

Abstract
The unique polarization and high-energy demand of neurons necessitates specialized mechanisms to maintain energy homeostasis throughout the cell, particularly in the distal axon. Mitochondria play a key role in meeting axonal energy demand by generating ATP through oxidative phosphorylation. Recent evidence demonstrates how axonal mitochondrial trafficking and anchoring are coordinated to sense and respond to altered energy requirements. If and when these mechanisms are impacted in pathological conditions, such as injury and neurodegenerative disease, is an emerging research frontier. Recent evidence also suggests that axonal energy demand may be supplemented by local glial cells, including astrocytes and oligodendrocytes. In this review, we provide an updated discussion of how oxidative phosphorylation, aerobic glycolysis, and oligodendrocyte-derived metabolic support contribute to the maintenance of axonal energy homeostasis.
Keywords: mitochondria, neuronal energetics, oligodendrocytes, axonal transport, glia, metabolic transporters, axonal energy metabolism
INTRODUCTION
The human brain is comprised of ~86 billion neuronal cells and ~84 billion glial cells (Azevedo et al, 2009), all wired in intricate networks from which our thoughts, movements, and experiences emerge. Considering the elaborate structure and function of neural circuitry, it is not surprising that the brain consumes ~20-25% of our daily energy budget (Herculano-Houzel, 2012; Mink et al, 1981; Rolfe & Brown, 1997). Action potentials, maintenance of ionic gradients, organelle transport, neurotransmitter uptake, synaptic vesicle recycling, and axonal growth require continual energy production within the brain. Energy substrates, such as glucose, ketone bodies, and lactate (Camandola & Mattson, 2017), are supplied to the brain via adaptive changes in blood flow (Ogawa et al, 1992). These substrates are metabolized to generate adenosine triphosphate (ATP), the primary energy currency for all living organisms. As ATP diffusion is not effective over the long distances of neuronal processes (Hubley et al, 1996; Sun et al, 2013), neurons require specialized mechanisms to maintain energy homeostasis throughout the cell, particularly in distal axons that can extend several centimeters to several feet long for some peripheral nerves (Saxton & Hollenbeck, 2012). Precisely how neurons sense and respond to dynamic changes in energy demand during rest, activation, growth, and in pathological conditions is still being elucidated (Sheng, 2017). However, considering that mitochondrial dysfunction is associated with numerous neurodegenerative diseases (Pathak et al, 2013; Sheng & Cai, 2012), determining if and when bioenergetic failure in distal axons contributes to neuronal pathology is highly clinically relevant. For these reasons, the study of axonal energy metabolism is an important emerging frontier.
A continuous supply of ATP is essential for cell growth, survival, and function in the brain, with synapses being the primary sites of ATP consumption (Attwell & Laughlin, 2001; Harris et al, 2012; Lennie, 2003; Nicholls & Budd, 2000; Sheng, 2017). Within neurons, ATP is primarily generated by the complete oxidation of glucose to carbon dioxide (CO2) and water. Glucose is first catabolized into pyruvate through glycolysis, a ten-step enzymatic process occurring in the cytosol. Pyruvate is subsequently transported into the mitochondria and metabolized into acetyl-coenzyme A (acetyl-CoA). In the presence of oxygen, acetyl-CoA is processed in the mitochondrial matrix via the citric acid cycle, generating the reducing equivalents NADH and FADH. Electrons from NADH and FADH are then passed through the electron transport chain complexes, thereby establishing an electrochemical gradient across the inner mitochondrial membrane. This gradient allows H+ to flow back into the mitochondrial matrix via ATP synthase, powering the regeneration of 30-36 ATP (per one glucose molecule) in a process known as oxidative phosphorylation (oxphos). During high levels of oxphos, glycolytic pyruvate production can exceed mitochondrial utilization. Under these circumstances, excess pyruvate is converted into lactate by lactate dehydrogenase (LDH), generating oxidized NAD+ and ~4 ATP molecules. This process of lactate production in the presence of oxygen is known as aerobic glycolysis. The first indication that neurons might undergo aerobic glycolysis was the observation that oxygen uptake is small relative to glucose uptake during brain activation (Fox et al, 1988; Madsen et al, 1999), suggesting that glycolysis is upregulated to a greater extent than oxphos during stimulation. Diaz-Garcia et al recently observed increased cytosolic NADH levels in the soma of stimulated neurons in acute hippocampal slices, demonstrating that neuronal glycolysis exceeds oxphos during activation (Diaz-Garcia et al, 2017). However, the differential upregulation of oxphos and aerobic glycolysis within specific intracellular compartments and during various activation states remains controversial.
In this review, we focus on energy supply and maintenance in the axonal process. Axons serve as the conduit for action potential generation and propagation, transport of synaptic machinery, and delivery of intracellular organelles. Following the generation of a single action potential, an estimated 400-800×106 ATP molecules are required to restore the electrochemical gradient via Na+/K+ pumping (Attwell & Laughlin, 2001; Hallermann et al, 2012; Lennie, 2003). For comparison, it has been estimated that a single bacterial cell requires ~3×106 ATP molecules per second to meet its energy needs (Pommerville, 2013). Thus, efficient regulation of energy production and maintanence within axons is critical for neurons to meet their dynamic energy requirements. Elucidating the mechanisms by which axons maintain this energy balance is crucial, as white matter regions are often the first to succumb to energy deficits and neurodegenerative pathology. This review provides an updated overview as to how neurons maintain energy homeostasis in distal axons. We discuss exciting new evidence demonstrating how axonal mitochondrial trafficking and anchoring are coordinated to sense and respond to altered energy requirements under physiological and pathological stress conditions. In addition, we discuss the emerging hypothesis that oligodendrocytes support axonal energy metabolism, review the evidence surrounding this hypothesis, and highlight questions that remain to be answered. For additional insights and alternative perspectives of neuronal energy metabolism, we refer readers to the following articles (Ashrafi & Ryan, 2017; Attwell & Laughlin, 2001; Chih & Roberts, 2003; Dienel, 2012; Harris & Attwell, 2012; Magistretti & Allaman, 2015; Pathak et al, 2013; Pellerin et al, 2007; Riske et al, 2017; Sheng, 2017; Yellen, 2018).
AXONAL ATP PRODUCTION AND MAINTENANCE
Presynaptic ATP production in maintaining synaptic transmission
Several studies demonstrate the importance of local, mitochondrial-derived ATP in maintaining synaptic function. In Drosophila, depletion of presynaptic mitochondria results in defective mobilization of synaptic vesicle reserve pool and failed neurotransmission following high frequency stimulation, which can be rescued by the addition of ATP (Verstreken et al, 2005). In another study, depletion of synaptic mitochondria inhibited mobilization of synaptic vesicles in the readily releasable pool in mature neurons (Ma et al, 2009). Even in hippocampal synaptosomes, inhibition of oxphos reduces ATP levels and evoked synaptic vesicle release, directly demonstrating that synaptic physiology is affected by local, mitochondrial ATP production (Ivannikov et al, 2013). By 3D EM analysis of hippocampi, the Harris group recently demonstrated that only 33% of total presynapses retain mitochondria and sustained synaptic activity is restricted to these mitochondria-containing presynapses during long-term potentiation (Smith et al, 2016). However, the mechanisms retaining and anchoring mitochondria at presynapses remain elusive.
In cultured mature neurons, approximately 20-30% of axonal mitochondria move bi-directionally, some of which pass through, pause briefly, or remain at presynapses (Saxton & Hollenbeck, 2012; Sheng, 2014). Our lab has characterized mitochondrial motility patterns in axons by imaging of live hippocampal neurons: while 16.29% of mitochondria are stationed within presynapses, 14.77% and 15.31% of mitochondria quickly pass through or pause briefly at presynapses, respectively (Sun et al, 2013). These patterns are consistent with a previous study in cortical neurons (Chang et al, 2006a). These studies support the model that anchored presynaptic mitochondria continuously supply ATP to sustain prolonged synaptic transmission and presynapses lacking an anchored mitochondrion must rely on ATP via local glycolysis or diffusion from mitochondria outside of synapses, which may not be adequate for meeting the high energy requirements during repetitive synaptic transmission. To test this model, our lab utilized syntaphilin (snph) KO neurons, in which SNPH-mediated axonal mitochondrial anchoring is abolished (Kang et al, 2008a). We demonstrated that enhanced axonal mitochondrial transport dynamically alters presynaptic ATP levels and thus influences ATP-dependent presynaptic functions, contributing to the pulse-to-pulse variability of presynaptic strength (Sun et al, 2013). These studies reveal a key role for anchored mitochondria as an ideal energy station by constantly supplying ATP to fuel synaptic function, converging on a role for mitochondrial oxphos in sustaining synaptic vesicle release and recycling.
As mitochondria appear to generate ~93% of ATP in presynaptic terminals, glycolysis appears to contribute the remaining 7% (Harris et al, 2012). This supplementary role for glycolysis in supplying presynaptic ATP was suggested in the 1980’s, when several biochemical studies reported an enrichment of glycolytic enzymes at the synapse (Knull, 1978; Knull et al, 1980; Knull & Fillmore, 1985; Lipton & Robacker, 1983). Synaptic clustering of a “glycolytic metabolon” has also been observed during energetic stress (Jang et al, 2016). A combined role for oxphos and glycolysis has been observed by the Ryan Lab, which utilized a fluorescent sensor for presynaptic ATP to demonstrate that neuronal activity and the associated calcium influx drives ATP synthesis through both glycolysis and oxphos (Rangaraju et al, 2014). Further, activity recruits the glucose transporter GLUT4 to the plasma membrane in order to upregulate local glycolysis (Ashrafi et al, 2017). Interestingly, while glycolytic inhibition only reduced ATP levels by 19%, there was a 50% reduction in synaptic vesicle endocytosis (Rangaraju et al, 2014). Inhibition of mitochondrial oxphos also reduced endocytosis, but much higher stimulation conditions were required to achieve complete arrest (Rangaraju et al, 2014). These studies demonstrate that both glycolysis and oxphos contribute to presynaptic function by generating ATP in an activity-dependent manner. Similarly, Sobieski et al found that simultaneous inhibition of both glycolysis and oxphos is required to achieve complete inhibition of evoked synaptic transmission (Sobieski et al, 2017). Therefore, it is likely that both oxphos and aerobic glycolysis contribute to synaptic energy metabolism and offer redundancies that confer energetic flexibility for synaptic activity.
Mechanisms controlling mitochondrial positioning in axons for local energy supply
As the large majority of mitochondrial proteins are encoded in the nucleus, mitochondrial biogenesis is thought to occur primarily in the soma. Considering that it would take an estimated 10 years for ATP to diffuse from the cell body down a 1-m long axon (Harris et al, 2012), a crucial component of fulfilling local axonal ATP demand is the transport of energy-producing machinery from the cell body to the distal axons (anterograde movement). Axonal delivery of mitochondria is driven by ATP-hydrolyzing motor proteins along microtubule tracks. In neurons, motor proteins of the kinesin-1 family (KIF5A, KIF5B, KIF5C) drive anterograde movement from the soma toward distal regions (Cai et al, 2005; MacAskill et al, 2009). Mitochondria return to the cell body via retrograde transport driven by dynein motor proteins (Hirokawa et al, 2010) (Figure 1). Current evidence suggests that motor proteins carrying mitochondria hydrolyze ATP derived locally from mitochondrial oxphos to power transport (Zala et al, 2013). Interestingly, approximately 70-80% of mitochondria are held stationary at any given time, suggesting that mitochondrial positioning is crucial for local energy supply. Our lab previously demonstrated that axonal mitochondria are held static by a molecular anchoring protein known as syntaphilin (SNPH) via microtubule interaction (Chen & Sheng, 2013; Kang et al, 2008b). SNPH targets axonal mitochondria through its carboxyl-terminal mitochondria-targeting domain and axon-sorting sequence. Deleting snph results in a robust increase of axonal mitochondria in the motile pool both in vitro and in vivo. In contrast, overexpressing SNPH abolishes mitochondrial transport in axons. Thus, the snph knockout mouse serves as a genetic model for investigating the impact of altered axonal mitochondrial trafficking and anchoring on synaptic transmission, nerve regeneration and degeneration (Sheng, 2017). In addition, Myosin VI (Pathak et al, 2010) and GlcNAcytlation of the mitochondrial protein Milton (Pekkurnaz et al, 2014) also arrest mitochondrial motility. More thorough reviews of mechanisms of mitochondrial trafficking and anchoring can be found in other articles (MacAskill & Kittler, 2010; Misgeld & Schwarz, 2017; Schwarz, 2013; Sheng, 2014; Sheng & Cai, 2012).
Figure 1. Mitochondrial trafficking and anchoring in the axonal compartment.
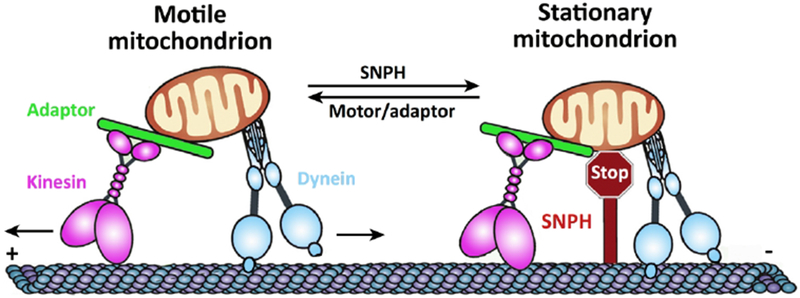
Mitochondria are transported down the axon by microtubule-based motor proteins, with kinesin moving towards the plus-end (anterograde) and dynein moving towards the minus-end (retrograde) of the uniformly arranged microtubules. Kinesin-1 motor proteins attach to mitochondria via adaptor proteins. Static mitochondria are anchored to microtubules via Syntaphilin (SNPH), which holds mitochondria stationary. Adapted with modification from Sheng et al., JCB, 2017.
Mitochondria anchored at distal axons and synapses ideally serve as local energy sources; thus, regulating the trafficking and anchoring status of axonal mitochondria ensures that metabolically active areas are constantly supplied with ATP. The directionality of mitochondrial transport also plays a crucial role in quality control, as aged and dysfunctional mitochondria are preferentially transported back to the cell body to maintain health of the overall mitochondrial pool in axons (Lin et al., 2017). Mitochondrial dysfunction and impaired transport are hallmark features of several major neurodegenerative diseases, thus elucidating the mechanisms regulating mitochondrial trafficking and anchoring represents an important emerging field. One study demonstrated that the vast majority of mitochondria with high membrane potential are transported towards the growth cone, while those dysfunctional mitochondria with low membrane potential are transported back towards the cell body (Miller & Sheetz, 2004). However, other studies using alternative labeling dyes and mitochondrial uncouplers have observed no bias in transport direction (Baqri et al, 2009; Verburg & Hollenbeck, 2008). Our lab recently set out to address this controversy by providing mechanistic insight of biased retrograde transport for stressed mitochondria (Lin et al, 2017). As elevated SNPH expression in mature neurons (Kang et al, 2008a; Zhou et al, 2016) immobilizes the majority of axonal mitochondria in vitro and in vivo, we asked whether mature neurons have an intrinsic mechanism for turning off the SNPH anchoring mechanism in order to remobilize stressed mitochondria in distal axons under chronic stress conditions. To test this, we recapitulated chronic mitochondrial dysfunction by treating neurons with a very mild stress (5 nM Antimycin-A), a 1000× lower dosage than previously reported in the literature. This mild treatment induced chronic and reversible mitochondrial stress. Surprisingly, axonal mitochondria responded to this mild stress by biased directional transport, favoring the removal of stressed mitochondria out of axons (Lin et al, 2017). We further demonstrated that this transport regulation was triggered by bulk release of the anchor protein SNPH in the form of cargo vesicles budding from stressed mitochondria. Consistently, dysfunctional mitochondria robustly released SNPH cargo vesicles in the axons of spinal ventral root motor neurons during the early asymptomatic stages of fALS-linked SOD1G93A mice and of AD-related hAPP mutant cortical neurons in the early pathological stages. Releasing SNPH remobilizes stressed mitochondria and enhances their retrograde transport to the soma for repair or degradation via lysosomes. Progressive mitochondrial damage depleted SNPH after disease onset in both fALS- and hAPP-linked mouse brains and postmortem human brains from AD patients. This pathway is likely the first-line of surveillance in maintaining axonal mitochondrial integrity and thus energy homeostasis in response to chronic pathological stress (Lin et al, 2017).
Regulation of mitochondrial trafficking and anchoring to meet energy requirements
In order to meet the dynamic energy requirements in the distal axonal compartment, mitochondria must be trafficked and positioned appropriately. Precisely how motor proteins are recruited to and released from transport cargo remains unknown. It is assumed that localized reductions in energy availability serve as a signal to nearby mitochondria, which may then be recruited and positioned to meet local energy demand. Emerging evidence suggests that neurons may also coordinate mitochondrial biogenesis by sensing local energy levels. While the chemical mediator of this signaling mechanism has not been identified, several have been suggested. The ATP/ADP ratio was implicated as a signaling mechanism because increased ADP levels halt mitochondrial motility in respiratory neurons (Mironov, 2007). Additional studies investigated the ATP/ADP ratio in the context of growing axons, which require a considerable amount of energy to power the synthesis of raw building materials and the delivery of these materials to growing tips (Vaarmann et al, 2016). AMPK, an AMP-activated protein kinase, is a master regulator of cellular energy homeostasis and is activated upon AMP/ADP elevation (Mihaylova & Shaw, 2012). Activation of mitochondrial biogenesis through the AMPK–PGC-1α–NRF1 axis accelerates generation of new mitochondria and increases mitochondrial density and the ATP/ADP ratio in axonal terminals, thereby ensuring sufficient energy-production for axonal growth. Our lab recently examined this issue using developing cortical neurons and found a strong correlation of mitochondrial distribution patterns and the ATP/ADP ratio in the distal tips of growing axons (Zhou et al, 2016). Over-expressing SNPH in developing neurons restricted axonal mitochondria within the proximal axons due to reduced axonal flux, which correlated with a reduced ATP/ADP ratio in the distal axon segment and small growth cones. Conversely, overexpressing Miro1 increased mitochondrial density in distal axons and the average size of growth cones. These results demonstrate that alterations in the ATP/ADP ratio are tightly correlated with mitochondrial density and positioning in growing tips. However, precisely how mitochondria sense and respond to changes in the ATP/ADP ratio via immobilization or remobilization will be a significant discovery.
The Polleux lab provided mechanistic insights as to how the balance between motile and stationary mitochondria alters axonal growth status (Courchet et al, 2013). This study identifies two kinases, LKB1 and the AMPK-related kinase NUAK1, as important mediators of axon branching and mitochondria capture at nascent presynaptic sites. Intriguingly, this study also highlighted a critical role for SNPH, by demonstrating that SNPH-mediated mitochondrial anchoring promotes axon branching both in vitro and in vivo. A second study provided evidence that AMP activation is required for axonal branch formation in an ATP-dependent manner (Tao et al, 2014). Activation of AMPK increased anterograde flux of mitochondria toward axonal terminals and increased mitochondrial positioning before new axonal branches were emerged. A third study revealed an intriguing mechanism underlying the role of mitochondria in determining axon branch sites (Spillane et al, 2013). Anchored mitochondria promote the maturation of axonal filopodia into axon branches through ATP generation; blocking mitochondrial ATP production impairs maturation of axonal branches, even when mitochondria remain anchored at these hot spots. Together, these studies support the notion that regulation of mitochondrial trafficking and anchoring, likely through activation of AMPK, is critical to maintain the necessary ATP supply for localized axonal growth and branching.
Local Ca2+ concentration has also been implicated as an indicator of energy availability, as elevated intracellular calcium levels recruit mitochondria to active synapses via the mitochondrial outer membrane proteins Miro1/2 (Chang et al, 2006a; Rintoul et al, 2003; Yi et al, 2004). As mitochondrial motor receptors, Miro1/2 link mitochondria to kinesin KIF5 motors. Upon sensing cytosolic Ca2+, Miro1/2 change conformation and disrupts its coupling to transport motors (Fransson et al, 2003; MacAskill et al, 2009; Wang & Schwarz, 2009). We refer the reader to more thorough reviews of Miro-Ca2+ signaling regulation of mitochondrial transport (MacAskill & Kittler, 2010; Saxton & Hollenbeck, 2012; Schwarz, 2013; Sheng, 2014; Sheng & Cai, 2012). Our lab recently revealed that mitochondrial anchoring is also regulated by the Ca2+ signaling associated with synaptic activity via modulation of the KIF5-SNPH motor-anchor interaction (Chen & Sheng, 2013). SNPH competes with the KIF5 adaptor Trak2 to bind with KIF5 motors and inhibits motor ATPase activity. Consistently, deleting the snph gene abolished activity-dependent mitochondrial anchoring in axons. These findings suggest an “engine-switch and brake” model: when a motile mitochondrion passes by an activated presynapse, SNPH responds to elevated Ca2+ levels (stop sign) by switching off the engine (motor) and places a brake on the motile mitochondrion, thus arresting the mitochondrion at an active synapse. When the Ca2+ signal is removed, the motor-adaptor complexes are quickly reactivated and thus mitochondria are remobilized.
The Schwarz lab recently revealed an alterative pathway for regulating mitochondrial transport by sensing local supply of glucose, the main carbon source for ATP production in mitochondria. Activity level of the enzyme O-GlcNAc Transferase (OGT) is dependent on glucose availability and its activation arrests mitochondrial motility via modification of Milton (Pekkurnaz et al, 2014). Through this pathway, mitochondrial transport is arrested in areas where glucose level is elevated, thus ensuring rapid ATP production. Further work is needed to understand how the glucose signaling pathway works in conjunction with the complex regulation of mitochondrial transport in response to changes in the local ATP/ADP ratio, synaptic activity, and Ca2+ signals under physiological and pathological conditions.
Axonal energy deficits in disease and injury
While energy deficits are associated with many neurodegenerative diseases (Pathak et al, 2013), the fundamental question of whether bioenergetic failure in axons and synapses contributes to early disease pathology remains unknown. Mitochondrial dysfunction and transport deficits are prominent features of major neurodegenerative diseases including Alzheimer’s Disease (AD), Parkinson’s Disease (PD), Huntington’s Disease (HD), ALS, Charcot-Marie-Tooth Disease (CMT), and Stroke (for extensive reviews see (Chen & Chan, 2009; Raefsky & Mattson, 2017; Sheng & Cai, 2012). Axonal swellings containing accumulated organelles and pathological proteins may inhibit mitochondrial transport. These axonal swellings are prevalent in the early stages of disease in both mouse models of AD and human AD samples (Stokin et al, 2005). Using the Aβ precursor protein transgenic (AβPP) mouse model for AD, Calkins et al (2011) demonstrated reduced anterograde motility of mitochondria into axons of primary neurons (Calkins et al, 2011). Mutant Huntington protein also causes alterations in mitochondrial transport (Chang et al, 2006b; Trushina et al, 2004), suggesting that lack of axonal mitochondria may play a role in the degeneration of striatal neurons in Huntington’s Disease. Several studies have reported alterations of axonal mitochondrial transport in models of ALS (Bilsland et al, 2010; Collard et al, 1995; Sasaki et al, 2005; Warita et al, 1999; Williamson & Cleveland, 1999). Interestingly, work from our lab demonstrated that rescuing mitochondrial motility in hSOD1G93A mice by crossing with snph KO mice does not slow ALS-like pathological processes (Zhu & Sheng, 2011), suggesting that impaired mitochondrial transport is likely a manifestation of a more generalized transport defect. This is further supported by our recent study showing that imparied axonal trafficking and maturation of the endolysosomal system interferes with the removal of damaged mitochondria in distal axons through the autophagic-lysosomal system in fALS-linked hSOD1G93A mice (Xie et al, 2015).
Overall, impaired trafficking in neurodegeneration is likely the result of combined disruptions in mitochondrial activity, transport machinery, and associations with microtubules (Gunawardena & Goldstein, 2001; Trushina et al, 2004). This model is further complicated by the contribution of normal aging, as we and others have demonstrated that axonal transport is decreased in aged neurons (Lewis et al, 2016; Milde et al, 2015; Morsci et al, 2016; Takihara et al, 2015; Vagnoni & Bullock, 2018). Aging is the primary risk factor for the development of neurodegenerative diseases, thus the challenge for future studies is to isolate the specific effects of disease pathology from normal aging on mitochondrial trafficking.
Energy deficit is defined as insufficient ATP supply due to mitochondrial damage and/or during bouts of increased energy consumption, such as axonal regeneration. Mitochondrial damage by axonal injury, mature neuron-associated decline of axonal mitochondrial transport (Zhou et al, 2016), and enhanced energy consumption collectively contribute to energy deficits in injured axons. Regenerating axons appear to adapt to this increased energy demand by altering mitochondrial dynamics (Kiryu-Seo & Kiyama, 2018). Misgeld et al (2007) demonstrated that following transection of the intercostal nerve, anterograde transport of axonal mitochondria proximal to the injury site increased by more than 80% to populate regenerating axonal tips (Misgeld et al, 2007). Another study reported increased velocity of anterogradely-moving mitochondria following sciatic nerve injury (Kiryu-Seo et al, 2016). Interestingly, mitochondrial response is not the same across all neurons and axons that adaptively increase their mitochondria exhibit a higher regenerative capacity (Cartoni et al, 2017; Han et al, 2016). In agreement, increasing axonal mitochondrial transport via knockout of the mitochondrial anchoring protein SNPH (Zhou et al, 2016) or by overexpression of the mitochondrial protein Armcx1 (Cartoni et al, 2016) improves axonal regeneration. Enhanced mitochondrial transport rescues energy deficits by replenishing more healthy mitochondria to injured axons (Zhou et al, 2016). Thus, activating an intrinsic “regeneration program” requires the coordinated recovery of energy supply through enhanced mitochondrial transport. Such coordinated regulation may represent a valid therapeutic strategy to facilitate nerve regeneration and recovery after injury and diseases. Future development of safe and effective small molecule compounds will be an attractive strategy to selectively increase mitochondrial motility and rescue the local energy deficits within injured axons (Kaasik, 2016; Sheng, 2017).
Regulation of axonal glycolysis
Considering the ongoing debate about the role of neuronal glycolysis, we find it worthwhile to summarize what little is known about the glycolytic capacity of the axonal compartment. Unlike mitochondria, soluble glycolytic enzymes are transported to the axon with other cytosolic proteins via slow axonal transport component b (SCb) (Brady et al, 2013). It is presumed that kinesins and dyneins mediate SCb motility, although the precise mechanisms underlying the recruitment of motor proteins to SCb cargoes remains to be elucidated (Maday et al, 2014). Interestingly, the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) localizes on vesicles via a Huntington-dependent mechanism and provides “on-board” energy for fast axonal transport of fast moving vesicles, including BDNF, APP, and TrkB (Zala et al, 2013). While very little is known about glycolysis in healthy axons, it appears that this process might play an important role in protecting axons from Wallerian degeneration. Rescue of basal glycolysis rather than oxphos appears to underlie the protective effect of the Sarml−/− mutation in a model of Wallerian degeneration (Godzik & Coleman, 2015). Similarly, the protective effect of nicotinamide mononucleotide adenylytransferase (NMNAT) against Wallerian degeneration is not mediated by mitochondria, as axons depleted of mitochondria via Milton loss-of-function are still protected (Kitay et al, 2013). While these studies implicate glycolysis in axonal energetic homeostasis and recovery from injury, more work is needed to determine if and when glycolysis contributes to axonal energy metabolism.
ENERGETIC COUPLING OF GLIA AND AXONS
A comprehensive model of energy metabolism within axons must include the influence of surrounding glial cells, including astrocytes, oligodendrocytes, and microglia. Most CNS axons exist in white matter, in which oligodendrocyte glial cells wrap lipid-rich myelin sheathes in concentric spirals around the axon. Myelinated segments (internodes) are separated by nodes of Ranvier, unmyelinated regions where ion channels are clustered at high densities. This organization is key for saltatory propagation, the process by which action potentials are regenerated from node to node, effectively speeding up impulse propagation (Bhatt et al, 2014; Frankenhaeuser, 1952; Hartline & Colman, 2007; McDougall et al, 2018). Myelin sheaths physically isolate axonal internodes from the extracellular milieu, whereas nodes of Ranvier are exposed to the extracellular environment and are frequently contacted by fibrous astrocytes (Black & Waxman, 1988; Butt et al, 1994; Raine, 1984). This arrangement suggests that white matter astrocytes and oligodendrocytes are well positioned to supplement axons with energy metabolites. Several studies have described an astrocyte-to-neuron L-lactate shuttle (ANLS), in which glycolytic astrocytes export lactate to be taken up by neurons, where it is converted into pyruvate and shuttled through the mitochondria for ATP production (Magistretti et al, 1993; Pellerin & Magistretti, 1994; Pellerin & Magistretti, 2012). The Ransom lab published several studies examining the transfer of energy substrates from white matter astrocytes to axons in optic nerve preparations. Their model suggests that glycogen, which is primarily stored within astrocytes in the CNS, is converted into lactate and exported from astrocytes into neuronal axons during aglycemia and increased metabolic demand (Brown et al, 2004; Brown & Ransom, 2015; Brown et al, 2005; Brown et al, 2003; Tekkok et al, 2005). These studies have been thoroughly discussed in a recent review (Baltan, 2015). As the vast majority of ANLS literature is not focused on white matter per se, we refer the reader to alternative sources for more exhaustive consideration of this hypothesis (Alberini et al, 2018; Barros, 2013; Dienel & McKenna, 2014; Pellerin & Magistretti, 2012; Riske et al, 2017; Tang, 2018; Weber & Barros, 2015). It is important to note the widespread controversy over the existence of the ANLS, with substantial debate around whether neurons actually export lactate during heightened activity, leading to substantial lactate efflux from the brain (Chih et al, 2001; Chih & Roberts, 2003; Dienel, 2017; Dienel & Cruz, 2015; Ferguson et al, 2018; Riske et al, 2017; Yellen, 2018). Overall, significantly more work is needed in order to parse the influence of white matter astrocytes on modulating axonal energy metabolism.
Considering the physical isolation of myelinated axons from the extracellular milieu, there has been growing interest in the hypothesis that oligodendrocytes may support axons via supply of energy substrates. It has long been known that oligodendrocytes are necessary for neuronal survival and maintenance of the axonal compartment (Edgar et al, 2009; Griffiths et al, 1998; Lappe-Siefke et al, 2003; Nave & Trapp, 2008; Saab et al, 2013). Interestingly, several groups have demonstrated a functional uncoupling of myelination and oligodendrocyte-derived axonal support. Mice harboring mutations in oligodendrocyte-specific proteins, such as proteolipid protein (PLP1) or 2’,3’-cyclic nucleotide 3’-phosphodiesterase (CNP), exhibit axonal pathology and degeneration despite no overt signs of myelin abnormality (Edgar et al, 2010; Edgar et al, 2009; Griffiths et al, 1998; Lappe-Siefke et al, 2003; Yin et al, 2016). In addition, targeted ablation of oligodendrocyte cell bodies leads to axonal pathology well before myelin structure is affected (Oluich et al, 2012; Traka et al, 2010). Together, these studies demonstrate that axonal integrity and long-term survival are dependent on oligodendrocyte-derived protective functions independent of the effect of myelination on conduction velocity. Perhaps these alternative functions for oligodendrocytes could shed light on the role of non-myelinating perineuronal oligodendrocytes. Along the same lines, the Arlotta lab reported a seminal study describing partial myelination profiles of somatosensory neurons in the superficial cortex (Tomassy et al, 2014). These neurons exhibited myelinated segments intermixed with long sections of unmyelinated axon, unlike the all-or-none scheme necessary for saltatory conduction. Although this pattern cannot be generalized to all neurons, it does suggest that oligodendrocytes may serve a purpose other than saltatory conduction in some regions of the brain. Parvalbumin-positive inhibitory basket cells are also myelinated in a patchy fashion (Micheva et al, 2016). This finding was especially surprising, as partial myelination of such short-range axons is unlikely to speed up impulse propagation in any meaningful way. Aside from promoting saltatory conduction, what oligodendrocyte function(s) are critical for axonal health? Oligodendrocyte support of axonal energy metabolism has emerged as a leading hypothesis. Here, we review the evidence surrounding this hypothesis and highlight questions that remain to be answered.
Theoretical basis for oligodendrocyte-derived metabolic support
Axons can extend several feet in length (Saxton & Hollenbeck, 2012) and thus require an enormous amount of energy to maintain their integrity and function (Attwell & Laughlin, 2001; Hallermann et al, 2012; Lennie, 2003; Sheng, 2017). Due to this energetic vulnerability, it was suggested that axons might require additional metabolic support from neighboring glia. To probe the veracity of this supposition, Harris and Atwell performed a detailed energy budget investigating whether axonal mitochondria in the fully myelinated optic nerve could theoretically provide enough ATP to power action potential firing and maintenance of the resting membrane potential. They found that for optic nerve axons with a diameter larger than 0.9μm, local mitochondria produce more ATP than used for action potentials. They concluded that large-diameter axons can sustain their energy demand via mitochondrial oxphos with glucose import at the nodes. Interestingly, they also reported that small diameter fibers have a 38% shortfall in ATP production compared to ATP usage (Harris & Attwell, 2012), suggesting that small axons may utilize glial-derived metabolites for energetic support. ‘Myelinic channels’, tube-like cytosolic spaces that run parallel to the periaxonal space, could serve as a transport site for oligodendrocyte-derived soluble components to the axonal compartment (Snaidero & Simons, 2014). Additional tools and experiments are needed to test this assumption in vivo.
Experimental evidence for oligodendrocyte-derived metabolic support
Axonal mitochondrial pathology is associated with oligodendrocyte dysfunction. A predominant factor in the emergence of the metabolic support hypothesis is the presence of mitochondrial abnormalities in a wide array of disease models characterized by primary oligodendrocyte dysfunction. These changes primarily fall into three categories: alterations in axonal mitochondrial activity, density, and transport.
1. Mitochondrial Activity:
Bristow et al demonstrated that axonal mitochondrial enzyme activity is significantly reduced following developmental myelination of human and rabbit optic nerve tissue (Bristow et al, 2002). The opposing situation occurs in Shiverer mice, which harbor a mutation in myelin basic protein (MBP) and exhibit widespread myelination failure accompanied by twofold higher cytochrome c oxidase expression in white matter tracts at 3 months of age (Andrews et al, 2006). These studies suggest that myelinated axons are more energy efficient or perhaps less reliant on mitochondrial oxphos. If this assumption were true, then demyelinated axons would be expected to compensate by upregulating energy production. Indeed, large diameter axons within inactive chronic multiple sclerosis (MS) lesions contain higher complex IV activity, increased mitochondrial mass, and elevated expression of the axonal mitochondrial anchoring protein syntaphilin (SNPH) (Mahad et al, 2009). Interestingly, small diameter axons do not exhibit adaptive changes in mitochondria, which the authors suggest could underlie the predominant loss of small diameter demyelinated axons (Mahad et al, 2009). This notion is also theoretically supported by a white matter energy budget demonstrating that small diameter fibers have a 38% shortfall in ATP production compared to ATP usage (Harris & Attwell, 2012). Importantly, a subsequent study of human biopsies obtained from actively demyelinating MS lesions, demonstrated that mitochondrial damage and loss of membrane potential is the first pathological change to occur prior to demyelination or focal axonal degeneration (Nikic et al, 2011). A recent study also emphasized mitochondrial pathology as an early effect of oligodendrocyte dysfunction in a mouse model of hereditary spastic paraplegia, in which PLP in the CNS was replaced with the peripheral myelin protein Po (Yin et al, 2016). By 1 month of age, mitochondrial cristae are reduced, ATP levels are low, and axonal degeneration is prevalent, strongly suggesting that oligodendrocyte dysfunction leads to mitochondrial damage and energy failure (Yin et al, 2016).
2. Mitochondrial Density:
Oligodendrocytes also appear to regulate the density of axonal mitochondria. In the developing cat, myelinated afferent fibers contain significantly less mitochondria than unmyelinated fibers (Heppelmann et al, 1994). Oppositely, experimentally demyelinated axons in the cat optic nerve exhibit increased mitochondrial density, which returns to control levels once remyelination is complete (Mutsaers & Carroll, 1998). Models of dysmyelination, including the myelin-deficient Wistar rat and the Shiverer mouse, also exhibit increased axonal mitochondria (Andrews et al, 2006; Dentinger et al, 1985). Hemizygous Plp1-overexpressing mice, which exhibit developmentally normal myelination followed by partial demyelination later in life (Anderson et al, 1998), serve as a model for adaptive changes to myelin status. These mice harbor increased axonal mitochondrial density in the spinal cord and optic nerve (Hogan et al, 2009). Importantly, these findings are recapitulated in human disease, as demyelinated axons contain significantly more axonal mitochondria compared to control axons in MS issue (Witte et al, 2009; Zambonin et al, 2011). Collectively, these findings provide additional support for the hypothesis that oligodendrocytes regulate axonal mitochondrial density and the associated energetics.
3. Mitochondrial Transport:
One possible explanation for increased mitochondrial density in demyelinated and dysmyelinated axons is compensatory changes in axonal transport. Interestingly, oligodendrocytes regulate the development and maintenance of the axolemma (Davis et al, 1996; Einheber et al, 1997; Kordeli et al, 1990; Menegoz et al, 1997; Rasband et al, 1999), providing a clear link to microtubule arrangement and organelle motility. The presence of bulbous axonal swellings containing a wide array of organelles in models of oligodendrocyte dysfunction supports the hypothesis that oligodendrocytes regulate transport of axonal organelles. For example, although PLP/DM20-null mice develop normally and exhibit compact myelination, axonal swellings containing mitochondria, dense bodies, and multivesicular bodies begin appearing between 6-8 weeks (Griffiths et al, 1998). A later study of PLP-deficient mice and tissue from human patients also demonstrated axonal accumulations of mitochondria accompanied by length-dependent axonal degeneration (Garbern et al, 2002). Similarly, CNP-null mice exhibit axonal swellings containing mitochondria, dense bodies, and multivesicular bodies that later progress to neurodegeneration and premature death (Lappe-Siefke et al, 2003). Mitochondria also accumulate in the bulbous swellings of demyelinated spinal cord axons in DTA mice, in which oligodendrocyte cell bodies are targeted by diphtheria toxin and selectively ablated (Traka et al, 2010). Interestingly, it appears that axonal accumulations frequently localize to juxtaparanodal regions, leading to speculation that retrograde transport might be preferentially impacted by oligodendrocyte dysfunction. However, using the PLP-null model, the Griffiths lab employed cholera toxin B tracing to demonstrate that early deficits in fast axonal transport occur in both the anterograde and retrograde directions (Edgar & Garbern, 2004). As it appears that mitochondrial accumulation is not due to preferential defects in retrograde transport, an alternate hypothesis is increased mitochondrial anchoring within axons affected by oligodendrocyte dysfunction. Indeed, the Trapp lab demonstrated that demyelination increases axonal mitochondrial anchoring and that remyelination restores anchoring to control levels (Kiryu-Seo et al, 2010; Ohno et al, 2014). The authors demonstrate that upregulation of the anchoring protein SNPH and subsequent immobilization of mitochondria protects demyelinated axons from degeneration (Ohno et al, 2014). Another study demonstrated that deleting SNPH from the Shiverer mouse prolonged survival and reduced cerebellar degeneration, presumably by replenishing healthy mitochondria from the soma (Joshi et al, 2015).
Collectively, these studies demonstrate enhanced mitochondrial activity and elevated mitochondrial density in unmyelinated, demyelinated, and dysmyelinated axons. It has long been hypothesized that these adaptive changes in mitochondria reflect the increased energy demand of unsheathed axons, along which ion channels are distributed homogenously rather than accumulated at the nodes. It is possible that additional mitochondria, stationed in close proximity to these newly distributed channels, would be necessary to adequately fuel ion channel function. However, this hypothesis does not explain why mitochondrial abnormalities and transport deficits are observed in models of oligodendrocyte dysfunction in which compact myelination is preserved. Rather, these findings suggest a more complex and active participation of oligodendrocytes in regulating axonal energy metabolism.
Lactate shuttling as a mechanism of oligodendrocyte-derived energetic support
Oligodendrocytes express N-methyl-D-aspartate receptors (NMDAR) on their membranes and exhibit waves of calcium influx in response to glutamate (Karadottir et al, 2005; Micu et al, 2006; Salter & Fern, 2005), demonstrating that oligodendrocytes harbor mechanisms for sensing the activity of nearby neurons. One hypothesis is that this sensing mechanism might allow oligodendrocytes to provide on-demand metabolic support to the axonal compartment. The Nave Lab proposes that activity-dependent neuronal glutamate release into the periaxonal space leads to NMDAR activation that relays neuronal energy demand to nearby oligodendrocytes (Saab et al, 2016). As oligodendrocyte NMDAR activation stimulates mobilization and incorporation of glucose transporter 1 (GLUT1) into the myelin sheath, the authors suggest that activity-dependent neuronal glutamate release increases oligodendrocyte glucose uptake, glycolytic activity, and the associated lactate build-up. Their model suggests that lactate is subsequently exported out of oligodendrocytes via monocarboxylate transporter 1 (MCT1) and into axons via MCT2, where it is presumably converted to pyruvate and metabolized via mitochondrial oxphos (Funfschilling et al, 2012) (Figure 2A). In agreement, the Rothstein Lab utilized lentiviral knockdown to demonstrate that functional oligodendrocyte MCT1 is critical for axonal health and survival (Lee et al, 2012). While these studies certainly highlight the importance of lactate uptake in brain function, additional work is needed to parse the directionality of lactate transport and underlying mechanisms of neuroprotection. Considering the widespread controversy over the existence of the ANLS (Chih et al, 2001; Chih & Roberts, 2003; Dienel, 2017; Dienel & Cruz, 2015; Ferguson et al, 2018; Riske et al, 2017; Yellen, 2018), it is worthwhile to consider the underlying assumptions and open questions regarding the hypothesized model in which axons rely on oligodendrocyte-derived lactate. Specifically, the current model rests on three primary assumptions: (1) post-myelination oligodendrocytes favor aerobic glycolysis, leading to a high intracellular lactate concentration relative to the periaxonal space; (2) myelinated axons exhibit a low intracellular lactate concentration relative to the periaxonal space; and (3) myelinated axons metabolize oligodendrocyte-derived lactate. We examine these assumptions by reviewing the relevant literature.
Figure 2. Hypothesized mechanisms of oligodendrocyte-axon energetic coupling.
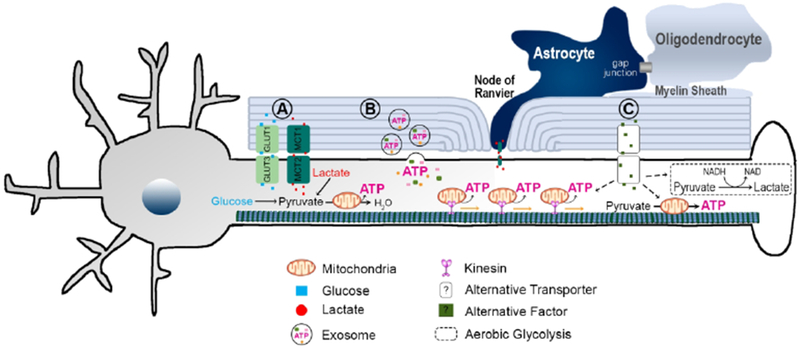
Oligodendrocytes contribute to axonal energy metabolism and this metabolic coupling is critical for maintaining axonal integrity. Although the mechanisms underlying this coupling remain to be fully elucidated, several pathways have been hypothesized. (A) Direct transport of metabolic substrates, such as glucose, lactate, or pyruvate may proceed via adjacent transporters located on the axonal membrane and myelin sheath. (B) Metabolic enzymes or proteins may be delivered to the axon via uptake of oligodendrocyte-derived exosomes. (C) Additional pathways of energy regulation are likely to be discovered, as the mechanism(s) through which oligodendrocytes regulate axonal mitochondrial transport have not been elucidated. In addition, there may be alternative pathways through which oligodendrocytes directly enhance mitochondrial oxphos or stimulate glycolysis within the axonal compartment.
1. Aerobic glycolysis in oligodendrocytes:
The directionality of lactate transport through MCTs is based on the concentration gradient across the plasma membrane (Hertz & Dienel, 2005). Therefore, a necessary assumption of the oligodendrocyte-to-neuron lactate shuttle model is that oligodendrocytes primarily metabolize glucose via aerobic glycolysis, leading to lactate production and export into the extracellular space. Current evidence suggests that during development, newly formed oligodendrocytes actually import lactate. By measuring the intracellular pH of oligodendrocytes in the mouse corpus callosum, the Attwell lab demonstrated that lactate is taken up into developing oligodendrocytes in early postnatal brain slices (Rinholm et al, 2011). Considering that cultured oligodendrocytes readily oxidize lactate for lipid biosynthesis (Sanchez-Abarca et al, 2001), it is likely that lactate import helps fuel myelin sheath formation during development. Once myelination is complete, there may be a metabolic shift towards enhanced glycolysis in mature oligodendrocytes. The Nave lab demonstrated that post-myelination oligodendrocytes rendered incapable of oxphos can survive by upregulating aerobic glycolysis (Funfschilling et al, 2012). Although this data demonstrates the metabolic flexibility of oligodendrocytes, it does not necessitate that they favor glycolysis under basal conditions. More direct evidence comes from oligodendrocyte progenitor cells (OPCs) and mature oligodendrocytes isolated directly from adult human brains, as both were found to utilize aerobic glycolysis for the majority of ATP production (Rone et al, 2016). Oligodendrocytes isolated from adult rats also preferentially utilize glycolysis under basal conditions, but switch to oxphos under metabolic stress (Rao et al, 2017). Of note, the composition of culture media must be considered when interpreting these results, as a recent study demonstrated a strong effect of extracellular lactate on oligodendrocyte metabolism (Amaral et al, 2016). In the absence of extracellular lactate, the ratio of oligodendrocyte lactate production to pyruvate oxidation was 60:40. However, in the presence of extracellular lactate, the ratio of oligodendrocyte lactate production to pyruvate oxidation shifted to 30:70 (Amaral et al, 2016), indicating that extracellular lactate might signal oligodendrocytes to upregulate their mitochondrial activity. If this finding holds true in vivo, then lactate production and export from neighboring cells could influence oligodendrocyte glycolytic rate in real time. One final consideration is mitochondrial transport within oligodendrocytes, as mitochondrial positioning is necessary for local ATP generation. A recent study utilized electron microscopy to show that there are small numbers of mitochondria in the cytoplasmic ridges of the sheath, although the low surface area of cristae suggests low ATP production (Rinholm et al, 2016). Overall, current evidence suggests that adult mature oligodendrocytes favor glycolysis, but also exhibit metabolic flexibility that may be regulated by local, environmental cues.
2. Aerobic glycolysis in axons:
In order for lactate to be imported into myelinated axons, axonal lactate concentration must be low relative to the periaxonal space. As discussed previously, whether neurons primarily utilize oxphos or glycolysis and under what activation states is a highly controversial matter (Chih et al, 2001; Chih & Roberts, 2003; Dienel, 2017; Dienel & Cruz, 2015; Ferguson et al, 2018; Riske et al, 2017; Yellen, 2018). Considering the heterogenous findings between intracellular compartments and resting versus stimulated nerve cells, it seems imperative to interpret results only within the context measured. For example, in axonal terminals, where presynapses are located, synaptic activity imposes large energetic demands that are met by local ATP synthesis via glycolysis and oxidative phosphorylation (Ashrafi & Ryan, 2017; Ivannikov et al, 2013; Rangaraju et al, 2014; Sheng, 2017; Sobieski et al, 2017; Sun et al, 2013; Verstreken et al, 2005). To date, the relative contribution of oxphos versus glycolysis within the axonal compartment has not been reported. This issue is clearly relevant when evaluating whether axons rely on oligodendrocyte-derived lactate. For example, if the recent report of upregulated glycolysis and lactate production in stimulated neuronal somas (Diaz-Garcia et al, 2017) holds true in the axonal compartment, then the hypothesized model could work in reverse-such that axonal release of lactate might serve to alter the metabolism of neighboring oligodendrocytes (Amaral et al, 2016).
3. Neuronal metabolism of oligodendrocyte-derived lactate:
So far, the evidence that neurons metabolize oligodendrocyte-derived lactate is indirect. Using a conditional Cox10 mutant mouse, in which oligodendrocytes and Schwann cells are rendered incapable of oxphos, Funfschilling et al demonstrated that mature oligodendrocytes survive well in the absence of cytochrome C oxidase (COX) activity and that this mutation does not induce demyelination, axonal degeneration, or inflammation (Funfschilling et al, 2012). The authors argue that oligodendrocytes survive by relying on aerobic glycolysis, evidenced by increased lactate levels in the cortex and corpus callosum of mutant mice. Extracellular lactate could only be detected under isoflurane anesthesia in both wild type and mutant mice, which was interpreted as evidence for axonal metabolism of oligodendrocyte-derived lactate (Funfschilling et al, 2012). While this is certainly possible, an alternative hypothesis is reduced clearance of lactate from the extracellular milieu, as decreased CSF clearance under anesthesia has been reported (Gakuba et al, 2018; Lorenzo et al, 1968). A study from the Rothstein lab also suggested that axons rely on oligodendrocyte-derived lactate, as oligodendrocyte-specific loss of MCT1 increases the number of degenerative axons in the optic nerve and corpus callosum (Lee et al, 2012). However, the directionality of lactate transport in these experiments remains unknown. It is possible that lactate deprivation through knockdown of oligodendrocyte MCT1 may cause cellular dysfunction through unknown mechanisms. We have previously discussed how dysfunctional oligodendrocytes can induce axonal degeneration, even when myelin sheaths remain intact (Edgar et al, 2010; Edgar et al, 2009; Griffiths et al, 1998; Lappe-Siefke et al, 2003; Yin et al, 2016). Overall, additional experiments in which oligodendrocyte-derived lactate is directly labeled and traced are needed in order to prove the proposed model.
Alternative mechanisms of oligodendrocyte-derived metabolic support
1. Glucose Shuttling:
In their white matter energy budget, Harris and Atwell conclude that direct transport of glucose from oligodendrocytes to axons may be an efficient means of feeding axonal mitochondria with the necessary substrates for oxphos, particularly during sudden increases in firing rates or in axons with long internodes (Harris & Attwell, 2012). Evidence for this glucose shuttling pathway was recently obtained using coronal slices of mouse corpus callosum and a model of glucose deprivation (aglycemia) (Meyer et al, 2018). Meyer et al demonstrate that filling single oligodendrocytes with glucose is sufficient to maintain compound action potentials (CAPs) in aglycemic axons, presumably by allowing glucose to be delivered locally to myelinated axons (Figure 2A). Importantly, oligodendrocyte networks established via gap junctions must be intact in order for single cell glucose filling to successfully abrogate loss of CAPs under aglycemic conditions (Meyer et al, 2018). These results demonstrate that oligodendrocyte networks are well positioned to provide metabolic support during energetic stress. However, because this paradigm severely alters glucose concentration gradients, which drive glucose transport into and out of neurons and oligodendrocytes, future experiments are needed to determine whether oligodendrocyte-derived glucose is metabolized by neurons under basal conditions.
2. Pyruvate Shuttling:
Pyruvate is transported via the same mechanism as lactate, gradient-dependent flux through MCTs. Thus, the effects reported by Lee et al (2012) and Funfschilling et al (2012) could be due to loss of pyruvate transport through MCT1. Further study is needed to elucidate the role of intercellular pyruvate transport and metabolism.
3. Exosome Delivery:
Oligodendrocytes synthesize and release exosomes containing a wide array of proteins, many of which are involved in energy metabolism (Kramer-Albers et al, 2007). Neurons can uptake oligodendrocyte-derived exosomes and retrieve their internal contents, which increases neuronal firing rates and alters gene expression (Frohlich et al, 2014). In addition, oligodendrocyte-derived exosomes increase neuronal metabolic activity in response to oxygen glucose deprivation (Frohlich et al, 2014). Schwann cells, which myelinate the peripheral nervous system, also release exosomes that are internalized by peripheral axons and aid in regeneration following sciatic nerve injury (Lopez-Verrilli et al, 2013). While these studies imply that myelinating glial cells might support axonal energy metabolism via exosomal delivery (Figure 2B), more work is needed in order to identify signaling molecules that are carried by oligodendrocyte-derived exosomes and may regulate axonal energy metabolism.
CONCLUDING REMARKS
Understanding how axons generate and utilize energy under various physiological and pathological conditions will be important for the development of therapeutic interventions designed to maintain axon health and neuronal survival in disease and injury. Energy production and maintenance in extended axonal processes present unique challenges: (1) energetic machinery (such as mitochondria) must be transported from the cell body toward distal axons and synapses, and (2) many axons are isolated from energy substrates in the extracellular environment by myelin sheaths. It is clear that mitochondria play a major role in meeting axonal energy demand and that their transport to and anchoring along the axon is crucial for maintaining axonal energy homeostasis. Going forward, it will be important to determine when axonal mitochondrial trafficking and anchoring is disrupted in various neurodegenerative diseases (Lin et al, 2017) and whether trafficking or anchoring is directly impaired by pathological proteins or as a secondary effect of concurrent pathological processes or aging. Although mitochondrial oxphos likely supplies most axonal energy, future experiments should address if and when aerobic glycolysis is uprelated to supplement energy production in the axonal compartment.
Current evidence suggests that oligodendrocytes contribute to axonal energy metabolism and that this metabolic coupling is critical for maintaining axonal integrity. However, more work is needed to determine whether oligodendrocytes aid axons directly, through provision of energy substrates such as lactate, or indirectly by altering metabolic activity in axons, regulating axonal mitochondrial transport and anchoring, or through unknown mechanisms (Figure 2C). Interestingly, at least in larger axons, the ability to adapt to myelin defects appears crucial for axonal survival. Such adaption does not occur in many models of oligodendrocyte dysfunction associated with axonal degeneration, possibly due to disrupted metabolic support. Elucidating the role of oligodendrocytes in supporting axonal energy metabolism could have broad impacts, as oligodendrocyte dysfunction is a prominent feature of many neurological diseases, including AD, PD, HD, ALS, and MS.
SIGNIFICANCE STATEMENT.
Central nervous system energy metabolism and maintenance are of great interest to the neurobiology field at large, as energetic failure is implicated in many neurological disorders and in CNS regeneration. Elucidating mechanisms for maintaining neuronal energy supply within specific intracellular compartments and during various activation states remains an active research area. Here, we focus in on axonal energy metabolism- describing major contributions from axonal mitochondria and surrounding glial cells. Emphasis is placed on discussing how axonal mitochondrial trafficking and anchoring is regulated to meet local energy demand, as well as how oligodendrocytes are hypothesized to supply axons with energy substrates.
Open Questions:
-
1)
Why does oligodendrocyte dysfunction lead to abnormal mitochondrial activity and distribution, even when ion channels remain clustered at the nodes?
-
2)
Does oligodendrocyte dysfunction impact mitochondrial activity or transport first?
-
2)
Which oligodendrocyte-derived signals are responsible for the regulation of axonal transport?
-
3)
Which axonal signaling pathways are activated downstream of the oligodendrocyte-derived signal?
-
4)
Do oligodendrocytes influence axonal transport in a generalized manner or via differential targeting of specific organelles and proteins?
Open Questions:
-
1)
How is oligodendrocyte metabolism altered during development and in response to neuronal activity?
-
2)
How do oxphos and glycolysis contribute to energy production in myelinated axons during rest and neuronal activation?
-
3)
Is oligodendrocyte-derived lactate exported to axons and metabolized for energy production?
Acknowledgements
We thank members of the Sheng lab for constructive discussion.
Funding: This work is supported by the Intramural Research Program of NINDS, NIH ZIA NS003029 and ZIA NS002946 (Z-H. Sheng) and the NINDS Competitive Postdoctoral Fellowship Award (K. A. C).
Footnotes
Conflict of Interest Statement
The authors have no conflict of interest to declare.
Citations
- Alberini CM, Cruz E, Descalzi G, Bessieres B, Gao V. 2018. Astrocyte glycogen and lactate: New insights into learning and memory mechanisms. Glia 66: 1244–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral AI, Hadera MG, Tavares JM, Kotter MR, Sonnewald U. 2016. Characterization of glucose-related metabolic pathways in differentiated rat oligodendrocyte lineage cells. Glia 64: 21–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson T, Schneider A, Barrie J, Klugmann M, McCulloch M, et al. 1998. Late-Onset Neurodegeneration in Mice With Increased Dosage of the Proteolipid Protein Gene. J Comp Neurol 394: 506–19 [DOI] [PubMed] [Google Scholar]
- Andrews H, White K, Thomson C, Edgar J, Bates D, et al. 2006. Increased Axonal Mitochondrial Activity as an Adaptation to Myelin Deficiency in the Shiverer Mouse. J Neurosci Res 83: 1533–9 [DOI] [PubMed] [Google Scholar]
- Ashrafi G, Ryan TA. 2017. Glucose metabolism in nerve terminals. Curr Opin Neurobiol 45: 156–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi G, Wu Z, Farrell RJ, Ryan TA. 2017. GLUT4 Mobilization Supports Energetic Demands of Active Synapses. Neuron 93: 606–15 e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Laughlin S. 2001. An Energy Budget for Signaling in the Grey Matter of the Brain. J Cereb Blood Flow Metab 21: 1133–45 [DOI] [PubMed] [Google Scholar]
- Azevedo FA, Carvalho LR, Grinberg LT, Farfel JM, Ferretti RE, et al. 2009. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J Comp Neurol 513: 532–41 [DOI] [PubMed] [Google Scholar]
- Baltan S 2015. Can lactate serve as an energy substrate for axons in good times and in bad, in sickness and in health? Metab Brain Dis 30: 25–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baqri RM, Turner BA, Rheuben MB, Hammond BD, Kaguni LS, Miller KE. 2009. Disruption of mitochondrial DNA replication in Drosophila increases mitochondrial fast axonal transport in vivo. PLoS One 4: e7874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros LF. 2013. Metabolic signaling by lactate in the brain. Trends Neurosci 36: 396–404 [DOI] [PubMed] [Google Scholar]
- Bhatt A, Fan LW, Pang Y. 2014. Strategies for myelin regeneration: lessons learned from development. Neural Regen Res 9: 1347–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilsland LG, Sahai E, Kelly G, Golding M, Greensmith L, Schiavo G. 2010. Deficits in axonal transport precede ALS symptoms in vivo. Proc Natl Acad Sci U S A 107: 20523–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black J, Waxman S 1988. The Perinodal Astrocyte. Glia 1: 169–83 [DOI] [PubMed] [Google Scholar]
- Brady ST, Colman DR, Brophy PJ. 2013. Subcellular Organization of the Nervous System: Organelles and Their Functions In Fundamental Neuroscience, pp. 61–92 [Google Scholar]
- Bristow E, Griffiths P, Andrews R, Johnson M, Turnbull D. 2002. The Distribution of Mitochondrial Activity in Relation to Optic Nerve Structure. Arch Ophthalmol 120: 791–6 [DOI] [PubMed] [Google Scholar]
- Brown AM, Baltan Tekkok S, Ransom BR. 2004. Energy transfer from astrocytes to axons: the role of CNS glycogen. Neurochem Int 45: 529–36 [DOI] [PubMed] [Google Scholar]
- Brown AM, Ransom BR. 2015. Astrocyte glycogen as an emergency fuel under conditions of glucose deprivation or intense neural activity. Metab Brain Dis 30: 233–9 [DOI] [PubMed] [Google Scholar]
- Brown AM, Sickmann HM, Fosgerau K, Lund TM, Schousboe A, et al. 2005. Astrocyte glycogen metabolism is required for neural activity during aglycemia or intense stimulation in mouse white matter. J Neurosci Res 79: 74–80 [DOI] [PubMed] [Google Scholar]
- Brown AM, Tekkok SB, Ransom BR. 2003. Glycogen regulation and functional role in mouse white matter. J Physiol 549: 501–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butt A, Duncan A, Berry M. 1994. Astrocyte associations with nodes of Ranvier: ultrastructural analysis of HRP-filled astrocytes in the mouse optic nerve. Journal of Neurocytology 23: 486–99 [DOI] [PubMed] [Google Scholar]
- Cai Q, Gerwin C, Sheng ZH. 2005. Syntabulin-mediated anterograde transport of mitochondria along neuronal processes. J Cell Biol 170: 959–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH. 2011. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum Mol Genet 20: 4515–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camandola S, Mattson MP. 2017. Brain metabolism in health, aging, and neurodegeneration. EMBO J 36: 1474–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartoni R, Norsworthy MW, Bei F, Wang C, Li S, et al. 2016. The Mammalian-Specific Protein Armcx1 Regulates Mitochondrial Transport during Axon Regeneration. Neuron 92: 1294–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartoni R, Pekkurnaz G, Wang C, Schwarz TL, He Z. 2017. A high mitochondrial transport rate characterizes CNS neurons with high axonal regeneration capacity. PLoS One 12: e0184672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DT, Honick AS, Reynolds IJ. 2006a. Mitochondrial trafficking to synapses in cultured primary cortical neurons. J Neurosci 26: 7035–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DT, Rintoul GL, Pandipati S, Reynolds IJ. 2006b. Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol Dis 22: 388–400 [DOI] [PubMed] [Google Scholar]
- Chen H, Chan DC. 2009. Mitochondrial dynamics--fusion, fission, movement, and mitophagy--in neurodegenerative diseases. Hum Mol Genet 18: R169–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Sheng ZH. 2013. Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. J Cell Biol 202: 351–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chih C-P, Lipton P, Roberts ELJ. 2001. Do active cerebral neurons really use lactate rather than glucose? TRENDS in Neuroscience 24: 573–8 [DOI] [PubMed] [Google Scholar]
- Chih CP, Roberts EL Jr. 2003. Energy substrates for neurons during neural activity: a critical review of the astrocyte-neuron lactate shuttle hypothesis. J Cereb Blood Flow Metab 23: 1263–81 [DOI] [PubMed] [Google Scholar]
- Collard J, Côté F, Julien J. 1995. Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Neuron 375: 61–4 [DOI] [PubMed] [Google Scholar]
- Courchet J, Lewis TL Jr, Lee S, Courchet V, Liou DY, et al. 2013. Terminal axon branching is regulated by the LKB1-NUAK1 kinase pathway via presynaptic mitochondrial capture. Cell 153: 1510–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J, Lambert S, Bennett V. 1996. Molecular composition of the node of Ranvier: identification of ankyrin-binding cell adhesion molecules neurofascin (mucin+/third FNIII domain−) and NrCAM at nodal axon segments. J Cell Biol 135: 1355–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dentinger M, Barron K, Csiza C. 1985. Glial and Axonal Development in Optic Nerve of Myelin Deficient Rat Mutant. Brain Res 344: 255–66 [DOI] [PubMed] [Google Scholar]
- Diaz-Garcia CM, Mongeon R, Lahmann C, Koveal D, Zucker H, Yellen G. 2017. Neuronal Stimulation Triggers Neuronal Glycolysis and Not Lactate Uptake. Cell Metab 26: 361–74 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienel GA. 2012. Brain lactate metabolism: the discoveries and the controversies. J Cereb Blood Flow Metab 32: 1107–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienel GA. 2017. Lack of appropriate stoichiometry: Strong evidence against an energetically important astrocyte-neuron lactate shuttle in brain. J Neurosci Res 95: 2103–25 [DOI] [PubMed] [Google Scholar]
- Dienel GA, Cruz NF. 2015. Contributions of glycogen to astrocytic energetics during brain activation. Metab Brain Dis 30: 281–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienel GA, McKenna MC. 2014. A dogma-breaking concept: glutamate oxidation in astrocytes is the source of lactate during aerobic glycolysis in resting subjects. J Neurochem 131: 395–8 [DOI] [PubMed] [Google Scholar]
- Edgar JM, Garbern J. 2004. The myelinated axon is dependent on the myelinating cell for support and maintenance: molecules involved. J Neurosci Res 76: 593–8 [DOI] [PubMed] [Google Scholar]
- Edgar JM, McCulloch MC, Montague P, Brown AM, Thilemann S, et al. 2010. Demyelination and axonal preservation in a transgenic mouse model of Pelizaeus-Merzbacher disease. EMBO Mol Med 2: 42–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar JM, McLaughlin M, Werner HB, McCulloch MC, Barrie JA, et al. 2009. Early ultrastructural defects of axons and axon-glia junctions in mice lacking expression of Cnp1. Glia 57: 1815–24 [DOI] [PubMed] [Google Scholar]
- Einheber S, Zanazzi G, Ching W, Scherer S, Milner T, et al. 1997. The Axonal Membrane Protein Caspr, a Homologue of Neurexin IV, Is a Component of the Septate-like Paranodal Junctions That Assemble during Myelination. J Cell Biol 139: 1495–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson BS, Rogatzki MJ, Goodwin ML, Kane DA, Rightmire Z, Gladden LB. 2018. Lactate metabolism: historical context, prior misinterpretations, and current understanding. Eur J Appl Physiol 118: 691–728 [DOI] [PubMed] [Google Scholar]
- Fox P, Raichle M, Mintun M, Dence C. 1988. Nonoxidative glucose consumption during focal physiologic neural activity. Science 241: 462–4 [DOI] [PubMed] [Google Scholar]
- Frankenhaeuser B 1952. Saltatory conduction in myelinated nerve fibres. J Physiol 118: 107–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransson A, Ruusala A, Aspenstrom P. 2003. Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis. J Biol Chem 278: 6495–502 [DOI] [PubMed] [Google Scholar]
- Frohlich D, Kuo WP, Fruhbeis C, Sun JJ, Zehendner CM, et al. 2014. Multifaceted effects of oligodendroglial exosomes on neurons: impact on neuronal firing rate, signal transduction and gene regulation. Philos Trans R Soc Lond B Biol Sci 369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, et al. 2012. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485: 517–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gakuba C, Gaberel T, Goursaud S, Bourges J, Di Palma C, et al. 2018. General Anesthesia Inhibits the Activity of the “Glymphatic System”. Theranostics 8: 710–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbern J, Yool D, Moore G, Wilds I, Faulk M, et al. 2002. Patients lacking the major CNS myelin protein, proteolipid protein 1, develop length-dependent axonal degeneration in the absence of demyelination and inflammation. Brain 125: 551–61 [DOI] [PubMed] [Google Scholar]
- Godzik K, Coleman MP. 2015. The axon-protective WLD(S) protein partially rescues mitochondrial respiration and glycolysis after axonal injury. J Mol Neurosci 55: 865–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, et al. 1998. Axonal Swellings and Degeneration in Mice Lacking the Major Proteolipid of Myelin. Science 280: 1610–3 [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Goldstein L. 2001. Disruption of Axonal Transport and Neuronal Viability by Amyloid Precursor Protein Mutations in Drosophila. Neuron 32: 389–401 [DOI] [PubMed] [Google Scholar]
- Hallermann S, de Kock CP, Stuart GJ, Kole MH. 2012. State and location dependence of action potential metabolic cost in cortical pyramidal neurons. Nat Neurosci 15: 1007–14 [DOI] [PubMed] [Google Scholar]
- Han SM, Baig HS, Hammarlund M. 2016. Mitochondria Localize to Injured Axons to Support Regeneration. Neuron 92: 1308–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JJ, Attwell D. 2012. The energetics of CNS white matter. J Neurosci 32: 356–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JJ, Jolivet R, Attwell D. 2012. Synaptic energy use and supply. Neuron 75: 762–77 [DOI] [PubMed] [Google Scholar]
- Hartline DK, Colman DR. 2007. Rapid conduction and the evolution of giant axons and myelinated fibers. Curr Biol 17: R29–35 [DOI] [PubMed] [Google Scholar]
- Heppelmann B, Messlinger K, Neiss W, Schmidt R. 1994. Mitochondria in fine afferent nerve fibres of the knee joint in the cat: a quantitative electron-microscopical examination. Cell Tissue Res 275: 493–501 [DOI] [PubMed] [Google Scholar]
- Herculano-Houzel S 2012. The remarkable, yet not extraordinary, human brain as a scaled-up primate brain and its associated cost. Proc Natl Acad Sci U S A 109 Suppl 1: 10661–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz L, Dienel GA. 2005. Lactate transport and transporters: general principles and functional roles in brain cells. J Neurosci Res 79: 11–8 [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Niwa S, Tanaka Y. 2010. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron 68: 610–38 [DOI] [PubMed] [Google Scholar]
- Hogan V, White K, Edgar J, McGill A, Karim S, et al. 2009. Increase in mitochondrial density within axons and supporting cells in response to demyelination in the Plp1 mouse model. J Neurosci Res 87: 452–9 [DOI] [PubMed] [Google Scholar]
- Hubley M, Locke B, Moerland T. 1996. The effects of temperature, pH, and magnesium on the diffusion coefficient of ATP in solutions of physiological ionic strength. Biochim Biophys Acta 1291: 115–21 [DOI] [PubMed] [Google Scholar]
- Ivannikov MV, Sugimori M, Llinas RR. 2013. Synaptic vesicle exocytosis in hippocampal synaptosomes correlates directly with total mitochondrial volume. J Mol Neurosci 49: 223–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang S, Nelson JC, Bend EG, Rodriguez-Laureano L, Tueros FG, et al. 2016. Glycolytic Enzymes Localize to Synapses under Energy Stress to Support Synaptic Function. Neuron 90: 278–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi DC, Zhang CL, Lin TM, Gusain A, Harris MG, et al. 2015. Deletion of mitochondrial anchoring protects dysmyelinating shiverer: implications for progressive MS. J Neurosci 35: 5293–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaasik A 2016. Mitochondrial Mobility and Neuronal Recovery. N Engl J Med 375: 1295–6 [DOI] [PubMed] [Google Scholar]
- Kang J, Tian J, Pan P, Zald P, Li C, et al. 2008a. Docking of Axonal Mitochondria by Syntaphilin Controls their Mobility and Affects Short-term Facilitation. Cell 132: 137–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JS, Tian JH, Pan PY, Zald P, Li C, et al. 2008b. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 132: 137–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karadottir R, Cavelier P, Bergersen LH, Attwell D. 2005. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature 438: 1162–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryu-Seo S, Kiyama H. 2018. Mitochondrial behavior during axon regeneration/degeneration in vivo. Neurosci Res [DOI] [PubMed] [Google Scholar]
- Kiryu-Seo S, Ohno N, Kidd GJ, Komuro H, Trapp BD. 2010. Demyelination increases axonal stationary mitochondrial size and the speed of axonal mitochondrial transport. J Neurosci 30: 6658–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryu-Seo S, Tamada H, Kato Y, Yasuda K, Ishihara N, et al. 2016. Mitochondrial fission is an acute and adaptive response in injured motor neurons. Sci Rep 6: 28331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitay BM, McCormack R, Wang Y, Tsoulfas P, Zhai RG. 2013. Mislocalization of neuronal mitochondria reveals regulation of Wallerian degeneration and NMNAT/WLD(S)-mediated axon protection independent of axonal mitochondria. Hum Mol Genet 22: 1601–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knull H 1978. Association of glycolytic enzymes with particulate fractions from nerve endings. Biochim Biophys Acta 522: 1–9 [DOI] [PubMed] [Google Scholar]
- Knull H, Bronstein W, DesJardins P, Niehaus WJ. 1980. Interaction of selected brain glycolytic enzymes with an F-actin-tropomyosin complex. J Neurochem 34: 222–5 [DOI] [PubMed] [Google Scholar]
- Knull H, Fillmore S. 1985. Glycolytic enzyme levels in synaptosomes. Comp Biochem Physiol B 81: 349–51 [DOI] [PubMed] [Google Scholar]
- Kordeli E, Davis J, Trapp B, Bennett V. 1990. An Isoform of Ankyrin Is Localized at Nodes of Ranvier in Myelinated Axons of Central and Peripheral Nerves. The Journal of Cell Biology 110: 1341–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer-Albers EM, Bretz N, Tenzer S, Winterstein C, Mobius W, et al. 2007. Oligodendrocytes secrete exosomes containing major myelin and stress-protective proteins: Trophic support for axons? Proteomics Clin Appl 1: 1446–61 [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, et al. 2003. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet 33: 366–74 [DOI] [PubMed] [Google Scholar]
- Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, et al. 2012. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487: 443–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennie P 2003. The Cost of Cortical Computation. Current Biology 13: 493–97 [DOI] [PubMed] [Google Scholar]
- Lewis TL Jr., Turi GF, Kwon SK, Losonczy A, Polleux F. 2016. Progressive Decrease of Mitochondrial Motility during Maturation of Cortical Axons In Vitro and In Vivo. Curr Biol 26: 2602–08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MY, Cheng XT, Tammineni P, Xie Y, Zhou B, et al. 2017. Releasing Syntaphilin Removes Stressed Mitochondria from Axons Independent of Mitophagy under Pathophysiological Conditions. Neuron 94: 595–610 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton P, Robacker K. 1983. Glycolysis and brain function: [K+]o stimulation of protein synthesis and K+ uptake require glycolysis. Fed Proc. 42: 2875–80 [PubMed] [Google Scholar]
- Lopez-Verrilli MA, Picou F, Court FA. 2013. Schwann cell-derived exosomes enhance axonal regeneration in the peripheral nervous system. Glia 61: 1795–806 [DOI] [PubMed] [Google Scholar]
- Lorenzo AV, Hammerstad JP, Cutler RWP. 1968. The effect of anaesthetic agents on the cerebrospinal fluid clearance of 35 S-Sulphate and 125 I-Iodide. Biochemical Pharmacology 12: 1279–83 [DOI] [PubMed] [Google Scholar]
- Ma H, Cai Q, Lu W, Sheng ZH, Mochida S. 2009. KIF5B motor adaptor syntabulin maintains synaptic transmission in sympathetic neurons. J Neurosci 29: 13019–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAskill AF, Brickley K, Stephenson FA, Kittler JT. 2009. GTPase dependent recruitment of Grif-1 by Miro1 regulates mitochondrial trafficking in hippocampal neurons. Mol Cell Neurosci 40:301–12 [DOI] [PubMed] [Google Scholar]
- MacAskill AF, Kittler JT. 2010. Control of mitochondrial transport and localization in neurons. Trends Cell Biol 20: 102–12 [DOI] [PubMed] [Google Scholar]
- Maday S, Twelvetrees AE, Moughamian AJ, Holzbaur EL. 2014. Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron 84: 292–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen P, Cruz N, Sokoloff L, Dienel G. 1999. Cerebral oxygen/glucose ratio is low during sensory stimulation and rises above normal during recovery: excess glucose consumption during stimulation is not accounted for by lactate efflux from or accumulation in brain tissue. J Cereb Blood Flow Metab 19: 393–400 [DOI] [PubMed] [Google Scholar]
- Magistretti P, Sorg O, Yu N, Martin J, Pellerin L. 1993. Neurotransmitters Regulate Energy Metabolism in Astrocytes: Implications for the Metabolic Trafficking between Neural Cells. Dev Neurosci 15: 306–12 [DOI] [PubMed] [Google Scholar]
- Magistretti PJ, Allaman I. 2015. A cellular perspective on brain energy metabolism and functional imaging. Neuron 86: 883–901 [DOI] [PubMed] [Google Scholar]
- Mahad DJ, Ziabreva I, Campbell G, Lax N, White K, et al. 2009. Mitochondrial changes within axons in multiple sclerosis. Brain 132: 1161–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDougall S, Vargas Riad W, Silva-Gotay A, Tavares ER, Harpalani D, et al. 2018. Myelination of Axons Corresponds with Faster Transmission Speed in the Prefrontal Cortex of Developing Male Rats. eNeuro 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menegoz M, Gaspar P, Le Bert M, Galvez T, Burgaya F, et al. 1997. Paranodin, a Glycoprotein of Neuronal Paranodal Membranes. Neuron 19: 319–31 [DOI] [PubMed] [Google Scholar]
- Meyer N, Richter N, Fan Z, Siemonsmeier G, Pivneva T, et al. 2018. Oligodendrocytes in the Mouse Corpus Callosum Maintain Axonal Function by Delivery of Glucose. Cell Rep 22: 2383–94 [DOI] [PubMed] [Google Scholar]
- Micheva KD, Wolman D, Mensh BD, Pax E, Buchanan J, et al. 2016. A large fraction of neocortical myelin ensheathes axons of local inhibitory neurons. Elife 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micu I, Jiang Q, Coderre E, Ridsdale A, Zhang L, et al. 2006. NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature 439: 988–92 [DOI] [PubMed] [Google Scholar]
- Mihaylova MM, Shaw RJ. 2012. The AMPK signalling pathway coordinates cell growth, autophagy, & metabolism. Nature Cell Biology 13: 1016–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milde S, Adalbert R, Elaman MH, Coleman MP. 2015. Axonal transport declines with age in two distinct phases separated by a period of relative stability. Neurobiol Aging 36: 971–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KE, Sheetz MP. 2004. Axonal mitochondrial transport and potential are correlated. J Cell Sci 117: 2791–804 [DOI] [PubMed] [Google Scholar]
- Mink J, Blumenschine R, Adams D. 1981. Ratio of central nervous system to body metabolism in vertebrates: its constancy and functional basis. Am J Physiol 241: R203–12 [DOI] [PubMed] [Google Scholar]
- Mironov SL. 2007. ADP regulates movements of mitochondria in neurons. Biophys J 92: 2944–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld T, Kerschensteiner M, Bareyre FM, Burgess RW, Lichtman JW. 2007. Imaging axonal transport of mitochondria in vivo. Nat Methods 4: 559–61 [DOI] [PubMed] [Google Scholar]
- Misgeld T, Schwarz T. 2017. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 96: 651–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsci NS, Hall DH, Driscoll M, Sheng ZH. 2016. Age-Related Phasic Patterns of Mitochondrial Maintenance in Adult Caenorhabditis elegans Neurons. J Neurosci 36: 1373–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutsaers S, Carroll W. 1998. Focal accumulation of intra-axonal mitochondria in demyelination of the cat optic nerve. Acta Neuropathol 96: 139–43 [DOI] [PubMed] [Google Scholar]
- Nave KA, Trapp BD. 2008. Axon-glial signaling and the glial support of axon function. Annu Rev Neurosci 31: 535–61 [DOI] [PubMed] [Google Scholar]
- Nicholls D, Budd S. 2000. Mitochondria and Neuronal Survival. Physiological Reviews 2000: 1. [DOI] [PubMed] [Google Scholar]
- Nikic I, Merkler D, Sorbara C, Brinkoetter M, Kreutzfeldt M, et al. 2011. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med 17:495–9 [DOI] [PubMed] [Google Scholar]
- Ogawa S, Tank DW, Menon R, Ellermann JM, Kim S-G, et al. 1992. Intrinsic signal changes accompanying sensory stimulation: Functional brain mapping with magnetic resonance imaging. PNAS 89: 5951–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno N, Chiang H, Mahad DJ, Kidd GJ, Liu L, et al. 2014. Mitochondrial immobilization mediated by syntaphilin facilitates survival of demyelinated axons. Proc Natl Acad Sci U S A 111: 9953–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oluich LJ, Stratton JA, Xing YL, Ng SW, Cate HS, et al. 2012. Targeted ablation of oligodendrocytes induces axonal pathology independent of overt demyelination. J Neurosci 32: 8317–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak D, Berthet A, Nakamura K. 2013. Energy failure: does it contribute to neurodegeneration? Ann Neurol 74: 506–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak D, Sepp KJ, Hollenbeck PJ. 2010. Evidence That Myosin Activity Opposes Microtubule-Based Axonal Transport of Mitochondria. Journal of Neuroscience 30: 8984–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekkurnaz G, Trinidad JC, Wang X, Kong D, Schwarz TL. 2014. Glucose regulates mitochondrial motility via Milton modification by O-GlcNAc transferase. Cell 158: 54–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Bouzier-Sore AK, Aubert A, Serres S, Merle M, et al. 2007. Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia 55: 1251–62 [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti P. 1994. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. PNAS 91: 10625–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. 2012. Sweet sixteen for ANLS. J Cereb Blood Flow Metab 32: 1152–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommerville JC. 2013. Metabolism of Microorganisms In Alcamo’s Fundamentals of Neurobiology, ed. Johnson M, pp. 157–87: Jones & Bartlett Learning [Google Scholar]
- Raefsky SM, Mattson MP. 2017. Adaptive responses of neuronal mitochondria to bioenergetic challenges: Roles in neuroplasticity and disease resistance. Free Radic Biol Med 102: 203–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raine C 1984. On the association between perinodal astrocytic processes and the node of Ranvier in the C.N.S. Journal of Neurocytology 13: 21–27 [DOI] [PubMed] [Google Scholar]
- Rangaraju V, Calloway N, Ryan TA. 2014. Activity-driven local ATP synthesis is required for synaptic function. Cell 156: 825–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao VTS, Khan D, Cui QL, Fuh SC, Hossain S, et al. 2017. Distinct age and differentiation-state dependent metabolic profiles of oligodendrocytes under optimal and stress conditions. PLoS One 12: e0182372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN, Peles E, Trimmer JS, Levinson SR, Lux SE, Shrager P. 1999. Dependence of Nodal Sodium Channel Clustering on Paranodal Axoglial Contact in the Developing CNS. The Journal of Neuroscience 19: 7516–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinholm JE, Hamilton NB, Kessaris N, Richardson WD, Bergersen LH, Attwell D. 2011. Regulation of oligodendrocyte development and myelination by glucose and lactate. J Neurosci 31: 538–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinholm JE, Vervaeke K, Tadross MR, Tkachuk AN, Kopek BG, et al. 2016. Movement and structure of mitochondria in oligodendrocytes and their myelin sheaths. Glia 64: 810–25 [DOI] [PubMed] [Google Scholar]
- Rintoul GL, Filiano AJ, Brocard JB, Kress GJ, Reynolds IJ. 2003. Glutamate Decreases Mitochondrial Size and Movement in Primary Forebrain Neurons. The Journal of Neuroscience 23: 7881–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riske L, Thomas R, Baker G, Dursun S. 2017. Lactate in the brain: an update on its relevance to brain energy, neurons, glia and panic disorder. Ther Adv Psychopharmacol 7: 85–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolfe DFS, Brown GC. 1997. Cellular Energy Utilization and Molecular Origin of Standard Metabolic Rate in Mammals. Physiological Reviews 77: 731–58 [DOI] [PubMed] [Google Scholar]
- Rone MB, Cui QL, Fang J, Wang LC, Zhang J, et al. 2016. Oligodendrogliopathy in Multiple Sclerosis: Low Glycolytic Metabolic Rate Promotes Oligodendrocyte Survival. J Neurosci 36: 4698–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saab AS, Tzvetanova ID, Nave KA. 2013. The role of myelin and oligodendrocytes in axonal energy metabolism. Curr Opin Neurobiol 23: 1065–72 [DOI] [PubMed] [Google Scholar]
- Saab AS, Tzvetavona ID, Trevisiol A, Baltan S, Dibaj P, et al. 2016. Oligodendroglial NMDA Receptors Regulate Glucose Import and Axonal Energy Metabolism. Neuron 91: 119–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter MG, Fern R. 2005. NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature 438: 1167–71 [DOI] [PubMed] [Google Scholar]
- Sanchez-Abarca LI, Tabernero A, Medina JM. 2001. Oligodendrocytes use lactate as a source of energy and as a precursor of lipids. Glia 36: 321–9 [DOI] [PubMed] [Google Scholar]
- Sasaki S, Warita H, Abe K, Iwata M. 2005. Impairment of axonal transport in the axon hillock and the initial segment of anterior horn neurons in transgenic mice with a G93A mutant SOD1 gene. Acta Neuropathol 110: 48–56 [DOI] [PubMed] [Google Scholar]
- Saxton WM, Hollenbeck PJ. 2012. The axonal transport of mitochondria. J CellSci 125: 2095–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz TL. 2013. Mitochondrial trafficking in neurons. Cold Spring Harb Perspect Biol 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng ZH. 2014. Mitochondrial trafficking and anchoring in neurons: New insight and implications. J Cell Biol 204: 1087–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng ZH. 2017. The Interplay of Axonal Energy Homeostasis and Mitochondrial Trafficking and Anchoring. Trends Cell Biol 27: 403–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng ZH, Cai Q. 2012. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci 13: 77–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HL, Bourne JN, Cao G, Chirillo MA, Ostroff LE, et al. 2016. Mitochondrial support of persistent presynaptic vesicle mobilization with age-dependent synaptic growth after LTP. eLife 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snaidero N, Simons M. 2014. Myelination at a glance. J Cell Sci 127: 2999–3004 [DOI] [PubMed] [Google Scholar]
- Sobieski C, Fitzpatrick MJ, Mennerick SJ. 2017. Differential Presynaptic ATP Supply for Basal and High-Demand Transmission. J Neurosci 37: 1888–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillane M, Ketschek A, Merianda TT, Twiss JL, Gallo G. 2013. Mitochondria coordinate sites of axon branching through localized intra-axonal protein synthesis. Cell Rep 5: 1564–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokin G, Lillo C, Falzone T, Brusch R, Rockenstein E, et al. 2005. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science 307: 1282–8 [DOI] [PubMed] [Google Scholar]
- Sun T, Qiao H, Pan PY, Chen Y, Sheng ZH. 2013. Motile axonal mitochondria contribute to the variability of presynaptic strength. Cell Rep 4: 413–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takihara Y, Inatani M, Eto K, Inoue T, Kreymerman A, et al. 2015. In vivo imaging of axonal transport of mitochondria in the diseased and aged mammalian CNS. Proc Natl Acad Sci U S A 112:10515–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang BL. 2018. Brain activity-induced neuronal glucose uptake/glycolysis: Is the lactate shuttle not required? Brain Res Bull 137: 225–28 [DOI] [PubMed] [Google Scholar]
- Tao K, Matsuki N, Koyama R. 2014. AMP-activated protein kinase mediates activity-dependent axon branching by recruiting mitochondria to axon. Dev Neurobiol 74: 557–73 [DOI] [PubMed] [Google Scholar]
- Tekkok SB, Brown AM, Westenbroek R, Pellerin L, Ransom BR. 2005. Transfer of glycogen-derived lactate from astrocytes to axons via specific monocarboxylate transporters supports mouse optic nerve activity. J Neurosci Res 81: 644–52 [DOI] [PubMed] [Google Scholar]
- Tomassy GS, Berger DR, Chen HH, Kasthuri N, Hayworth KJ, et al. 2014. Distinct profiles of myelin distribution along single axons of pyramidal neurons in the neocortex. Science 344: 31924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traka M, Arasi K, Avila RL, Podojil JR, Christakos A, et al. 2010. A genetic mouse model of adult-onset, pervasive central nervous system demyelination with robust remyelination. Brain 133: 3017–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trushina E, Dyer RB, Badger JD, 2nd, Ure D, Eide L, et al. 2004. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol Cell Biol 24: 8195–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaarmann A, Mandel M, Zeb A, Wareski P, Liiv J, et al. 2016. Mitochondrial biogenesis is required for axonal growth. Development 143: 1981–92 [DOI] [PubMed] [Google Scholar]
- Vagnoni A, Bullock SL. 2018. A cAMP/PKA/Kinesin-1 Axis Promotes the Axonal Transport of Mitochondria in Aging Drosophila Neurons. Current Biology 28: 1265–72.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verburg J, Hollenbeck PJ. 2008. Mitochondrial membrane potential in axons increases with local nerve growth factor or semaphorin signaling. J Neurosci 28: 8306–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstreken P, Ly CV, Venken KJ, Koh TW, Zhou Y, Bellen HJ. 2005. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 47: 365–78 [DOI] [PubMed] [Google Scholar]
- Wang X, Schwarz TL. 2009. The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell 136: 163–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warita H, Itoyama Y, Abe K. 1999. Selective impairment of fast anterograde axonal transport in the peripheral nerves of asymptomatic transgenic mice with a G93A mutant SOD1 gene. Brain Res 819: 120–31 [DOI] [PubMed] [Google Scholar]
- Weber B, Barros LF. 2015. The Astrocyte: Powerhouse and Recycling Center. Cold Spring Harb Perspect Biol 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson TL, Cleveland DW. 1999. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nature Neuroscience 2: 50–56 [DOI] [PubMed] [Google Scholar]
- Witte ME, Bo L, Rodenburg RJ, Belien JA, Musters R, et al. 2009. Enhanced number and activity of mitochondria in multiple sclerosis lesions. J Pathol 219: 193–204 [DOI] [PubMed] [Google Scholar]
- Xie Y, Zhou B, Lin MY, Wang S, Foust KD, Sheng ZH. 2015. Endolysosomal Deficits Augment Mitochondria Pathology in Spinal Motor Neurons of Asymptomatic fALS Mice. Neuron 87: 355–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G 2018. Fueling thought: Management of glycolysis and oxidative phosphorylation in neuronal metabolism. J Cell Biol 217: 2235–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M, Weaver D, Hajnoczky G. 2004. Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J Cell Biol 167: 661–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Kidd GJ, Ohno N, Perkins GA, Ellisman MH, et al. 2016. Proteolipid protein-deficient myelin promotes axonal mitochondrial dysfunction via altered metabolic coupling. J Cell Biol 215: 531–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zala D, Hinckelmann MV, Yu H, Lyra da Cunha MM, Liot G, et al. 2013. Vesicular glycolysis provides on-board energy for fast axonal transport. Cell 152: 479–91 [DOI] [PubMed] [Google Scholar]
- Zambonin JL, Zhao C, Ohno N, Campbell GR, Engeham S, et al. 2011. Increased mitochondrial content in remyelinated axons: implications for multiple sclerosis. Brain 134: 1901–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Yu P, Lin MY, Sun T, Chen Y, Sheng ZH. 2016. Facilitation of axon regeneration by enhancing mitochondrial transport and rescuing energy deficits. J Cell Biol 214: 103–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YB, Sheng ZH. 2011. Increased axonal mitochondrial mobility does not slow amyotrophic lateral sclerosis (ALS)-like disease in mutant SOD1 mice. J Biol Chem 286: 23432–40 [DOI] [PMC free article] [PubMed] [Google Scholar]