

Abstract
Brain tumors such as adult glioblastomas and pediatric high-grade gliomas or medulloblastomas are among the leading causes of cancer-related deaths, exhibiting poor prognoses with little improvement in outcomes in the past several decades. These tumors are heterogeneous and can be initiated from various neural cell types, contributing to therapy resistance. How such heterogeneity arises is linked to the tumor cell of origin and their genetic alterations. Brain tumorigenesis and progression recapitulate key features associated with normal neurogenesis; however, the underlying mechanisms are quite dysregulated as tumor cells grow and divide in an uncontrolled manner. Recent comprehensive genomic, transcriptomic, and epigenomic studies at single-cell resolution have shed new light onto diverse tumor-driving events, cellular heterogeneity, and cells of origin in different brain tumors. Primary and secondary glioblastomas develop through different genetic alterations and pathways, such as EGFR amplification and IDH1/2 or TP53 mutation, respectively. Mutations such as histone H3K27M impacting epigenetic modifications define a distinct group of pediatric high-grade gliomas such as diffuse intrinsic pontine glioma. The identification of distinct genetic, epigenomic profiles and cellular heterogeneity has led to new classifications of adult and pediatric brain tumor subtypes, affording insights into molecular and lineage-specific vulnerabilities for treatment stratification. This review discusses our current understanding of tumor cells of origin, heterogeneity, recurring genetic and epigenetic alterations, oncogenic drivers and signaling pathways for adult glioblastomas, pediatric high-grade gliomas, and medulloblastomas, the genetically heterogeneous groups of malignant brain tumors.
Keywords: glioma, medulloblastoma, neural stem cells, glial progenitor cells, oligodendrocyte progenitors, oncogenic signaling network, Tumor suppressors, epigenetic regulation
Graphical Abstract
INTRODUCTION
Brain tumors are primary tumors of the central nervous system. They include high-grade gliomas such as glioblastoma (GBM), low-grade gliomas, pediatric diffuse intrinsic pontine glioma (DIPG), as well as medulloblastoma in both children and adults (Liu et al., 2017; Sampetrean and Saya, 2018). The prognosis for GBM remains poor with less than 5.5% of patients surviving past the 5-year mark (Brennan et al., 2013; Jansen et al., 2010; Ostrom et al., 2017). DIPG outcomes are even poorer, with an average overall survival of less than 1 year (Lapin et al., 2017). Treatment of high-grade gliomas poses many challenges due to the infiltrative and resistant nature of tumors to conventional radiotherapy and chemotherapies.
Gliomas are classified by the WHO according to the glial tissue lineage they most resemble: astrocytomas, oligodendrogliomas, mixed oligoastrocytomas, and other diffuse gliomas. Tumors are also graded from I to IV based on malignancy of the tumor with I being benign (Louis, 2007; Verhaak et al., 2010; Vitucci et al., 2011). Grade IV astrocytomas and oligoastrocytomas are classified as GBM (Louis, 2007; Vitucci et al., 2011). Gliomas are highly infiltrative; tumor cells often migrate along white matter tracts to infiltrate and invade regions outside the original location, making surgical removal difficult (Holland, 2001). These tumors display significant cellular and genetic heterogeneity (Filbin et al., 2018; Parsons, 2008; Patel et al., 2014; Tirosh et al., 2016), which may contribute to tumor recurrence and therapy resistance (Mackay et al., 2017; Morrissy et al., 2016). The impossibility of complete surgical resection along with resistance to both radiation therapy and chemotherapy make GBM and DIPG almost impossible to cure with current standard treatment methods (Lapin et al., 2017; Maher et al., 2001). Additionally, tumors that recur after resection often progress to higher grades of glioma and become treatment resistance, which ultimately causes the mortality in patients (Ohgaki and Kleihues, 2009; van den Boom et al., 2003). Various efforts have been made recently to better understand brain tumors including investigations of cell of origin or tumor stem/initiating cells, transcriptome profiling, exome, genome, and epigenome sequencing, single-cell RNA sequencing, and use of genetically engineered mouse models. This review focuses on the convergence of developmental processes and brain tumorigenesis at molecular and cellular levels, highlighting potential vulnerabilities and targets for treating distinct subtypes of brain tumors.
CELLS OF ORIGIN AND GLIOMA STEM CELLS
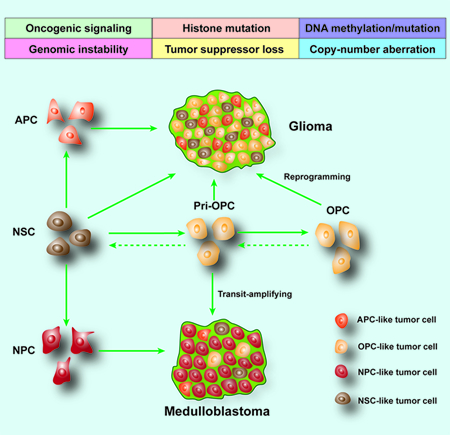
A better understanding the cell of origin of brain tumors may allow diagnosis at a pre-malignant stage and identification of lineage-specific vulnerabilities for therapeutic targeting. Neural stem cells are candidates for targets of transformation because of their inherent self-renewing capacity and their longevity; however, recent studies demonstrated that, upon oncogenic alterations, progenitors or committed cells can serve as tumor-initiating cells by dedifferentiating and reacquiring stem-cell-like traits (Friedmann-Morvinski et al., 2012; Schwitalla et al., 2013). The cell of origin of gliomas, defined by Visvader as tumor-initiating cells that undergo genetic mutations leading to the initiation of cancer (Visvader, 2011) are controversial. Investigations into which cells can undergo mutagenesis to initiate cancer indicate that neural stem cells (NSCs), oligodendrocyte precursor cells (OPCs), and astrocytes all have the ability, upon pathological insult, to proliferate—a prerequisite for the accumulation of mutations that induce tumorigenesis (Figure 1A).
Figure 1. Cells of origin and tumor-driving pathways in brain tumors.

A) Multipotent NSCs have capability to self-renew and differentiate into different fate-restricted progenitors. Under genetic and epigenetic alterations, these progenitor cells could be transformed into malignant tumors including gliomas from NSC, OPC or APC as well as medulloblastomas from NPCs.
B) A hierarchical model for tumor cell evolution and plasticity to generate distinct tumor-forming cells and tumor heterogeneity gained after tumor initiation.
C) Malignant transformation of GBM is the result of different driver pathway alterations. RTK signaling pathway activation mediated by extracellular signal (growth factors, cytokines, and hormones) promotes cell cycling for tumorigenesis. Besides, point mutation of histone 3 (H3K27M, H3G34R/V) and up-regulation of oncogenic factors, like MYC(N) and AURK, which lead to tumor cell proliferation. Moreover, IDH mutations later Krebs cycle in mitochondria, which further alter activity of downstream epigenetic regulators, including TET1/2 and KDM, leading to methylation alterations for DNA and nucleosome states susceptible to for tumorigenesis. GBM, glioblastoma; NSCs, neural stem cells; NPC, neuron progenitor cell; APC, astrocyte progenitor cell; OPC, oligodendrocyte progenitor cell; RTK, receptor tyrosine kinase; MYC, myelocytomatosis viral oncogene; AURK, aurora kinase; IDH, Isocitrate dehydrogenase; TET, Tet oncogene; KDM, lysine(K)-specific demethylase.
Neural stem/progenitor cells:
Neural stem/progenitor cells were an early contender for the cell of origin for gliomas; studies done using high-penetrance glioma mouse models showed that cells within the neural stem cell niche were more readily transformed than differentiated cell types outside of the niche (Alcantara Llaguno et al., 2009; Holland, 2000) (Figure 1A). Targeted deletion of tumor suppressor genes e.g. Nf1, Trp53, and Pten, in cells of the nestin- or GFAP-expressing neural progenitor cells led to fully penetrant GBM in mice, demonstrating the tumor-forming potential of NSCs in mice (Alcantara Llaguno et al., 2009; Zheng et al., 2008). The GFP reporter in the nestin-thymidine kinase-GFP transgenic mice was found to label quiescent neural stem cells in the adult subventricular zone as well as stem-like cells in tumor tissues of an animal model of glioma, suggesting the presence of stem-like cells in gliomas (Chen et al., 2012). Notably, ablation of the nestin-positive stem-like cells through activation of the thymidine kinase with ganciclovir attenuated tumor growth, although it failed to halt tumor progression (Chen et al., 2012), indicating that other nestin-negative progenitors may also contribute to the glioma growth. A recent study of a cohort of human GBMs using deep genomic sequencing indicated that normal subventricular zone tissue contained low-level GBM driver mutations, suggesting that NSCs migrate from the subventricular zone and lead to the development of high-grade malignant gliomas (Lee et al., 2018). Although these studies show that mutations in NSCs are sufficient for glioma formation, they did not demonstrate that NSCs are necessary or exclusive players in gliomagenesis.
Astrocyte lineage cells:
Astrocytes are contenders as the cell of origin because proliferating astrocyte progenitors may exist in the postnatal brain (Ge et al., 2012), although whether differentiated astrocytes can undergo cell division in the postnatal brain still remains controversial. In addition, astrocyte precursor cells have yet been identified or defined (Figure 1A). Upon traumatic brain injury, we and others have shown that some astrocytes re-enter the cell cycle (Bardehle et al., 2013; Chen et al., 2008). Implantation of astrocytes that express oncogenes into mouse brains or ablation of tumor suppressor genes from mouse brains led to malignant gliomas, therefore these cells can form gliomas (Bachoo et al., 2002; Endersby et al., 2011; Ghazi et al., 2012; Liu et al., 2011; Munoz et al., 2013; Paugh et al., 2013; Radke et al., 2013). These experiments are not definitive, however, as the typical astrocyte marker GFAP is also expressed by NSCs (Fraser et al., 2004; Lee et al., 2000; Molofsky et al., 2012). Tamoxifen-induced human GFAP promoter driven-CreER (GFAP-CreER) deletion of tumor suppressors Trp53, Pten, and Rb in adult mice led to high-grade astrocytoma formation in or contiguous to the adult proliferative niches in SVZ (as expected from the ability of NSCs to form gliomas) as well as in non-proliferative zones (Chow et al., 2011). GFAP-expressing astrocytes could be the cells of origin in these zones. In addition, a recent study of astrocyte diversity in the adult brain suggests that a subpopulation of astrocytes is the malignant analog of glioma (Lin et al., 2017).
Oligodendrocyte precursor cells:
OPCs are the most abundant cycling cell population of the adult central nervous system (Dawson et al., 2003; Imamoto et al., 1978) and represent the main pool of proliferative progenitor cells (~ 70%) in the normal adult rodent brain (Dawson et al., 2003; Dimou et al., 2008). Almost all mitotic cells co-express the OPC markers OLIG2 or NG2 in the human hippocampus (Geha et al., 2010). Their prevalence and mitotic characteristics throughout brain development make them possible cells of origin in brain tumorigenesis (Figure 1A). OPCs can be transformed and form malignant gliomas through overexpression of PDGF, the mitogen for OPCs (Dai, 2001; Lindberg et al., 2009; Uhrbom et al., 1998). Inactivation of p53 and Nf1 specifically in adult OPCs directed by NG2-CreER gives rise to malignant gliomas. (Galvao et al., 2014). Similarly, deletion of Nf1, Trp53, and Pten in the NG2-expressing OPCs in adult mice leads to GMB formation, although the tumors are more restricted to the ventral brain regions (Alcantara Llaguno et al., 2015).
In human brain tumors, OLIG2 is present, to various extents, in all grades of pediatric and adult diffuse gliomas including astrocytomas, oligodendrogliomas, and GBMs (Ligon et al., 2004; Lu et al., 2001; Otero et al., 2011). OLIG2, an essential transcription factor for OPC specification during central nervous system development, is expressed in OPCs and their primitive progenitors and controls the OPC-astrocyte fate switch in the developing brain (Lu et al., 2002; Takebayashi et al., 2002; Zhang et al., 2016b; Zhou and Anderson, 2002; Zhu et al., 2012). Notably, we and others showed that a large population of OLIG2+ cells in human gliomas, particularly proneural GBMs, expresses the proliferative marker Ki67 and the stem-cell marker CD133, suggesting that proliferative OLIG2+ cells are tumor-propagating cells (Ligon et al., 2007; Lu et al., 2016; Singh et al., 2016).
Strikingly, mosaic analysis with double markers (MADM) at a single-cell level revealed a critical role of OPCs in proliferation and expansion of glioma cells (Zong et al., 2005). Introduction of glioma-initiating mutations in Trp53/Nf1 in NSCs results in an expansion of OPC-like cells rather than proliferation of NSCs themselves prior to malignancy (Liu et al., 2011), suggesting that OPCs are a cell of origin, or transit-amplifying cells, for this model of glioma even when the initial mutations are in NSCs. In addition, in the OPC-expressing NG2-Cre-driven MADM, Trp53/Nf1 deletion initiated in OPCs results reactivation and subsequent expansion of mutant OPCs prior to their malignant transformation (Liu et al., 2011), suggesting that OPCs themselves can be directly transformed into malignant tumor cells likely through step-wise genetic and epigenetic reprogramming.
Recent single-cell transcriptomics analyses of different human gliomas with distinct driver mutations, including oligodendrogliomas, astrocytomas, GBMs, and DIPGs, revealed a prominent primitive OPC-like progenitor population that has a stemness-associated signature (Filbin et al., 2018; Patel et al., 2014; Tirosh et al., 2016; Venteicher et al., 2017). These observations indicate that human gliomas that arise from distinct genetic mutations may originate from the primitive OPC-like progenitors (pri-OPCs), the early progenitor cells preceding OPC commitment (Weng et al., 2019). The highly proliferative pri-OPCs may function as transit-amplifying cells during the onset of tumorigenesis and recurrence. The analyses of different tumorigenic phases at the single-cell level in a murine glioma model indicate that reprogramming of the OPC intermediates into a stem-like state, rather than direct stem-cell proliferation, resulted in their malignant transformation (Weng et al., 2019).
Once tumor development is initiated, how do tumor cells sustain or self-renew? The cancer stem cell theory posits that a small population of cells expressing CD133, a marker of neural stem cells, can sustain tumorigenesis through self-renewal and are capable of regenerating the tumor when transplanted (Singh et al., 2004). These CD133+ cells can differentiate into neurons, astrocytes, and oligodendrocytes when exposed to serum, that match the heterogeneous signatures of glioma tumors (Furnari et al., 2007; Nunes et al., 2003). This supports the notion that CD133+ NSC-like cells serve as glioma stem cells.
However, the existence of any dedicated cancer stem cell in brain tumors has yet to be defined. Controversy over the definition of glioma stem cells exists in part because there is evidence that certain CD133− cells are also capable of forming tumors in transplantation models (Beier et al., 2007; Joo et al., 2008). Additionally, there is evidence suggesting that OPC-like progenitors are the tumor-sustaining glioma stem cells: Transplantation of as few as 50 tumor cells matching an OPC profile cause glioma in immunocompromised host mice, whereas cells with an NSC profile do not (Persson et al., 2010). Similarly, under certain pathological conditions such as Nf1 and Trp53 mutations, fate-committed cells like OPCs can dedifferentiate and behave as facultative stem cells (Galvao et al., 2014; Liu et al., 2011). Given that malignant gliomas are highly heterogeneous, their cells of origin may derive from NSCs, OPCs and/or astrocytes in a context-dependent manner. Specific cell types may have distinct susceptibility to oncogenic signaling and reprogramming, which lead to their ultimate transformation. Future single-cell fate mapping at temporal and spatial resolution would require to define the cell of the origin in gliomas. Thus, the tumor cells of origin may encompass a range of the plastic cell types or progenitor cells that are susceptible to malignant transformation (Figure 1A).
GLIOMA HETEROGENEITY
Brain tumors are highly heterogeneous tumors, and precise cells of origin and driver oncogenes are difficult to define. Tumorigenesis is ascribed to a succession of genetic and epigenetic alterations that ultimately lead to the formation of a cell population characterized by functional and phenotypic heterogeneity (Hanahan and Weinberg, 2011). With the advancement of molecular technology, high-throughput and single-cell sequencing have allowed an unprecedented level of genomic analysis that has increased our understanding of gliomas and their heterogeneities. Intratumor heterogeneity could be the result of distinct tumor-initiating cells within tumor tissues (the stochastic model) or a set of cancer stem-like cells that can give rise to distinct tumor-forming cells (the hierarchy model), reflecting the heterogeneity gained after tumor initiation (Figure 1B).
The Cancer Genome Atlas Project (The Cancer Genome Atlas Research, 2008), a collaborative effort to increase understanding of the molecular and genetic basis of different cancers, was the first to complete a comprehensive genomic characterization of glioblastomas. This effort collected confirmed the relevance of the receptor tyrosine kinases (RTKs), tumor suppressor p53, and RB pathways in GBM, with 88%, 87%, and 78% of GBMs having altered signaling through these pathways, respectively (Parsons, 2008; The Cancer Genome Atlas Research, 2008).
Based on gene expression profiles in tumor tissues, GBMs have been classified into four distinct molecular subtypes: proneural, classical, neural, and mesenchymal (Verhaak et al., 2010). The proneural GBM subtype has OPC gene signatures and amplification of PDGFRA (the gene encoding platelet-derived growth factor), TP53 mutations, and/or mutations in IDH genes (which encode isocitrate dehydrogenases) (Brennan et al., 2013; Verhaak et al., 2010). The classical subtype is characterized by an astrocytic signature and activation or amplification of the gene encoding epidermal growth factor receptor (EGFR) and/or loss of the CDKN2A locus (Hayden, 2010; Verhaak et al., 2010). It is worth noting that the mesenchymal subtype is characterized by reactive astrocyte (CD44+) and microglia signatures, NF1 deletion, and elevation of TNF-α and NF-κB signaling (Verhaak et al., 2010). Consistently, TNF-α treatment could lead to a shift of proneural GSCs towards a mesenchymal state with CD44 expression and NF-κB activation (Bhat et al., 2013), suggesting the tumor microenvironment plays a role in this transition. However, it is worth noting that mesenchymal genetic signatures may potentially result from tumor stromal components, as opposed to the tumor cells of the neoplastic tissues, which rather exhibit expression profiles similar to stem-like cells and proneural tumor cells (Calon et al., 2015; Isella et al., 2015; Kim and Verhaak, 2015; Martinez et al., 2015), suggesting that mesenchymal subtype tumors consist of a mixture of tumor stem-like cells and tumor stromal microenvironment components. In addition, recent studies suggest that the neural GBM signature may be attributable to the contamination of normal brain tissue profiles (Wang et al., 2018b).
Single-cell sequencing, which allows profiling of single cells as opposed to bulk tumor tissue, has also been utilized to define the heterogeneity of gliomas (Khoo et al., 2016; Levitin et al., 2018; Ortega et al., 2017). This approach has enabled the dissection of heterogeneous cellular populations within tumors by identifying potential malignantly transformed cells (e.g. stem-like subpopulations and sublineages of developmentally related cells) and cell types comprising tumor microenvironments (e.g. infiltrating immune populations and other stromal populations) in different types of brain tumors (Filbin et al., 2018; Patel et al., 2014; Tirosh and Suva, 2018). In addition, single-cell analysis may further reveal diverse physiological states of tumor cells such as cycling or quiescent phases, stressed, hypoxic or DNA damage states (Patel et al., 2014; Tirosh and Suva, 2018). Single-cell profiling of IDH1 and IDH2 mutant human oligodendrogliomas revealed that cancer cells consist of a mix of oligodendrocyte- and astrocyte-lineage progenitor cells as well as undifferentiated stem-like progenitors, which are highly proliferative and responsible for fueling the tumor growth (Tirosh et al., 2016). Similarly, the analysis of H3K27M-glioma indicated that these tumors mainly contain OPC-like progenitors with stemness signatures (Filbin et al., 2018). Thus, the malignant populations within tumor tissues appear to be superimposed on aberrant developmental processes, consisting of stem-like cells, OPC-like and astrocyte-lineage-like cells.
By fate mapping of primary barcoded human GBM cells following serial xenografts, Lan et al. showed that tumorigenesis progresses through a developmental-like hierarchy rooted in GBM stem cells from slow-cycling stem-like cells to rapidly cycling progenitors, which in turn generate post-mitotic cells (Lan et al., 2017). Single-cell analysis of the tumor core and surrounding tissues revealed that infiltrating cells share gene signatures of tumor cells preexisted in heterogeneous GBM samples, suggesting that a common cellular mechanism drives tumor infiltration (Darmanis et al., 2017). These studies at the single-cell level have resulted in a detailed view of the molecular landscape of gliomas, characterizing the heterogeneity that exists within and between tumors, which is likely due to distinct patterns of somatic mutations, copy number alterations and DNA methylation status in tumor cells. Ultimately, single-cell fate tracing studies, e.g. through CRISPR–Cas9 mediated genetic barcoding (Kalhor et al., 2017; McKenna et al., 2016) or the mosaic lineage tracing MADM system (Zong et al., 2005), will need to test whether GBM tumor cells are derived from common or distinct progenitors.
GLIOMA CORE REGULATORY PATHWAYS
Cooperative actions of lineage-determining and signal-dependent transcription factors dictates the spatio-temporal pattern of gene expression and cellular reprogramming that predisposes post-mitotic cells to acquire stem cell-like properties, giving rise to tumor-initiating cells endowed with long-term tumorigenic capacity (Friedmann-Morvinski et al., 2012; Vogelstein et al., 2013). Given their pivotal roles in the determination of cell identity and cell growth, decommissioning of the lineage differentiation program and activation of oncogenic transcriptional programs are critical steps toward cell reprogramming and oncogenic transformation (Vogelstein et al., 2013).
Core signaling pathways:
Analysis of the large-scale molecular and genomic information present in the TCGA database indicated that the main signaling pathways involved in GBM tumorigenesis are the p53 pathway (p53/MDM2/4/p14ARF), the PTEN/NF1/RTK pathway (EGFR/RAS/NF1/PTEN/PI3K), , and the RB pathway (p16INK4a/CDK4/RB1) (Ohgaki and Kleihues, 2007, 2009) (Figure 1B). Glioblastomas are divided into primary and secondary classifications. It is believed that primary GBM arises de novo and secondary GBM arises from lower grade gliomas and progresses to stage IV (Holland, 2001; Maher et al., 2001; Ohgaki and Kleihues, 2007, 2009). Although both forms have the same histological appearance and clinically grim prognosis, they exhibit distinct genetic or oncogenic mutations (Furnari et al., 2007; Ohgaki and Kleihues, 2007, 2009). Primary GBMs are genetically associated with loss of heterozygosity in 10q (70%), EGFR amplification (35%), p16INK4a deletion (31%), and PTEN mutations (25%), and occur mainly in adults, while secondary GBMs exhibit the most frequent mutations in the TP53 gene and affect younger populations (Furnari et al., 2007; Ohgaki and Kleihues, 2007, 2009) (Table 1). Notably, different tumors, and even different cells within the same tumors, can have mutations in different parts of these pathways (Patel et al., 2014).
Table 1.
Genetic alterations and pathways in primary and secondary GBM
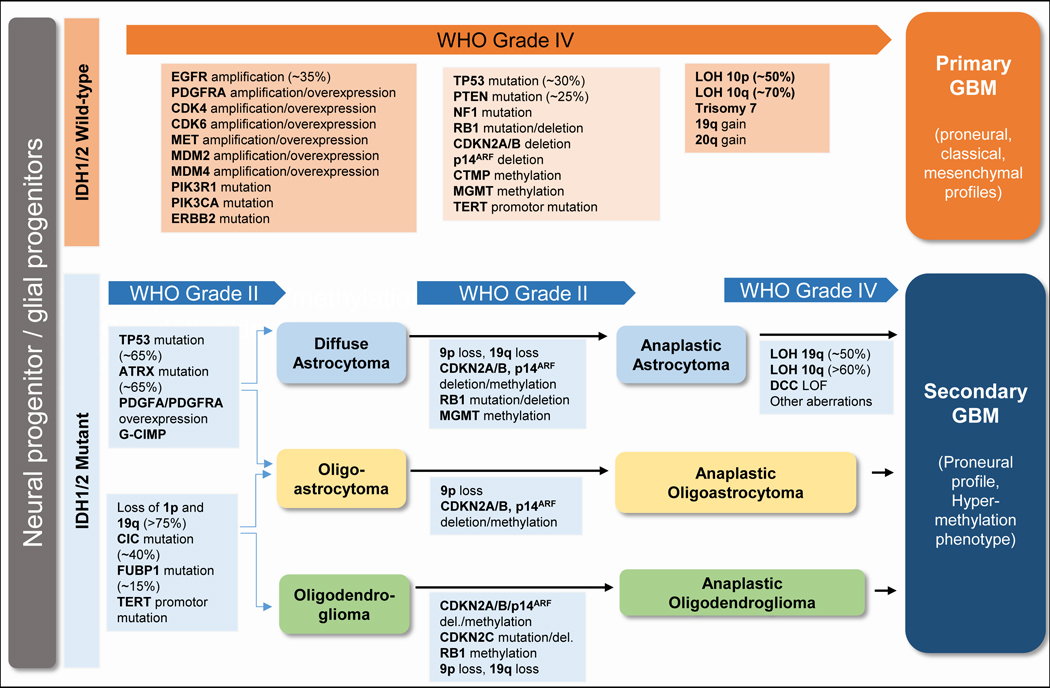 |
TP53 encodes the tumor suppressor p53; this protein pauses the cell cycle in response to DNA damage and can initiate apoptosis (Meletis et al., 2006; Vogelstein and Kinzler, 2004). Mutations in TP53 are found in astrocytomas of multiple grades (Louis et al., 1993; van Meyel et al., 1994) and may allow early astrocytomas to evade apoptosis (Zhu et al., 2005). Introduction of germline mutations into tumor suppressor genes Nf1 and Trp53 in mice is sufficient to induce development of various grades of astrocytomas (Reilly et al., 2000).
The PTEN/NF1/RTK pathway controls cell growth and involves RTKs such as EGFR and PDGFR and their downstream cascade proteins that promote cell growth (e.g., RAS, MAPK, and PI3K). The tumor suppressors PTEN and NF1 are the negative regulators of PI3K and RAS activity, respectively, and are the modulators of cell cycle entry in neural stem cells (Fraser et al., 2004; Groszer et al., 2006) (Figure 1C). Inactivation of both Trp53 and Pten, but not either gene alone, in murine neural stem cells leads to high-grade malignant glioma by driving MYC oncoprotein expression (Zheng et al., 2008). Furthermore, ablation of Pten in the Nf1/Trp53 model generates highly penetrant GBMs (Kwon et al., 2008), suggesting a cooperative effect of these oncogenic- and tumor-suppressive pathways (Table 1).
RB is a tumor suppressor protein that regulates the G1/S checkpoint through sequestering E2F and inhibiting its oncogenic activity (Malumbres and Barbacid, 2001). Mutations in genes in the RB pathway (e.g., RB, p16INK4a, and CDK4) result in increased mitotic activity, leading to malignant transformation (Knudsen and Wang, 2010; Sharpless and DePinho, 1999).
Activation of Hippo-Taz/Yap signaling has also been linked to GBM formation (Zhang et al., 2016a). Silencing of Taz and its co-transcriptional regulator Tead in mesenchymal GBM cells decreases self-renewal, invasion, and tumor formation, whereas overexpression of TAZ in proneural GBM cells induces mesenchymal signature gene expression (Bhat et al., 2011). In summary, mutations in these oncogenic and tumor suppressive signaling pathways lead to apoptosis resistance, increased cell growth, and unchecked proliferation (Figure 1B).
Core transcriptional regulators of neurodevelopment and tumorigenesis crosstalk:
Transcriptional regulators play key roles in both normal development and tumorigenesis, which share similar cellular hierarchies. Recent studies identify a core set of neurodevelopmental transcriptional regulators, including POU3F2, SOX2, SALL2, and OLIG2, which are essential for GBM propagation (Suva et al., 2014). Activation of these factors is sufficient to reprogram differentiated cells into tumor-propagating cells (Suva et al., 2014).
Among these core regulatory factors, OLIG2 is a central nervous system-restricted transcription factor that plays a critical role in OPC specification and proliferation (Lu et al., 2002; Takebayashi et al., 2002; Zhang et al., 2016b; Zhou and Anderson, 2002). During early embryogenesis, Olig2 is strongly expressed in the progenitors of medial ganglionic eminence (MGE) but weakly in the lateral ganglionic eminence (LGE). In the developing cortex of later embryonic stages, Olig2 is mainly expressed in the transit-amplifying progenitors in the SVZ and oligodendrocyte lineage cells (Lu et al., 2002; Miyoshi et al., 2007; Yue et al., 2006; Zhu et al., 2012), but not neural stem cells (e.g. nestin+ cells) in the VZ. Interestingly, OLIG2 appears to be ubiquitously expressed in human gliomas (Ligon et al., 2004; Lu et al., 2001) and has a regulatory role in glioma initiation and tumor phenotype plasticity (Kupp et al., 2016; Ligon et al., 2007; Lu et al., 2016; Singh et al., 2016).
An early study showed that overexpression of oncogenic EGFRvIII does not induce transformation of Olig2−/− neural stem cells carrying Cdkn2a−/− alleles into glioma in allografts (Ligon et al., 2007). In contrast, in a mouse model of proneural GBM with Trp53/Pten deletions and PDGFB expression, Olig2 deletion delays, but does not completely block, tumor growth (Lu et al., 2016). The distinct effect of Olig2 deletion on glioma growth inhibition under different oncogenic mutations suggest that OLIG2 regulates gliomagenesis in a context (e.g. p53 mutation status)-dependent manner.
Olig2 deletion leads to a shift to a slower-growing phenotype with an astroglial gene expression pattern (Lu et al., 2016). Olig2-deletion in a murine proneural GBM model leads to decreases in expression of PDGFRα (Lu et al., 2016), a key regulator of OPC proliferation. Similarly, in human proneural GBM cell lines, OLIG2-knockdown leads to downregulation of PDGFRα signaling (Kupp et al., 2016; Lu et al., 2016), suggesting the OLIG2 is a key regulator of PDGFRα signaling (Figure 1B). It is worth noting that Olig2-deficient tumor cells in a Pten/Trp53 negative mouse proneural glioma model appear to utilize other signaling pathways (e.g., the EGFR pathway) as an alternative to sustain tumor cell growth, despite downregulated PDGFRα signaling (Lu et al., 2016). This is in contrast to the essential role of OLIG2 glioma formation in p53-expressing backgrounds (Ligon et al., 2007). This suggests that OLIG2 function is likely dependent on the mutation status of p53 given that Olig2 negatively regulates p53 and p21 tumor suppressors (Mehta et al., 2011).
Core transcriptional regulators such as SOX2 and OLIG2, which also mark OPCs, can be elevated by oncogenic RTK signaling such as that mediated by EGFRvIII. Interestingly, once activated, these factors can induce the transformation of tumor-suppressor-deficient astrocytes into glioma-initiating cells independent of RTK signaling (Singh et al., 2017). In addition, given the intratumoral heterogeneity of these tumors, a subpopulation of tumor-initiating cells within the tumor may not rely on these core factors e.g. Olig2, for tumor growth eventually (Lu et al., 2016; Yu et al., 2017). This suggests that additional growth regulators other than these core transcription factors may support tumor cell growth.
Olig2 deletion in the proneural GBM tumor model leads to a transcriptional program shift with downregulation of the OPC-associated proneural signature and an up-regulation of astrocyte-associated classical tumor signature (Lu et al., 2016). This is in keeping with the role of OLIG2 in the OPC-astrocyte fate switch during development (Cai et al., 2007; Lu et al., 2002; Zhu et al., 2012). Similarly, the opposing transcription factors SOX10 and NFIA, which control the balance of oligodendrocyte and astrocyte fate during development, also regulate oligodendroglioma and astrocytoma formation (Glasgow et al., 2014), suggesting a common theme for the control of glial lineage diversification during development and tumorigenesis, and that tumor phenotypes (oligodendrocyte- versus astrocyte-like phenotypes) may be delineated by the lineage-driving transcriptional regulators. In summary, glial lineage specification factors such as OLIG2 are key players in oncogenic reprogramming, particularly for proneural subtype of gliomas, as their activation in cooperation with other oncogenic insults predisposes glial progenitors to neoplastic transformation and triggers tumor initiation through the modulation of the epigenetic state of enhancers.
EPIGENETIC REGULATION IN GLIOBLASTOMA
Cell reprogramming requires overcoming epigenetic barriers that are involved in maintaining cell-specific transcriptional programs that preserve cell identity (Apostolou and Hochedlinger, 2013; Feinberg et al., 2016). Epigenetic modifications such as DNA methylation, histone modifications, chromatin remodeling, and three-dimensional nucleosome organization are not only crucial for normal development but also for tumor initiation and progression (Apostolou and Hochedlinger, 2013; Feinberg et al., 2016). Recent discoveries of cellular and genomic reprogramming in glioblastomas have highlighted the importance of the chromatin modification landscape in the regulation of gliomagenesis and progression. Defining the specific chromatin modification landscapes of distinct tumors may identify specific vulnerabilities that can be harnessed for therapy.
DNA methylation:
Epigenetic silencing through promoter hypermethylation of tumor suppressor genes such as those controlling the cell cycle and DNA repair, PTEN, RB, RASSF1A, CDKN2A/2B, and MGMT, is commonly observed in cancers including brain tumors (Baylin and Jones, 2011; Malzkorn et al., 2011; Martinez and Esteller, 2010; Schmidt et al., 2012; Veeriah et al., 2009). The analysis of cancer methylome not only reveals somatically acquired DNA methylation alterations but also identifies the tumor cell of origin (Capper et al., 2018; Hovestadt et al., 2014), which allows tracing the primary site of metastatic tumors (Moran et al., 2016). DNA methylation is regulated by DNA methyltransferases and TET (the ten-eleven translocation) demethylation enzymes, the latter leads to DNA hydroxymethylation and demyelination (Ito et al., 2010; Tahiliani et al., 2009). DNA methylation represses gene expression directly or by recruitment of repressive chromatin remodelers including methyl-CpG-binding domain (MBD) proteins such as including MeCP2, MBD1, MBD2 and MBD4 (Bhattacharjee et al., 2016; Ikegami et al., 2009). Global DNA hypermethylation promotes chromosomal instability, reactivation of transposable elements, and loss of imprinting (Deleris et al., 2012; Estecio and Issa, 2011). Local hypermethylation silences expression specific tumor suppressor genes, and hypomethylation induces oncogene activation.
Dysregulation of DNA methylation patterns has been linked to the initiation and progression of GBM. A well-studied example is hypermethylation of a CpG island that results in the silencing of the expression of a DNA repair enzyme MGMT, encoding O6-methylguanine-DNA methyltransferase. Brain tumors with hypermethylated MGMT, are susceptible to genetic mutations in critical genes such as TP53 or KRAS (Baylin and Jones, 2011; Malzkorn et al., 2011; Martinez and Esteller, 2010; Schmidt et al., 2012; Veeriah et al., 2009). Epigenetic silencing of MGMT leads to a defect in the removal of carcinogen-induced O6-methylguanine adducts from DNA, resulting in G to A transition mutations (Baylin and Jones, 2011; Malzkorn et al., 2011; Martinez and Esteller, 2010).
Interestingly, the defect in DNA repair enzyme MGMT is associated with a better response to treatment with alkylating agents (Bobola et al., 2015). MGMT methylation status is now used as a biomarker for predicting drug responsiveness in GBM patients. Epigenetic changes like DNA methylation likely regulate expression of GBM-associated genes. Resetting of DNA methylation in GBM-initiating cells by reprogramming induced pluripotent stem cells results in suppression of malignant cellular behavior when directed toward nonneural lineages (Stricker et al., 2013). Although TET2 mutations have been linked to acute myeloid leukemia (Liu et al., 2014), currently, the functions of TET and hydroxymethylation in brain tumorigenesis remain to be determined.
Histone modifications:
Malignant transformation of brain tumors is associated with aberrant alterations of histone modifications. The histone methyltransferase LSD1 (also known as HDM1A) can decrease trimethylation of histone 3 at K4 over the MYC locus and elevates its expression (Kozono et al., 2015). MYC, in turn, regulates the expression of a core set of transcription factors, including OLIG2, SOX2, and POU3F2, which are required for the stemness maintenance of GBM cells (Kozono et al., 2015; Suva et al., 2014). LSD1 also suppresses the p53 signaling pathway, thereby promoting GBM cell tumorigenesis and metastasis (Yi et al., 2016). In contrast, overexpression of JMJD3 (also known as KDM6B), which demethylates histone 3 at K27, induces a p53-mediated differentiation of glioblastoma stem cells, suggesting that JMJD3 functions as a tumor suppressor by regulating p53 protein nuclear stabilization (Ene et al., 2012)
The type II arginine methyltransferase PRMT5, that catalyzes monomethylation and symmetric dimethylation of arginine residues (Huang et al., 2011), is also critical for self-renewal capacity and proliferation of GBM cells as it regulates PTEN expression and controls AKT and ERK activity (Banasavadi-Siddegowda et al., 2017). Expression of PRMT5 correlates with malignant grade in gliomas and is critical for tumor growth (Banasavadi-Siddegowda et al., 2018; Han et al., 2014). These observations indicate that histone methylation and demethylation may play important roles in GBM growth.
Histone acetylation and deacetylation are usually regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs). HATs are divided into two groups based on subcellular localization [42]. Type A HATs (e.g., GCN5, p300/CBP, and TAFII250) are localized in the nucleus and acetylate nucleosomal histones whereas type B HATs (e.g., HAT1) are localized in the cytoplasm and acetylate newly synthesized histones (Lee and Workman, 2007). Histone acetylation levels are often correlated with glioma progression (Huang et al., 2015; Lee et al., 2014). Histone acetyltransferase KAT6A acetylates histone 3 at K23 and recruits the nuclear receptor binding protein TRIM24 to activate the transcription of PIK3CA, thereby promoting PI3K/AKT signaling and GBM progression (Lv et al., 2017). KAT6A silencing suppresses cell proliferation, cell migration, colony formation, and tumor development in an orthotopic mouse xenograft model system (Lv et al., 2017). The specific role of individual histone acetyltransferase in gliomagenesis has not yet been fully defined at present.
HDACs also play important roles in the regulation of GBM tumorigenesis. HDAC inhibitors have been shown to induce re-expression of silenced tumor suppressor genes (Dickinson et al., 2010; Newbold et al., 2013). HDAC inhibition can sensitize EGFR/EGFRvIII-overexpressing, erlotinib-resistant GBM cells to RTK inhibition (Liffers et al., 2016). Inhibition of HDACs and blockade of the glycolytic pathway synergistically induce GBM cell death (Egler et al., 2008). Inactivation of class IIa HDACs by mTORC2 leads to acetylation of FOXO1 and FOXO3, which increases c-MYC levels by suppression of miR-34c expression, leading to an increase in glycolytic metabolism and GBM cell growth (Masui et al., 2013). Thus, HATs and HDACs may coordinate to regulate GBM growth likely in a context-specific manner.
Oncometabolites:
Mutations in metabolic regulatory enzymes can affect DNA and histone methylation. For instance, the mutations in isocitrate dehydrogenase (IDH) 1 and 2, which occur early in gliomagenesis, lead to the accumulation of 2-hydroxyglutarate, known as a “oncometabolite” (Cohen et al., 2013; Yan et al., 2009). The oncometabolite, 2-hydroxyglutarate (2-HG), is a competitive inhibitor of α-ketoglutarate (α-KG)-dependent epigenetic enzymes such as histone lysine demethylases and TET hydroxylases (Feinberg et al., 2016; Li et al., 2018; Lu et al., 2012). Mutations in IDH1 and IDH2 impair DNA demethylation by inhibiting TET family enzymes and causes hypermethylation in glioma (Turcan et al., 2012). Intriguingly, IDH1/2 mutations also disrupt histone demethylation and cause the progenitor cells to become susceptible to transformation (Lu et al., 2012). Similarly, mutations in fumarate hydratase and succinate dehydrogenase can lead to accumulation of fumarate and succinate, respectively, which also function as competitive inhibitors of histone demethylases and TET enzymes to alter DNA and histone methylation state and cause genomic instability (Vogelstein et al., 2013; Xiao et al., 2012) (Figure 1C). A recent study indicates that metabolic enzymes such as α-ketoglutarate dehydrogenase can be localized in the nucleus and bind to lysine acetyltransferase KAT2A (also known as GCN5) together with succinyl-coenzyme A. The KAT2A complex then acts as a succinyltransferase to catalyze lysine succinylation on histone 3 at K79 to regulate gene transcription and tumor cell proliferation (Wang et al., 2017).
In summary, epigenetic alterations such as promoter hypermethylation or oncometabolites can not only cause dysregulation of gene expression but also predispose to genetic mutations (Choi and Lee, 2013; You and Jones, 2012), which can lead to transformation and tumor progression. Thus, epigenetic regulatory mechanisms are prevalent in tumorigenesis, the regulatory and genetic events can cooperate and co-opt each other to impinge on neoplastic transformation during brain tumorigenesis.
PEDIATRIC BRAIN TUMORS
Pediatric brain tumors include pediatric high-grade gliomas (HGGs) and medulloblastomas. Genetic and epigenetic profiling experiments have revealed molecular characteristics of these diseases and have allowed subclassifications useful for treatment stratification. Pediatric HGGs are the most common malignant pediatric brain tumors and include GBMs, anaplastic astrocytomas, and DIPGs (Liu et al., 2017; Ostrom et al., 2014). Based on distinct DNA methylation patterns in adult GBMs and pediatric HGGs, six biological HGG subgroups have been identified: IDH (also called G-CIMP), mesenchymal, RTK-I, RTK-II, H3K27, and H3G34 tumors (Liu et al., 2017; Sturm et al., 2012). IDH, K27, and RTK-I tumor clusters exhibit a correlation with proneural GBM gene expression patterns, whereas H3G34 and RTK-II subgroups possess classical gene expression signatures (Sturm et al., 2014).
Cells of origin in DIPG:
DIPG is an especially malignant type of glioma due to its diffuse nature and location within the pons, making surgical resection virtually impossible, leading to very poor prognosis (Lapin et al., 2017; Warren, 2012). DIPGs could be derived from neural stem cells (Filbin et al., 2018; Misuraca et al., 2015) or OLIG2+ OPC-like progenitors (Filbin et al., 2018; Monje et al., 2011) (Figure 2A).
Figure 2. Developmental origins and key signaling pathways in MB tumorigenesis.

A) Potential progenitor sources for distinct MB in the posterior fossa. Progenitor cells in the ventricular zone (SHH, Group 3 and Group 4), EGL (SHH), anterior brainstem progenitors (SHH or other groups), and dorsal brainstem (WNT) contribute to distinct MB subtype tumorigenesis.
B) Distinct MB subtypes (WNT, SHH, Group 3 and Group 4) show different signaling driving pathways for promoting cell cycling and growth. In WNT MB, activated β-catenin cooperates with transcription factor LEF/TCF to promote cell proliferation. In SHH MB, transmembrane proteins PTCH, SMO and GPCR mediated transcriptional factor Gli activation for tumorigenesis. In Group 3 MB, oncogene MYC activates transcriptional factor GFI1 and promote tumorigenesis. Group 4 MB shows activation of ERBB4 and EGFR signaling. MB, medulloblastoma; LEF/TCF, lymphoid enhancer binding factor/T-cell specific transcription factor; PTCH, patched; SMO, smoothed; GPCR, G protein coupled receptor; Gli, glioma-associated oncogene; MYC, myelocytomatosis viral oncogene; GFI1, growth factor independent 1 transcriptional repressor.
Genetic alterations in DIPG:
Whole genome, exome, and methylome sequencing of biopsy and autopsy tissues have revealed the most common mutations and chromosomal and epigenetic abnormalities in DIPG (Mathew and Rutka, 2018; Taylor et al., 2014b). Surprisingly, histone mutations are the most common mutation in DIPG, seen in up to 80% of cases (Khuong-Quang et al., 2012; Lapin et al., 2017; Lewis et al., 2013; Saratsis et al., 2014). The most common histone mutations in DIPG are lysine 27 (K27) to methionine (M) of histone 3 variants H3F3A and HIST1H3B (H3.3 or H3.1) to form H3.3K27M or H3.1K27M mutant proteins (Lapin et al., 2017). In addition, G34R or G34V mutations, which are restricted to H3.3, are mostly found in hemispheric GBM-like tumors in adolescents (Korshunov et al., 2016). These mutations to histone 3 are unique to HGGs and are not found in adult GBMs (Castel et al., 2015; Lewis et al., 2013), suggesting that DIPG behaves distinctly from adult gliomas.
Mutations in histone 3 variants (e.g., H3.3K27M or H3.1K27M) lead to an inhibition of PRC2, the polycomb repressive complex, activity, causing a global decrease in trimethylation of histone 3 at K27 and abnormal epigenetic silencing of tumor suppressors such as p16INK4a (Cohen et al., 2017). TP53 mutations have been identified in up to 40% of DIPGs and often co-occur with PDGFRA amplification (Grill et al., 2012; Puget et al., 2012; Taylor et al., 2014a). Although H3K27M alone is insufficient to drive tumorigenesis, it does induce glioma formation when combined with Trp53 mutations and activating PDGFRAD842V by orthotopic implantation of transformed human astrocytes or NSCs (Funato et al., 2014), in utero focal delivery into the embryonic cortex (Pathania et al., 2017) or through genetically engineered mice (Larson et al., 2019). Mutations in histone 3 at G34 are closely linked to ATRX mutations, which could promote telomere lengthening (Heaphy et al., 2011).
PDGFRA amplification or activating mutations (Mackay et al., 2017; Paugh et al., 2013) (Filbin et al., 2018), which trigger the RTK-RAS-PI3K-AKT pathway, are the most commonly observed alterations in DIPG (~32% of patients) (Monje et al., 2011; Paugh et al., 2011; Puget et al., 2012; Wu et al., 2014). Amplifications or mutations in other factors involved in the RTK/RAS pathway (such as PI3KCA) are also common in DIPG, and the pathway itself is active in approximately 69% of DIPG tumors (Monje et al., 2011; Paugh et al., 2011; Puget et al., 2012; Wu et al., 2014).
Another gene commonly mutated in DIPG is ACVR1 (~25% of patients). ACVR1 encodes ALK2 the receptor for bone morphogenetic protein (BMP) (Buczkowicz et al., 2014; Fontebasso et al., 2014; Hoffman et al., 2016; Mackay et al., 2017; Taylor et al., 2014a; Wu et al., 2014). These ACVR1 mutations often co-segregate with H3.1 mutations and are usually are found in tumors with wild-type p53 (~90%) or PI3K pathway alterations (~56%) (Buczkowicz et al., 2014; Fontebasso et al., 2014; Taylor et al., 2014a; Wu et al., 2014). The ACVR1 mutation results in a slight activation of the BMP signaling pathway, which promotes a differentiated phenotype and could possibly account for the increased survival advantage conferred by ACVR1 mutations (Piccirillo et al., 2006). In addition to the mutations in histone 3, ACVR1, TP53, PDGFRA, PIK3CA, and MYC (Lapin et al., 2017), chromosomal abnormalities are especially frequent in DIPG and include gains of 1q, 2q, 8q, and 9q and losses in 11p, 17p13.1, 14q, 18p, and 22q (Castel et al., 2015; Puget et al., 2012; Zarghooni et al., 2010) (Table 1).
Increasing methylation of histone 3 at K27 (H3K27me3) by inhibition of JMJD3 can prolong survival of the mice with DIPG xenografts (Hashizume et al., 2014). In contrast, inhibition of a PRC2 component EZH2 activity suppressed tumor cell growth in a mouse model of DIPG and in patient-derived DIPG cell lines (Mohammad et al., 2017). The latter observation suggests that residual PRC2 activity, which likely selectively regulates expression of tumor-suppressor protein p16 (also known as INK4A) is critical for the growth of H3K27M-DIPGs. Intriguingly, EZH2 inactivation in a mouse model of group 3 medulloblastoma accelerated tumorigenesis by derepressing the proto-oncogene GFI1 in collaboration with MYC (Vo et al., 2017). Thus, whether EZH2 acts as an oncogene or tumor suppressor appears to be context dependent.
Combined treatment with the HDAC inhibitor panobinostat and the histone demethylase inhibitor GSK-J4 has been found to effectively block tumor cell growth in vitro and in orthotopic H3K27M-DIPG xenograft models (Grasso et al., 2015). Moreover, inhibitors of PRC2-EZH2 and CDK4/6 blocks H3K27M-DIPG cell growth due to the unique repression of CDKN2A transcription by H3.3K27M mutations (Cordero et al., 2017; Mohammad et al., 2017). The presence of unique genetic and epigenetic abnormalities indicates that DIPGs are not a subset of GBMs. Recent studies have furthered our understanding of driver mutations and of specific epigenetic vulnerabilities in DIPG, which should allow guiding development of treatments for these deadly pediatric brain tumors.
Medulloblastomas:
Medulloblastomas (MBs) account for 20% of malignant pediatric brain tumors (Ostrom et al., 2017; Yi and Wu, 2018). Unique gene mutations are found in each case, in contrast to hotspot mutations seen in DIPGs (Jones et al., 2012; Schwartzentruber et al., 2012). Based on genetic and epigenetic profiling, medulloblastoma can be classified into WNT (Wingless), SHH (Sonic Hedgehog), Group 3, and Group 4 subtypes (Northcott et al., 2011; Taylor et al., 2012). WNT-MB accounts for about 10% of medulloblastoma cases and has the best prognosis of the four subtypes with long-term survival exceeding 90% (Ellison et al., 2011). SHH-MB tumors are present in about 30% of MB cases, and these tumors are driven by mutations in the SHH signaling pathway (He et al., 2014; Jones et al., 2012; Northcott et al., 2012). Group 3 MB (G3-MB) accounts for about 25% of medulloblastomas and has the worst prognosis of the four subgroups (Liu et al., 2017). Group 4 MB (G4-MB) is the most prevalent MB subtype at 35%; the molecular pathogenesis of the subtype is the least understood among MBs (Gajjar and Robinson, 2014; Northcott et al., 2012).
The cells of origin in MB:
Although MBs are localized in the posterior fossa or cerebellum, they exhibit extensive intertumoral heterogeneity in primary, recurrent and metastatic tumors (Cavalli et al., 2017). WNT-MB is believed to originate in the lower rhombic lip or embryonic dorsal brainstem. Activation of Ctnnb1 in the dorsal brainstem progenitors (Zic1+ cells) concurrent with Trp53 deletion results in abnormal hyperproliferation and tumors with a WNT-MB profile (Gibson et al., 2010) (Figure 2A).
In SHH-MB, although granule neuron precursor cells may very well serve as the cell of origin for tumor cell propagation (Gibson et al., 2010; Schuller et al., 2008; Yang et al., 2008), the identity of tumor-initiating cells is not known. Deletion of Ptch1 and GNAS or activation of SMO in a variety of progenitor cells including cerebellar neuroepithelial (nestin+/GFAP+ neural stem/progenitor) cells (Schuller et al., 2008; Yang et al., 2008), OPC-related Olig1+ progenitors in the anterior brainstem (He et al., 2014), or brainstem progenitors (Grammel et al., 2012) results in SHH-MB in animal models. This suggests that various progenitors are susceptible to the activation of SHH signaling and undergo malignant transformation into SHH-MB (Figure 2A).
The G3-MB tumors are often located near the fourth ventricle, suggesting that cerebellar stem/progenitor cells or granule neuron precursor cells are likely the source of G3-MB (Kawauchi et al., 2012; Pei et al., 2012). Similar to G3-MBs, G4-MBs are often detected close to the fourth ventricle or related to Tbr2+ unipolar brush cells (UBCs) through single-cell analysis (Wang et al., 2018a)(Vladoiu et al., bioRxiv, 2018; http://dx.doi.org/10.1101/350280). The progenitors of the nuclear transitory zone at the upper rhombic lip are thought to be a cell of origin for G4-MBs (Lin et al., 2016), although the precise cells of origin for G4-MB are not known (Figure 2A).
A recent whole-genome analysis demonstrated substantial genetic divergence of the dominant clones between primary and post-therapy human MBs. The dominant clone at recurrence arose through clonal selection of a pre-existing minor clone present at diagnosis (Morrissy et al., 2016). The developmental hierarchy and cellular heterogeneity during tumorigenesis and recurrence in each MB subtypes needs to be further defined by single-cell transcriptomics, clonal mapping, and proteomics analyses. In summary, although the precise cells of origin remain unclear, MBs can arise from multiple cell types from different locations at stem cell or intermediate progenitor stages during tumor initiation and recurrence (Figure 2A).
Genetic mechanisms underlying distinct MB subgroups:
In WNT-MB, activating mutations in the CTNNB1 gene encoding β-catenin, or mutations in DDX3X, an ATP-dependent RNA helicase, and in chromatin remodelers SMARCA4 and CREBBP have been proposed to play an oncogenic role in tumorigenesis (Jones et al., 2012; Northcott et al., 2012; Robinson et al., 2012).
The SHH-MB subgroup tumors are characterized by a variety of mutations in the SHH pathway including loss-of-function mutations in PTCH1 and SUFU, activating mutations in SMO, and amplifications of SHH, GLI2, and MYCN (Jones et al., 2012; Liu et al., 2017; Pugh et al., 2012; Robinson et al., 2012). Recently, nonsense mutations in GNAS, which encodes a G-protein Gαs, were detected in an aggressive form of SHH-MB (He et al., 2014). SHH signaling is mediated through activation of the G protein-coupled receptor-like seven-transmembrane protein SMO. The Gαs protein may couple other G protein-coupled receptors, including GPR161, to inhibit SHH signaling (Shimada et al., 2018). Activation of Gαs protein not only stimulates cyclic adenosine monophosphate (cAMP)-dependent signaling but also inhibits ciliary trafficking of hedgehog components. Elevation of cAMP inhibits MB growth by blocking SMO signaling to decrease tumor cell proliferation (He et al., 2014) (Figure 2B). A recent study identifies the protein kinase CK2 as a driver of phosphorylation events to promote GNP growth during tumorigenesis, and inhibition of CK2 blocks the growth of murine SHH-MB tumors (Purzner et al., 2018), suggesting that CK2 may mediate the downstream event of SHH-SMO signaling. In addition, SHH MBs can be highly heterogeneous and further divided into four subtypes: divided into four subtypes: α, β, γ and δ, which correlate with genetic alterations and prognosis. For instance, SHH α subtype is associated with very poor prognosis and enriched with TP53 mutations and MYCN and GLI2 amplification, while SHH β subtype is often metastatic and affects mainly infants (Cavalli et al., 2017) (Figure 2B).
The most common genetic alteration in G3-MB is MYC amplification (Liu et al., 2017). G3-MB also has characteristic large-scale genomic instability, including somatic copy number alterations, fusion of MYC and PVT1 genes, and loss of chromosome 17p and gain of 17q (known as i17q) (Jones et al., 2012; Liu et al., 2017; Northcott et al., 2012). Genomic structural rearrangement in a subset of G3-MBs could lead to super-enhancer relocation and activation of GFI1 family oncogenes to promote tumorigenesis (Northcott et al., 2014). Activation of Notch and TGF-β signaling pathways as well as in-frame insertions in the KBTBD4 locus were also identified (Kool, 2008; Northcott et al., 2017), although their functional significance in G3-MB tumorigenesis remains unknown. G3-MB can be also classified into other distinct subgroups based on the distinct gene expression profiles and alterations (Cavalli et al., 2017) (Table 2).
Table 2.
Genetic, clinical features and modeling of medulloblastoma subgroups
| Subgroups | WNT | SHH | Group 3 | Group4 |
|---|---|---|---|---|
| Percentage | ~ 10% | ~ 30% | ~ 25% | ~ 35% |
| Histology | Classic, rarely LCA | Classic; Desmoplastic, LCA | Classic; LCA | Classic; rarely LCA |
| Metastatic rate | Low (5–10%) | Low (10–15%) | 40–45% | 35–40% |
| Prognosis | Very good | Intermediate | Poor | Intermediate |
| Recurrence | N/A | Local | Metastatic | Metastatic |
| Diagnostics | Nuclear β-catenin; CTNNB1 mut; monosomy 6; DKK | SFRP1, YAP1; 9q loss; GLI2 gain | NPR3; MYC gain | KCNA1 |
| Cytogenetics | Chr 6 loss | Chr 3q, 9p gain; 9q, 10q, 14q, 17p loss | Chr 1q, 7, 17q, 18 gain; 8, 10q, 11, 16q, 17p loss; iso17q | Chr 4, 7, 17q, 18 gain; 8, 10, 11, 17p, X loss; iso17q |
| Mutations, SNVs or indels | CTNNB1, DDX3X, SMARCA4, TP53, CSNK2B, CREBBP, MLL2 | PTCH1, SMO, SUFU, GNAS, MLL2, DDX3X, TP53, BCOR, LDB1 | SMARCA4, MLL2, SPTB, CTDNEP1, LRP1B, TNXB, GPS2 | KDM6A, MML3, HDAC2, ZMYM3, CBFA2T2, CTDNEP1 |
| Somatic alterations | No recurrent alteration | GLI1/2, MYCN, CCND2 amplification; PTCH1, TP53 deletion; GNAS mutation (LOF) | MYC, OTX2 amplification; PVT1-MYC fusion | SNCAIP duplication; CDK6, MYCN, OTX2 amplification |
| Pathways | WNT signaling | SHH signaling | MYC and TGFβ signaling | ERBB4-SRC signaling; NFĸB signaling |
| Cells of origin | Lower rhombic lip progenitors; mossy-fiber neuron precursors | Cerebellar NSCs or GNPs of EGL; brainstem progenitors in the 4th ventricle | Cerebellar NSCs or GNPs | Cerebellar NTZ progenitors, unipolar brush cells, other progenitors |
| Mouse models of medulloblastoma | Blbp-Cre;Ctnnb1F/+ Trp53F/F Pik3caE545K | 1. Ptch+/− or Ptch+/−; Trp53+/− 2. SmoA1;Atoh1/GFAP/Nes-Cre 3. Ptch1F/F;Atoh1/GFAP/Nes-Cre 4. GNASF/F;Atoh1/GFAP/Nes-Cre |
Cerebellar transplants: 1. NSC (MycT58A;DNp53) 2. GNP (Myc;Trp53−/−) 3. NSC (MycT58A; Gfi1/1b) 4. Glt-tTA;TRE-MycN |
Cerebellar progenitors transfected with SRCO/E with DNp53 |
G4-MBs share similarities in mutations with G3-MBs, with a higher rate of i17q than G3-MB (Gajjar and Robinson, 2014; Northcott et al., 2011; Northcott et al., 2012). The most frequently seen genetic alterations in G4-MB include amplification of MYCN and CDK6, mutations in KDM6A, which encodes a histone demethylase, and tandem duplication of SNCAIP, and overexpression of its downstream gene PRDM6 (Northcott et al., 2017; Pugh et al., 2012). Mutations to other histone modifying genes MLL3 and HDAC2 are also seen in G4-MB (Pugh et al., 2012). The functional consequence of these genomic alterations remains to be defined. Recent proteomic and phosphoproteomic analyses identified aberrant activation of ERBB4-SRC signaling in G4-MB (Archer et al., 2018; Forget et al., 2018). The combination of activated SRC signaling and p53 inactivation induces murine tumors (Forget et al., 2018), suggesting that ERBB4-SRC pathway is oncogenic in G4-MB (Figure 2B)(Table 2).
MBs also contain high rates of alterations in epigenetic regulators, with each subtype displaying a distinct epigenetic signature, including distinct DNA methylation patterns (Batora et al., 2014; Jones et al., 2013; Schwalbe et al., 2013) (Figure 2B). It is possible that epigenetic alterations serve as drivers of oncogenesis in MB (Yi and Wu, 2018). Common epigenetic dysregulations seen in MB include aberrant histone 3 methylations including hypermethylation of K4 and K27 (H3K4me3 and H3K27me3) and hypomethylation of K9 (Lindsey et al., 2004; Liu et al., 2017). Suppression of the hypermethylated phenotype results in a differentiated phenotype that leads to prolonged survival (Lawlor and Thiele, 2012). Analyses of MBs at the levels of transcriptome, genome, epigenome, and proteome and a better understanding of the microenvironment of these tumors will reveal the mechanisms of MB tumorigenesis and guide development of subtype-specific animal models and treatment strategies.
FINAL REMARKS AND PERSPECTIVES
Over the past several decades, considerable efforts have been dedicated to the identification of actionable targets for treatment of malignant brain tumors; however, the prognosis of brain tumor patients remains grim due to complex entities of malignant glioma in which diverse genetic and nongenetic effects determine tumor heterogeneity and clinical course. The relationship between the cells of origin and glioma stem cells is not well defined. Knowledge of the earliest events of gliomagenesis and would allow for earlier diagnoses.
Recent discoveries that brain tumorigenesis and development of the nervous system are linked at the root of stem cells revealed important cellular targets for cancer treatment. More extensive small-molecule and genetic screening to identify methods for inhibiting proliferation of tumor-initiating cells and for blocking shared or core signaling pathways will contribute to the development of the next generation of prognostic and therapeutic approaches for cancer. Advances in transgenic technologies and animal models have allowed the establishment of mouse models that mirror the human disease with increasing fidelity; these models facilitate identification of the pathways involved in the pathogenesis of brain tumors at single-cell resolution. The core transcriptional regulators such as OLIG2 and SOX2 are predominantly expressed in glioma-initiating cells and make appealing targets for therapy in primary and recurrent tumors. A better understanding of the mechanism of tumor stem/progenitor cell lineage progression and identification of the key fulcrum that balances tumor cell proliferation with differentiation should allow us to reactivate or reprogram of tumor-propagating cells to differentiate into slow-growing or post-mitotic cell types. Additionally, further investigation is necessary into the role of epigenetic regulation and dysregulation in pediatric brain cancers. Understanding the epigenetic vulnerabilities may provide insights into novel therapeutic approaches, and indeed many novel agents targeting chromatin modifiers such as the HDAC, BRD4 and EZH2 inhibitors are currently in development or in early clinical trials. Moreover, further characterization of complex glioma stem-cell niches, interactions with their tumor microenvironment, and tumor heterogeneity will lead to development of targeted therapies such as anti-tumor immunotherapies, which have not yet been explored fully in brain tumors. These types of therapies may one day lead to treatments that induce full remission.
Acknowledgments
The authors would like to thank Dr. Kalen Berry and Rohit Rao for comments. This study was funded in part by grants from the NIH (R01 NS078092 and R01 NS075243) to Q.R.L.
Footnotes
Declaration of Interests
The authors declare no competing interests.
References:
- Alcantara Llaguno S, Chen J, Kwon CH, Jackson EL, Li Y, Burns DK, Alvarez-Buylla A, Parada LF, 2009. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell 15, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcantara Llaguno SR, Wang Z, Sun D, Chen J, Xu J, Kim E, Hatanpaa KJ, Raisanen JM, Burns DK, Johnson JE, Parada LF, 2015. Adult Lineage-Restricted CNS Progenitors Specify Distinct Glioblastoma Subtypes. Cancer Cell 28, 429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolou E, Hochedlinger K, 2013. Chromatin dynamics during cellular reprogramming. Nature 502, 462–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer TC, Ehrenberger T, Mundt F, Gold MP, Krug K, Mah CK, Mahoney EL, Daniel CJ, LeNail A, Ramamoorthy D, Mertins P, Mani DR, Zhang H, Gillette MA, Clauser K, Noble M, Tang LC, Pierre-Francois J, Silterra J, Jensen J, Tamayo P, Korshunov A, Pfister SM, Kool M, Northcott PA, Sears RC, Lipton JO, Carr SA, Mesirov JP, Pomeroy SL, Fraenkel E, 2018. Proteomics, Post-translational Modifications, and Integrative Analyses Reveal Molecular Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 34, 396–410 e398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachoo RM, Maher EA, Ligon KL, Sharpless NE, Chan SS, You MJ, Tang Y, DeFrances J, Stover E, Weissleder R, Rowitch DH, Louis DN, DePinho RA, 2002. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell 1, 269–277. [DOI] [PubMed] [Google Scholar]
- Banasavadi-Siddegowda YK, Russell L, Frair E, Karkhanis VA, Relation T, Yoo JY, Zhang J, Sif S, Imitola J, Baiocchi R, Kaur B, 2017. PRMT5-PTEN molecular pathway regulates senescence and self-renewal of primary glioblastoma neurosphere cells. Oncogene 36, 263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banasavadi-Siddegowda YK, Welker AM, An M, Yang X, Zhou W, Shi G, Imitola J, Li C, Hsu S, Wang J, Phelps M, Zhang J, Beattie CE, Baiocchi R, Kaur B, 2018. PRMT5 as a druggable target for glioblastoma therapy. Neuro Oncol 20, 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardehle S, Kruger M, Buggenthin F, Schwausch J, Ninkovic J, Clevers H, Snippert HJ, Theis FJ, Meyer-Luehmann M, Bechmann I, Dimou L, Gotz M, 2013. Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nat Neurosci 16, 580–586. [DOI] [PubMed] [Google Scholar]
- Batora NV, Sturm D, Jones DT, Kool M, Pfister SM, Northcott PA, 2014. Transitioning from genotypes to epigenotypes: why the time has come for medulloblastoma epigenomics. Neuroscience 264, 171–185. [DOI] [PubMed] [Google Scholar]
- Baylin SB, Jones PA, 2011. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer 11, 726–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ, Aigner L, Brawanski A, Bogdahn U, Beier CP, 2007. CD133(+) and CD133(−) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res 67, 4010–4015. [DOI] [PubMed] [Google Scholar]
- Bhat KP, Balasubramaniyan V, Vaillant B, Ezhilarasan R, Hummelink K, Hollingsworth F, Wani K, Heathcock L, James JD, Goodman LD, Conroy S, Long L, Lelic N, Wang S, Gumin J, Raj D, Kodama Y, Raghunathan A, Olar A, Joshi K, Pelloski CE, Heimberger A, Kim SH, Cahill DP, Rao G, Den Dunnen WF, Boddeke HW, Phillips HS, Nakano I, Lang FF, Colman H, Sulman EP, Aldape K, 2013. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell 24, 331–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat KP, Salazar KL, Balasubramaniyan V, Wani K, Heathcock L, Hollingsworth F, James JD, Gumin J, Diefes KL, Kim SH, Turski A, Azodi Y, Yang Y, Doucette T, Colman H, Sulman EP, Lang FF, Rao G, Copray S, Vaillant BD, Aldape KD, 2011. The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes Dev 25, 2594–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee D, Shenoy S, Bairy KL, 2016. DNA Methylation and Chromatin Remodeling: The Blueprint of Cancer Epigenetics. Scientifica (Cairo) 2016, 6072357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobola MS, Alnoor M, Chen JY, Kolstoe DD, Silbergeld DL, Rostomily RC, Blank A, Chamberlain MC, Silber JR, 2015. O(6)-methylguanine-DNA methyltransferase activity is associated with response to alkylating agent therapy and with MGMT promoter methylation in glioblastoma and anaplastic glioma. BBA Clin 3, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, Genovese G, Shmulevich I, Barnholtz-Sloan J, Zou L, Vegesna R, Shukla SA, Ciriello G, Yung WK, Zhang W, Sougnez C, Mikkelsen T, Aldape K, Bigner DD, Van Meir EG, Prados M, Sloan A, Black KL, Eschbacher J, Finocchiaro G, Friedman W, Andrews DW, Guha A, Iacocca M, O’Neill BP, Foltz G, Myers J, Weisenberger DJ, Penny R, Kucherlapati R, Perou CM, Hayes DN, Gibbs R, Marra M, Mills GB, Lander E, Spellman P, Wilson R, Sander C, Weinstein J, Meyerson M, Gabriel S, Laird PW, Haussler D, Getz G, Chin L, 2013. The somatic genomic landscape of glioblastoma. Cell 155, 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, Morrison A, Lewis P, Bouffet E, Bartels U, Zuccaro J, Agnihotri S, Ryall S, Barszczyk M, Chornenkyy Y, Bourgey M, Bourque G, Montpetit A, Cordero F, Castelo-Branco P, Mangerel J, Tabori U, Ho KC, Huang A, Taylor KR, Mackay A, Bendel AE, Nazarian J, Fangusaro JR, Karajannis MA, Zagzag D, Foreman NK, Donson A, Hegert JV, Smith A, Chan J, Lafay-Cousin L, Dunn S, Hukin J, Dunham C, Scheinemann K, Michaud J, Zelcer S, Ramsay D, Cain J, Brennan C, Souweidane MM, Jones C, Allis CD, Brudno M, Becher O, Hawkins C, 2014. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nature genetics 46, 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Chen Y, Cai WH, Hurlock EC, Wu H, Kernie SG, Parada LF, Lu QR, 2007. A crucial role for Olig2 in white matter astrocyte development. Development 134, 1887–1899. [DOI] [PubMed] [Google Scholar]
- Calon A, Lonardo E, Berenguer-Llergo A, Espinet E, Hernando-Momblona X, Iglesias M, Sevillano M, Palomo-Ponce S, Tauriello DV, Byrom D, Cortina C, Morral C, Barcelo C, Tosi S, Riera A, Attolini CS, Rossell D, Sancho E, Batlle E, 2015. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nature genetics 47, 320–329. [DOI] [PubMed] [Google Scholar]
- Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, Koelsche C, Sahm F, Chavez L, Reuss DE, Kratz A, Wefers AK, Huang K, Pajtler KW, Schweizer L, Stichel D, Olar A, Engel NW, Lindenberg K, Harter PN, Braczynski AK, Plate KH, Dohmen H, Garvalov BK, Coras R, Holsken A, Hewer E, Bewerunge-Hudler M, Schick M, Fischer R, Beschorner R, Schittenhelm J, Staszewski O, Wani K, Varlet P, Pages M, Temming P, Lohmann D, Selt F, Witt H, Milde T, Witt O, Aronica E, Giangaspero F, Rushing E, Scheurlen W, Geisenberger C, Rodriguez FJ, Becker A, Preusser M, Haberler C, Bjerkvig R, Cryan J, Farrell M, Deckert M, Hench J, Frank S, Serrano J, Kannan K, Tsirigos A, Bruck W, Hofer S, Brehmer S, Seiz-Rosenhagen M, Hanggi D, Hans V, Rozsnoki S, Hansford JR, Kohlhof P, Kristensen BW, Lechner M, Lopes B, Mawrin C, Ketter R, Kulozik A, Khatib Z, Heppner F, Koch A, Jouvet A, Keohane C, Muhleisen H, Mueller W, Pohl U, Prinz M, Benner A, Zapatka M, Gottardo NG, Driever PH, Kramm CM, Muller HL, Rutkowski S, von Hoff K, Fruhwald MC, Gnekow A, Fleischhack G, Tippelt S, Calaminus G, Monoranu CM, Perry A, Jones C, Jacques TS, Radlwimmer B, Gessi M, Pietsch T, Schramm J, Schackert G, Westphal M, Reifenberger G, Wesseling P, Weller M, Collins VP, Blumcke I, Bendszus M, Debus J, Huang A, Jabado N, Northcott PA, Paulus W, Gajjar A, Robinson GW, Taylor MD, Jaunmuktane Z, Ryzhova M, Platten M, Unterberg A, Wick W, Karajannis MA, Mittelbronn M, Acker T, Hartmann C, Aldape K, Schuller U, Buslei R, Lichter P, Kool M, Herold-Mende C, Ellison DW, Hasselblatt M, Snuderl M, Brandner S, Korshunov A, von Deimling A, Pfister SM, 2018. DNA methylation-based classification of central nervous system tumours. Nature 555, 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castel D, Philippe C, Calmon R, Le Dret L, Truffaux N, Boddaert N, Pages M, Taylor KR, Saulnier P, Lacroix L, Mackay A, Jones C, Sainte-Rose C, Blauwblomme T, Andreiuolo F, Puget S, Grill J, Varlet P, Debily MA, 2015. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol 130, 815–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli FMG, Remke M, Rampasek L, Peacock J, Shih DJH, Luu B, Garzia L, Torchia J, Nor C, Morrissy AS, Agnihotri S, Thompson YY, Kuzan-Fischer CM, Farooq H, Isaev K, Daniels C, Cho BK, Kim SK, Wang KC, Lee JY, Grajkowska WA, Perek-Polnik M, Vasiljevic A, Faure-Conter C, Jouvet A, Giannini C, Nageswara Rao AA, Li KKW, Ng HK, Eberhart CG, Pollack IF, Hamilton RL, Gillespie GY, Olson JM, Leary S, Weiss WA, Lach B, Chambless LB, Thompson RC, Cooper MK, Vibhakar R, Hauser P, van Veelen MC, Kros JM, French PJ, Ra YS, Kumabe T, Lopez-Aguilar E, Zitterbart K, Sterba J, Finocchiaro G, Massimino M, Van Meir EG, Osuka S, Shofuda T, Klekner A, Zollo M, Leonard JR, Rubin JB, Jabado N, Albrecht S, Mora J, Van Meter TE, Jung S, Moore AS, Hallahan AR, Chan JA, Tirapelli DPC, Carlotti CG, Fouladi M, Pimentel J, Faria CC, Saad AG, Massimi L, Liau LM, Wheeler H, Nakamura H, Elbabaa SK, Perezpena-Diazconti M, Chico Ponce de Leon F, Robinson S, Zapotocky M, Lassaletta A, Huang A, Hawkins CE, Tabori U, Bouffet E, Bartels U, Dirks PB, Rutka JT, Bader GD, Reimand J, Goldenberg A, Ramaswamy V, Taylor MD, 2017. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 31, 737–754 e736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, Parada LF, 2012. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488, 522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Miles DK, Hoang T, Shi J, Hurlock E, Kernie SG, Lu QR, 2008. The basic helix-loop-helix transcription factor olig2 is critical for reactive astrocyte proliferation after cortical injury. J Neurosci 28, 10983–10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JD, Lee JS, 2013. Interplay between Epigenetics and Genetics in Cancer. Genomics Inform 11, 164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow LM, Endersby R, Zhu X, Rankin S, Qu C, Zhang J, Broniscer A, Ellison DW, Baker SJ, 2011. Cooperativity within and among Pten, p53, and Rb pathways induces high-grade astrocytoma in adult brain. Cancer Cell 19, 305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AL, Holmen SL, Colman H, 2013. IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep 13, 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen KJ, Jabado N, Grill J, 2017. Diffuse intrinsic pontine gliomas-current management and new biologic insights. Is there a glimmer of hope? Neuro Oncol 19, 1025–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero FJ, Huang Z, Grenier C, He X, Hu G, McLendon RE, Murphy SK, Hashizume R, Becher OJ, 2017. Histone H3.3K27M Represses p16 to Accelerate Gliomagenesis in a Murine Model of DIPG. Mol Cancer Res 15, 1243–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C, 2001. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev 15, 1913–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmanis S, Sloan SA, Croote D, Mignardi M, Chernikova S, Samghababi P, Zhang Y, Neff N, Kowarsky M, Caneda C, Li G, Chang SD, Connolly ID, Li Y, Barres BA, Gephart MH, Quake SR, 2017. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep 21, 1399–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MR, Polito A, Levine JM, Reynolds R, 2003. NG2-expressing glial progenitor cells: an abundant and widespread population of cycling cells in the adult rat CNS. Mol Cell Neurosci 24, 476–488. [DOI] [PubMed] [Google Scholar]
- Deleris A, Stroud H, Bernatavichute Y, Johnson E, Klein G, Schubert D, Jacobsen SE, 2012. Loss of the DNA methyltransferase MET1 Induces H3K9 hypermethylation at PcG target genes and redistribution of H3K27 trimethylation to transposons in Arabidopsis thaliana. PLoS Genet 8, e1003062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson M, Johnstone RW, Prince HM, 2010. Histone deacetylase inhibitors: potential targets responsible for their anti-cancer effect. Invest New Drugs 28 Suppl 1, S3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimou L, Simon C, Kirchhoff F, Takebayashi H, Gotz M, 2008. Progeny of Olig2-expressing progenitors in the gray and white matter of the adult mouse cerebral cortex. J Neurosci 28, 10434–10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egler V, Korur S, Failly M, Boulay JL, Imber R, Lino MM, Merlo A, 2008. Histone deacetylase inhibition and blockade of the glycolytic pathway synergistically induce glioblastoma cell death. Clin Cancer Res 14, 3132–3140. [DOI] [PubMed] [Google Scholar]
- Ellison DW, Kocak M, Dalton J, Megahed H, Lusher ME, Ryan SL, Zhao W, Nicholson SL, Taylor RE, Bailey S, Clifford SC, 2011. Definition of disease-risk stratification groups in childhood medulloblastoma using combined clinical, pathologic, and molecular variables. J Clin Oncol 29, 1400–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endersby R, Zhu X, Hay N, Ellison DW, Baker SJ, 2011. Nonredundant functions for Akt isoforms in astrocyte growth and gliomagenesis in an orthotopic transplantation model. Cancer Res 71, 4106–4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ene CI, Edwards L, Riddick G, Baysan M, Woolard K, Kotliarova S, Lai C, Belova G, Cam M, Walling J, Zhou M, Stevenson H, Kim HS, Killian K, Veenstra T, Bailey R, Song H, Zhang W, Fine HA, 2012. Histone demethylase Jumonji D3 (JMJD3) as a tumor suppressor by regulating p53 protein nuclear stabilization. PLoS One 7, e51407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estecio MR, Issa JP, 2011. Dissecting DNA hypermethylation in cancer. FEBS Lett 585, 2078–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg AP, Koldobskiy MA, Gondor A, 2016. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet 17, 284–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filbin MG, Tirosh I, Hovestadt V, Shaw ML, Escalante LE, Mathewson ND, Neftel C, Frank N, Pelton K, Hebert CM, Haberler C, Yizhak K, Gojo J, Egervari K, Mount C, van Galen P, Bonal DM, Nguyen QD, Beck A, Sinai C, Czech T, Dorfer C, Goumnerova L, Lavarino C, Carcaboso AM, Mora J, Mylvaganam R, Luo CC, Peyrl A, Popovic M, Azizi A, Batchelor TT, Frosch MP, Martinez-Lage M, Kieran MW, Bandopadhayay P, Beroukhim R, Fritsch G, Getz G, Rozenblatt-Rosen O, Wucherpfennig KW, Louis DN, Monje M, Slavc I, Ligon KL, Golub TR, Regev A, Bernstein BE, Suva ML, 2018. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 360, 331–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset PO, Bechet D, Faury D, De Jay N, Ramkissoon LA, Corcoran A, Jones DT, Sturm D, Johann P, Tomita T, Goldman S, Nagib M, Bendel A, Goumnerova L, Bowers DC, Leonard JR, Rubin JB, Alden T, Browd S, Geyer JR, Leary S, Jallo G, Cohen K, Gupta N, Prados MD, Carret AS, Ellezam B, Crevier L, Klekner A, Bognar L, Hauser P, Garami M, Myseros J, Dong Z, Siegel PM, Malkin H, Ligon AH, Albrecht S, Pfister SM, Ligon KL, Majewski J, Jabado N, Kieran MW, 2014. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nature genetics 46, 462–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forget A, Martignetti L, Puget S, Calzone L, Brabetz S, Picard D, Montagud A, Liva S, Sta A, Dingli F, Arras G, Rivera J, Loew D, Besnard A, Lacombe J, Pages M, Varlet P, Dufour C, Yu H, Mercier AL, Indersie E, Chivet A, Leboucher S, Sieber L, Beccaria K, Gombert M, Meyer FD, Qin N, Bartl J, Chavez L, Okonechnikov K, Sharma T, Thatikonda V, Bourdeaut F, Pouponnot C, Ramaswamy V, Korshunov A, Borkhardt A, Reifenberger G, Poullet P, Taylor MD, Kool M, Pfister SM, Kawauchi D, Barillot E, Remke M, Ayrault O, 2018. Aberrant ERBB4-SRC Signaling as a Hallmark of Group 4 Medulloblastoma Revealed by Integrative Phosphoproteomic Profiling. Cancer Cell 34, 379–395 e377. [DOI] [PubMed] [Google Scholar]
- Fraser MM, Zhu X, Kwon CH, Uhlmann EJ, Gutmann DH, Baker SJ, 2004. Pten loss causes hypertrophy and increased proliferation of astrocytes in vivo. Cancer Res 64, 7773–7779. [DOI] [PubMed] [Google Scholar]
- Friedmann-Morvinski D, Bushong EA, Ke E, Soda Y, Marumoto T, Singer O, Ellisman MH, Verma IM, 2012. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science 338, 1080–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato K, Major T, Lewis PW, Allis CD, Tabar V, 2014. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 346, 1529–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, Chin L, DePinho RA, Cavenee WK, 2007. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev 21, 2683–2710. [DOI] [PubMed] [Google Scholar]
- Gajjar AJ, Robinson GW, 2014. Medulloblastoma-translating discoveries from the bench to the bedside. Nat Rev Clin Oncol 11, 714–722. [DOI] [PubMed] [Google Scholar]
- Galvao RP, Kasina A, McNeill RS, Harbin JE, Foreman O, Verhaak RGW, Nishiyama A, Miller CR, Zong H, 2014. Transformation of quiescent adult oligodendrocyte precursor cells into malignant glioma through a multistep reactivation process. Proceedings of the National Academy of Sciences of the United States of America 111, E4214–E4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge WP, Miyawaki A, Gage FH, Jan YN, Jan LY, 2012. Local generation of glia is a major astrocyte source in postnatal cortex. Nature 484, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geha S, Pallud J, Junier MP, Devaux B, Leonard N, Chassoux F, Chneiweiss H, Daumas-Duport C, Varlet P, 2010. NG2+/Olig2+ cells are the major cycle-related cell population of the adult human normal brain. Brain Pathol 20, 399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazi SO, Stark M, Zhao Z, Mobley BC, Munden A, Hover L, Abel TW, 2012. Cell of origin determines tumor phenotype in an oncogenic Ras/p53 knockout transgenic model of high-grade glioma. J Neuropathol Exp Neurol 71, 729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C, Kranenburg TA, Hogg T, Poppleton H, Martin J, Finkelstein D, Pounds S, Weiss A, Patay Z, Scoggins M, Ogg R, Pei Y, Yang ZJ, Brun S, Lee Y, Zindy F, Lindsey JC, Taketo MM, Boop FA, Sanford RA, Gajjar A, Clifford SC, Roussel MF, McKinnon PJ, Gutmann DH, Ellison DW, Wechsler-Reya R, Gilbertson RJ, 2010. Subtypes of medulloblastoma have distinct developmental origins. Nature 468, 1095–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasgow SM, Zhu W, Stolt CC, Huang TW, Chen F, LoTurco JJ, Neul JL, Wegner M, Mohila C, Deneen B, 2014. Mutual antagonism between Sox10 and NFIA regulates diversification of glial lineages and glioma subtypes. Nat Neurosci 17, 1322–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grammel D, Warmuth-Metz M, von Bueren AO, Kool M, Pietsch T, Kretzschmar HA, Rowitch DH, Rutkowski S, Pfister SM, Schuller U, 2012. Sonic hedgehog-associated medulloblastoma arising from the cochlear nuclei of the brainstem. Acta Neuropathol 123, 601–614. [DOI] [PubMed] [Google Scholar]
- Grasso CS, Tang Y, Truffaux N, Berlow NE, Liu L, Debily MA, Quist MJ, Davis LE, Huang EC, Woo PJ, Ponnuswami A, Chen S, Johung TB, Sun W, Kogiso M, Du Y, Qi L, Huang Y, Hutt-Cabezas M, Warren KE, Le Dret L, Meltzer PS, Mao H, Quezado M, van Vuurden DG, Abraham J, Fouladi M, Svalina MN, Wang N, Hawkins C, Nazarian J, Alonso MM, Raabe EH, Hulleman E, Spellman PT, Li XN, Keller C, Pal R, Grill J, Monje M, 2015. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med 21, 555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grill J, Puget S, Andreiuolo F, Philippe C, MacConaill L, Kieran MW, 2012. Critical oncogenic mutations in newly diagnosed pediatric diffuse intrinsic pontine glioma. Pediatric blood & cancer 58, 489–491. [DOI] [PubMed] [Google Scholar]
- Groszer M, Erickson R, Scripture-Adams DD, Dougherty JD, Le Belle J, Zack JA, Geschwind DH, Liu X, Kornblum HI, Wu H, 2006. PTEN negatively regulates neural stem cell self-renewal by modulating G0-G1 cell cycle entry. Proc Natl Acad Sci U S A 103, 111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Li R, Zhang W, Yang X, Wheeler CG, Friedman GK, Province P, Ding Q, You Z, Fathallah-Shaykh HM, Gillespie GY, Zhao X, King PH, Nabors LB, 2014. Expression of PRMT5 correlates with malignant grade in gliomas and plays a pivotal role in tumor growth in vitro. J Neurooncol 118, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA, 2011. Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hashizume R, Andor N, Ihara Y, Lerner R, Gan H, Chen X, Fang D, Huang X, Tom MW, Ngo V, Solomon D, Mueller S, Paris PL, Zhang Z, Petritsch C, Gupta N, Waldman TA, James CD, 2014. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat Med 20, 1394–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden EC, 2010. Genomics boosts brain-cancer work. Nature 463, 278. [DOI] [PubMed] [Google Scholar]
- He X, Zhang L, Chen Y, Remke M, Shih D, Lu F, Wang H, Deng Y, Yu Y, Xia Y, Wu X, Ramaswamy V, Hu T, Wang F, Zhou W, Burns DK, Kim SH, Kool M, Pfister SM, Weinstein LS, Pomeroy SL, Gilbertson RJ, Rubin JB, Hou Y, Wechsler-Reya R, Taylor MD, Lu QR, 2014. The G protein alpha subunit Galphas is a tumor suppressor in Sonic hedgehog-driven medulloblastoma. Nat Med 20, 1035–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, Bettegowda C, Rodriguez FJ, Eberhart CG, Hebbar S, Offerhaus GJ, McLendon R, Rasheed BA, He Y, Yan H, Bigner DD, Oba-Shinjo SM, Marie SK, Riggins GJ, Kinzler KW, Vogelstein B, Hruban RH, Maitra A, Papadopoulos N, Meeker AK, 2011. Altered telomeres in tumors with ATRX and DAXX mutations. Science 333, 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman LM, DeWire M, Ryall S, Buczkowicz P, Leach J, Miles L, Ramani A, Brudno M, Kumar SS, Drissi R, Dexheimer P, Salloum R, Chow L, Hummel T, Stevenson C, Lu QR, Jones B, Witte D, Aronow B, Hawkins CE, Fouladi M, 2016. Spatial genomic heterogeneity in diffuse intrinsic pontine and midline high-grade glioma: implications for diagnostic biopsy and targeted therapeutics. Acta Neuropathol Commun 4, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland EC, 2000. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nature Genet 25, 55–57. [DOI] [PubMed] [Google Scholar]
- Holland EC, 2001. Gliomagenesis: genetic alterations and mouse models. Nat Rev Genet 2, 120–129. [DOI] [PubMed] [Google Scholar]
- Hovestadt V, Jones DT, Picelli S, Wang W, Kool M, Northcott PA, Sultan M, Stachurski K, Ryzhova M, Warnatz HJ, Ralser M, Brun S, Bunt J, Jager N, Kleinheinz K, Erkek S, Weber UD, Bartholomae CC, von Kalle C, Lawerenz C, Eils J, Koster J, Versteeg R, Milde T, Witt O, Schmidt S, Wolf S, Pietsch T, Rutkowski S, Scheurlen W, Taylor MD, Brors B, Felsberg J, Reifenberger G, Borkhardt A, Lehrach H, Wechsler-Reya RJ, Eils R, Yaspo ML, Landgraf P, Korshunov A, Zapatka M, Radlwimmer B, Pfister SM, Lichter P, 2014. Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature 510, 537–541. [DOI] [PubMed] [Google Scholar]
- Huang J, Vogel G, Yu Z, Almazan G, Richard S, 2011. Type II arginine methyltransferase PRMT5 regulates gene expression of inhibitors of differentiation/DNA binding Id2 and Id4 during glial cell differentiation. J Biol Chem 286, 44424–44432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R, Qian D, Hu M, Zhang X, Song J, Li L, Chen H, Wang B, 2015. Association between human cytomegalovirus infection and histone acetylation level in various histological types of glioma. Oncol Lett 10, 2812–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami K, Ohgane J, Tanaka S, Yagi S, Shiota K, 2009. Interplay between DNA methylation, histone modification and chromatin remodeling in stem cells and during development. Int J Dev Biol 53, 203–214. [DOI] [PubMed] [Google Scholar]
- Imamoto K, Paterson JA, Leblond CP, 1978. Radioautographic investigation of gliogenesis in the corpus callosum of young rats. I. Sequential changes in oligodendrocytes. The Journal of comparative neurology 180, 115–128, 132–117. [DOI] [PubMed] [Google Scholar]
- Isella C, Terrasi A, Bellomo SE, Petti C, Galatola G, Muratore A, Mellano A, Senetta R, Cassenti A, Sonetto C, Inghirami G, Trusolino L, Fekete Z, De Ridder M, Cassoni P, Storme G, Bertotti A, Medico E, 2015. Stromal contribution to the colorectal cancer transcriptome. Nature genetics 47, 312–319. [DOI] [PubMed] [Google Scholar]
- Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y, 2010. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466, 1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen M, Yip S, Louis DN, 2010. Molecular pathology in adult gliomas: diagnostic, prognostic, and predictive markers. Lancet Neurol 9, 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT, Jager N, Kool M, Zichner T, Hutter B, Sultan M, Cho YJ, Pugh TJ, Hovestadt V, Stutz AM, Rausch T, Warnatz HJ, Ryzhova M, Bender S, Sturm D, Pleier S, Cin H, Pfaff E, Sieber L, Wittmann A, Remke M, Witt H, Hutter S, Tzaridis T, Weischenfeldt J, Raeder B, Avci M, Amstislavskiy V, Zapatka M, Weber UD, Wang Q, Lasitschka B, Bartholomae CC, Schmidt M, von Kalle C, Ast V, Lawerenz C, Eils J, Kabbe R, Benes V, van Sluis P, Koster J, Volckmann R, Shih D, Betts MJ, Russell RB, Coco S, Tonini GP, Schuller U, Hans V, Graf N, Kim YJ, Monoranu C, Roggendorf W, Unterberg A, Herold-Mende C, Milde T, Kulozik AE, von Deimling A, Witt O, Maass E, Rossler J, Ebinger M, Schuhmann MU, Fruhwald MC, Hasselblatt M, Jabado N, Rutkowski S, von Bueren AO, Williamson D, Clifford SC, McCabe MG, Collins VP, Wolf S, Wiemann S, Lehrach H, Brors B, Scheurlen W, Felsberg J, Reifenberger G, Northcott PA, Taylor MD, Meyerson M, Pomeroy SL, Yaspo ML, Korbel JO, Korshunov A, Eils R, Pfister SM, Lichter P, 2012. Dissecting the genomic complexity underlying medulloblastoma. Nature 488, 100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT, Northcott PA, Kool M, Pfister SM, 2013. The role of chromatin remodeling in medulloblastoma. Brain Pathol 23, 193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo KM, Kim SY, Jin X, Song SY, Kong DS, Lee JI, Jeon JW, Kim MH, Kang BG, Jung Y, Jin J, Hong SC, Park WY, Lee DS, Kim H, Nam DH, 2008. Clinical and biological implications of CD133-positive and CD133-negative cells in glioblastomas. Laboratory investigation; a journal of technical methods and pathology 88, 808–815. [DOI] [PubMed] [Google Scholar]
- Kalhor R, Mali P, Church GM, 2017. Rapidly evolving homing CRISPR barcodes. Nat Methods 14, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi D, Robinson G, Uziel T, Gibson P, Rehg J, Gao C, Finkelstein D, Qu C, Pounds S, Ellison DW, Gilbertson RJ, Roussel MF, 2012. A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell 21, 168–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoo BL, Chaudhuri PK, Ramalingam N, Tan DS, Lim CT, Warkiani ME, 2016. Single-cell profiling approaches to probing tumor heterogeneity. Int J Cancer 139, 243–255. [DOI] [PubMed] [Google Scholar]
- Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, Bartels U, Albrecht S, Schwartzentruber J, Letourneau L, Bourgey M, Bourque G, Montpetit A, Bourret G, Lepage P, Fleming A, Lichter P, Kool M, von Deimling A, Sturm D, Korshunov A, Faury D, Jones DT, Majewski J, Pfister SM, Jabado N, Hawkins C, 2012. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 124, 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Verhaak RG, 2015. Transcriptional mimicry by tumor-associated stroma. Nature genetics 47, 307–309. [DOI] [PubMed] [Google Scholar]
- Knudsen ES, Wang JY, 2010. Targeting the RB-pathway in cancer therapy. Clin Cancer Res 16, 1094–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kool M, 2008. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One 3, e3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korshunov A, Capper D, Reuss D, Schrimpf D, Ryzhova M, Hovestadt V, Sturm D, Meyer J, Jones C, Zheludkova O, Kumirova E, Golanov A, Kool M, Schuller U, Mittelbronn M, Hasselblatt M, Schittenhelm J, Reifenberger G, Herold-Mende C, Lichter P, von Deimling A, Pfister SM, Jones DT, 2016. Histologically distinct neuroepithelial tumors with histone 3 G34 mutation are molecularly similar and comprise a single nosologic entity. Acta Neuropathol 131, 137–146. [DOI] [PubMed] [Google Scholar]
- Kozono D, Li J, Nitta M, Sampetrean O, Gonda D, Kushwaha DS, Merzon D, Ramakrishnan V, Zhu S, Zhu K, Matsui H, Harismendy O, Hua W, Mao Y, Kwon CH, Saya H, Nakano I, Pizzo DP, VandenBerg SR, Chen CC, 2015. Dynamic epigenetic regulation of glioblastoma tumorigenicity through LSD1 modulation of MYC expression. Proc Natl Acad Sci U S A 112, E4055–4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupp R, Shtayer L, Tien AC, Szeto E, Sanai N, Rowitch DH, Mehta S, 2016. Lineage-Restricted OLIG2-RTK Signaling Governs the Molecular Subtype of Glioma Stem-like Cells. Cell Rep 16, 2838–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CH, Zhao D, Chen J, Alcantara S, Li Y, Burns DK, Mason RP, Lee EY, Wu H, Parada LF, 2008. Pten haploinsufficiency accelerates formation of high-grade astrocytomas. Cancer Res 68, 3286–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan X, Jorg DJ, Cavalli FMG, Richards LM, Nguyen LV, Vanner RJ, Guilhamon P, Lee L, Kushida MM, Pellacani D, Park NI, Coutinho FJ, Whetstone H, Selvadurai HJ, Che C, Luu B, Carles A, Moksa M, Rastegar N, Head R, Dolma S, Prinos P, Cusimano MD, Das S, Bernstein M, Arrowsmith CH, Mungall AJ, Moore RA, Ma Y, Gallo M, Lupien M, Pugh TJ, Taylor MD, Hirst M, Eaves CJ, Simons BD, Dirks PB, 2017. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature 549, 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapin DH, Tsoli M, Ziegler DS, 2017. Genomic Insights into Diffuse Intrinsic Pontine Glioma. Front Oncol 7, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson JD, Kasper LH, Paugh BS, Jin H, Wu G, Kwon CH, Fan Y, Shaw TI, Silveira AB, Qu C, Xu R, Zhu X, Zhang J, Russell HR, Peters JL, Finkelstein D, Xu B, Lin T, Tinkle CL, Patay Z, Onar-Thomas A, Pounds SB, McKinnon PJ, Ellison DW, Zhang J, Baker SJ, 2019. Histone H3.3 K27M Accelerates Spontaneous Brainstem Glioma and Drives Restricted Changes in Bivalent Gene Expression. Cancer Cell 35, 140–155 e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor ER, Thiele CJ, 2012. Epigenetic changes in pediatric solid tumors: promising new targets. Clin Cancer Res 18, 2768–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JC, Mayer-Proschel M, Rao MS, 2000. Gliogenesis in the central nervous system. Glia 30, 105–121. [DOI] [PubMed] [Google Scholar]
- Lee JH, Lee JE, Kahng JY, Kim SH, Park JS, Yoon SJ, Um JY, Kim WK, Lee JK, Park J, Kim EH, Lee JH, Lee JH, Chung WS, Ju YS, Park SH, Chang JH, Kang SG, Lee JH, 2018. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 560, 243–247. [DOI] [PubMed] [Google Scholar]
- Lee JV, Carrer A, Shah S, Snyder NW, Wei S, Venneti S, Worth AJ, Yuan ZF, Lim HW, Liu S, Jackson E, Aiello NM, Haas NB, Rebbeck TR, Judkins A, Won KJ, Chodosh LA, Garcia BA, Stanger BZ, Feldman MD, Blair IA, Wellen KE, 2014. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab 20, 306–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KK, Workman JL, 2007. Histone acetyltransferase complexes: one size doesn’t fit all. Nat Rev Mol Cell Biol 8, 284–295. [DOI] [PubMed] [Google Scholar]
- Levitin HM, Yuan J, Sims PA, 2018. Single-Cell Transcriptomic Analysis of Tumor Heterogeneity. Trends Cancer 4, 264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD, 2013. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Egervari G, Wang Y, Berger SL, Lu Z, 2018. Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat Rev Mol Cell Biol [DOI] [PMC free article] [PubMed]
- Liffers K, Kolbe K, Westphal M, Lamszus K, Schulte A, 2016. Histone Deacetylase Inhibitors Resensitize EGFR/EGFRvIII-Overexpressing, Erlotinib-Resistant Glioblastoma Cells to Tyrosine Kinase Inhibition. Target Oncol 11, 29–40. [DOI] [PubMed] [Google Scholar]
- Ligon KL, Alberta JA, Kho AT, Weiss J, Kwaan MR, Nutt CL, Louis DN, Stiles CD, Rowitch DH, 2004. The oligodendroglial lineage marker OLIG2 is universally expressed in diffuse gliomas. J Neuropathol Exp Neurol 63, 499–509. [DOI] [PubMed] [Google Scholar]
- Ligon KL, Huillard E, Mehta S, Kesari S, Liu H, Alberta JA, Bachoo RM, Kane M, Louis DN, Depinho RA, Anderson DJ, Stiles CD, Rowitch DH, 2007. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron 53, 503–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Erkek S, Tong Y, Yin L, Federation AJ, Zapatka M, Haldipur P, Kawauchi D, Risch T, Warnatz HJ, Worst BC, Ju B, Orr BA, Zeid R, Polaski DR, Segura-Wang M, Waszak SM, Jones DT, Kool M, Hovestadt V, Buchhalter I, Sieber L, Johann P, Chavez L, Groschel S, Ryzhova M, Korshunov A, Chen W, Chizhikov VV, Millen KJ, Amstislavskiy V, Lehrach H, Yaspo ML, Eils R, Lichter P, Korbel JO, Pfister SM, Bradner JE, Northcott PA, 2016. Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature 530, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JCC, Yu K, Hatcher A, Huang TW, Lee HK, Carlson J, Weston MC, Chen F, Zhang Y, Zhu W, Mohila CA, Ahmed N, Patel AJ, Arenkiel BR, Noebels JL, Creighton CJ, Deneen B, 2017. Identification of diverse astrocyte populations and their malignant analogs. Nat Neurosci 20, 396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg N, Kastemar M, Olofsson T, Smits A, Uhrbom L, 2009. Oligodendrocyte progenitor cells can act as cell of origin for experimental glioma. Oncogene 28, 2266–2275. [DOI] [PubMed] [Google Scholar]
- Lindsey JC, Lusher ME, Anderton JA, Bailey S, Gilbertson RJ, Pearson AD, Ellison DW, Clifford SC, 2004. Identification of tumour-specific epigenetic events in medulloblastoma development by hypermethylation profiling. Carcinogenesis 25, 661–668. [DOI] [PubMed] [Google Scholar]
- Liu C, Sage JC, Miller MR, Verhaak RG, Hippenmeyer S, Vogel H, Foreman O, Bronson RT, Nishiyama A, Luo L, Zong H, 2011. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 146, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu KW, Pajtler KW, Worst BC, Pfister SM, Wechsler-Reya RJ, 2017. Molecular mechanisms and therapeutic targets in pediatric brain tumors. Sci Signal 10. [DOI] [PubMed] [Google Scholar]
- Liu WJ, Tan XH, Luo XP, Guo BP, Wei ZJ, Ke Q, He S, Cen H, 2014. Prognostic significance of Tet methylcytosine dioxygenase 2 (TET2) gene mutations in adult patients with acute myeloid leukemia: a meta-analysis. Leuk Lymphoma 55, 2691–2698. [DOI] [PubMed] [Google Scholar]
- Louis DN, 2007. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114, 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis DN, von Deimling A, Chung RY, Rubio MP, Whaley JM, Eibl RH, Ohgaki H, Wiestler OD, Thor AD, Seizinger BR, 1993. Comparative study of p53 gene and protein alterations in human astrocytic tumors. J Neuropathol Exp Neurol 52, 31–38. [DOI] [PubMed] [Google Scholar]
- Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, Wellen KE, O’Rourke DM, Berger SL, Chan TA, Levine RL, Mellinghoff IK, Thompson CB, 2012. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Chen Y, Zhao C, Wang H, He D, Xu L, Wang J, He X, Deng Y, Lu EE, Liu X, Verma R, Bu H, Drissi R, Fouladi M, Stemmer-Rachamimov AO, Burns D, Xin M, Rubin JB, Bahassi EM, Canoll P, Holland EC, Lu QR, 2016. Olig2-Dependent Reciprocal Shift in PDGF and EGF Receptor Signaling Regulates Tumor Phenotype and Mitotic Growth in Malignant Glioma. Cancer Cell 29, 669–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu QR, Park JK, Noll E, Chan JA, Alberta J, Yuk D, Alzamora MG, Louis DN, Stiles CD, Rowitch DH, Black PM, 2001. Oligodendrocyte lineage genes (OLIG) as molecular markers for human glial brain tumors. Proc Natl Acad Sci U S A 98, 10851–10856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu QR, Sun T, Zhu Z, Ma N, Garcia M, Stiles CD, Rowitch DH, 2002. Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell 109, 75–86. [DOI] [PubMed] [Google Scholar]
- Lv D, Jia F, Hou Y, Sang Y, Alvarez AA, Zhang W, Gao WQ, Hu B, Cheng SY, Ge J, Li Y, Feng H, 2017. Histone Acetyltransferase KAT6A Upregulates PI3K/AKT Signaling through TRIM24 Binding. Cancer Res 77, 6190–6201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, Bjerke L, Clarke M, Vinci M, Nandhabalan M, Temelso S, Popov S, Molinari V, Raman P, Waanders AJ, Han HJ, Gupta S, Marshall L, Zacharoulis S, Vaidya S, Mandeville HC, Bridges LR, Martin AJ, Al-Sarraj S, Chandler C, Ng HK, Li X, Mu K, Trabelsi S, Brahim DH, Kisljakov AN, Konovalov DM, Moore AS, Carcaboso AM, Sunol M, de Torres C, Cruz O, Mora J, Shats LI, Stavale JN, Bidinotto LT, Reis RM, Entz-Werle N, Farrell M, Cryan J, Crimmins D, Caird J, Pears J, Monje M, Debily MA, Castel D, Grill J, Hawkins C, Nikbakht H, Jabado N, Baker SJ, Pfister SM, Jones DTW, Fouladi M, von Bueren AO, Baudis M, Resnick A, Jones C, 2017. Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 32, 520–537 e525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher EA, Furnari FB, Bachoo RM, Rowitch DH, Louis DN, Cavenee WK, DePinho RA, 2001. Malignant glioma: genetics and biology of a grave matter. Genes Dev 15, 1311–1333. [DOI] [PubMed] [Google Scholar]
- Malumbres M, Barbacid M, 2001. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer 1, 222–231. [DOI] [PubMed] [Google Scholar]
- Malzkorn B, Wolter M, Riemenschneider MJ, Reifenberger G, 2011. Unraveling the glioma epigenome: from molecular mechanisms to novel biomarkers and therapeutic targets. Brain Pathol 21, 619–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez E, Yoshihara K, Kim H, Mills GM, Trevino V, Verhaak RG, 2015. Comparison of gene expression patterns across 12 tumor types identifies a cancer supercluster characterized by TP53 mutations and cell cycle defects. Oncogene 34, 2732–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez R, Esteller M, 2010. The DNA methylome of glioblastoma multiforme. Neurobiol Dis 39, 40–46. [DOI] [PubMed] [Google Scholar]
- Masui K, Tanaka K, Akhavan D, Babic I, Gini B, Matsutani T, Iwanami A, Liu F, Villa GR, Gu Y, Campos C, Zhu S, Yang H, Yong WH, Cloughesy TF, Mellinghoff IK, Cavenee WK, Shaw RJ, Mischel PS, 2013. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab 18, 726–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew RK, Rutka JT, 2018. Diffuse Intrinsic Pontine Glioma : Clinical Features, Molecular Genetics, and Novel Targeted Therapeutics. J Korean Neurosurg Soc 61, 343–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Findlay GM, Gagnon JA, Horwitz MS, Schier AF, Shendure J, 2016. Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science 353, aaf7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta S, Huillard E, Kesari S, Maire CL, Golebiowski D, Harrington EP, Alberta JA, Kane MF, Theisen M, Ligon KL, Rowitch DH, Stiles CD, 2011. The central nervous system-restricted transcription factor Olig2 opposes p53 responses to genotoxic damage in neural progenitors and malignant glioma. Cancer Cell 19, 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meletis K, Wirta V, Hede SM, Nistér M, Lundeberg J, Frisén J, 2006. p53 suppresses the self-renewal of adult neural stem cells. Development 133, 363–369. [DOI] [PubMed] [Google Scholar]
- Misuraca KL, Cordero FJ, Becher OJ, 2015. Pre-Clinical Models of Diffuse Intrinsic Pontine Glioma. Front Oncol 5, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi G, Butt SJ, Takebayashi H, Fishell G, 2007. Physiologically distinct temporal cohorts of cortical interneurons arise from telencephalic Olig2-expressing precursors. J Neurosci 27, 7786–7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad F, Weissmann S, Leblanc B, Pandey DP, Hojfeldt JW, Comet I, Zheng C, Johansen JV, Rapin N, Porse BT, Tvardovskiy A, Jensen ON, Olaciregui NG, Lavarino C, Sunol M, de Torres C, Mora J, Carcaboso AM, Helin K, 2017. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med 23, 483–492. [DOI] [PubMed] [Google Scholar]
- Molofsky AV, Krencik R, Ullian EM, Tsai HH, Deneen B, Richardson WD, Barres BA, Rowitch DH, 2012. Astrocytes and disease: a neurodevelopmental perspective. Genes Dev 26, 891–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monje M, Mitra SS, Freret ME, Raveh TB, Kim J, Masek M, Attema JL, Li G, Haddix T, Edwards MS, Fisher PG, Weissman IL, Rowitch DH, Vogel H, Wong AJ, Beachy PA, 2011. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc Natl Acad Sci U S A 108, 4453–4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran S, Martinez-Cardus A, Sayols S, Musulen E, Balana C, Estival-Gonzalez A, Moutinho C, Heyn H, Diaz-Lagares A, de Moura MC, Stella GM, Comoglio PM, Ruiz-Miro M, Matias-Guiu X, Pazo-Cid R, Anton A, Lopez-Lopez R, Soler G, Longo F, Guerra I, Fernandez S, Assenov Y, Plass C, Morales R, Carles J, Bowtell D, Mileshkin L, Sia D, Tothill R, Tabernero J, Llovet JM, Esteller M, 2016. Epigenetic profiling to classify cancer of unknown primary: a multicentre, retrospective analysis. Lancet Oncol 17, 1386–1395. [DOI] [PubMed] [Google Scholar]
- Morrissy AS, Garzia L, Shih DJ, Zuyderduyn S, Huang X, Skowron P, Remke M, Cavalli FM, Ramaswamy V, Lindsay PE, Jelveh S, Donovan LK, Wang X, Luu B, Zayne K, Li Y, Mayoh C, Thiessen N, Mercier E, Mungall KL, Ma Y, Tse K, Zeng T, Shumansky K, Roth AJ, Shah S, Farooq H, Kijima N, Holgado BL, Lee JJ, Matan-Lithwick S, Liu J, Mack SC, Manno A, Michealraj KA, Nor C, Peacock J, Qin L, Reimand J, Rolider A, Thompson YY, Wu X, Pugh T, Ally A, Bilenky M, Butterfield YS, Carlsen R, Cheng Y, Chuah E, Corbett RD, Dhalla N, He A, Lee D, Li HI, Long W, Mayo M, Plettner P, Qian JQ, Schein JE, Tam A, Wong T, Birol I, Zhao Y, Faria CC, Pimentel J, Nunes S, Shalaby T, Grotzer M, Pollack IF, Hamilton RL, Li XN, Bendel AE, Fults DW, Walter AW, Kumabe T, Tominaga T, Collins VP, Cho YJ, Hoffman C, Lyden D, Wisoff JH, Garvin JH Jr., Stearns DS, Massimi L, Schuller U, Sterba J, Zitterbart K, Puget S, Ayrault O, Dunn SE, Tirapelli DP, Carlotti CG, Wheeler H, Hallahan AR, Ingram W, MacDonald TJ, Olson JJ, Van Meir EG, Lee JY, Wang KC, Kim SK, Cho BK, Pietsch T, Fleischhack G, Tippelt S, Ra YS, Bailey S, Lindsey JC, Clifford SC, Eberhart CG, Cooper MK, Packer RJ, Massimino M, Garre ML, Bartels U, Tabori U, Hawkins CE, Dirks P, Bouffet E, Rutka JT, Wechsler-Reya RJ, Weiss WA, Collier LS, Dupuy AJ, Korshunov A, Jones DT, Kool M, Northcott PA, Pfister SM, Largaespada DA, Mungall AJ, Moore RA, Jabado N, Bader GD, Jones SJ, Malkin D, Marra MA, Taylor MD, 2016. Divergent clonal selection dominates medulloblastoma at recurrence. Nature 529, 351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz DM, Singh S, Tung T, Agnihotri S, Nagy A, Guha A, Zadeh G, Hawkins C, 2013. Differential transformation capacity of neuro-glial progenitors during development. Proc Natl Acad Sci U S A 110, 14378–14383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newbold A, Matthews GM, Bots M, Cluse LA, Clarke CJ, Banks KM, Cullinane C, Bolden JE, Christiansen AJ, Dickins RA, Miccolo C, Chiocca S, Kral AM, Ozerova ND, Miller TA, Methot JL, Richon VM, Secrist JP, Minucci S, Johnstone RW, 2013. Molecular and biologic analysis of histone deacetylase inhibitors with diverse specificities. Mol Cancer Ther 12, 2709–2721. [DOI] [PubMed] [Google Scholar]
- Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, Grobner S, Segura-Wang M, Zichner T, Rudneva VA, Warnatz HJ, Sidiropoulos N, Phillips AH, Schumacher S, Kleinheinz K, Waszak SM, Erkek S, Jones DTW, Worst BC, Kool M, Zapatka M, Jager N, Chavez L, Hutter B, Bieg M, Paramasivam N, Heinold M, Gu Z, Ishaque N, Jager-Schmidt C, Imbusch CD, Jugold A, Hubschmann D, Risch T, Amstislavskiy V, Gonzalez FGR, Weber UD, Wolf S, Robinson GW, Zhou X, Wu G, Finkelstein D, Liu Y, Cavalli FMG, Luu B, Ramaswamy V, Wu X, Koster J, Ryzhova M, Cho YJ, Pomeroy SL, Herold-Mende C, Schuhmann M, Ebinger M, Liau LM, Mora J, McLendon RE, Jabado N, Kumabe T, Chuah E, Ma Y, Moore RA, Mungall AJ, Mungall KL, Thiessen N, Tse K, Wong T, Jones SJM, Witt O, Milde T, Von Deimling A, Capper D, Korshunov A, Yaspo ML, Kriwacki R, Gajjar A, Zhang J, Beroukhim R, Fraenkel E, Korbel JO, Brors B, Schlesner M, Eils R, Marra MA, Pfister SM, Taylor MD, Lichter P, 2017. The whole-genome landscape of medulloblastoma subtypes. Nature 547, 311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S, Bouffet E, Clifford SC, Hawkins CE, French P, Rutka JT, Pfister S, Taylor MD, 2011. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 29, 1408–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott PA, Lee C, Zichner T, Stutz AM, Erkek S, Kawauchi D, Shih DJ, Hovestadt V, Zapatka M, Sturm D, Jones DT, Kool M, Remke M, Cavalli FM, Zuyderduyn S, Bader GD, VandenBerg S, Esparza LA, Ryzhova M, Wang W, Wittmann A, Stark S, Sieber L, Seker-Cin H, Linke L, Kratochwil F, Jager N, Buchhalter I, Imbusch CD, Zipprich G, Raeder B, Schmidt S, Diessl N, Wolf S, Wiemann S, Brors B, Lawerenz C, Eils J, Warnatz HJ, Risch T, Yaspo ML, Weber UD, Bartholomae CC, von Kalle C, Turanyi E, Hauser P, Sanden E, Darabi A, Siesjo P, Sterba J, Zitterbart K, Sumerauer D, van Sluis P, Versteeg R, Volckmann R, Koster J, Schuhmann MU, Ebinger M, Grimes HL, Robinson GW, Gajjar A, Mynarek M, von Hoff K, Rutkowski S, Pietsch T, Scheurlen W, Felsberg J, Reifenberger G, Kulozik AE, von Deimling A, Witt O, Eils R, Gilbertson RJ, Korshunov A, Taylor MD, Lichter P, Korbel JO, Wechsler-Reya RJ, Pfister SM, 2014. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 511, 428–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, Stutz AM, Korshunov A, Reimand J, Schumacher SE, Beroukhim R, Ellison DW, Marshall CR, Lionel AC, Mack S, Dubuc A, Yao Y, Ramaswamy V, Luu B, Rolider A, Cavalli FM, Wang X, Remke M, Wu X, Chiu RY, Chu A, Chuah E, Corbett RD, Hoad GR, Jackman SD, Li Y, Lo A, Mungall KL, Nip KM, Qian JQ, Raymond AG, Thiessen NT, Varhol RJ, Birol I, Moore RA, Mungall AJ, Holt R, Kawauchi D, Roussel MF, Kool M, Jones DT, Witt H, Fernandez LA, Kenney AM, Wechsler-Reya RJ, Dirks P, Aviv T, Grajkowska WA, Perek-Polnik M, Haberler CC, Delattre O, Reynaud SS, Doz FF, Pernet-Fattet SS, Cho BK, Kim SK, Wang KC, Scheurlen W, Eberhart CG, Fevre-Montange M, Jouvet A, Pollack IF, Fan X, Muraszko KM, Gillespie GY, Di Rocco C, Massimi L, Michiels EM, Kloosterhof NK, French PJ, Kros JM, Olson JM, Ellenbogen RG, Zitterbart K, Kren L, Thompson RC, Cooper MK, Lach B, McLendon RE, Bigner DD, Fontebasso A, Albrecht S, Jabado N, Lindsey JC, Bailey S, Gupta N, Weiss WA, Bognar L, Klekner A, Van Meter TE, Kumabe T, Tominaga T, Elbabaa SK, Leonard JR, Rubin JB, Liau LM, Van Meir EG, Fouladi M, Nakamura H, Cinalli G, Garami M, Hauser P, Saad AG, Iolascon A, Jung S, Carlotti CG, Vibhakar R, Ra YS, Robinson S, Zollo M, Faria CC, Chan JA, Levy ML, Sorensen PH, Meyerson M, Pomeroy SL, Cho YJ, Bader GD, Tabori U, Hawkins CE, Bouffet E, Scherer SW, Rutka JT, Malkin D, Clifford SC, Jones SJ, Korbel JO, Pfister SM, Marra MA, Taylor MD, 2012. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature 488, 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes MC, Roy NS, Keyoung HM, Goodman RR, McKhann G 2nd, Jiang L, Kang J, Nedergaard M, Goldman SA, 2003. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat Med 9, 439–447. [DOI] [PubMed] [Google Scholar]
- Ohgaki H, Kleihues P, 2007. Genetic pathways to primary and secondary glioblastoma. Am J Pathol 170, 1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohgaki H, Kleihues P, 2009. Genetic alterations and signaling pathways in the evolution of gliomas. Cancer Sci 100, 2235–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega MA, Poirion O, Zhu X, Huang S, Wolfgruber TK, Sebra R, Garmire LX, 2017. Using single-cell multiple omics approaches to resolve tumor heterogeneity. Clin Transl Med 6, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J, Wolinsky Y, Kruchko C, Barnholtz-Sloan J, 2014. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol 16 Suppl 4, iv1–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrom QT, Gittleman H, Liao P, Vecchione-Koval T, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS, 2017. CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro Oncol 19, v1–v88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero JJ, Rowitch D, Vandenberg S, 2011. OLIG2 is differentially expressed in pediatric astrocytic and in ependymal neoplasms. J Neurooncol 104, 423–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, 2008. An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, Louis DN, Rozenblatt-Rosen O, Suva ML, Regev A, Bernstein BE, 2014. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania M, De Jay N, Maestro N, Harutyunyan AS, Nitarska J, Pahlavan P, Henderson S, Mikael LG, Richard-Londt A, Zhang Y, Costa JR, Hebert S, Khazaei S, Ibrahim NS, Herrero J, Riccio A, Albrecht S, Ketteler R, Brandner S, Kleinman CL, Jabado N, Salomoni P, 2017. H3.3(K27M) Cooperates with Trp53 Loss and PDGFRA Gain in Mouse Embryonic Neural Progenitor Cells to Induce Invasive High-Grade Gliomas. Cancer Cell 32, 684–700 e689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paugh BS, Broniscer A, Qu C, Miller CP, Zhang J, Tatevossian RG, Olson JM, Geyer JR, Chi SN, da Silva NS, Onar-Thomas A, Baker JN, Gajjar A, Ellison DW, Baker SJ, 2011. Genome-wide analyses identify recurrent amplifications of receptor tyrosine kinases and cell-cycle regulatory genes in diffuse intrinsic pontine glioma. J Clin Oncol 29, 3999–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paugh BS, Zhu X, Qu C, Endersby R, Diaz AK, Zhang J, Bax DA, Carvalho D, Reis RM, Onar-Thomas A, Broniscer A, Wetmore C, Jones C, Ellison DW, Baker SJ, 2013. Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res 73, 6219–6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei Y, Moore CE, Wang J, Tewari AK, Eroshkin A, Cho YJ, Witt H, Korshunov A, Read TA, Sun JL, Schmitt EM, Miller CR, Buckley AF, McLendon RE, Westbrook TF, Northcott PA, Taylor MD, Pfister SM, Febbo PG, Wechsler-Reya RJ, 2012. An animal model of MYC-driven medulloblastoma. Cancer Cell 21, 155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson AI, Petritsch C, Swartling FJ, Itsara M, Sim FJ, Auvergne R, Goldenberg DD, Vandenberg SR, Nguyen KN, Yakovenko S, Ayers-Ringler J, Nishiyama A, Stallcup WB, Berger MS, Bergers G, McKnight TR, Goldman SA, Weiss WA, 2010. Non-stem cell origin for oligodendroglioma. Cancer Cell 18, 669–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G, Brem H, Olivi A, Dimeco F, Vescovi AL, 2006. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 444, 761–765. [DOI] [PubMed] [Google Scholar]
- Puget S, Philippe C, Bax DA, Job B, Varlet P, Junier MP, Andreiuolo F, Carvalho D, Reis R, Guerrini-Rousseau L, Roujeau T, Dessen P, Richon C, Lazar V, Le Teuff G, Sainte-Rose C, Geoerger B, Vassal G, Jones C, Grill J, 2012. Mesenchymal transition and PDGFRA amplification/mutation are key distinct oncogenic events in pediatric diffuse intrinsic pontine gliomas. PLoS One 7, e30313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, Carneiro MO, Carter SL, Cibulskis K, Erlich RL, Greulich H, Lawrence MS, Lennon NJ, McKenna A, Meldrim J, Ramos AH, Ross MG, Russ C, Shefler E, Sivachenko A, Sogoloff B, Stojanov P, Tamayo P, Mesirov JP, Amani V, Teider N, Sengupta S, Francois JP, Northcott PA, Taylor MD, Yu F, Crabtree GR, Kautzman AG, Gabriel SB, Getz G, Jager N, Jones DT, Lichter P, Pfister SM, Roberts TM, Meyerson M, Pomeroy SL, Cho YJ, 2012. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 488, 106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purzner T, Purzner J, Buckstaff T, Cozza G, Gholamin S, Rusert JM, Hartl TA, Sanders J, Conley N, Ge X, Langan M, Ramaswamy V, Ellis L, Litzenburger U, Bolin S, Theruvath J, Nitta R, Qi L, Li XN, Li G, Taylor MD, Wechsler-Reya RJ, Pinna LA, Cho YJ, Fuller MT, Elias JE, Scott MP, 2018. Developmental phosphoproteomics identifies the kinase CK2 as a driver of Hedgehog signaling and a therapeutic target in medulloblastoma. Sci Signal 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radke J, Bortolussi G, Pagenstecher A, 2013. Akt and c-Myc induce stem-cell markers in mature primary p53(−)/(−) astrocytes and render these cells gliomagenic in the brain of immunocompetent mice. PLoS One 8, e56691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks T, 2000. Nf1; Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nature Genet 26, 109–113. [DOI] [PubMed] [Google Scholar]
- Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L, Phoenix TN, Hedlund E, Wei L, Zhu X, Chalhoub N, Baker SJ, Huether R, Kriwacki R, Curley N, Thiruvenkatam R, Wang J, Wu G, Rusch M, Hong X, Becksfort J, Gupta P, Ma J, Easton J, Vadodaria B, Onar-Thomas A, Lin T, Li S, Pounds S, Paugh S, Zhao D, Kawauchi D, Roussel MF, Finkelstein D, Ellison DW, Lau CC, Bouffet E, Hassall T, Gururangan S, Cohn R, Fulton RS, Fulton LL, Dooling DJ, Ochoa K, Gajjar A, Mardis ER, Wilson RK, Downing JR, Zhang J, Gilbertson RJ, 2012. Novel mutations target distinct subgroups of medulloblastoma. Nature 488, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampetrean O, Saya H, 2018. Modeling phenotypes of malignant gliomas. Cancer Sci 109, 6–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saratsis AM, Kambhampati M, Snyder K, Yadavilli S, Devaney JM, Harmon B, Hall J, Raabe EH, An P, Weingart M, Rood BR, Magge SN, MacDonald TJ, Packer RJ, Nazarian J, 2014. Comparative multidimensional molecular analyses of pediatric diffuse intrinsic pontine glioma reveals distinct molecular subtypes. Acta Neuropathol 127, 881–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt N, Windmann S, Reifenberger G, Riemenschneider MJ, 2012. DNA hypermethylation and histone modifications downregulate the candidate tumor suppressor gene RRP22 on 22q12 in human gliomas. Brain Pathol 22, 17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, Huillard E, Sun T, Ligon AH, Qian Y, Ma Q, Alvarez-Buylla A, McMahon AP, Rowitch DH, Ligon KL, 2008. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell 14, 123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalbe EC, Williamson D, Lindsey JC, Hamilton D, Ryan SL, Megahed H, Garami M, Hauser P, Dembowska-Baginska B, Perek D, Northcott PA, Taylor MD, Taylor RE, Ellison DW, Bailey S, Clifford SC, 2013. DNA methylation profiling of medulloblastoma allows robust subclassification and improved outcome prediction using formalin-fixed biopsies. Acta Neuropathol 125, 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, Hovestadt V, Albrecht S, Kool M, Nantel A, Konermann C, Lindroth A, Jager N, Rausch T, Ryzhova M, Korbel JO, Hielscher T, Hauser P, Garami M, Klekner A, Bognar L, Ebinger M, Schuhmann MU, Scheurlen W, Pekrun A, Fruhwald MC, Roggendorf W, Kramm C, Durken M, Atkinson J, Lepage P, Montpetit A, Zakrzewska M, Zakrzewski K, Liberski PP, Dong Z, Siegel P, Kulozik AE, Zapatka M, Guha A, Malkin D, Felsberg J, Reifenberger G, von Deimling A, Ichimura K, Collins VP, Witt H, Milde T, Witt O, Zhang C, Castelo-Branco P, Lichter P, Faury D, Tabori U, Plass C, Majewski J, Pfister SM, Jabado N, 2012. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231. [DOI] [PubMed] [Google Scholar]
- Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, Ziegler PK, Canli O, Heijmans J, Huels DJ, Moreaux G, Rupec RA, Gerhard M, Schmid R, Barker N, Clevers H, Lang R, Neumann J, Kirchner T, Taketo MM, van den Brink GR, Sansom OJ, Arkan MC, Greten FR, 2013. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 152, 25–38. [DOI] [PubMed] [Google Scholar]
- Sharpless NE, DePinho RA, 1999. The INK4A/ARF locus and its two gene products. Curr Opin Genet Dev 9, 22–30. [DOI] [PubMed] [Google Scholar]
- Shimada IS, Hwang SH, Somatilaka BN, Wang X, Skowron P, Kim J, Kim M, Shelton JM, Rajaram V, Xuan Z, Taylor MD, Mukhopadhyay S, 2018. Basal Suppression of the Sonic Hedgehog Pathway by the G-Protein-Coupled Receptor Gpr161 Restricts Medulloblastoma Pathogenesis. Cell Rep 22, 1169–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh DK, Kollipara RK, Vemireddy V, Yang XL, Sun Y, Regmi N, Klingler S, Hatanpaa KJ, Raisanen J, Cho SK, Sirasanagandla S, Nannepaga S, Piccirillo S, Mashimo T, Wang S, Humphries CG, Mickey B, Maher EA, Zheng H, Kim RS, Kittler R, Bachoo RM, 2017. Oncogenes Activate an Autonomous Transcriptional Regulatory Circuit That Drives Glioblastoma. Cell Rep 18, 961–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Fiorelli R, Kupp R, Rajan S, Szeto E, Lo Cascio C, Maire CL, Sun Y, Alberta JA, Eschbacher JM, Ligon KL, Berens ME, Sanai N, Mehta S, 2016. Post-translational Modifications of OLIG2 Regulate Glioma Invasion through the TGF-beta Pathway. Cell Rep 16, 950–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB, 2004. Identification of human brain tumour initiating cells. Nature 432, 396–401. [DOI] [PubMed] [Google Scholar]
- Stricker SH, Feber A, Engstrom PG, Caren H, Kurian KM, Takashima Y, Watts C, Way M, Dirks P, Bertone P, Smith A, Beck S, Pollard SM, 2013. Widespread resetting of DNA methylation in glioblastoma-initiating cells suppresses malignant cellular behavior in a lineage-dependent manner. Genes Dev 27, 654–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm D, Bender S, Jones DT, Lichter P, Grill J, Becher O, Hawkins C, Majewski J, Jones C, Costello JF, Iavarone A, Aldape K, Brennan CW, Jabado N, Pfister SM, 2014. Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat Rev Cancer 14, 92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tonjes M, Sill M, Bender S, Kool M, Zapatka M, Becker N, Zucknick M, Hielscher T, Liu XY, Fontebasso AM, Ryzhova M, Albrecht S, Jacob K, Wolter M, Ebinger M, Schuhmann MU, van Meter T, Fruhwald MC, Hauch H, Pekrun A, Radlwimmer B, Niehues T, von Komorowski G, Durken M, Kulozik AE, Madden J, Donson A, Foreman NK, Drissi R, Fouladi M, Scheurlen W, von Deimling A, Monoranu C, Roggendorf W, Herold-Mende C, Unterberg A, Kramm CM, Felsberg J, Hartmann C, Wiestler B, Wick W, Milde T, Witt O, Lindroth AM, Schwartzentruber J, Faury D, Fleming A, Zakrzewska M, Liberski PP, Zakrzewski K, Hauser P, Garami M, Klekner A, Bognar L, Morrissy S, Cavalli F, Taylor MD, van Sluis P, Koster J, Versteeg R, Volckmann R, Mikkelsen T, Aldape K, Reifenberger G, Collins VP, Majewski J, Korshunov A, Lichter P, Plass C, Jabado N, Pfister SM, 2012. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22, 425–437. [DOI] [PubMed] [Google Scholar]
- Suva ML, Rheinbay E, Gillespie SM, Patel AP, Wakimoto H, Rabkin SD, Riggi N, Chi AS, Cahill DP, Nahed BV, Curry WT, Martuza RL, Rivera MN, Rossetti N, Kasif S, Beik S, Kadri S, Tirosh I, Wortman I, Shalek AK, Rozenblatt-Rosen O, Regev A, Louis DN, Bernstein BE, 2014. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 157, 580–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A, 2009. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takebayashi H, Nabeshima Y, Yoshida S, Chisaka O, Ikenaka K, Nabeshima Y, 2002. The basic helix-loop-helix factor olig2 is essential for the development of motoneuron and oligodendrocyte lineages. Curr Biol 12, 1157–1163. [DOI] [PubMed] [Google Scholar]
- Taylor KR, Mackay A, Truffaux N, Butterfield Y, Morozova O, Philippe C, Castel D, Grasso CS, Vinci M, Carvalho D, Carcaboso AM, de Torres C, Cruz O, Mora J, Entz-Werle N, Ingram WJ, Monje M, Hargrave D, Bullock AN, Puget S, Yip S, Jones C, Grill J, 2014a. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nature genetics 46, 457–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor KR, Vinci M, Bullock AN, Jones C, 2014b. ACVR1 mutations in DIPG: lessons learned from FOP. Cancer Res 74, 4565–4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, Eberhart CG, Parsons DW, Rutkowski S, Gajjar A, Ellison DW, Lichter P, Gilbertson RJ, Pomeroy SL, Kool M, Pfister SM, 2012. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 123, 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research, N., 2008. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Suva ML, 2018. Dissecting human gliomas by single-cell RNA sequencing. Neuro Oncol 20, 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, Fisher JM, Rodman C, Mount C, Filbin MG, Neftel C, Desai N, Nyman J, Izar B, Luo CC, Francis JM, Patel AA, Onozato ML, Riggi N, Livak KJ, Gennert D, Satija R, Nahed BV, Curry WT, Martuza RL, Mylvaganam R, Iafrate AJ, Frosch MP, Golub TR, Rivera MN, Getz G, Rozenblatt-Rosen O, Cahill DP, Monje M, Bernstein BE, Louis DN, Regev A, Suva ML, 2016. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, Thompson CB, Kaufman A, Guryanova O, Levine R, Heguy A, Viale A, Morris LG, Huse JT, Mellinghoff IK, Chan TA, 2012. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhrbom L, Hesselager G, Nister M, Westermark B, 1998. Induction of brain tumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res 58, 5275–5279. [PubMed] [Google Scholar]
- van den Boom J, Wolter M, Kuick R, Misek DE, Youkilis AS, Wechsler DS, Sommer C, Reifenberger G, Hanash SM, 2003. Characterization of gene expression profiles associated with glioma progression using oligonucleotide-based microarray analysis and real-time reverse transcription-polymerase chain reaction. Am J Pathol 163, 1033–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meyel DJ, Ramsay DA, Casson AG, Keeney M, Chambers AF, Cairncross JG, 1994. p53 mutation, expression, and DNA ploidy in evolving gliomas: evidence for two pathways of progression. Journal of the National Cancer Institute 86, 1011–1017. [DOI] [PubMed] [Google Scholar]
- Veeriah S, Brennan C, Meng S, Singh B, Fagin JA, Solit DB, Paty PB, Rohle D, Vivanco I, Chmielecki J, Pao W, Ladanyi M, Gerald WL, Liau L, Cloughesy TC, Mischel PS, Sander C, Taylor B, Schultz N, Major J, Heguy A, Fang F, Mellinghoff IK, Chan TA, 2009. The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers. Proc Natl Acad Sci U S A 106, 9435–9440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, Hovestadt V, Escalante LE, Shaw ML, Rodman C, Gillespie SM, Dionne D, Luo CC, Ravichandran H, Mylvaganam R, Mount C, Onozato ML, Nahed BV, Wakimoto H, Curry WT, Iafrate AJ, Rivera MN, Frosch MP, Golub TR, Brastianos PK, Getz G, Patel AP, Monje M, Cahill DP, Rozenblatt-Rosen O, Louis DN, Bernstein BE, Regev A, Suva ML, 2017. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, Hayes DN, 2010. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvader JE, 2011. Cells of origin in cancer. Nature 469, 314–322. [DOI] [PubMed] [Google Scholar]
- Vitucci M, Hayes DN, Miller CR, 2011. Gene expression profiling of gliomas: merging genomic and histopathological classification for personalised therapy. British journal of cancer 104, 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo BT, Li C, Morgan MA, Theurillat I, Finkelstein D, Wright S, Hyle J, Smith SMC, Fan Y, Wang YD, Wu G, Orr BA, Northcott PA, Shilatifard A, Sherr CJ, Roussel MF, 2017. Inactivation of Ezh2 Upregulates Gfi1 and Drives Aggressive Myc-Driven Group 3 Medulloblastoma. Cell Rep 18, 2907–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Kinzler KW, 2004. Cancer genes and the pathways they control. Nat Med 10, 789–799. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., Kinzler KW, 2013. Cancer genome landscapes. Science 339, 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Garancher A, Ramaswamy V, Wechsler-Reya RJ, 2018a. Medulloblastoma: From Molecular Subgroups to Molecular Targeted Therapies. Annu Rev Neurosci 41, 207–232. [DOI] [PubMed] [Google Scholar]
- Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, deCarvalho AC, Lyu S, Li P, Li Y, Barthel F, Cho HJ, Lin YH, Satani N, Martinez-Ledesma E, Zheng S, Chang E, Gabriel Sauve CE, Olar A, Lan ZD, Finocchiaro G, Phillips JJ, Berger MS, Gabrusiewicz KR, Wang G, Eskilsson E, Hu J, Mikkelsen T, DePinho RA, Muller F, Heimberger AB, Sulman EP, Nam DH, Verhaak RGW, 2018b. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 33, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Guo YR, Liu K, Yin Z, Liu R, Xia Y, Tan L, Yang P, Lee JH, Li XJ, Hawke D, Zheng Y, Qian X, Lyu J, He J, Xing D, Tao YJ, Lu Z, 2017. KAT2A coupled with the alpha-KGDH complex acts as a histone H3 succinyltransferase. Nature 552, 273–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren KE, 2012. Diffuse intrinsic pontine glioma: poised for progress. Front Oncol 2, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng Q, Wang J, Wang J, He D, Cheng Z, Zhang F, Verma R, Xu L, Dong X, Liao Y, He X, Potter A, Zhang L, Zhao C, Xin M, Zhou Q, Aronow JB, Blackshear PJ, Rich JN, He Q, Zhou W, Suvà ML, Waclaw R, Potter SS, Yu G, Lu QR, 2019. Single-cell Transcriptomics Resolves Intermediate Glial Progenitors and Uncovers a Pivotal Determinant of Cell Fate and Gliomagenesis. Cell Stem Cell In press [DOI] [PMC free article] [PubMed]
- Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, Zhu X, Qu C, Chen X, Zhang J, Easton J, Edmonson M, Ma X, Lu C, Nagahawatte P, Hedlund E, Rusch M, Pounds S, Lin T, Onar-Thomas A, Huether R, Kriwacki R, Parker M, Gupta P, Becksfort J, Wei L, Mulder HL, Boggs K, Vadodaria B, Yergeau D, Russell JC, Ochoa K, Fulton RS, Fulton LL, Jones C, Boop FA, Broniscer A, Wetmore C, Gajjar A, Ding L, Mardis ER, Wilson RK, Taylor MR, Downing JR, Ellison DW, Zhang J, Baker SJ, 2014. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet 46, 444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, Zhao S, Ye D, Xiong Y, Guan KL, 2012. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev 26, 1326–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD, 2009. IDH1 and IDH2 mutations in gliomas. N Engl J Med 360, 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZJ, Ellis T, Markant SL, Read TA, Kessler JD, Bourboulas M, Schuller U, Machold R, Fishell G, Rowitch DH, Wainwright BJ, Wechsler-Reya RJ, 2008. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell 14, 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi J, Wu J, 2018. Epigenetic regulation in medulloblastoma. Mol Cell Neurosci 87, 65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi L, Cui Y, Xu Q, Jiang Y, 2016. Stabilization of LSD1 by deubiquitinating enzyme USP7 promotes glioblastoma cell tumorigenesis and metastasis through suppression of the p53 signaling pathway. Oncol Rep 36, 2935–2945. [DOI] [PubMed] [Google Scholar]
- You JS, Jones PA, 2012. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell 22, 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Khan OF, Suva ML, Dong B, Panek WK, Xiao T, Wu M, Han Y, Ahmed AU, Balyasnikova IV, Zhang HF, Sun C, Langer R, Anderson DG, Lesniak MS, 2017. Multiplexed RNAi therapy against brain tumor-initiating cells via lipopolymeric nanoparticle infusion delays glioblastoma progression. Proc Natl Acad Sci U S A 114, E6147–E6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue T, Xian K, Hurlock E, Xin M, Kernie SG, Parada LF, Lu QR, 2006. A critical role for dorsal progenitors in cortical myelination. J Neurosci 26, 1275–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarghooni M, Bartels U, Lee E, Buczkowicz P, Morrison A, Huang A, Bouffet E, Hawkins C, 2010. Whole-genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase as potential therapeutic targets. J Clin Oncol 28, 1337–1344. [DOI] [PubMed] [Google Scholar]
- Zhang H, Geng D, Gao J, Qi Y, Shi Y, Wang Y, Jiang Y, Zhang Y, Fu J, Dong Y, Gao S, Yu R, Zhou X, 2016a. Expression and significance of Hippo/YAP signaling in glioma progression. Tumour Biol [DOI] [PubMed]
- Zhang L, He X, Liu L, Jiang M, Zhao C, Wang H, He D, Zheng T, Zhou X, Hassan A, Ma Z, Xin M, Sun Z, Lazar MA, Goldman SA, Olson EN, Lu QR, 2016b. Hdac3 Interaction with p300 Histone Acetyltransferase Regulates the Oligodendrocyte and Astrocyte Lineage Fate Switch. Dev Cell 36, 316–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, Perry SR, Tonon G, Chu GC, Ding Z, Stommel JM, Dunn KL, Wiedemeyer R, You MJ, Brennan C, Wang YA, Ligon KL, Wong WH, Chin L, DePinho RA, 2008. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 455, 1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Anderson DJ, 2002. The bHLH transcription factors OLIG2 and OLIG1 couple neuronal and glial subtype specification. Cell 109, 61–73. [DOI] [PubMed] [Google Scholar]
- Zhu X, Zuo H, Maher BJ, Serwanski DR, LoTurco JJ, Lu QR, Nishiyama A, 2012. Olig2-dependent developmental fate switch of NG2 cells. Development 139, 2299–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Guignard F, Zhao D, Liu L, Burns DK, Mason RP, Messing A, Parada LF, 2005. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell 8, 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong H, Espinosa JS, Su HH, Muzumdar MD, Luo L, 2005. Mosaic analysis with double markers in mice. Cell 121, 479–492. [DOI] [PubMed] [Google Scholar]