at the cell edge similar to that observed for the full-length EF1α (Figure 8H). These rat carcinoma cells generally exhibited a heterogeneous phenotype as previously shown (Edmonds et al,. 1996; Chan et al., 1998; Edmonds et al., 1999). Nonetheless, within this variety of morphologies, full-length EF1α-GFP and domain III-GFP showed similar localizations. These results indicated that the C-terminal actin-binding site was the dominant binding site in vivo and it was sufficient for localization of EF1α on F-actin to the cell edge.
Domain III of EF1α Inhibits the Binding of β-Actin mRNA to EF1α–F-Actin Bundles In Vitro and β-Actin mRNA Localization In Vivo
The mapping of two actin-binding sites and the demonstration that the C terminus of domain III is the dominant actin-binding site of EF1α have enabled us to study these sites further in terms of mRNA binding. To locate which domain of EF1α interacted with β-actin mRNA, we used the recombinant truncates of EF1α for an mRNA-binding study. Interestingly, both domains I and III, but not domain II, bound to the mRNA (Figure 9).
Figure 9.

Domains I and III, but not domain II of EF1α, bind to mRNA. In the absence of actin, the proteins were allowed to attach to the wells before BSA blocking. Each protein (100 nM) interacts with 3 nM biotin-labeled β-actin mRNA. Lane 1, GST-domain I; lane 2, GST-domain II; lane 3, GST-domain III; lane 4, GST. Bars, SD (n = 3).
Because domain III of EF1α was able to bind to β-actin mRNA, we tested whether domain III can compete with full-length EF1α for mRNA binding. As shown in Figure 10 (lanes 1 and 2), incubation of domain III with β-actin mRNA in microplate wells containing EF1α-F-actin bundles reduced the amount of the mRNA bound to EF1α-F-actin bundles, suggesting that domain III competes with full-length EF1α for binding to the mRNA. To test the effect of domain III of EF1α on β-actin mRNA localization in vivo, it was expressed in CEFs as a dominant negative to disrupt the interaction of EF1α with F-actin and the mRNA. As shown in Figure 11A, cells expressing GFP-domain III were inhibited in β-actin mRNA localization to protrusions compared with the untransfected cells. Expression of GFP protein alone in CEFs did not alter the normal level of β-actin mRNA localization, demonstrating that domain III of EF1α specifically causes delocalization of β-actin mRNA in the cells. Furthermore, this inhibitory effect caused by expression of GFP-domain III is significant because localization of the β-actin mRNA in these cells was suppressed to almost background level represented by the localizations of α-tubulin mRNA in normally cultured CEFs and β-actin mRNA in CEFs in serum-depleted culture (Figure 11A, lanes 5 and 6). Localization of β-actin mRNA has been known to depend on the presence of serum in cell culture (Latham et al. 1994), whereas α-tubulin mRNA was shown not to localize in cells (Shestakova et al., 1999).
Figure 10.

Both domain III of EF1α and EBS of Bni1p inhibit EF1α/F-actin binding to mRNA in vitro. Bundles containing 100 nM EF1α and 200 nM F-actin were immobilized in the wells of an ELISA plate (lanes 1–3). GST-EBS of Bni1p (100 nM, lane 4) or GST (100 nM, lane 5) was immobilized in the wells as controls. The wells were then blocked with BSA. After blocking, buffer alone (for lanes 1, 4, and 5) or 5 μM recombinant domain III of EF1α (lane 2) or GST-EBS of Bni1p (lane 3) was added and incubated for 1 h. Biotin-labeled β-actin mRNA (3 nM) was then added and processed as usual. Lanes 4 and 5 show that neither GST-EBS of Bni1p nor GST alone binds to the mRNA. Bars, SD (n = 3).
Figure 11.

Expression of GFP fusion of domain III of EF1α or EBS of Bni1p inhibits the localization of β-actin mRNA to the protrusions in vivo. Transfected cells were identified by their green fluorescence. Cells with concentrated mRNA in protrusions were scored as localized, and those with perinuclearly or uniformly distributed mRNA were scored as nonlocalized. The percentage of cells with localized mRNA was calculated for each experiment and summarized in A. Lane 1, β-actin mRNA in untransfected (275 cells); lane 2, β-actin mRNA in cells expressing GFP (231 cells); lane 3, β-actin mRNA in cells expressing GFP-domain III (308 cells); lane 4, β-actin mRNA in cells expressing GFP-EBS of Bni1p (349 cells); lane 5, α-tubulin mRNA (a nonlocalized mRNA) in untransfected cells (557 cells); lane 6, β-actin mRNA in untransfected but serum-starved cells (2460 cells). Bars, SD; lanes 1–3 and 5, n = 3; lane 4, n = 2; lane 6, n = 10. (B) Representative cells expressing GFP-EBS of Bni1p or GFP-domain III of EF1α have a uniform distribution of β-actin mRNA, whereas most of untransfected cells localize β-actin mRNA to protrusions. (a and c) GFP fluorescence indicating transfected cells (marked with asterisks); (b and d) β-actin mRNA (arrowheads indicate β-actin mRNA localization to the protrusions). Bar, 20 μm.
EBS of Bni1p Inhibits Binding of β-Actin mRNA to EF1α–F-Actin Bundles In Vitro and β-Actin mRNA Localization In Vivo
The above data suggested an anchoring role for EF1α in β-actin mRNA localization in the protrusions by the binding of EF1α to both F-actin and the mRNA simultaneously. The ability to anchor mRNA is proposed to require the assembly of a targeting scaffold composed of F-actin cross-linked by EF1α, a bivalent interaction. Inhibition of cross-linking of actin filaments by EF1α would prevent assembly of this scaffold and, therefore, is predicted to inhibit β-actin mRNA targeting. Bni1p has been shown to bind to EF1α and inhibit actin binding. In Bni1p, the EBS has been localized to a short sequence (amino acids 1328–1513) between the FH1 and FH2 domains. The sequence is sufficient to block F-actin bundling but not binding by EF1α (Umikawa et al., 1998). These results suggest that Bni1p, in the absence of RhoA, inhibited the interaction between EF1α and F-actin sufficiently to convert it from a bivalent to a monovalent interaction, which selectively disrupts EF1α-F-actin bundles in vivo. Thus, this action of the EBS of Bni1p is predicted to have the same consequences as the dominant negative action of domain III of EF1α. To test whether the interaction between Bni1p and EF1α can inhibit the binding of mRNA to the EF1α–F-actin bundle, we expressed the EBS of Bni1p as a GST fusion protein and confirmed its EF1α-binding ability in vitro (Liu, Grant, Persky, Latham, Singer, and Condeelis, unpublished results). This recombinant EBS was used in the ELISA mRNA-binding assay, where it inhibited binding of β-actin mRNA to EF1α–F-actin bundles (Figure 10, lane 3). A GFP-EBS fusion protein was then expressed in CEFs to determine its effect on anchoring of β-actin mRNA to the EF1α–F-actin compartment in protrusions (Figure 11). Compared with control cells either untransfected or expressing GFP alone, the number of cells with β-actin mRNA localized in protrusions was reduced significantly only in cells expressing the recombinant EBS of Bni1p.
DISCUSSION
The results of this study implicate EF1α as part of the molecular machinery for anchoring mRNA to actin filaments in vivo. The key properties of EF1α that contribute to this role as elucidated by this study are: 1) EF1α binds actin at the extreme N- and C-terminal regions of the protein, allowing one EF1α molecule to cross-link two actin filaments; 2) the C-terminal actin-binding site is required for the cytoskeletal location of EF1α in cell protrusions; 3) EF1α can bind to β-actin mRNA with high affinity even while bound to F-actin; 4) the localization of EF1α, and its binding to actin filaments in protrusions, is spatially correlated with the targeting of β-actin mRNA; and 5) EF1α binding to β-actin mRNA can be inhibited by EBS of Bni1p.
The Role of EF1α in Anchoring versus Transport of mRNA
The ability of EF1α to bind to F-actin and β-actin mRNA simultaneously indicates that EF1α can act as a linker between mRNA and the cytoskeleton to anchor mRNA at the cell cortex and protrusions. Because EF1α itself is an elongation factor, which is essential for protein synthesis, it is conceivable that eukaryotic cells have evolved to localize both EF1α and mRNA to sites where synthesis of a subset of functionally related proteins is essential for sustainable cell polarity. One example would be localized β-actin mRNA, where translation would supply β-actin protein near sites of polymerization. Because β-actin is the preferred isoform of actin in protrusions (Hoock et al., 1991), localized synthesis may be crucial to maintaining this distribution. However, the binding of EF1α to mRNA appears not to be selective for β-actin mRNA. Therefore, EF1α may act as a nonselective anchor for any mRNA that is asymmetric. The binding of EF1α to antisense RNA suggests that it may be the secondary structure and not the precise sequence that is recognized by EF1α. It is known that the stem loop secondary structures in the 3′-UTR of β-actin mRNA and in ASH1 mRNA are important for interaction with their corresponding binding proteins that are needed for mRNA localization (Oleynikov and Singer, 1998; Chartrand et al., 1999). Possibly EF1α recognizes only structured mRNAs.
Targeting of mRNA in vivo is the sum of its transport and anchoring. Anchoring is defined here as the ability to bind mRNA to actin filaments in the correct location in vivo (Sundell and Singer, 1991). Either the targeting or the anchoring requires a 3′-UTR zipcode (Kislauskis et al., 1993, 1994). The results of this study indicate that the binding of mRNA to the EF1α–F-actin complex does not require a 3′-UTR. These results also suggest that EF1α, when bound to actin filaments, can act as an anchor by binding to mRNA that is transported to the cell cortex and protrusions by other mechanisms such as myosin-mediated transport (Latham et al., 2001). We have observed that both domains I and III, but not domain II, bind to β-actin mRNA. Interestingly, although domain I binds to F-actin significantly more weakly than domain III, the mRNA-binding abilities of domains I and III appear similar. It is tempting to speculate that, because of their differences in binding to actin and mRNA, domain I may tend to bind to mRNA, whereas domain III prefers to bind to F-actin, thereby allowing EF1α to bind to (but not bundle) actin filaments and mRNA simultaneously.
The responsibility for selective localization of certain mRNAs in protrusions may result from the transport apparatus. Proteins such as ZBP-1, a 3′-UTR zipcode-binding protein in CEFs, may recognize specific mRNAs for transport to protrusions (Oleynikov and Singer, 1998) where they would be nonselectively anchored on the EF1α-F-actin complex. It is not clear whether EF1α is cotransported with β-actin mRNA or whether EF1α is localized in protrusions by an independent mechanism before the mRNA is transported there. In oligodendrocytes and neurons, mRNA is transported along microtubules in particles containing EF1α, suggesting that higher order structures may be involved in transport (Barbarese et al., 1995; Bassell et al., 1998). It is interesting to note that EF1α localizes with actin filaments in protrusions but not with stress fibers. In chicken fibroblasts, serum stimulation leads to rapid localization of β-actin mRNA at the cell front similar to EFIα (Latham et al., 1994). Electron microscopy studies have shown that EF1α molecules colocalize with mRNA on actin filaments (Bassell et al., 1994). The underlying mechanism as to why EF1α binds to the actin filaments in protrusions but not to the actin filaments in stress fibers remains to be elucidated. One possible explanation is that, because stress fibers are rich in actin-binding proteins such as α-actinin and fimbrin that have a hexagonal bundling rule, EF1α may be excluded from the stress fibers because of its unique bonding rule that favors square-packed bundles (Owen et al., 1992).
A Single EF1α Molecule Is Sufficient to Cross-link Two Actin Filaments
Bundling of actin filaments by EF1α has been observed by investigators using EF1α isolated from different sources. Kurasawa et al. (1996) proposed that F-actin is cross-linked by a dimer of EF1α in which each monomer contains one actin-binding site. Our previous and current data, however, consistently indicate that a single EF1α molecule can cross-link two actin filaments. There is no indication that EF1α forms an oligomer because analytical gel filtration and chemical cross-linking indicate that EF1α is a monomer in solution (Edmonds et al., 1998). In addition, electron microscopy studies of EF1α crossbridges in two-dimensional rafts of actin filaments indicate an interfilament spacing of 120 Å, in agreement with the measured radius of a single globular 50-kDa polypeptide (Edmonds et al., 1999). As demonstrated in this report, there are two separate actin-binding sites on each EF1α molecule, which can be clearly illustrated on a three-dimensional model of EF1α (Figure 12).
Although the actin-binding activity of domain I is relatively weak compared with domain III when it was analyzed alone as a recombinant truncate, it is possible that domain I may exhibit tighter actin-binding activity in the native protein because cooperativity takes place during F-actin cross-linking involving both domains I and III. This prediction is consistent with the ability of EF1α to cross-link actin filaments in vitro and in vivo but not lead to large bundles in the cell, unlike fimbrin for instance, which has two high-affinity–binding sites. This prediction is also in agreement with electron microscope analyses of fibroblasts and Dictyostelium cells in which only cross-linked filaments but not bundles were found in association with EF1α molecules in situ (Bassell et al., 1994; Liu et al., 1996a).
Regulation of the Binding of EF1α to β-Actin mRNA
The implication of EF1α in β-actin mRNA anchoring raises a question as to how the binding of EF1α to β-actin mRNA is regulated. Furthermore, the binding of EF1α to the open reading frame (ORF) of β-actin mRNA appears to conflict with the idea that the localized mRNA is translation competent. As mentioned in RESULTS, the binding of EF1α to β-actin mRNA appeared not to be affected by pH, the guanine nucleotide-binding state of EF1α, and KCl up to 200 mM, leaving the first question unanswered. Because the mRNA has to dissociate from EF1α to participate in translation, one can only speculate that, at some point by unknown mechanisms, EF1α may dissociate from the ORF and allow the ribosome to ride through it. Such translation-competent ORFs may lack the secondary structure that is required for EF1α binding. In the mean time, the 3′-UTR may be still tethered by unknown mechanisms, including binding to EF1α.
Regulation of the Binding of EF1α to Actin
A key issue is the mechanism by which the EF1α-F-actin interaction is regulated. We have shown previously that small changes in pH that are correlated with increases in protein synthesis in vivo cause the dissociation of the domain I actin-binding site on EF1α from F-actin to allow binding of aminoacyl-tRNA to EF1α (Liu et al., 1996b). Although a pH change probably potentiates the disassembly of EF1α–F-actin bundles to free EF1α for participation in protein synthesis, regulation by pH is suspected to lack the kind of specificity required for fine spatial and temporal control of mRNA anchoring that appears necessary for regulating the behavior of crawling cells (Kislauskis et al., 1997). However, an additional regulatory mechanism has been found that might provide finer spatial control of the EF1α–F-actin interaction. Downstream targets of RhoA include Bnilp in yeast, p140mDia in mammals, and diaphanous in Drosophila (Evangelista et al., 1997; Imamura et al., 1997; Watanabe et al., 1997). These proteins are members of the formin family and share conserved formin homology (FH) domains (Castrillon and Wasserman, 1994). The functions of these proteins are related to the regulation of cell polarity. Diaphanous is required for cytokinesis, Bni1p is concentrated at the tips of yeast mating projections and is required for normal bud growth, and p140mDia is enriched in the leading edge of crawling cells. Structural comparisons indicate that p140mDia, Bni1p, and diaphanous are the most highly related formins with more distantly related members, including cappuccino and formin (Watanabe et al., 1997). All contain FH1 and FH2 domains, and p140mDia, Bnilp, and diaphanous contain a RhoA-binding site at the N terminus. The FH1 domains of p140mDia (Watanabe et al., 1997) and Bnilp (Evangelista et al., 1997) are polyproline rich and bind profilin in vitro and may mediate colocalization of formins with profilin in vivo.
BNI1 deletion mutants in yeast show abnormal morphology and unpolarized distribution of cortical actin patches (Evangelista et al., 1997). Bni1p binds to EF1α, and the EBS on Bni1p has been identified (Umikawa et al., 1998). Deletion of the EBS from Bni1p results in a protein that fails to suppress the bni/bnr synthetic phenotype (e.g., temperature-sensitive growth and loss of polarity. Bnr1p is another formin protein with at least partial functional overlap with Bni1p). These results indicate that the interaction between EF1α and Bni1p is important for the function of Bni1p (Umikawa et al., 1998). Furthermore, the BNI1 mutant, she5, prevents bud localization of ASH1 mRNA localization coding for Ash1p (Long et al., 1997), an important regulator of mating-type switching (Jansen et al., 1996). These results indicate that the EF1α-Bni1p interaction is important for ASH1 mRNA localization and cell polarity. These results are reminiscent of a similar study in which overexpression of p140mDia in COS-7 cells resulted in the disassembly of F-actin bundles and loss of cell polarity (Watanabe et al., 1997). Finally, mammalian cells transfected with active but not with inactive RhoA localize β-actin mRNA more efficiently to the leading edge of crawling cells, whereas transfection with all other small G-proteins has little effect compared with controls (Latham et al., 2001). These results suggest that one downstream target of RhoA, the formins, could regulate mRNA targeting to the actin cytoskeleton by maintaining the stability of the EF1α-F-actin complex. These results and the conclusion are consistent with the EBS-Bni1p–mediated loss of β-actin mRNA localization to protrusions in CEFs in vivo and inhibition of binding of the mRNA to EF1α in vitro as reported in this study.
ACKNOWLEDGMENTS
We thank Mr. Michael Cammer, Mr. Jeffery Wyckoff, and Dr. Maryse Bailly for help with imaging techniques and tissue culture; Dr. Fumio Matsumora for fascin; Dr. Paul Matsudaira for fimbrin; Mr. Steve Braut for probe synthesis; the Analytical Imaging Facility of the Albert Einstein College of Medicine for use of light microscopes; Mr. Sergei Levin for help in manipulating the three-dimensional model of EF1α; and Miss Jamie Mandac for help with the development of the ELISA assays. This work was supported by grants to R.H.S. and J.C. from the National Institutes of Health.
Abbreviations used:
- BSA
bovine serum albumin
- CEF
chicken embryo fibroblast
- DTT
dithiothreitol
- EBS
EF1α-binding site
- EF1α
elongation factor 1α
- EGF
epidermal growth factor
- ELISA
enzyme-linked immunosorbent assay
- FH
formin homology
- FISH
fluorescence in situ hybridization
- GFP
green fluorescence protein
- GST
glutathione S-transferase
- Ig
immunoglobulin
- ORF
open reading frame
- PBS
phosphate-buffered saline
- PIPES
1,4-piperazinediethanesulfonic acid
- 3′-UTR
3′-untranslated region
Footnotes
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.01–03-0140. Article and publication date are at www.molbiolcell.org/cgi/10.1091/mbc.01–03-0140.
REFERENCES
- Barbarese E, Koppel DE, Deutscher MP, Smith CL, Ainger K, Morgan F, Carson JH. Protein translation components are colocalized in granules in oligodendrocytes. J Cell Sci. 1995;108:2781–2790. doi: 10.1242/jcs.108.8.2781. [DOI] [PubMed] [Google Scholar]
- Bassell GJ. High resolution distribution of mRNA within the cytoskeleton. J Cell Biochem. 1993;52:127–133. doi: 10.1002/jcb.240520203. [DOI] [PubMed] [Google Scholar]
- Bassell GJ, Powers CM, Taneja KL, Singer RH. Single mRNAs visualized by ultrastructural in situ hybridization are principally localized at actin filament intersections in fibroblasts. J Cell Biol. 1994;126:863–876. doi: 10.1083/jcb.126.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassell GJ, Zhang H, Byrd AL, Femino AM, Singer RH, Taneja KL, Lifshitz LM, Herman IM, Kosik KS. Sorting of beta-actin mRNA and protein to neurites and growth cones in culture. J Neurosci. 1998;18:251–265. doi: 10.1523/JNEUROSCI.18-01-00251.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresnick AR, Janmey PA, Condeelis J. Evidence that a 27-residue sequence is the actin-binding site of ABP-120. J Biol Chem. 1991;266:12989–12993. [PubMed] [Google Scholar]
- Carson JH, Worboys K, Ainger K, Barbarese E. Translocation of myelin basic protein mRNA in oligodendrocytes requires microtubules and kinesin. Cell Motil Cytoskeleton. 1997;38:318–328. doi: 10.1002/(SICI)1097-0169(1997)38:4<318::AID-CM2>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Castrillon DH, Wasserman SA. Diaphanous is required for cytokinesis in Drosophila and shares domains of similarity with the products of the limb deformity gene. Development. 1994;120:3367–3377. doi: 10.1242/dev.120.12.3367. [DOI] [PubMed] [Google Scholar]
- Cervera M, Dreyfuss G, Penman S. Messenger RNA is translated when associated with the cytoskeletal framework in normal and VSV-infected HeLa cells. Cell. 1981;23:113–120. doi: 10.1016/0092-8674(81)90276-2. [DOI] [PubMed] [Google Scholar]
- Chan AY, Raft S, Bailly M, Wyckoff JB, Segall JE, Condeelis JS. EGF stimulates an increase in actin nucleation and filament number at the leading edge of the lamellipod in mammary adenocarcinoma cells. J Cell Sci. 1998;111:199–211. doi: 10.1242/jcs.111.2.199. [DOI] [PubMed] [Google Scholar]
- Chartrand P, Meng XH, Singer RH, Long RM. Structural elements required for the localization of ASH1 mRNA and of a green fluorescent protein reporter particle in vivo. Curr Biol. 1999;9:333–336. doi: 10.1016/s0960-9822(99)80144-4. [DOI] [PubMed] [Google Scholar]
- Condeelis J. Elongation factor 1 alpha, translation and the cytoskeleton. Trends Biochem Sci. 1995;20:169–170. doi: 10.1016/s0968-0004(00)88998-7. [DOI] [PubMed] [Google Scholar]
- Dharmawardhane S, Demma M, Yang F, Condeelis J. Compartmentalization and actin binding properties of ABP-50: the elongation factor-1 alpha of Dictyostelium. Cell Motil Cytoskeleton. 1991;20:279–288. doi: 10.1002/cm.970200404. [DOI] [PubMed] [Google Scholar]
- Durso NA, Cyr RJ. Beyond translation: elongation factor-1alpha and the cytoskeleton. Protoplasma. 1994;180:99–105. [Google Scholar]
- Edmonds BT, Bell A, Wyckoff J, Condeelis J, Leyh TS. The effect of F-actin on the binding and hydrolysis of guanine nucleotide by Dictyostelium elongation factor 1A. J Biol Chem. 1998;273:10288–10295. doi: 10.1074/jbc.273.17.10288. [DOI] [PubMed] [Google Scholar]
- Edmonds BT, Liu G, Condeelis J. Eukaryotic elongation factor 1A (ABP-50) In: Kreis T, Vale R, editors. Guidebook to the Cytoskeletal and Motor Proteins. New York: Oxford University Press; 1999. [Google Scholar]
- Edmonds BT, Wyckoff J, Yeung YG, Wang Y, Stanley ER, Jones J, Segall J, Condeelis J. Elongation factor-1 alpha is an overexpressed actin binding protein in metastatic rat mammary adenocarcinoma. J Cell Sci. 1996;109:2705–2714. doi: 10.1242/jcs.109.11.2705. [DOI] [PubMed] [Google Scholar]
- Evangelista M, Blundell K, Longtine MS, Chow CJ, Adames N, Pringle JR, Peter M, Boone C. Bni1p, a yeast formin linking cdc42p and the actin cytoskeleton during polarized morphogenesis. Science. 1997;276:118–122. doi: 10.1126/science.276.5309.118. [DOI] [PubMed] [Google Scholar]
- Guan KL, Dixon JE. Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal Biochem. 1991;192:262–267. doi: 10.1016/0003-2697(91)90534-z. [DOI] [PubMed] [Google Scholar]
- Hill MA, Schedlich L, Gunning P. Serum-induced signal transduction determines the peripheral location of beta-actin mRNA within the cell. J Cell Biol. 1994;126:1221–1229. doi: 10.1083/jcb.126.5.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoock TC, Newcomb PM, Herman IM. Beta actin and its mRNA are localized at the plasma membrane and the regions of moving cytoplasm during the cellular response to injury. J Cell Biol. 1991;112:653–664. doi: 10.1083/jcb.112.4.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme EC, Birdsall NJM. Receptor-Ligand Interactions: A Practical Approach. New York: Oxford University Press; 1992. Strategy and tactics in receptor-binding studies. [Google Scholar]
- Imamura H, Tanaka K, Hihara T, Umikawa M, Kamei T, Takahashi K, Sasaki T, Takai Y. Bni1p and Bnr1p: downstream targets of the Rho family small G-proteins which interact with profilin and regulate actin cytoskeleton in Saccharomyces cerevisiae. EMBO J. 1997;16:2745–2755. doi: 10.1093/emboj/16.10.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen RP, Dowzer C, Michaelis C, Galova M, Nasmyth K. Mother cell-specific HO expression in budding yeast depends on the unconventional myosin myo4p and other cytoplasmic proteins. Cell. 1996;84:687–697. doi: 10.1016/s0092-8674(00)81047-8. [DOI] [PubMed] [Google Scholar]
- Kislauskis EH, Li Z, Singer RH, Taneja KL. Isoform-specific 3′-untranslated sequences sort alpha-cardiac and beta-cytoplasmic actin messenger RNAs to different cytoplasmic compartments. J Cell Biol. 1993;123:165–172. doi: 10.1083/jcb.123.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kislauskis EH, Zhu X, Singer RH. Sequences responsible for intracellular localization of beta-actin messenger RNA also affect cell phenotype. J Cell Biol. 1994;127:441–451. doi: 10.1083/jcb.127.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kislauskis EH, Zhu X, Singer RH. beta-Actin messenger RNA localization and protein synthesis augment cell motility. J Cell Biol. 1997;136:1263–1270. doi: 10.1083/jcb.136.6.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurasawa Y, Hanyu K, Watanabe Y, Numata O. F-actin bundling activity of Tetrahymena elongation factor 1 alpha is regulated by Ca2+/calmodulin. J Biochem (Tokyo) 1996;119:791–798. doi: 10.1093/oxfordjournals.jbchem.a021309. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural protein during the assembly of the head of bateriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Latham VM, Jr, Kislauskis EH, Singer RH, Ross AF. Beta-actin mRNA localization is regulated by signal transduction mechanisms. J Cell Biol. 1994;126:1211–1219. doi: 10.1083/jcb.126.5.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latham VM, Yu EH, Tullio AN, Adelstein RS, Singer RH. A Rho-dependent signaling pathway operating through myosin localizes beta-actin mRNA in fibroblasts. Curr Biol. 2001;11:1010–1016. doi: 10.1016/s0960-9822(01)00291-3. [DOI] [PubMed] [Google Scholar]
- Lawrence JB, Singer RH. Intracellular localization of messenger RNAs for cytoskeletal proteins. Cell. 1986;45:407–415. doi: 10.1016/0092-8674(86)90326-0. [DOI] [PubMed] [Google Scholar]
- Lenk R, Ransom L, Kaufmann Y, Penman S. A cytoskeletal structure with associated polyribosomes obtained from HeLa cells. Cell. 1977;10:67–78. doi: 10.1016/0092-8674(77)90141-6. [DOI] [PubMed] [Google Scholar]
- Liu G, Edmonds BT, Condeelis J. pH, EF1α and the actin cytoskeleton. Trends Cell Biol. 1996a;6:168–171. doi: 10.1016/0962-8924(96)20013-3. [DOI] [PubMed] [Google Scholar]
- Liu G, Tang J, Edmonds BT, Murray J, Levin S, Condeelis J. F-actin sequesters elongation factor 1alpha from interaction with aminoacyl-tRNA in a pH-dependent reaction. J Cell Biol. 1996b;135:953–963. doi: 10.1083/jcb.135.4.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long RM, Singer RH, Meng X, Gonzalez I, Nasmyth K, Jansen RP. Mating type switching in yeast controlled by asymmetric localization of ASH1 mRNA. Science. 1997;277:383–387. doi: 10.1126/science.277.5324.383. [DOI] [PubMed] [Google Scholar]
- Minich WB, Ovchinnikov LP. Role of cytoplasmic mRNP proteins in translation. Biochimie. 1992;74:477–483. doi: 10.1016/0300-9084(92)90088-v. [DOI] [PubMed] [Google Scholar]
- Murray JW, Edmonds BT, Liu G, Condeelis J. Bundling of actin filaments by elongation factor 1 alpha inhibits polymerization at filament ends. J Cell Biol. 1996;135:1309–1321. doi: 10.1083/jcb.135.5.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki K, Yumura S. Differential association of three actin-bundling proteins with microfilaments in Dictyostelium amoebae. Eur J Cell Biol. 1995;66:75–81. [PubMed] [Google Scholar]
- Oleynikov, Singer RH. ZBP1: a novel highly conserved RNA-binding protein involved in the localization of beta-actin mRNA. Mol Biol Cell. 1998;9:189a. [Google Scholar]
- Owen CH, DeRosier DJ, Condeelis J. Actin crosslinking protein EF-1α of Dictyostelium discoideum has a unique bonding rule that allows square-packed bundles. J Struct Biol. 1992;109:248–254. doi: 10.1016/1047-8477(92)90037-b. [DOI] [PubMed] [Google Scholar]
- Ruzanov PV, Evdokimova VM, Korneeva NL, Hershey JW, Ovchinnikov LP. Interaction of the universal mRNA-binding protein, p50, with actin: a possible link between mRNA and microfilaments. J Cell Sci. 1999;112:3487–3496. doi: 10.1242/jcs.112.20.3487. [DOI] [PubMed] [Google Scholar]
- Segall JE, Tyerech S, Boselli L, Masseling S, Helft J, Chan A, Jones J, Condeelis J. EGF stimulates lamellipod extension in metastatic mammary adenocarcinoma cells by an actin-dependent mechanism. Clin Exp Metastasis. 1996;14:61–72. doi: 10.1007/BF00157687. [DOI] [PubMed] [Google Scholar]
- Shestakova EA, Singer RH, Condeelis J. The physiological significance of β-actin mRNA localization in determining cell polarity, and directional motility. Proc Natl Acad Sci USA. 2001;98:7045–7050. doi: 10.1073/pnas.121146098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuster CB, Lin AY, Nayak R, Herman IM. Beta cap73: a novel beta actin-specific binding protein. Cell Motil Cytoskeleton. 1996;35:175–187. doi: 10.1002/(SICI)1097-0169(1996)35:3<175::AID-CM1>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Sundell CL, Singer RH. Requirement of microfilaments in sorting of actin messenger RNA. Science. 1991;253:1275–1277. doi: 10.1126/science.1891715. [DOI] [PubMed] [Google Scholar]
- Taneja KL, Lifshitz LM, Fay FS, Singer RH. Poly(A) RNA codistribution with microfilaments: evaluation by in situ hybridization and quantitative digital imaging microscopy. J Cell Biol. 1992;119:1245–1260. doi: 10.1083/jcb.119.5.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilney LG, Bonder EM, DeRosier DJ. Actin filaments elongate from their membrane-associated ends. J Cell Biol. 1981;90:485–494. doi: 10.1083/jcb.90.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umikawa M, Tanaka K, Kamei T, Shimizu K, Imamura H, Sasaki T, Takai Y. Interaction of Rho1p target Bni1p with F-actin-binding elongation factor 1alpha: implication in Rho1p-regulated reorganization of the actin cytoskeleton in Saccharomyces cerevisiae. Oncogene. 1998;16:2011–2016. doi: 10.1038/sj.onc.1201724. [DOI] [PubMed] [Google Scholar]
- van Venrooij WJ, Sillekens PT, van Eekelen CA, Reinders RJ. On the association of mRNA with the cytoskeleton in uninfected and adenovirus-infected human KB cells. Exp Cell Res. 1981;135:79–91. doi: 10.1016/0014-4827(81)90301-3. [DOI] [PubMed] [Google Scholar]
- Wang YL. Exchange of actin subunits at the leading edge of living fibroblasts: possible role of treadmilling. J Cell Biol. 1985;101:597–602. doi: 10.1083/jcb.101.2.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Madaule P, Reid T, Ishizaki T, Watanabe G, Kakizuka A, Saito Y, Nakao K, Jockusch BM, Narumiya S. p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J. 1997;16:3044–3056. doi: 10.1093/emboj/16.11.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]