

Abstract
Macrophages promote both injury and repair following myocardial infarction, but discriminating functions within mixed populations remains challenging. Here we used fate mapping and single-cell transcriptomics to demonstrate that at steady state, TIMD4+LYVE1+MHC-IIloCCR2− resident cardiac macrophages self-renew with negligible blood monocyte input. Monocytes partially replaced resident TIMD4−LYVE1−MHC-IIhiCCR2− macrophages and fully replaced TIMD4−LYVE1−MHC-IIhiCCR2+ macrophages, revealing a hierarchy of monocyte contribution to functionally distinct macrophage subsets. Ischemic injury reduced TIMD4+ and TIMD4− resident macrophage abundance within infarcted tissue while recruited, CCR2+ monocyte-derived macrophages adopted multiple cell fates, including those nearly indistinguishable from resident macrophages. Despite this similarity, inducible depletion of resident macrophages using a Cx3cr1-based system led to impaired cardiac function and promoted adverse remodeling primarily within the peri-infarct zone, highlighting a non-redundant, cardioprotective role of resident cardiac macrophages. Lastly, we demonstrate the ability of TIMD4 to be used as a durable lineage marker of a subset of resident cardiac macrophages.
Ischemic cardiovascular disease remains the leading cause of mortality worldwide1. After a myocardial infarction, an inflammatory response is directed to the site of injury resulting in structural and biochemical changes that can lead to adverse cardiac remodeling and dysfunction, manifesting clinically as heart failure2. Macrophages are specialized mononuclear phagocytes that reside in all tissues from the earliest stages of development, with particularly important roles in the response to injury3, 4, 5, 6. The mammalian neonatal heart has a remarkable capacity to fully regenerate after injury7. Clodronate liposomal depletion of neonatal macrophages prevents regeneration, in part through macrophage specific expression of growth factors, such as IGF-1 and other mechanisms yet to be characterized8, 9, 10. Following ischemic injury in adult mice, both excessive numerical expansion of macrophages8, 11 and non-selective depletion of macrophages12 limits cardiac repair through excessive inflammatory tissue damage and impaired scar formation, respectively. Macrophage composition in tissue is both heterogeneous in steady state and highly dynamic after injury. Within the adult heart, the majority of resident cardiac macrophages originate from embryonic development, both from early E8.5 erythromyeloid progenitors (EMP), and also later from fetal monocytes5. The relative contribution of EMP-derived fetal liver monocytes and emerging hematopoietic stem cell (HSC)-derived monocytes to the adult CD64+ cardiac macrophage pool is not known. However after birth, the slow recruitment of invading monocyte-derived macrophages from circulation leads to competition for tissue residency, and it is not clear whether monocyte-independent populations remain13.
Circulating monocytes express CCR2, a chemokine receptor important for migration14, and are enriched in components of the NLRP3 inflammasome. After tissue injury CCR2+ monocytes and macrophages can produce IL-1β5, a pro-inflammatory cytokine that promotes cardiovascular disease15. This is in contrast to resident CCR2− macrophages, which are primarily of embryonic origin and promote angiogenesis and cardiomyocyte proliferation, similar to neonatal macrophages8, 9, 16. However, beyond these observations, a more detailed understanding of cardiac macrophage heterogeneity is lacking.
Here, we investigated cardiac macrophage heterogeneity at steady state, plasticity post-infarct and the role of resident macrophages in repair. Through the combined use of genetic fate mapping, long-term parabiosis studies and single-cell RNA-sequencing (scRNA-seq) we observed that the healthy adult myocardium contains four populations of macrophages, including a subset that is maintained independent of blood monocytes (TIMD4+LYVE1+MHC-IIloCCR2−), a subset that is partially replaced by monocytes (TIMD4−LYVE1−MHC-IIhiCCR2−) and two CCR2+MHC-IIhi subsets that are fully replaced by monocytes. Ischemic injury induced tremendous diversification of these macrophage subsets. Post-myocardial infarction, the phosphatidylserine receptor TIMD417 and CCR2 emerged as mutually exclusive markers of resident and recruited macrophages, respectively. Resident macrophage abundance within the infarct zone was markedly reduced post infarct, and slowly increased through in situ proliferation. Depletion of diphtheria toxin receptor labeled, resident (Cx3cr1 expressing) macrophages promoted adverse cardiac remodeling primarily in the peri-infarct zone, a vulnerable region surrounding infarct tissue. Despite the ability of a small portion of recruited, monocyte-derived macrophages to assume a nearly identical transcriptional fate to the resident macrophage populations, resident cardiac macrophages possessed non-redundant, spatially localized cardioprotective functions.
Results
Single cell analyses reveal unique cardiac macrophage subsets
Flow cytometric analysis of cardiac CD45+CD64+CD11b+ macrophages revealed a single CCR2–MHC−IIlo macrophage subset in neonatal mice (P14), while in adult mice (>12 weeks) we detected additional CCR2+MHC-IIhi and CCR2–MHC-IIhi macrophage populations (Fig. 1a). To quantitate monocyte dependence we paired Ccr2−/− mice (Rosa26+/+), which lack circulating Ly6Chi monocytes to Ccr2+/+ mice, which carried a constitutively expressed membrane-localized TdTomato (Td) reporter gene in the Rosa26 locus (Rosa26mTmG/mTmG; R26 mTmG), for 6 months. Cardiac tissue from the Ccr2−/− mice indicated that most CCR2+ macrophages (>80%) and a smaller percentage of MHC-IIhi macrophages (~25%) within the heart were replaced with macrophages derived from Ccr2+/+ (Td+) monocytes in circulation, while MHC-IIlo macrophages had little replacement (~12% Td+; Fig. 1b).
Figure 1: Single cell RNA sequencing reveals distinct cardiac macrophage and dendritic cell subsets at steady state.
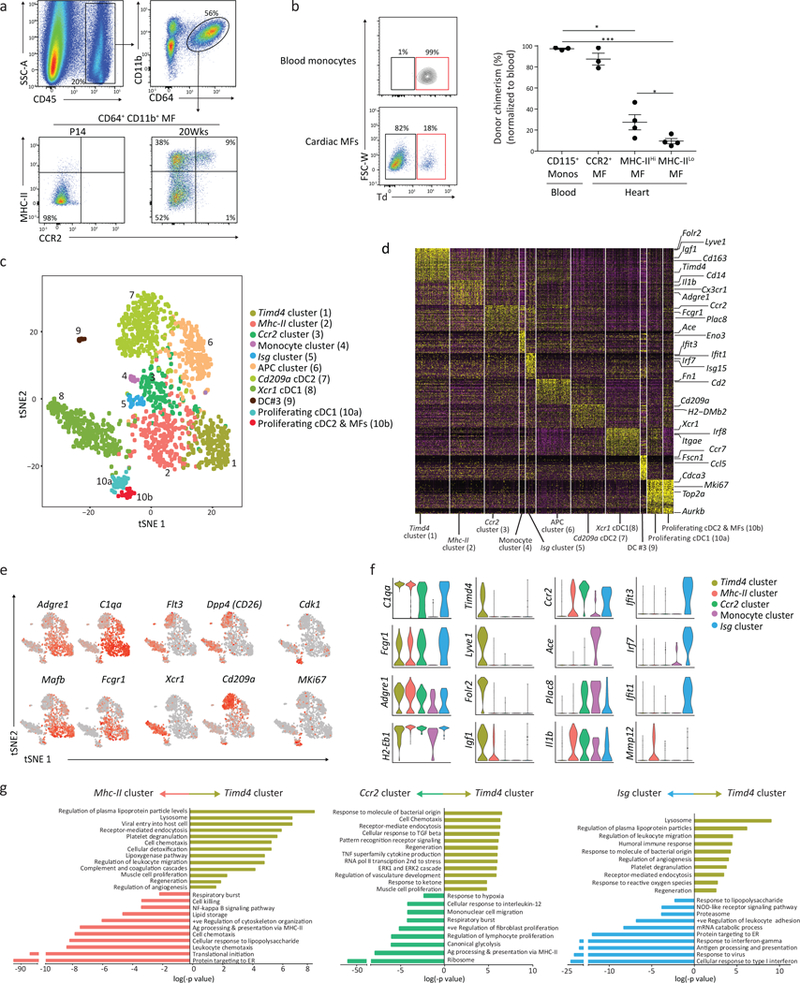
a) Representative flow plots of the gating scheme used to identify total cardiac macrophages (Live, CD45+CD64+CD11b+) were further parsed based on expression of CCR2 and MHC-II from neonate (P14) and adult (20 week old) mice; repeated independently 5 times with similar results. b) Parabiosis was performed on 8 week old recipient Ccr2−/− mice that were paired with R26mTmG/mTmG:Ccr2+/+ donor mice for 6 months. Donor chimerism was determined in blood CD115+ CD11b+ monocytes and CD64+CD11b+cardiac macrophages in the recipient mouse (Ccr2−/−). Chimerism (% Td+) was quantified and normalized to blood monocyte chimerism (graph; right). N=3,3,4,4. * p<0.05, ***p<0.001; two-tailed Student’s t test; center value, mean; error bars, SEM; repeated twice. c) Sorted cardiac CD45+CD64+CD11b+ macrophages and CD45+CD64−CD11c+MHC-IIlo-hi DCs from adult mice were pooled in a 1:1 ratio, transcriptomic analysis on 1780 individual cells was performed using the 10x Genomics platform. t-SNE dimensionality reduction analysis identified 11 major clusters. d) Heatmap of the 65 most differentially expressed genes in each cluster from Fig 1c. e) Feature plots depicting single cell gene expression of individual genes. f) Violin plots of cluster defining genes (cluster 1–5). g) Differentially expressed genes in Timd4 cluster were compared to the Mhc-II cluster, the Ccr2 cluster and the Isg cluster using gProfiler. Pathway enrichment is expressed as the log(-p value) – adjusted for multiple comparison.
The heart contains heterogeneous populations of conventional dendritic cells (cDC1 and cDC2)18, 19, which can express macrophage markers. To characterize mononuclear phagocyte heterogeneity we sorted cardiac CD45+CD64+CD11b+ macrophages and CD45+CD64−CD11c+MHC-IIlo-hi DCs from adult mice, pooled them in a 1:1 ratio (Supplementary Fig. 1a) and performed scRNA-seq on 1780 cells using the 10x Genomics platform. t-SNE dimensionality reduction analysis identified 11 major macrophage and DC clusters (Fig. 1c-e). To validate cluster identity we used the top ~30 cluster defining genes (Fig. 1d) to generate a similarity score to all mononuclear phagocyte populations found in the ImmGen20 data repository (Supplementary Fig. 1b). We confirmed 4 macrophage clusters (cluster 1, 2, 3 and 5), one monocyte cluster (cluster 4), two cDC clusters (CD209a+ cDC2, cluster 9 and XCR1+cDC1, cluster 8) and one cluster of DCs (DC#3, cluster 9) resembling DCs found in secondary lymphoid organs. Macrophages and DCs in cell cycle were defined by cell cycle specific genes21 in a sub-cluster containing cDC1s (cluster 10a) and a mixed sub-cluster containing both macrophages and DCs (cluster 10b). Lastly, we observed one myeloid antigen presenting cell (APC) cluster (cluster 6) that expressed some macrophage and DC genes (Adgre1, C1qc, C1qa, MHC-II genes, Itgax) but not lineage defining genes (Fcgr1, Maf, Mertk, Dpp4, Flt3, Zbtb46)22, 23; nor did this cluster have high expression of pDC genes (Siglech, Bst2, Ly6c2, Irf8) (Fig. 1d, Fig. Supplementary Fig. 1b,c). Of the macrophage clusters, cluster 1 expressed genes such as Timd4, Lyve1 and Igf1, and was the most transcriptionally unique macrophage cluster (termed Timd4 cluster hereafter; Fig. 1f). Cluster 2 had relatively higher expression of antigen presentation genes and correlated with CCR2–MHC-IIhi macrophages (termed Mhc-II cluster). Macrophages in cluster 3 were identified as CCR2+ macrophages (termed Ccr2 cluster). Cluster 5 was enriched in numerous interferon-stimulated genes (ISG) such as Irf7, Isg20 and Ifit1 (termed Isg cluster; Fig. 1d,f).
We performed pathway analysis individually comparing the Timd4 cluster to other clusters. Timd4 cluster pathways were focused on homeostatic and regenerative functions, i.e. endocytosis, lysosome, angiogenesis and regeneration (Fig. 1g). Relative to the Timd4 cluster, translational-ribosomal pathways were strongly upregulated in the Mhc-II, Ccr2 and Isg clusters, while classic inflammatory pathways, including respiratory burst, response to IL-12, IFN-γ and type I interferon were enriched in the Ccr2 and Isg clusters (Fig. 1g). These data reveal the existence of unique, cluster-specific functions in macrophages at steady-state.
Single cell trajectories reveal developmental relationships
To determine if hypothetical developmental relationships exist between monocytes and macrophage clusters we re-clustered these populations (clusters 1–5) to provide trajectories of single cell-based changes in gene expression (Fig. 2a). Seurat-defined clusters were superimposed on a pseudotime trajectory produced by the Monocle24 algorithm, which revealed that macrophages in the Timd4 cluster occupied a separate trajectory branch (B1; Fig. 2b). Monocytes and macrophages found in the Ccr2 and Isg clusters occupied a second branch (B2; Fig. 2b) while macrophages in the Mhc-II cluster straddled all three branches (B3; Fig. 2b). The Mpath25 algorithm demonstrated similar ordering, and suggested macrophages in the Isg cluster could arise from the Ccr2 cluster, rather than monocytes (Fig. 2c), which was confirmed by a restricted Monocle analysis (Fig. 2d).
Figure 2: High dimensional single cell mapping and trajectory analysis reveals 4 macrophage clusters that occupy distinct activation states.
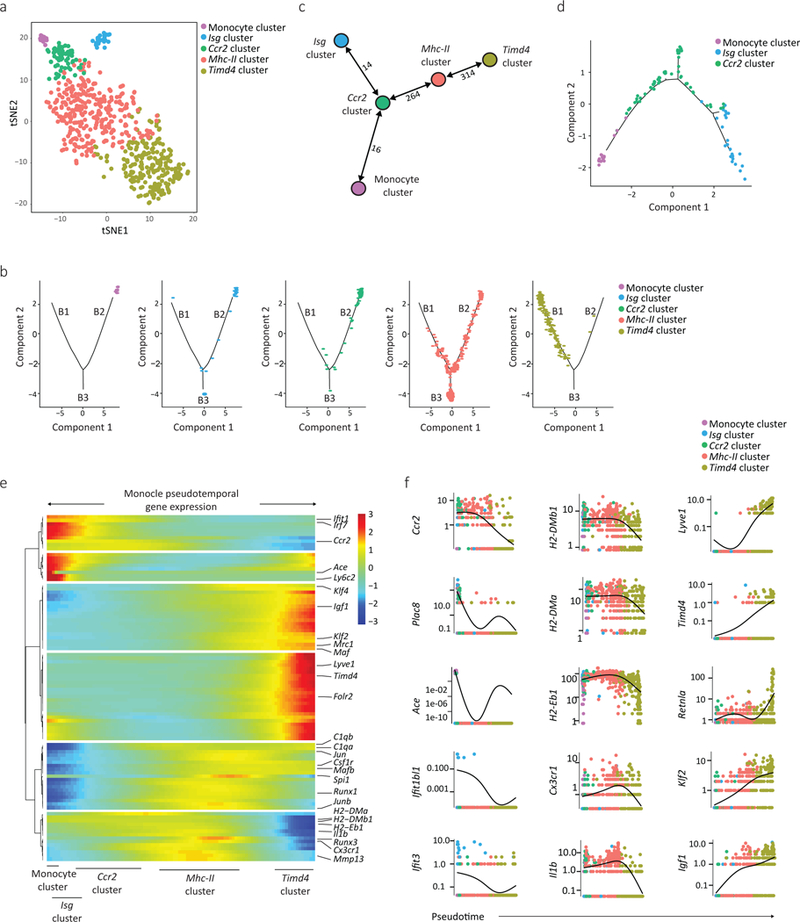
a) Cells found in clusters 1–5 (Fig. 1d, including Timd4 cluster, Mhc-II cluster, Ccr2 cluster, the Isg cluster and monocytes), were re-clustered using Seurat. Differentially expressed genes between clusters were used to generate hypothetical developmental relationships using both Monocle (b) and Mpath (c) algorithms. b) B1, B2, B3 represents individual branches in Monocle. c) Numbers between nodes are the absolute number of transitioning cells between two populations, and highlight the strength of relationship between two subsets. d) Focused Monocle trajectory analysis including only the monocyte cluster, Ccr2 cluster and Isg clusters. e) Heatmap of differentially expressed genes, ordered based on their common kinetics through pseudotime using Monocle. The relative position of individual Seurat-based clusters across Pseudotime is depicted below. f) Cluster defining gene expression plotted as a function of pseudotime with Seurat’s original cluster colours (a) superimposed.
Gene expression was then plotted as a function of pseudotime in Monocle to track changes across different macrophage states. Klf2, Mafb and Maf expression was low in the Ccr2 cluster, higher in the Mhc-II cluster and further increased in the Timd4 cluster (Fig. 2e,f) suggesting these transcription factors tracked with cardiac macrophage fate. Loss of MHC-II genes, Cx3cr1 and Il1b was associated with gain of Timd4, Lyve1 and Igf1 expression in the Timd4 cluster (Fig. 2e,f), which was the most transcriptionally distinct subset. In contrast, Ccr2, Mhc-II and Isg clusters appeared to be a spectrum of related lineages, with the Isg cluster adopting a unique activation state focused around type I IFN signaling. Tracking gene expression changes across macrophage states revealed coordinate patterns defining macrophage subset identification and functions.
Cx3cr1-based fate mapping tracks resident cardiac macrophages
To track cardiac macrophages that are maintained independent of blood monocytes we utilized the tamoxifen (TAM) inducible Cx3cr1CreERT2-IRES-YFP (termed Cx3cr1CreER-YFP) line controlled by the endogenous Cx3cr1 promoter26 crossed with Rosa26-stop-TdTomato reporter mice (termed Cx3cr1CreER-YFP:R26Td; Fig. 3a). Prior studies have shown robust TAM-inducible reporter gene expression in Cx3cr1-expressing resident macrophages, and transient labeling of blood monocytes13, 26, 27. We fed TAM-containing chow to 3 week old mice for 10 days, followed by normal chow for 1–10 weeks. Labeling of CD115+CD11b+Ly6c+ blood monocytes prior to TAM administration was negligible (<0.2% Td+). Ly6c+ blood monocytes were efficiently labeled (92–95% Td+) immediately after TAM administration and labelling returned to baseline 2–3 weeks after TAM discontinuation (<0.5%; Fig. 3b). More than 90% of total CD11b+CD64+ cardiac macrophages were Td+ immediately following TAM administration (Fig. 3b). Over a 5-week time period, CCR2– cardiac macrophages retained Td expression, while CCR2+ cardiac macrophages gradually lost Td expression after TAM discontinuation (Fig. 3b), consistent with replacement of CCR2+ macrophages by unlabeled monocytes.
Figure 3: TIMD4 and CCR2 are durable markers of resident and recruited macrophages.
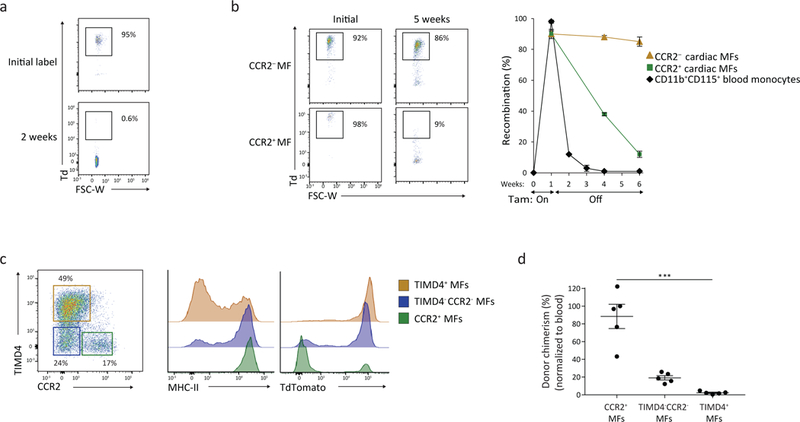
a-b) 3 week old Cx3cr1CreER-YFP:R26Td mice were administered fed tamoxifen (TAM) containing chow for 10 days, and then analyzed after TAM discontinuation (0–20 weeks). Recombination rates are expressed as %Td+ in blood CD115+ CD11b+ monocytes 2 weeks (a) and 5 weeks post TAM discontinuation in cardiac CD11b+CD64+CCR2+MHC-IIhi and CD11b+CD64+CCR2− macrophages (b). N=5; center value, mean; error bars, SEM. Repeated independently 2 times with similar results. c) 20 weeks after TAM discontinuation, total cardiac macrophages were defined into 3 populations based on expression of CCR2 and TIMD4, wherein MHC-II and TdTomato reporter expression was assessed. Repeated independently 5 times with similar results. d) Cardiac macrophages were isolated from parabiotic pairs (CD45.2:CD45.1) to determine donor chimerism after 9 weeks of pairing in the 3 populations outlined in Fig. 3c. Chimerism rates for cardiac macrophages were normalized for blood monocyte chimerism. N=5. ***p<0.001; two-tailed Student’s t test; center value, mean; error bars, SEM; one experiment.
To further assess macrophage turnover based on scRNA-seq cluster defining markers, resident cardiac macrophages were labelled (as in Fig. 3b) and analyzed 20 weeks post-TAM discontinuation. CCR2−MHC-IIloTIMD4+ cardiac macrophages retained high Td reporter expression (~90% Td+) after TAM discontinuation, and had very little donor chimerism (~2%) in parabiotic studies (Fig. 3c,d). The other two subsets had progressively higher turnover. CCR2−MHC-IIhiTIMD4− macrophages were ~75% Td+, with ~20% chimerism by parabiosis, while CCR2+MHC-IIhiTIMD4− macrophages were ~15–20% Td+, with almost complete chimerism (~90%) in parabiotic studies (Fig. 3c,d). Thus, CCR2 expression alone identified a subset of cardiac macrophages that was fully replaced by blood monocytes in adult mice, and stratifying remaining macrophages by TIMD4 and MHC-II identified macrophage subsets with limited monocyte-dependence.
Next we examined Td reporter expression in tissue macrophages in Cx3cr1CreER-YFP:R26Td mice from birth to adulthood without TAM or with TAM injected at E18.5. In the absence of TAM, Td+ macrophages were detected in resident macrophage populations that express Cx3cr1 immediately after birth (i.e., microglia and cardiac macrophages) (Supplementary Fig. 2a). Background Td expression reached a relative plateau of ~25% of total cardiac macrophages between 8–13 weeks of age. Tissue macrophages with low expression of Cx3cr1 (such as liver Kupffer cells) had very low Td expression (0–5%) and there was virtually no Td expression in blood monocytes at any time point examined (E18.5 – 35 weeks of age) in the absence of TAM. Eleven week old mice treated with TAM at E18.5 had high Td expression in brain microglia (~99%) and cardiac macrophages (~80%), and lower expression in liver Kupffer cells (~40%; Supplementary Fig. 2a). We also observed similar Td reporter expression in the absence of TAM in cardiac macrophages and brain microglia from an independently generated Cx3cr1CreERT2 line27 bred to the same R26Td reporter (Supplementary Fig. 2b).
To investigate whether monocyte-derived macrophages could be induced to express the Td reporter we isolated Td− bone marrow from adult CD45.2 Cx3cr1CreER-YFP:R26Td mice and reconstituted lethally irradiated CD45.1 recipients. Six weeks post-reconstitution, CD45.2+ monocytes and monocyte-derived cardiac CD45.2+ macrophages were Td– (~98%; Supplementary Fig. 2c), indicating that Cx3cr1CreER-YFP:R26Td monocytes do not induce the expression of the Td reporter upon macrophage differentiation in the absence of TAM. As such, while some macrophages populations in Cx3cr1CreER-YFP:R26Td express Td in the absence of TAM administration, Td reporter activity reliably distinguished resident cardiac macrophages from recruited, monocyte-derived macrophages.
Resident cardiac macrophages are lost within infarcted myocardium
To examine resident macrophage dynamics during sterile injury we utilized a model of myocardial infarction through surgical ligation of the left anterior descending artery5. Cardiac tissue was analyzed by flow cytometry at different times post-injury in the ischemic (infarct and peri-infarct tissue) and the remote (non-infarcted) zones of Cx3cr1CreER-YFP:R26Td mice fed TAM at 3 weeks of age for 10 days (Fig. 4a). TAM was discontinued for 6 weeks before mice were infarcted. The absolute number of resident Td+ macrophages in the ischemic zone was reduced by ~60% post-infarct (day 2) compared to non-infarcted mice (Fig. 4b), including both resident CCR2−MHC-IIloTIMD4+ and CCR2−MHC-IIhiTIMD4– macrophages (Supplementary Fig. 3a). Absolute numbers of resident Td+ macrophages slowly increased post-infarct (day 4–28), compared to day 2, while the number of recruited Td− macrophages decreased. However, the ratio of resident to recruited macrophages did not return to pre-infarct levels by 4 weeks post-infarct (Fig. 4c,d). Resident CCR2−MHC-IIhiTIMD4– macrophages were ~75% Td+ at steady state, but post-infarct only ~5% remained Td+, indicating recruited macrophages were the vast majority (Fig. 4e). In contrast, resident CCR2−MHC-IIloTIMD4+ macrophages were ~90% Td+ at baseline, and post-infarct, remained at ~75% Td+ at day 2 and at day 28 (Fig. 4e), indicating that TIMD4 expression was maintained on resident Td+ macrophages, and recruited macrophages did not become TIMD4+, even at 28 days post-infarct.
Figure 4: Resident Macrophages are lost in infarcted cardiac tissue and return via local proliferation.
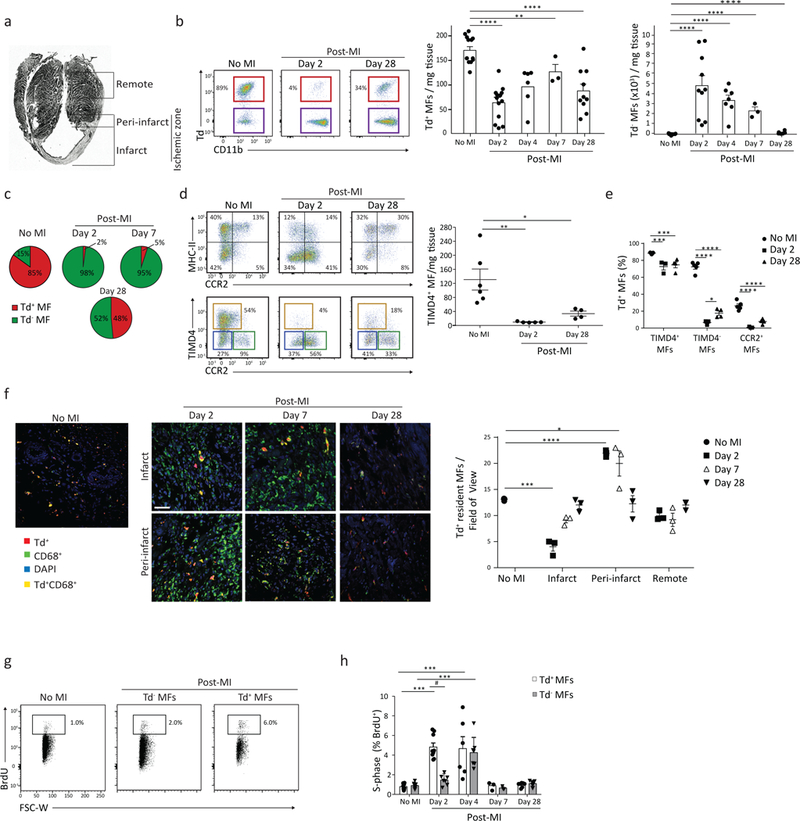
3 week old Cx3cr1CreER-YFP:R26Td mice were administered fed TAM-containing chow for 10 days, and then underwent left anterior descending artery ligation to induce a myocardial infarction (MI) 6 weeks post-TAM discontinuation. a) Paraffin-embedded section demonstrating the infarct zone, peri-infarct zone and the non-infarcted (remote) left ventricular myocardium. The ischemic zone, containing both infarct + peri-infarct regions, was analyzed in all flow cytometric experiments together. b) Cardiac tissue was isolated at different time points post-MI from the ischemic zone and representative flow plots cytometric analysis of resident CD64+CD11b+Td+ macrophages is shown (left). Adjacent graphs (right) quantify resident CD64+CD11b+Td+ and recruited CD64+CD11b+Td− macrophages CD64+CD11b+ Macrophages per mg of cardiac tissue. N(Td+)=13, 13, 6, 3, 10. N(Td−)= 12, 10, 7, 3, 6. Repeated 3 times for d2, 2 times for d4 and d28, 1 time for d7, 6 times for control with similar results. Two-tailed Student’s t test compared with control, corrected with Bonferroni. c) Td+ and Td− cardiac macrophages expressed as a % of total macrophages, at indicated times post-MI based on Fig 4b. d) Flow plots of CD64+CD11b+ cardiac macrophages stratified by MHC-II vs. CCR2, or TIMD4 vs. CCR2 post-MI; TIMD4+ Macrophages quantified per mg of tissue (graph, right). N=6, 5, 4. Repeated twice. One-way ANOVA adjusted for multiple comparisons. e) Percent of Td+ macrophages determined at each time point post-MI within 3 populations outlined in Fig. 3d. N=5, 3, 4. Repeated twice. Two-way ANOVA adjusted for multiple comparisons. f) Representative immunofluorescence images of cardiac tissue post-MI. Cardiac CD68+ macrophages were identified as Td+ resident macrophages, or Td− recruited macrophages, in the infarct, peri-infarct and remote regions; total number of such cells was quantified (graph, right) from 5 individual images/region/mouse. N=3. Two-tailed unpaired Student’s T test. One experiment. g-h) Representative flow cytometric plots of CD64+CD11b+ cardiac macrophages isolated 2 days post-MI from the ischemic zone. Mice were injected (i.p.) with BrdU 2 hours prior to isolation to label all cells in S-phase of cell cycle. The %BrdU+ was quantified in the Td+ resident MF and Td− recruited MF subsets were compared to animals with no MI (H). N= 10, 8, 9, 7, 7, 7, 3, 3, 7, 7. Repeated 3 times for d2, 2 times for d4 and d28, 1 time for d7, 6 times for control. Two-tailed Student’s T test compared with control, corrected with Bonferroni. For all: *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Center value, mean; error bars, SEM.
Immunofluorescence imaging also indicated a 70% loss of Td+ cardiac macrophages in the infarct zone at day 2 post-infarct compared to non-infarcted hearts, with a progressive increase in density over 28 days (Fig. 4f). Increased resolution of the infarct vs. peri-infarct region demonstrated a region-specific increase in the absolute number of Td+ cardiac macrophages in the peri-infarct zone at day 2 and day 7 post-infarct compared to control non-infarcted heart tissue. LYVE1 was co-expressed on TIMD4+ macrophages by flow cytometry (Supplementary Fig. 3b) and was highly expressed within the Timd4 cluster by scRNA-seq (Fig. 1f). We partitioned resident macrophages into CD68+LYVE1+Td+ and CD68+LYVE1–Td+ macrophages in the cardiac tissue sections (Supplementary Fig. 3c). LYVE1–Td+ macrophages increased in number in the peri-infarct zone at day 2 post-myocardial infarction compared to non-infarcted control, while the number of LYVE1+ Td+ macrophages did not change (Supplementary Fig. 3c), highlighting spatial enrichment of resident LYVE1− macrophages in the peri-infarct zone.
We assessed macrophage proliferation in vivo post-infarct by administering BrdU intra-peritoneally (i.p) 2 hours prior to isolation to label cells in S-phase5. We observed a proliferative burst in resident Td+ cardiac macrophages at day 2 and day 4 post-infarct compared to non-infarcted hearts, while recruited Td− macrophages only exhibited increased proliferation at day 4 post-infarct (Fig. 4g,h). When parsing resident Td+ macrophages into TIMD4+ and TIMD4− subsets we observed a slight proliferative advantage of resident Td+TIMD4− macrophages (Supplementary Fig. 3d), which supports the observation that the analogous resident Td+LYVE1− subset increased in cell number in the peri-infarct region (Supplementary Fig. 3c).
To ensure that Td– blood monocytes did not differentiate into Td+ macrophages following recruitment into the cardiac tissue post-infarct (in the absence of TAM) we produced chimeric mice in a CD45.1 background that contained CD45.2 Cx3cr1CreER-YFP:R26Td monocytes through both parabiosis (Supplementary Fig. 4a) or bone-marrow (BM) transfer following chest wall shielded irradiation (Supplementary Fig. 4b). Myocardial infarction was induced at 2 weeks post-parabiosis or 6 weeks post-BM transfer and cardiac tissue was analyzed by flow cytometry at 5 or 28 days post-MI, respectively. CD45.2+ monocytes and monocyte-derived macrophages isolated from CD45.1 mice did not induce Td expression following recruitment post-infarct in the absence of TAM (Supplementary Fig. 4a-b). To confirm TAM responsiveness in these two systems, CD45.2+ macrophages from donor parabiotic mice and BM chimeras injected with TAM prior to isolation were assessed and both demonstrated high numbers of Td+ macrophages. Thus, in the inflammatory environment following irradiation or myocardial infarction, the Cx3cr1CreER-YFP:R26Td line retained the ability to distinguish resident from recruited cardiac macrophages.
Recruited cardiac macrophage plasticity within infarcted tissue
To explore macrophage heterogeneity after ischemic injury, we performed scRNA-seq on CD45+CD11b+CD64lo-hi macrophages isolated from the ischemic zone of Cx3cr1CreER-YFP:R26Td mice treated with TAM at E18.5, infarcted at 7 weeks of age and macrophages isolated 11 days post-infarct (Fig. 5a). At 11 days post-MI the acute inflammatory response is resolved and recruited monocytes have differentiated into tissue macrophages28, 29, 30, making it an ideal time point to study macrophage heterogeneity. We sequenced 1806 cells from control non-infarcted mice and 4697 cells from the ischemic zone post-infarct. t-SNE analysis from non-infarcted mice revealed 6 clusters identical to those previously identified (Timd4 cluster, Mhc-II cluster, Ccr2 cluster, Isg cluster, proliferating macrophages and monocytes; Fig. 5b). In macrophages isolated from infarcted tissue, we identified 13 clusters, 6 of which precisely overlapped with the 6 clusters identified in non-infarcted hearts, and 7 clusters were unique to infarcted tissue (Fig. 5b). 64% of the macrophages sequenced post-infarct fell in unique clusters (Fig. 5b,c), indicating the majority of macrophages evolved distinct transcriptional signatures compared to non-infarcted.
Figure 5: High-dimensional analyses reveals plasticity of recruited macrophages to adopt both unique and resident-like transcriptional states within infarcted tissue.
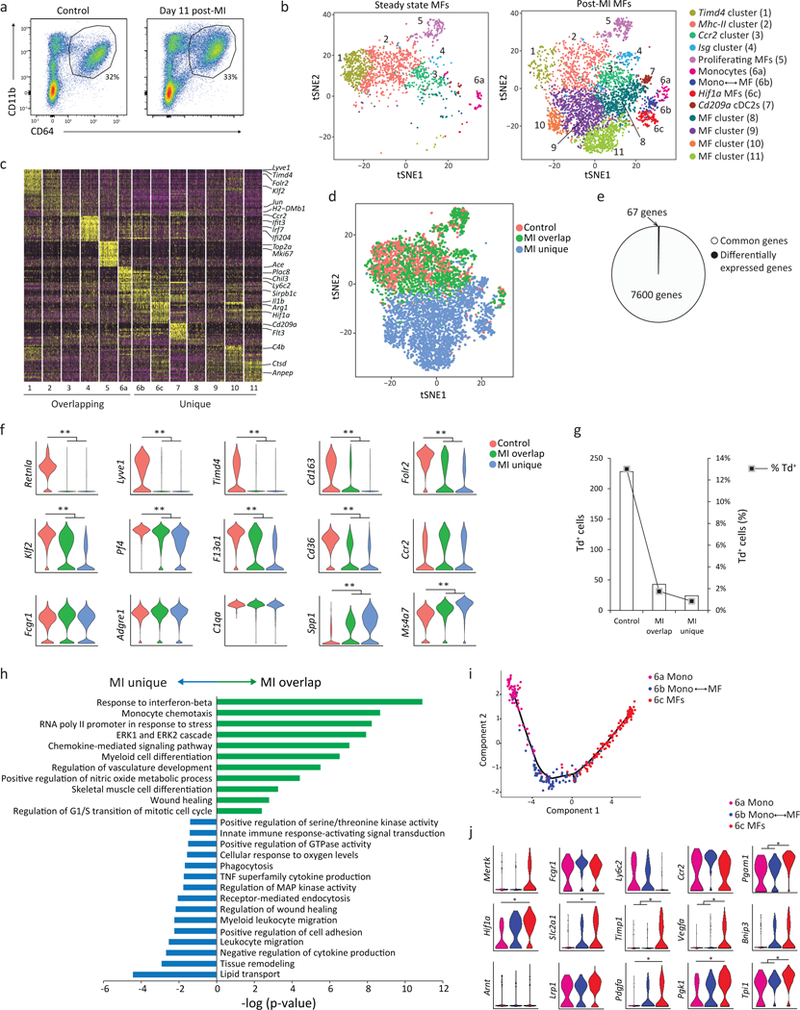
Cx3cr1CreER-YFP:R26Td mice were administered tamoxifen at E18.5. At 7 weeks of age, mice served as either controls (non-infarcted) or received a myocardial infarction. Cardiac tissue from control mice and from infarcted mice was isolated (the ischemic zone, 11 days post-MI), live cardiac CD45+CD64Dim-Hi CD11b+ macrophages were sorted and processed for scRNA-seq as in Fig 1c (1806 control cells; 4697 post-MI cells). One experiment, 3–4 pooled mice per group. b) Seurat-based tSNE analysis of merged control and post-MI samples revealed ‘Overlapping Clusters’, present in both control and post-MI samples, and ‘Unique Clusters’, present only post-MI. c) Heatmap of the top 20 differentially expressed genes per cluster post-MI. d) tSNE plot depicting cells in the control sample (pink), and cells in the MI sample that overlapped with control (MI-Overlap; green) and cells uniquely found post MI (MI-Unique; blue). e) Pie chart illustrating the number of differentially expressed genes between Control and MI-Overlap groups, relative to the number of commonly expressed genes between these two groups (see Supplementary Table 1 for differentially expressed genes per cluster). f) Representative violin plots of the differentially expressed genes between the 3 populations as defined in Fig. 5d; **p<10−13 (adjusted for multiple comparisons). g) Absolute number and percent of Td expressing cells in each cluster defined in Fig 5d. h) Pathway analysis (gProfiler) of differentially expressed genes in MI-Overlap vs MI-Unique populations. Pathway enrichment is expressed as the log(-p value) – adjusted for multiple comparison. i) A pseudotemporal trajectory (Monocle) analysis of cluster 6 subsets (Fig. 5b) demonstrates a linear developmental progression from cluster 6a (monocyte) to 6c (macrophage). j) Violin plots of differentially expressed genes in cluster 6, including HIF-1α / glycolysis pathway genes. *p<10-2. Statistical analysis for scRNA-seq data as described into the methods.
Post-infarct macrophages from individual overlapping clusters had nearly identical gene expression patterns compared to macrophages of the same cluster in the non-infarcted control sample, except for a total of 67 genes (sum across all overlapping clusters; Fig. 5d,e; Supplementary Fig. 3e and Table 1). Genes associated with mature macrophages (Lyve1, Timd4, Retnla, Cd163, Folr2 and Klf2) were down-regulated, while genes associated with monocytes (Ms4a7 and Spp1) were up-regulated in post-infarct macrophages compared to non-infarct control macrophages (Fig. 5f). Classic macrophage genes (Adgre1, Fcgr1 and C1qa) remained unchanged, as did the vast majority (7543) of other genes detected in each overlapping cluster (Fig. 5d,f). Macrophages expressing the Td transcript were found in control non-infarcted mice at a frequency much lower (~13%) than expected based on flow cytometric analysis (~80% Td+, Supplementary Fig. 2a), which limited our ability to track resident populations by scRNA-seq. However, similar to flow cytometric analysis, there was a decrease in both the absolute number and percent of Td expressing macrophages in both the post-infarct overlap and post-infarct unique clusters, compared to control (>90% loss of Td expressing macrophages; Fig. 5g). These data suggest that post-infarct, macrophages that were found in overlapping clusters (clusters 1–6) were mainly derived from Td– monocytes, yet they had nearly identical transcriptional signatures as macrophages in steady-state control mice with the exception of a very small number of core genes found in resident macrophages, such as Timd4, Lyve1, Igf1 and Folr2 (Fig. 4e).
Among the 7 unique post-infarct macrophage clusters, differential gene expression analysis and comparison to the ImmGen database identified four macrophage clusters (8–11) and one DC cluster (7; CD209a+ cDC2s) (Fig. 5b,c and Supplementary Fig. 3e,f). Compared to the overlapping clusters, macrophages in unique clusters 8–11 were enriched most strongly in inflammatory pathways (response to IFN-γ, monocyte chemotaxis), but also in some reparative pathways (Fig. 5h, Supplementary Table 2 for all cluster-specific genes). Cluster 6 was composed of 3 separate sub-clusters (6a, 6b and 6c) that clustered separately (Fig. 5b). Monocle trajectory analysis revealed three different fates along the same developmental trajectory, with a progressive increase in expression of macrophage genes and reciprocal loss of monocyte genes (Fig. 5i). We also observed the progressive upregulation of pathways involved in hypoxia (HIF-1α signaling p=1.4×10−3), glycolytic metabolism (p=0.02) and extracellular matrix interactions (p=1.6×10−3) as monocytes transitioned into macrophages (Fig. 5j), consistent with the anaerobic environment of ischemic tissue. As such, while recruited macrophages possessed a high degree of plasticity, they did not adopt the core transcriptional signature of resident populations, and could be distinguished by flow cytometry and scRNA-seq.
Cardiac macrophage heterogeneity in human cardiomyopathy
Next, we sorted human cardiac CD45+CD64+CD14+ macrophages from patients with cardiomyopathies. Based on MHC-II (HLA-DR) and CCR2 expression we identified 3 subsets, CCR2−MHC-IIhi, CCR2+MHC-IIhi and CCR2+MHC-IIlo subsets (Supplementary Fig. 5a-b), which appeared similar to mouse cardiac macrophages. Differentially expressed genes defined by murine t-SNE clusters were used to determine if sorted human cardiac macrophage populations expressed similar patterns. The human CCR2–MHC-IIhi macrophages expressed many genes found in the murine Timd4 cluster, including TIMD4, LYVE1, CD163, FOLR2, IGF1 and MAF (Supplementary Fig. 5c). Murine gene signatures associated with the Isg cluster, Ccr2 cluster and monocytes were found predominantly in human CCR2+ MHC-IIlo monocyte and CCR2+MHC-IIhi macrophages. Only 226 genes were differentially expressed between human CCR2–MHC-IIhi and human CCR2+MHC-IIhi macrophages, with the majority (179 genes) upregulated in CCR2+MHC-IIhi (Supplementary Fig. 5c). Pathway analysis indicated that human CCR2+MHC-IIhi macrophages were enriched in HSC lineage genes (p=2.8×10−6), cytokine-cytokine receptor interactions (p=4.6×10−5) and Toll-like receptor signaling (p=2.9 ×10−2), while human CCR2–MHC-IIhi cardiac macrophages were enriched in the complement cascade (47 genes upregulated; p=4.4×10−2). Thus, CCR2 defines recruited monocytes and monocyte-derived macrophages in both mouse models and in human cardiomyopathy.
Cx3cr1-based depletion post-infarct worsens cardiac function
Next, we used Cx3cr1CreER-YFP :R26Td/DTR mice to label resident macrophages with both the Td reporter and the diphtheria toxin receptor (DTR) to selectively deplete resident Td+ cardiac macrophages through diphtheria toxin (DT) injection, without affecting other cell types (Supplementary Fig. 6a,b). DT injection alone, in the absence of ischemic injury, did not cause any detectable recruitment of neutrophils and macrophages, nor cause cardiac fibrosis in wild-type mice that did not contain DTR (Supplementary Fig. 6c). To test the functional role of resident macrophages post-infarct, 3 week old Cx3cr1CreER-YFP:R26Td/DTR mice were fed TAM-chow for 10 days and TAM was discontinued for 6 weeks. Daily DT injections were started one day prior to myocardial infarction to deplete and suppress the numerical recovery of resident cardiac macrophages (Fig. 6a,b). There was no difference in the infarct size or cardiac function at day 7 post-infarct in DT-treated Cx3cr1CreER-YFP:R26Td/DTR mice compared to Cx3cr1CreER-YFP:R26Td/+ mice which lacked the DTR (Fig. 6c,d). However, global left ventricular systolic function deteriorated between day 7 and day 28 post-infarct, with increased cardiac hypertrophy, fibrosis and increased late mortality by day 35 post-infarct in DT-treated Cx3cr1CreER-YFP:R26Td/DTR mice compared to Cx3cr1CreER-YFP:R26Td/+ mice (Fig. 6e-i). There was no significant change in the magnitude of the inflammatory response in macrophage-depleted mice compared to non-depleted (Cx3cr1CreER-YFP:R26Td/+) mice at day 5 post-infarct (Supplementary Fig. 6d), indicating depletion of resident macrophages did not cause an exaggerated inflammatory response acutely.
Figure 6: Selective depletion of resident cardiac macrophages results in impaired infarct healing and adverse cardiac remodeling primarily within the peri-infarct region.
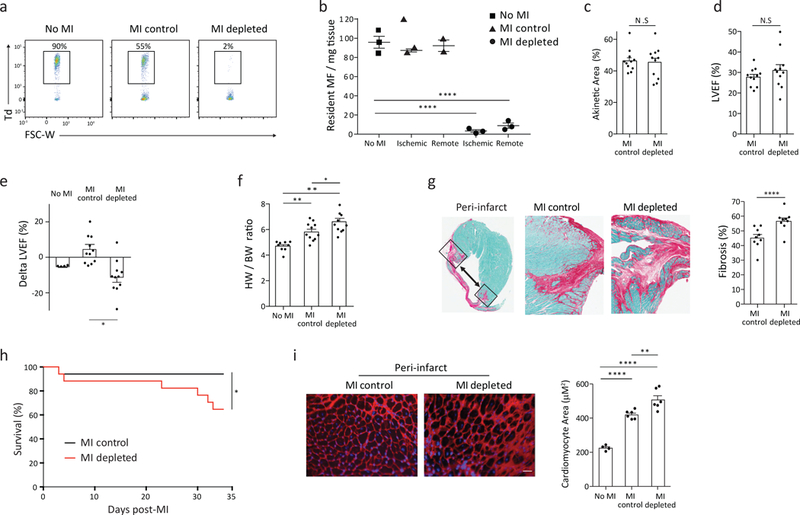
3 week old Cx3cr1CreER-YFP: R26Td/DTR were fed TAM-chow for 10 days, TAM was discontinued for 6 weeks, and mice either were non-infarcted (no MI) or received surgical myocardial infarction (MI). Mice that received a surgical MI were injected daily either PBS (MI-control) or 250ng diphtheria toxin (DT; MI-depleted), starting the day prior to surgery. a-b) Representative flow cytometric plots of CD45+CD64+CD11b+ macrophages demonstrating the number of Td+ cells remaining at day 35 post infarct within the ischemic zone, and quantitated (b) in both the ischemic zone and remote zone by flow cytometry per mg of tissue. Resident macrophages were identified as CD64+CD11b+Td+. N=3, 7. repeated twice, representative plot is shown. c) Infarct size was measured at day 7 post-MI by echocardiography, reported as % akinetic area. N=11. Repeated 2 times with similar results. d) Left ventricular ejection fraction (%LVEF) measured at day 7 post-MI. N=11. Repeated 2 times with similar results. e) Average change in ejection fraction from day 7 to day 28 post-MI (Delta LVEF). N=4, 11, 11. Repeated 2 times with similar results. f) Cardiac hypertrophy 35 days post-MI as measured by heart weight to body weight ratio. N=9, 11, 11. Repeated 2 times with similar results. g) Histological cross section (left) and quantification (right) of the cardiac peri-infarct zone at day 35 post-MI. Hearts were scanned at 10x magnification. Percentage fibrotic area was quantified using Fast Green staining and ImageJ analysis, 4 images/zone/heart in two sections, 150 μm apart. N=9. Repeated 1 time. h) Kaplan-Meier curve representing percent survival at day 35 post-MI. N=17. Repeated 2 times with similar results. I) Cardiomyocyte hypertrophy assessed with Wheat Germ Agglutinin staining in the peri-infarct zone. Hearts imaged at 20x magnification and cardiomyocyte area was measured in ImageJ, 8 images/zone/ heart at 2 cutting levels. N=4, 6, 6. Repeated 1 time. For all: *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; Two-tailed Student’s T test; center value, mean; error bars, SEM.
While the absolute number of resident CD64+Td+ cardiac macrophages was similar between non-infarcted controls and post-infarct hearts in the remote zone at all time points analyzed, there was increased recruitment of monocytes and monocyte-derived CD64+Td− macrophages in this area at day 2, 4 and 7 post-infarct (Supplementary Fig. 6e). However, CD64+Td+ resident macrophages proliferated robustly in this region, as determined by BrdU incorporation, at day 2 and day 4 post-infarct (Supplementary Fig. 6f). Depletion of resident cardiac macrophages led to a decrease in left ventricular systolic function from day 7 to day 28 post-infarct in the remote zone of DT-treated Cx3cr1CreER-YFP:R26Td/DTR mice, compared to no change in infarcted non-depleted Cx3cr1CreER-YFP:R26Td/+ mice (Supplementary Fig. 6g). We also observed an increase in cardiomyocyte hypertrophy, but no change in cardiac fibrosis, in the remote zone of depleted compared to non-depleted hearts 35 days post-infarct (Supplementary Fig. 6h,i). These data indicate a spatially restricted, primary role for resident Td+ macrophages in the peri-infarct zone to prevent pathological remodeling and development global left ventricular dysfunction in the surviving myocardium.
Discussion
Here we described the transcriptional and functional heterogeneity that exists within the innate immune cell compartment of the murine heart, in both steady state and following myocardial infarction. Through a combination of cell tracking and scRNA-seq we found that the healthy murine myocardium contains 4 transcriptionally-distinct cardiac macrophage subsets, with subset specific life-cycles and function. Resident cardiac macrophages accounted for only 2–5% of the total cardiac macrophages within the infarct zone during the first few weeks post-infarct, yet their depletion impaired cardiac function and worsened infarct healing. This is underscored by the observation that nearly identical, recruited macrophages were present at nearly the same time, yet did not compensate for resident macrophage depletion. A small number of cluster-specific, resident macrophage genes (Timd4, Lyve1, Igf1, etc), were not adopted by recruited macrophages, and these genes could have endowed resident macrophages with critical reparative functions. For instance, the phosphatidylserine receptor, TIMD4, is utilized by macrophages in efferocytosis17; without which ischemic injury is amplified31. LYVE1 binds hyaluronan expressed by smooth muscle cells and plays an integral role in vascular homeostasis32, therefore loss of LYVE1+ macrophages could disrupt adaptive healing mechanisms. The growth factor IGF1, secreted by cardiac macrophages, directly promotes angiogenesis10. Alternatively, by the time monocytes become peripherally-derived resident macrophages they might have missed the window of opportunity for providing effective cardioprotective functions. Additional studies, using Cx3cr1-based strategies to target resident macrophages would shed light on which, if any, of these potential mechanisms are involved.
During neonatal cardiac injury, cardiac macrophages increase in numbers through proliferation5, 8. In contrast, ischemic injury in adult mice reduced the abundance of resident Td+ cardiac macrophages within infarcted tissue, presumably through cell death due to anoxia and nutrient deprivation. Post-infarct, a robust proliferative program was triggered in the remaining resident macrophages, similar to neonatal cardiac macrophages. Importantly, the neonatal myocardium did not recruit monocytes in significant numbers, which is a fundamental difference compared to the adult8. The limited capacity to recruit monocytes in large numbers may be an adaptive mechanism that promotes reparative programs inherent to resident, embryonic-derived macrophages during development. The density of peripherally-derived CCR2+ cardiac macrophage correlates with cardiac dysfunction following heart transplantation in humans. In addition, preventing monocyte influx in adult mice has been demonstrated to have beneficial effects when excessive inflammation drives ischemic injury33, 34. Paradoxically, our data indicates that inhibiting monocyte influx alone could impair development of monocyte-derived macrophages with reparative functions and prevent replacement of reparative resident populations that are lost during the initial insult.
Understanding the tissue specific cues and the ability to guide monocyte fate selection during ischemic injury will be critical in influencing outcomes. However, it remains to be clarified whether the 9 macrophage clusters found post-infarct specify their fate when they enter the myocardium, whether these clusters are at their final transcriptional destinations, or are part of a continuum. Intriguingly, there is evidence that monocyte fate selection does not only occur in tissues, but also in the bone marrow. HSCs produce lineage-committed monocytes prior to release into circulation, and these monocyte lineages possess distinct tissue functions, presumably through epigenetic changes. For example, Ly6CloMSR1+CEACAM1+ blood monocytes enter the lung and promote tissue fibrosis, Ly6ChiCD209a+ monocytes preferentially differentiate into monocyte-derived DCs35, 36, and immunoregulatory Ym1+Ly6Chi monocytes are released only in the recovery phase following injury, and promote tissue repair37.
Prior studies have suggested that resident cardiac macrophages are slowly replaced by monocyte-derived populations13. However, our data suggest that cardiac macrophages must be stratified appropriately in order to understand their lifecycle. CCR2-MHC-IIloTIMD4+LYVE1+ macrophages, have negligible monocyte input in steady state, and even in chronically infarcted tissue. The identification of Timd4 and Lyve1 expression in resident macrophages from other tissues suggests that they may represent important resident macrophage markers38, 39, 40, and specifically within the myocardium – TIMD4 and LYVE1 may be used to identify resident macrophages in the absence of dedicated tracking approaches. It is plausible, that given enough time, tissue cues can prompt recruited macrophages to express genes such as Timd4 and Lyve1. This is evident by adoptive transfer experiments in the peritoneum and lung where the tissue guides the identity of the transferred macrophage population41, 42. Our study highlights the tremendous complexity and plasticity that underlies recruited and resident cardiac macrophages in steady state, and particularly within infarcted tissue. A greater understanding of this dynamic process is required to delineate the likely differential contribution of individual macrophage subsets to tissue repair and injury, so that sound therapeutic strategies can be generated.
Methods
Mice
All mice used in this study were purchased from The Jackson Laboratory and were bred in our animal facility prior to use. All mice were bred and housed in a pathogen-free environment at the University Health Network Animal Facility and were fed standard rodent chow. All experimental procedures were approved by the Animal Care Committee of the Toronto General Research Institute and were performed according to the guidelines of the Canadian Council on Animal Care.
Fate mapping.
To induce recombination in Cx3cr1CreER-YFP:R26Td mice tamoxifen was administered via tamoxifen-containing chow (Envigo) for 11 days or injected to pregnant females at E18.5 via intraperitoneal (i.p.) injection at 2mg per mouse. Tamoxifen preparation. 100mg of tamoxifen-free base (Sigma-Aldrich T5648) was added to 0.5mL 95% ethanol. 9.5 mL corn oil was then added to the mixture and vortexed/sonicated until dissolved.
BrdU incorporation.
For proliferation experiments, 2mg of BrdU (Sigma) was injected i.p. 2hr prior to organ harvest as previously described5. To detect intracellular BrdU, the BD Bioscience Cytofix/Cytoperm protocol was used. Cells were fixed after cell surface staining, washed and resuspended with perm buffer overnight. DNA was digested for 1hr with DNase (Sigma), cells were stained with FC block for 15min, and samples were labeled with anti-BrdU antibody.
Macrophage depletion.
Diphtheria Toxin (DT) (Sigma) was diluted in PBS and administered to mice i.p. acutely (500ng daily, 3 consecutive days) or chronically (500ng daily, 3 consecutive days then 250ng daily, 3x weekly for 2 additional weeks). For the long-term depletion and functional experiment, 500ng DT was administered i.p. daily for the first 7 days, followed by 250ng i.p. daily until the 35 days post-MI time point.
Human samples
Human cardiac tissue samples were obtained from the Washington School of Medicine from patients with end-stage cardiomyopathy during the time of implantation of the left ventricular assist device. Informed consent was received from all patients and approved by the Washington University IRB panel, with compliance to all relevant ethical regulations. After left ventricular apical core segment was removed, prior to placement of the inlet cannula, the cardiac tissue was immediately placed into cold RPMI media. Human cardiac tissue was minced and digested in a fashion identical to mouse cardiac tissue.
Cell visualization.
To visualize human macrophage subset cell morphology, hearts were harvested, digested, and prepared for flow cytometry. Sorted cells (AriaIII-CFI BRVY) were cytospun onto slides and stained with Hema3 (Fisher Scientific). Cells were imaged with a Zeiss Axioscope at 20x.
Gene Expression Analysis
RNA from human cardiac macrophages was extracted from Trizol samples using the Qiagen micro-kit (Qiagen) as previously described by us4. Total RNA concentration and quality was determined by Agilent 2100 Bioanalyzer (Agilent Technologies) according to manufacturer’s recommendations.
Transcriptional Array.
RNA transcripts were first amplified by WTA2 kit (Sigma Aldrich). 1 ng of total RNA was amplified according to manufacturer’s protocol. cDNAs were chemically labeled with Kreatech ULS labeling kit (Kreatech Diagnostics). Per reaction, 2.5 μg of DNA was mixed with Kreatech labeling buffer and Kreatech Cy5-ULS4. The reactions were incubated at 85oC for 15min in the dark and placed on ice for 3min. Labeled DNAs were purified with QIAquick PCR purification columns (Qiagen Sciences). Labeled DNAs were quantitated on a Nanodrop spectrophotometer. 2 μg of each labeled DNA was suspended in Agilent 2X Gene Expression hybridization buffer, Agilent 10X Blocking agent and Kreatech Kreablock. The hybridization solutions were applied to Agilent Human 4×44K V1 microarrays. Hybridization was carried out at 65oC for 20hrs. Washing procedures were carried out according to Agilent gene expression protocols. Slides were scanned on an Agilent SureScan microarray scanner to detect Cy5 fluorescence. Gridding and analysis of images was performed using Agilent Feature Extraction v10.7.3.1. Background subtracted, log transformed, quantile normalized data were analyzed using ANOVA testing with contrasts (Partek GS, Partek, St. Louis, MO). For exploratory analyses, we set the threshold at a two-fold change with an unadjusted p value = 0.001.
Tissue isolation and cell-surface staining
Mice were sacrificed by CO2 inhalation. Hearts were perfused with 20mL of cold PBS. Hearts, lungs, livers, and brains were chopped finely and digested, while shaking, for 1hr at 37°C in DMEM containing collagenase I (450 U/mL), DNase I (60 U/mL), and hyaluronidase (60 U/mL) enzymes (all Sigma)30. The digested material from heart, liver and brain was filtered through 40uM filters and pelleted by centrifugation (400xg for 5min at 4°C) in HBSS supplemented with 2% bovine serum + 0.2% BSA. Red blood cells (RBCs) were lysed in ACK lysis buffer (Invitrogen) for 5min at room temperature and resuspended in FACs buffer (PBS containing 2% FCS and 2mM EDTA). Liver was filtered and resuspended into 40% Percoll. Brain was filtered and resuspended into 40% Percoll mixed over top of 80% Percoll. Pellets were resuspended, RBCs were lysed, and samples were resuspended as above. Finally, blood was collected in syringes containing 50μL heparin, RBCs were lysed, and pellets were resuspended as above. Single-cell suspensions were then stained for cell-surface markers using antibodies listed in the ‘antibodies used’ section. Cells were stained in 50μL FACS buffer with 0.2μL antibody per sample, with the exception of 5μL of CCR2 antibody, for 30min at 4°C. Cells were washed and resuspended in 400μL FACs buffer to be run by flow cytometry (BD LSRII-OICR BGRV). For more information about tissue isolation, digestion and flow cytometric analysis please review uploaded video content [https://www.jove.com/video/58114/isolation-identification-extravascular-immune-cells] 43.
Flow Cytometry gating strategy and antibodies
After gating on CD45+ cells, doublets were excluded and live cells were analyzed using FSC/SSC live-dead exclusion. Single cell analysis software (FlowJo) was used to analyze cell cytometric data.
Gating Strategy (mouse):
Cardiac macrophages were identified as CD45+CD11b+CD64+ and were further parsed by CCR2, MHC-II, Timd4, Lyve1 and Td.
Infiltrating monocytes in the myocardium were identified as CD45+CD11b+CD64+Ly6C+.
Blood monocytes were identified as CD115+CD11b+Ly6C+.
Brain microglia were identified as CD45LowCD11b+F4/80+CD64+.
Liver macrophages were identified as CD45+F4/80+CD11b+MHC-IIhi.
Neutrophils were identified as CD45+CD11b+CD64−Ly6G+.
Lymphocytes were identified as CD45+CD64−CD11b−.
Gating Strategy (human):
Cardiac macrophages were identified as CD45+CD64+MerTK+CD14+ and were further parsed by CCR2 and HLA-DR.
Mouse antibodies use:
CD45 (30-F11); CD64 (X54–5/7.1); CD11c (N418); MHC-II (AF6–120.1); CD11b (M1/70); Timd4 (RMT4–54); Ly6C (HK1.4); CD115 (AF598); Ly6g (1A8); F4/80 (CI:A3–1); CD4 (RM4–5); CD8a (53–6.7); CD3e (145–2c11); BrdU (Bu20a); TruStain CD16/CD32 FC Block (93).
Human Antibody use:
CD45 (HI30); MerTK (2B10C42); CD14 (M5E2); CD64 (10.1); CCR2 (K036C2); HLA-DR (L243).
All antibodies listed above were purchased from Biolegend with the exception of CCR2 (475301) (R&D Systems), Lyve1 (ALY7) (eBiosciences) and B220 (RA3–6B2); BV421-SA (BD Biosciences). Antibody clone is provided in parentheses. For more details see the Life Sciences Reporting Summary
Myocardial infarction model
All left anterior descending (LAD) artery ligations were performed as previously described44. Briefly, following shaving of the chest and 2% isofluorane induction of anesthesia (0.8L/min oxygen), mice were intubated and ventilated with a pressure-control ventilator (Kent Scientific). Thorax and pericardium were opened and using a 7.0 silk suture (Deknatel) the LAD was ligated, the chest was closed, and the animal recovered with an immediate 0.1 mg/kg injection of buprenorphine s.q. followed by another injection 24hr after. Control mice are those with unopened chests.
Parabiosis
The lateral aspects of female donor mice (left) and recipient mice (right) were shaved and matching skin incisions were made from behind the ear to the tail of each mouse, as previously described by us5. The subcutaneous fascia was dissected to create ~0.5 cm of free skin. The olecranon and knee joints were attached by a mono-nylon 5.0 suture (Ethicon) and the dorsal and ventral skins were attached by continuous suture. Animals recovered with an immediate 0.1mg/kg injection of buprenorphine s.q. followed by another injection 24hr after. Maintenance saline injections s.q. were given for 1 week as well as 3% neomyocin antibiotics for 2 weeks. Mice were joined for 2–6 months. The percentage chimerism for each macrophage subset was normalized to blood monocytes in the recipient mouse (that originated from the donor) and expressed as a percentage (% normalized chimerism = (% donor cell in recipient / %Ly6Chi monocyte donor cells in recipient) *100).
Bone Marrow Chimeras
Adult wild-type mice (CD45.1) were sub-lethally irradiated twice with 628cGy with 3hrs in between. Chimeras were made with CD45.1 C57BL/6 hosts receiving ~5×106 BM cells from CD45.2 C57BL/6 mice (Cx3cr1creER-YFP:R26Td) on the day of irradiation. Irradiation was performed using a Gammacell 40 exactor. For shielded bone marrow chimeras, the same was performed with the addition of a 1cm thick lead shield covering the thoracic cavity of the mouse using an XRAD320 irradiator. All animals received 3% neomycin antibiotics for 2 weeks.
Cell sorting
Mouse hearts were isolated and enzymatically digested, as previously described 5, with the inclusion of 1mM Flavopiridol in the digestion buffer. Digestions were stopped after 20min and cells were processed into a single cell suspension on ice. Cells were stained with CD45 magnetic beads (Miltenyi Biotec) and subsequently fluorescently tagged antibodies against CD45, CD64 and CD11b. Hematopoietic cells were positively enriched using the AutoMacs instrument (Miltenyi Biotec) and live macrophages (CD45+, CD64+, CD11b+, DAPINeg) were sorted on the Aria Fusion (BD Bioscience) under low pressure into DMEM containing 50% FCS for cell visualization and 10X single cell RNA sequencing or RLT buffer (Qiagen) for RNA extraction.
Immunofluorescence
Hearts were isolated at reported times post-MI and cut longitudinally. They were fixed in 4% PFA overnight and followed by a 30% sucrose gradient overnight. Tissue was embedded in OCT media (Sakura Finetek) and flash frozen in isopentane suspended in liquid nitrogen. Tissue was sectioned in 10μm slices at two cutting levels. Sections were blocked for 1hr in 50:50 solution of perm block (5% BS, 0.01% Triton X in PBS) and normal serum block (Biolegend). Sections were washed with PBS and the primary antibody was diluted in FACs buffer and added overnight at 4°C in a hydrated chamber [Rat αCD68 (FA-11; ThermoFisher); Biotin αLyve1 (ALY7; eBiosciences)]. Sections were washed with PBS and stained with secondary antibody diluted in PBS for 1hr at 4°C [Goat αRat-IgG1-FITC (RTK2071); Goat αRat-IgG-AF647 (Poly4054) Biolegend]. For LYVE1 staining the TSA Fluorescein kit was used (PerkinElmer). Sections were mounted with SlowFade® Diamond Antifade Mountant with DAPI (ThermoFisher) and imaged on the LSM700 Confocal at 20x or 40x with immersion oil (AOmacrophage - Advanced Optical Microscopy Facility. Princess Margaret Cancer Research Tower, Toronto, ON, Canada). Five images were taken per heart in each of the remote (uninfarcted), peri-infarct, and infarct zones. A section without the primary antibody, however stained with the secondary IgG1 antibody, was used to control for background. All cells were counted blindly per field of view.
Echocardiography
Mouse echocardiography was performed using the VisualSonics Vevo® 2100 System using 1% isofluorane anesthetic. Temperature was held constant at 37°C and heart rate held between 450 and 500 beats/min. 2-dimensional B-mode images were obtained in the long and short axis views. VevoLab program was used for Simpson’s volume measurements. For ejection fraction calculation, the left ventricle was traced and divided into three segments (proximal, mid, and distal), as well as measurement of the length, in both diastole and systole, in triplicate. Measurements were made in a blinded fashion. Ejection fraction was obtained for both day 7 and day 28 post-MI time points, and a delta was calculated between the time points. For fractional shortening and end diastolic volume calculations, interventricular septum diameter, left interventricular diameter, and posterior wall diameter measurements were done in both diastole and systole, in triplicate. Uninfarcted control animals were used as a baseline.
Histology
Hearts were harvested day 35 post-MI. They were cut longitudinally to include a 4-chamber view of the heart and were fixed overnight in 10% buffered formalin. Hearts were then paraffin embedded and sectioned at two cutting levels at 8μm-thickness. After deparaffinization and rehydration steps, sections were stained with H&E, Picrosirius Red/Fast Green (Chondrex) for fibrosis, or wheat germ agglutinin (WGA) (Vector Labs) and mounted with SlowFade® Diamond Antifade Mountant with DAPI (ThermoFisher) for cardiomyocyte hypertrophy. Slides were imaged using the Aperio AT2 slide scanner or the Olympus Fluorescence Upright microscope at 20x (Advanced Optical Microscopy Facility. Princess Margaret Cancer Research Tower, Toronto, ON, Canada) in all three of the remote (uninfarcted), peri-infarct, and deep infarct zones. Fibrosis images were processed using Aperio ImageScope software and percent fibrosis was quantified on ImageJ software. WGA images were processed using Metamorph software. All cardiomyocytes in cross-section were counted and measured per field of view on ImageJ. All measurements were made in a blinded fashion.
Statistics
All data are presented as mean ± SEM. Sample size estimates were based on the variance seen in our previously published work5. Student’s T test was used for comparisons between experimental groups (Two-tailed, unpaired test was used unless otherwise indicated test was paired). No randomization was used in this study. Significant differences were defined at P < 0.05. If multiple comparisons were used, a Bonferonni correction was utilized. Survival analysis was performed using a Kaplan-Meier plot using a log-rank (Mantel-Cox) test. All statistical analyses were done using Prism software.
Single Cell droplet experiments (10x Genomics)
A total of 1780 cardiac macrophages and dendritic cells (1:1 ratio) for the macrophage-DC scRNA-Seq experiment, and 6503 cardiac mononuclear cells for the Control-MI scRNA-Seq experiment, were sequenced to a read depth of ~130,000 reads/cell and run through the 10x Genomics cell ranger platform.
Single cell suspensions were prepared as outlined by the 10x genomics Single Cell 3’ v2 Reagent Kit user guide. Briefly, samples were washed two times in PBS (Life Technologies) + 0.04% BSA (Sigma). Each wash was performed with a 6min centrifugation at 330 x g and re-suspension in 1 mL PBS + 0.04% BSA. Sample viability was assessed using a Trypan Blue (ThermoFisher) and a haemocytometer (ThermoFisher) and the appropriate volume for each sample was calculated. After droplet generation, samples were transferred onto a pre-chilled 96 well plate (Eppendorf), plates were heat-sealed and reverse transcription was performed using a Veriti 96-well thermal cycler (Thermo Fisher). After the reverse transcription, cDNA was recovered using Recovery Agent provided by 10x followed by a Silane DynaBead clean-up (ThermoFisher) as outlined by the user guide. Purified cDNA was amplified and cleaned up using SPRIselect beads (Beckman). Samples were diluted 4:1 (elution buffer (Qiagen):cDNA) and run on a Bioanalyzer (Agilent Technologies) to determine cDNA concentration. cDNA libraries were prepared as outlined by the Single Cell 3’ Reagent Kits v2 user guide with appropriate modifications to the PCR cycles based on the calculated cDNA concentration (as recommended by 10X Genomics).
Sequencing –
The molarity of each library was calculated based on library size as measured by bioanalyzer (Agilent Technologies) and qPCR amplification data (Kappa/Roche). Samples were pooled and normalized to 10nM, then diluted to 2nM using elution buffer (Qiagen) with 0.1% Tween20 (Sigma). Each 2nM pool was denatured using 0.1N NaOH at equal volumes for 5min at room temperature. Library pools were further diluted to 20 pM using HT-1 (Illumina) before being diluted to a final loading concentration of 14 pM. 150ul from the 14 pM pool was loaded into each well of an 8-well strip tube and loaded onto a cBot (Illumina) for cluster generation. Samples were sequenced on a HiSeq 2500 with the following run parameters: Read 1 – 26 cycles, read 2 – 98 cycles, index 1 – 8 cycles.
Single-cell RNA-seq Analysis
The sequenced data was processed into expression matrices with the Cell Ranger Single Cell Software Suite 1.3.1 by 10X Genomics (http://10xgenoics.come/). Raw base call files from HiSeq2500 sequencer were demultiplexed into library specific FASTQ files. Sequencing reads were aligned to the mouse transcriptome using STAR aligner45. Subsequently, cell barcodes and Unique Molecular Identifiers (UMIs), underwent filtering and correction. Reads associated with the retained barcodes were quantified and used to build a transcript count table.
The standard procedures of filtering, variable gene selection, dimensionality reduction and clustering were performed using the single cell RNA seq analysis R package, Seurat (v.2.3.0)46. To exclude low quality cells, in both single cell experiments, we filtered cells that expressed less than 200 genes/1000 UMIs and to exclude likely doublets cells with greater than 5800 UMIs were removed.
All genes that were not detected in at least three single cells were excluded. Based on these criteria, 12916 genes across 1590 samples in the macrophage-DC dataset and 15,147 genes across 6473 samples (1783 Control macrophages and 4690 post-infarct macrophages) in the Control-MI dataset remained for downstream analysis. In order to reduce biases caused by technical variation, sequencing depth and capture efficiency, normalization was performed using Lun et al’s deconvolution approach as implemented in R packages Scran (1.6.2)47 and Scater (1.6.0)48. This normalization method pools expression counts from linear size groups of cells. These group-specific size factors are than deconvolved into cell-specific size factors that are used to scale the counts of individual cells.
The expression matrix underwent dimensionality reduction using Principal Component Analysis (PCA). We first selected genes with high variance using the FindVariableGenes function with log-mean expression values between 0.06 and 2 for the macrophage-DC data and 0.0125 and 1.5 for the Control-MI data, and dispersion (variance/mean) between 0.5 and 30 for both datasets. The number of highly variable genes based on these criteria were 1344 genes for the macrophage-DC dataset, and 1242 genes for the Control-MI dataset. PCA was performed on the selected variable genes and 15 and 20 significant principle components were identified for downstream analysis of the macrophage-DC and Control-MI data respectively. The number of significant PCs to include in downstream analysis was determined based on the elbow point on the plot of standard deviations of PCs.
Clusters were identified by a graph based clustering approach implemented by the FindCluster function in Seurat. Cells were embedded in a k-nearest neighbors graph based on the euclidean distance matrix constructed on the significant PCs. The Louvian modularity optimization algorithm49 was applied to iteratively group cell together into clusters. We employed tSNE for dimensionality reduction and visualization of our datasets.
The modularity-based clustering is a sensitive method that can occasionally over-partition larger clusters in order to detect rare populations. We, therefore, merged together transcriptionally very similar clusters (no significant differential gene expression). In addition, two small clusters of contaminating B cells (enriched in Cd79b, Cd79a, Ly6d and Ptprcap) and stromal cells (enriched in Sparc, Fabp4, Igfbp7, Crip2) were removed from the Control-MI dataset before any subsequent analysis.
For all single cell differential expression tests, we used the MAST (Model-based Analysis of Single-cell Transcriptomics) test50. The MAST framework is a two-part generalized linear model that parametrizes both the discrete expression rate of each gene across cells and the continuous expression level conditional on the gene being expressed. To define a unique expression profile for each cluster, differential expression was tested between each cluster and all other clusters combined. MAST test as implemented in Seurat, returns an “adj_pval” and an “avg_logFC” for each gene. The expression profile of each cluster’s top 65 genes (based on avg_logFC) were visualized as a heatmap. Differential expression testing was also performed between two clusters, between control and post-infarct clusters and between overlapping and none-overlapping clusters as specified in figure 4. See Supplementary table 1 for differentially expressed genes between control and MI samples found within overlapping clusters. See supplementary table 2 for differentially expressed genes in each cluster found in post-infarct sample.
Pathway Enrichment Analysis
gProfiler (http://www.biit.cs.ut.ee/gprofiler/ )51 was applied to identify enriched pathways based on the differentially expressed genes between two groups. We implied pathway gene sets from biological processes of Gene Ontology (http://www.geneontology.org/) and molecular pathways of Reactome (http://www.reactome.org/).
Single Cell Trajectory Analysis
We used Monocle v.2.8.0 24, 52 to investigate inferred developmental trajectories between macrophage/monocyte subsets. Monocle uses the Reverse Graph Embedding to learn the sequence of gene expression changes that each cell undergoes within the dynamic biological sample provided. It than places each cell at an appropriate position along this trajectory of gene expression changes. The data, previously scaled/normalized/clustered by the Seurat tool, was loaded into a monocle object. Dimensionality reduction was performed using the reduceDimension command with parameters max_components = 2, reduction_method= “DDRTree”. The trajectory was than built using the plot_cell_trajectory command with default parameters. Cells were sorted in pseudotime order and placed along a trajectory based on a priori found differentially expressed genes. For each trajectory analysis, we defined the cluster with higher expression of monocytic genes as the “root_state” (the starting point) of the trajectory. Changes in genes that are significantly branch dependent were visualized by a heatmap using the “plot_pseudotime_heatmap”. The kinetic trend of the expression levels of significant genes through pseudotime was depicted by individual graphs using the plot_genes_in_pseudotime function.
The Mpath algorithm25 was employed as a secondary method for assessing developmental trajectories. Mpath constructs multibranching cell lineages and reorders individual cells along the branches. Scaled expression values of variable genes were extracted from Seurat pipeline and were used as input data to Mpath. The five clusters identified by Seurat were used as landmarks. Euclidean distance was used to calculate dissimilarities between cells and landmarks. Tree trimming is done by using trim_net function with method = “mst”.
Supplementary Material
Acknowledgements
This work was supported by the Canadian Institutes of Health Research (S.E. PJT364831, J.A.M., S.A.D.), Heart and Stroke Foundation (S.E.), March of Dimes (S.E.), Ted Rogers Centre for Heart Research (S.E., S.A.D., J.A.M.), the Peter Munk Cardiac Centre (S.E.) and the NIH (S.E. K08HL112826). We would like to thank D. Mann for his insight and advice, and S. Wilson for editorial assistance. Thanks to N. Winegarden and N. Khuu for help with processing single cell RNA sequencing samples.
Abbreviations:
- (CCR2)
C-C chemokine receptor 2
- (MHC-II)
Major Histocompatibility complex class II
- (MI)
Myocardial infarction
Footnotes
Data Availability Statement
Single Cell Sequence data that support the findings of this study is available through the NCBI GEO (GSE119355). The Agilent gene array data that support the findings of this study is available through the NCBI GEO (GSE119515).
Code Availability Statement
R Scripts for data processing is available through [https://github.com/snejat/EpelmanLab_scRNAseq_Cardiac_Macrophages]. There is no restriction to the use of the code or data. DOI 10.5281/zenodo.1407166
Competing financial interest: None
References:
- 1.Lozano R et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380, 2095–2128 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ismahil MA et al. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circ. Res 114, 266–282 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Epelman S, Lavine KJ & Randolph GJ Origin and Functions of Tissue Macrophages. Immunity 41, 21–35 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schulz C et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336, 86–90 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Epelman S et al. Embryonic and Adult-Derived Resident Cardiac Macrophages Are Maintained through Distinct Mechanisms at Steady State and during Inflammation. Immunity 40, 91–104 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ginhoux F & Guilliams M Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 44, 439–449 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Porrello ER et al. Transient regenerative potential of the neonatal mouse heart. Science 331, 1078–1080 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lavine K et al. Distinct Macrophage Lineages Contribute to Disparate Patterns of Cardiac Recovery and Remodeling in the Neonatal and Adult Heart. Proc. Natl. Acad. Sci 111, 16029–16034 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aurora AB et al. Macrophages are required for neonatal heart regeneration. J. Clin. Invest 124, 1382–1392 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leid JM et al. Primitive Embryonic Macrophages are Required for Coronary Development and Maturation. Circ. Res (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Panizzi P et al. Impaired infarct healing in atherosclerotic mice with Ly-6C(hi) monocytosis. J. Am. Coll. Cardiol 55, 1629–1638 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH & van Luyn MJ Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am. J. Pathol 170, 818–829 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Molawi K et al. Progressive replacement of embryo-derived cardiac macrophages with age. J. Exp. Med 211, 2151–2158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Serbina NV & Pamer EG Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol 7, 311–317 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Ridker PM et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med 377, 1119–1131 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Hulsmans M et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell 169, 510–522 e520 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miyanishi M et al. Identification of Tim4 as a phosphatidylserine receptor. Nature 450, 435–439 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Clemente-Casares X et al. A CD103(+) Conventional Dendritic Cell Surveillance System Prevents Development of Overt Heart Failure during Subclinical Viral Myocarditis. Immunity 47, 974–989 e978 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Van der Borght K et al. Myocardial Infarction Primes Autoreactive T Cells through Activation of Dendritic Cells. Cell Rep 18, 3005–3017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gautier EL et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol 13, 1118–1128 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tirosh I et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guilliams M et al. Unsupervised High-Dimensional Analysis Aligns Dendritic Cells across Tissues and Species. Immunity 45, 669–684 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Satpathy AT et al. Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J. Exp. Med 209, 1135–1152 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trapnell C et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J, Schlitzer A, Chakarov S, Ginhoux F & Poidinger M Mpath maps multi-branching single-cell trajectories revealing progenitor cell progression during development. Nat Commun 7, 11988 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parkhurst CN et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yona S et al. Fate Mapping Reveals Origins and Dynamics of Monocytes and Tissue Macrophages under Homeostasis. Immunity 38, 79–91 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leuschner F et al. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J. Exp. Med 209, 123–137 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leuschner F et al. Angiotensin-converting enzyme inhibition prevents the release of monocytes from their splenic reservoir in mice with myocardial infarction. Circ. Res 107, 1364–1373 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nahrendorf M et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med 204, 3037–3047 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wan E et al. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ. Res 113, 1004–1012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lim HY et al. Hyaluronan Receptor LYVE-1-Expressing Macrophages Maintain Arterial Tone through Hyaluronan-Mediated Regulation of Smooth Muscle Cell Collagen. Immunity 49, 326–341 e327 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Majmudar MD et al. Monocyte-directed RNAi targeting CCR2 improves infarct healing in atherosclerosis-prone mice. Circulation 127, 2038–2046 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bajpai G et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med 24, 1234–1245 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Satoh T et al. Identification of an atypical monocyte and committed progenitor involved in fibrosis. Nature 541, 96–101 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Menezes S et al. The Heterogeneity of Ly6Chi Monocytes Controls Their Differentiation into iNOS+ Macrophages or Monocyte-Derived Dendritic Cells. Immunity 45, 1205–1218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ikeda N et al. Emergence of immunoregulatory Ym1(+)Ly6C(hi) monocytes during recovery phase of tissue injury. Sci Immunol 3 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Mrdjen D et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 48, 599 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Han X et al. Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 172, 1091–1107 e1017 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Mass E et al. Specification of tissue-resident macrophages during organogenesis. Science 353 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van de Laar L et al. Yolk Sac Macrophages, Fetal Liver, and Adult Monocytes Can Colonize an Empty Niche and Develop into Functional Tissue-Resident Macrophages. Immunity 44, 755–768 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Gosselin D et al. An environment-dependent transcriptional network specifies human microglia identity. Science 356 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aronoff L, Epelman S & X C-C Isolation and Identification of Extravascular Immune Cells of the Heart. Journal of Visual Experiments 138, 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Noyan-Ashraf MH et al. GLP-1R agonist liraglutide activates cytoprotective pathways and improves outcomes after experimental myocardial infarction in mice. Diabetes 58, 975–983 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lun A, McCarthy D & Marioni J A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000 Res 5, 2122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCarthy D, Campbell K, Lun A & Wills Q Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 33, 1179–1186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blondel VD, Guillaume JL, Lambiotte R & Lefebvre E Fast unfolding of communities in large networks. J Stat Mech-Theory E (2008). [Google Scholar]
- 50.Finak G et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol 16, 278 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reimand J et al. g:Profiler—a web server for functional interpretation of gene lists (2016 update). Nucleic Acid Res 44, W83–W89 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qiu X et al. Reversed graph embedding resolves complex single-cell trajectories. Nature Methods 14, 979–982 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.