

Abstract
Objective:
The aim of this study was to investigate the key molecular alterations in small primary pancreatic neuroendocrine tumors (PanNETs) associated with the development of liver metastases.
Background:
Well-differentiated PanNETs with small size are typically indolent; however, a limited subset metastasize to the liver.
Methods:
A total of 87 small primary PanNETs (<3cm), including 32 metastatic cases and 55 nonmetastatic cases after a 5-year follow-up, were immunolabeled for DAXX/ATRX and analyzed for alternative lengthening of telomeres (ALT) by Fluorescence In Situ Hybridization. A subset of these cases, 24 that metastasized and 24 that did not metastasize, were assessed by targeted next-generation sequencing and whole-genome copy number variation.
Results:
In the entire cohort, high Ki-67 (OR 1.369; 95% CI 1.121–1.673; P = 0.002), N-stage (OR 4.568; 95%CI 1.458–14.312; P = 0.009), and ALT-positivity (OR 3.486;95%CI 1.093–11.115; P = 0.035) were independently associated with liver metastases. In the subset assessed by next-generation sequencing and copy number variation analysis, 3 molecular subtypes with differing risks of liver metastases were identified.Group 1 (n = 15; 73%metastasized) was characterized by recurrent chromosomal gains, CN-LOH, DAXX mutations, and ALT-positivity.Group 2 (n = 19; 42% metastasized, including 5 G1 tumors) was characterized by limited copy number alterations and mutations. Group 3 (n = 14; 35% metastasized) were defined by chromosome 11 loss.
Conclusions:
We identified genomic patterns of small PanNETs associated with a different risk for liver metastases. Molecular alterations, such as DAXX mutations, chromosomal gains, and ALT, are associated with an increased risk of metastasis in small PanNETs. Therefore, targeted sequencing and/or AsLT analysis may help in the clinical decisions for these small PanNETs.
Keywords: ALT, genetic subgroups, liver metastases, pancreatic neuroendocrine tumors (PanNETs)
The incidence of pancreatic neuroendocrine tumors (PanNETs) is 1 per 100,000 individuals,1 However, autopsy studies highlight a prevalence ranging from 1% to 10% in the general population, suggesting that most PanNETs do not progress to clinical diagnosis.2,3 In recent years, an increasing number of small and asymptomatic PanNETs have been diagnosed incidentally on routine abdominal imaging.4 As the risk for metastases has been associated with tumor size and high proliferative index,5–7 periodic observation without resection has been advocated for small tumors. However, such observational approaches have not been universally adopted, due to a small, but existing risk of liver metastases among small tumors, particularly with the absence of biomarkers predictive for progression.8,9
Recent whole genome sequencing and expression profiling suggest that an aggressive phenotype is associated with genetic changes in telomere maintenance and/or mTOR signaling.10–12 Loss of function of DAXX or ATRX genes promotes the activation of the alternative lengthening of telomeres (ALT) pathway.13,14 ALT is associated with chromosomal instability, larger tumor size, higher Ki67 and, in some studies, metastatic spread.10,15,16 Alterations in mTOR pathway genes are also associated with a poor prognosis and several therapeutic agents targeting the pathway are available for systemic treatment.10,17–19
Thus, there is an urgent need to define early biological mechanisms responsible for tumor progression that may serve as clinically useful biomarkers. Here, we analyzed molecular alterations of small sporadic well-differentiated PanNETs (<3 cm in size) to identify alterations associated with metastasis. In the entire cohort of 87 primary PanNETs, we assessed ATRX and DAXX protein expression, and ALT status. In a subset, 24 primary PanNETs that metastatized and 24 primary PanNETs that did not metastasize, matched for size and proliferation index, were analyzed by targeted next-generation sequencing and for copy number alterations. These genomic analyses identified 3 molecular subtypes with differing risks for liver metastases.
METHODS
Study Population
This study was approved by the Institutional Review Board (IRB) of the Johns Hopkins Hospital and tissue samples were retrieved from collaborating institutes with informed consent according to their local Institutional Review Boards. Well-differentiated (G1 and G2) PanNETs < 3 cm in maximum dimension were identified in the pancreatic resection databases of the Johns Hopkins Hospital, University of Verona, Italy; University of Utrecht, The Netherlands; Pederzoli Hospital, Peschiera, Italy. All PanNETs included were sporadic, unifocal, and nonfunctional. The study included patients who developed synchronous or metachronous liver metastases and patients who did not develop distant metastases after a surveillance period of at least 5 years. PanNETs were graded according to 2017 World Health Organization (WHO) classification based on mitotic rate and Ki-67: < 2 mitoses/10 high-power fields and Ki-67 < 3% for grade 1 (G1), and 2 to 20 mitoses/10 high-power fields, and Ki-67 of 3% to 20% for grade 2 (G2).20
Immunohistochemistry (IHC) and Fluorescence In Situ Hybridization (FISH)
IHC for ATRX and DAXX and telomere-specific FISH were performed on formalin-fixed paraffin-embedded (FFPE) tissues. Nuclear labeling was evaluated for DAXX and ATRX and cases were scored as negative if nuclear expression was completely lost (despite retaining cytoplasmic expression) and adequate internal positive controls were present (eg, nuclear labeling of endothelial cells, lymphocytes, islets of Langerhans).13,21,22 ALT-positive tumors were identified by cell-to-cell telomere length heterogeneity and the presence of large, ultrabright nuclear foci of FISH signals. As previously defined,13,14,22 cases were classified as ALT-positive when bright nuclear foci occurred in ≥1% of tumor cells with at least 500 neoplastic cells evaluated.
DNA Extraction
Five micrometer sections from FFPE tissues were manually macrodissected to enrich for neoplastic cellularity, DNA was extracted using QIAamp DNA FFPE Tissue Kit (Qiagen, German-town) and quantified by Quantifiler Human DNA Quantification kit (Applied Biosystems, Thermo Fisher Scientific, Waltham).
Targeted Next-generation Sequencing
An Ion AmpliSeq Custom Panel (AmpliSeq Designer version 4.4.7; Life Technologies, Thermo Fisher Scientific, Waltham) was used perform multiplex PCR and sequencing of 18 genes known to be targeted in pancreatic neuroendocrine or ductal neoplasms (MEN1, DAXX, ATRX, TSC1, TSC2, PTEN, PIK3CA, PHLDA3, TP53, KRAS, GNAS, SMAD4, CDKN2A, RNF43, TGFBR2, ARID1A, BRAF, MAP2K4).10,12,23,24 Briefly, DNA (4ng) was amplified using Ampliseq reagents for library preparation, which was then loaded and sequenced onto a 318v2 chips using an Ion Torrent Personal Genome Machine (PGM; Life Technologies). Postsequencing data analyses, including alignment to the hg19 human reference genome and variant calling, were performed using NextGENe software (v2.4; SoftGenetics, Chicago, IL). Alignments and putative mutations were visually verified using the Integrative Genomics Viewer (IGV, v2.3; Broad Institute, Cambridge) and the NextGENeViewer (v2.4; SoftGenetics).
SNP Array Analysis
Copy number analysis was performed at the JHU Microarray Core Facility using the Illumina HumanCytoSNP-12 v2.1 BeadChip, according to manufacturer’s protocols. Log R ratios (LRRs) and B allele frequencies for each sample were extracted using GenomeStudio software. Prior copy number analysis, LRRs were normalized performing a GC-content correction.25 We used the Circular Binary Segmentation algorithm to estimate mean LRR for distinct genomic segments, and we applied a mixture model to compute copy number alteration probabilities and to classify each segment accordingly, as implemented in the “CGHcall” and “CGHregion” R-Bioconductor packages.24,25 Sex chromosomes were not included in this analysis and tumor cellularity was estimated using the “qPure” algorithm.26 We used the hg19 human reference genome assembly (GRCh37) to annotate segments of copy number alteration with the associated genes. For regions of normal copy number, we further analyzed the B allele frequency distribution, identifying copy neutral loss of heterozygosis (CN-LOH, loss of 1 allele with duplication of the remaining allele). Chromothripsis was defined as the shattering and reassembly of 1 or more chromosomes and was detected using the CTLP Scanner algorithm.27
Statistical Analysis
Continuous variables were reported as median and interquartile range or mean and standard deviation according to the values distribution. Correlation between clinical-pathological characteristics with ALT phenotype and development of liver metastases was assessed by univariate analysis using chi-square test or Fisher exact tests for categorical variables, and Student t test or Mann-Whitney U for continuous variables. A forward stepwise logistic regression model was used to evaluate independent risk factors associated with development of liver metastasis. A 2-tailed P value ≤0.05 was considered significant. Data were analyzed using SPSS 24.0 for Windows v24.0.
RESULTS
As illustrated in Figure 1, 87 patients who underwent pancreatectomy for unifocal, well-differentiated primary PanNET < 3 cm in size met the inclusion criteria. These included 32 patients who developed liver metastases (19 synchronous, 13 metachronous), and 55 patients who remained disease-free after at least 5 years of follow-up (mean 117 mo, standard deviation 58 mo).
FIGURE 1.
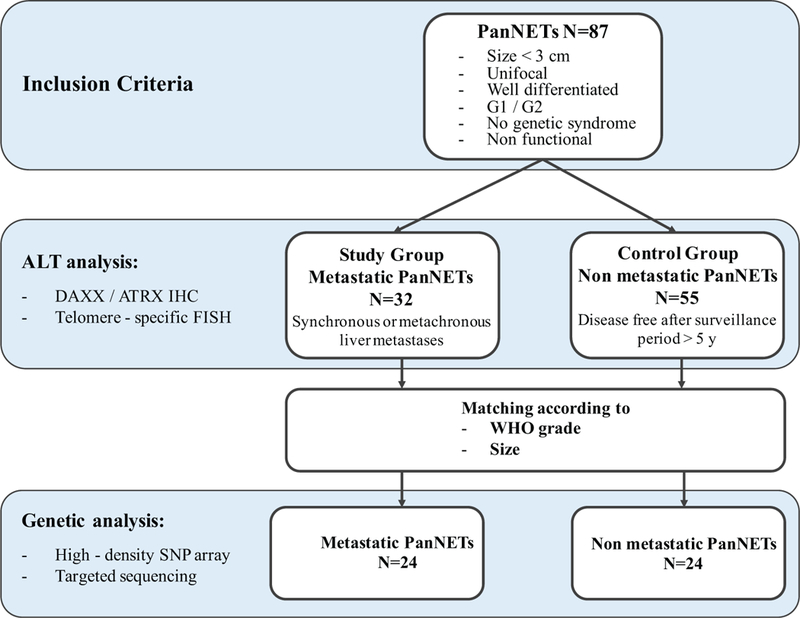
Study workflow.
PanNETs were treated by distal pancreatectomy in 47 patients, pancreatoduodenectomy in 36, and enucleation in 4. There were 54 (62.1%) G1 and 33 (37.9%) G2 tumors (Table 1). Lymphovascular and perineural invasion were present in 19 (21.8%) and 26 (29.9%) tumors, respectively; AJCC stage28 was T1 for 36 (41.4%), T2 for 31 (35.6%), and T3 for 20 (23.0%) tumors. Regional lymph node metastases were present in 29 (33.3%) tumors.
TABLE 1.
Univariate Analysis of Clinical, Pathological, and Biomarker Data Associated With Liver Metastases
| Variables | All Patients n = 87 | ALT n = 85 |
P | Liver Metastases n = 87 |
P | ||
|---|---|---|---|---|---|---|---|
| Negative n = 58 | Positive n = 27 | No n = 55 | Yes n = 32 | ||||
|
| |||||||
| Male, no. (%) | 44 (50.6) | 32 (55.2) | 12 (44.4) | 0.485 | 27 (49.1) | 17 (53.1) | 0.825 |
| Age, mean (SD), yr | 57 (12) | 57 (17) | 57 (19) | 0.884 | 58 (12) | 56 (13) | 0.483 |
| Location of tumor | 0.643 | 0.115 | |||||
| Head and uncinate, no. (%) | 36 (41.4) | 25 (43.1) | 10 (37.0) | 19 (34.5) | 17 (53.1) | ||
| Body and tail, no. (%) | 51 (58.6) | 33 (56.9) | 17 (63) | 15 (46.9) | 36 (65.5) | ||
| Tumor size, median (IQR), cm | 2.2 (1.1) | 2.0 (1.1) | 2.5 (0.9) | 0.070 | 2.0 (1.1) | 2.5 (0.8) | 0.010 |
| Size | 0.098 | 0.023 | |||||
| ≤ cm | 6 (6.9) | 6 (10.3) | 0 (0.0) | 6 (10.9) | 0 (0) | ||
| 1–2 cm | 36 (41.4) | 25 (43.1) | 9 (33.3) | 26 (47.3) | 10 (31.3) | ||
| >2 cm | 45 (51.7) | 27 (46.6) | 18 (66.7) | 23 (41.8) | 22 (68.8) | ||
| Ki67 index %, median (IQR) | 2.0 (3.3) | 1.2 (2.3) | 4.0 (4.3) | 0.001 | 1.0 (2.1) | 4.2 (6.7) | ≤0.001 |
| WHO grade | 0.004 | 0.001 | |||||
| G1, no. (%) | 54 (62.1) | 42 (72.4) | 10 (37.0) | 42 (76.4) | 12(37.5) | ||
| G2, no. (%) | 33 (37.9) | 16 (27.6) | 17 (63.0) | 13 (23.6) | 20 (62.5) | ||
| Lymphovascular invasion, no. (%) | 19 (21.8) | 8 (13.8) | 11 (40.7) | 0.010 | 8 (14.5) | 11 (34.4) | 0.057 |
| Perineural invasion, no. (%) | 26 (29.9) | 10 (17.2) | 15 (55.6) | 0.001 | 10 (18.2) | 16 (50) | 0.003 |
| T stage, no. (%) | 0.210 | 0.215 | |||||
| T1 stage, no. (%) | 36 (41.4) | 26 (44.8) | 10 (37.0) | 26 (47.3) | 10 (31.3) | ||
| T2 stage, no. (%) | 31 (35.6) | 17 (29.3) | 13 (48.1) | 16 (29.1) | 15 (46.9) | ||
| T3 stage, no. (%) | 20 (23.0) | 15 (25.9) | 4 (14.8) | 13 (23.6) | 7 (21.9) | ||
| N stage, no. (%) | 0.007 | ≤0.001 | |||||
| Nx-0 stage, no. (%) | 58 (66.7) | 44 (75.9) | 12 (44.4) | 45 (81.8) | 13 (40.6) | ||
| N1 stage, no. (%) | 29 (33.3) | 14 (24.1) | 15 (55.6) | 10 (18.2) | 19 (59.4) | ||
| Liver metastases, no. (%) | 32 (36.8) | 14 (24.1) | 18 (66.7) | ≤0.001 | |||
| ALT pos, no. (%) | 27/85 (31.8) | 9/53 (17) | 18/32 (56.3) | ≤0.001 | |||
| DAXX neg, no. (%) | 20/84 (23.8) | 1 (1.8) | 19 (70.4) | ≤0.001 | 7/52 (13.5) | 13/32 (40.6) | 0.008 |
| ATRX neg, no. (%) | 6/85 (7.1) | 0 (0) | 6 (22.2) | 0.001 | 3/53 (5.7) | 3/32 (9.4) | 0.668 |
P values in bold represent significant statistical differences.
DAXX/ATRX Immunolabeling and ALT Analysis
Immunolabeling for ATRX and DAXX and telomere-specific FISH analysis to detect ALT-positive tumors was performed on 85 of the 87 cases. For 2 cases, the material available for research purposes was limited and only used in the genomic analyses. Loss of DAXX or ATRX was detected respectively in 20/85 (23.5%, 13 metastatic and nonmetastatic) and 6/85 (7.1%, 3 metastatic and 3 nonmetastatic) tumors; the loss of protein expression in either ATRX or DAXX was mutually exclusive. Twenty-seven of the 85 tumors were ALT-positive (31.8%, 18 metastatic and 9 nonmetastatic). By univariate analysis, ALT was strongly associated with loss of DAXX or ATRX nuclear expression (P < 0.001 and P = 0.001, respectively), Ki-67 index (Fig. 2, P = 0.001) and WHO grade (P = 0.004). In all but 3 cases, results from IHC and ALT assessment were concordant. Discordant cases included 2 ALT-positive cases with retention of DAXX and ATRX protein expression, and 1 case with loss of nuclear DAXX expression without concurrent ALT.
FIGURE 2.
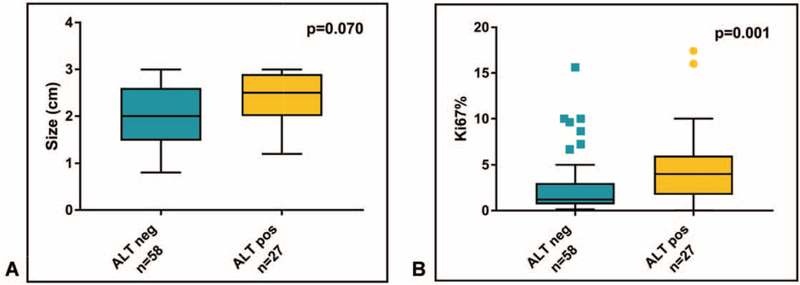
Tumor size (A) and Ki67% (B) stratified by ALT status. Box and whisker plots depict median and 10 to 90 percentile ranges.
Clinicopathological features associated with the development of liver metastases in univariate analysis were larger tumor size as a continuous (P = 0.010) and as a categorical (P = 0.023) variable, Ki-67 index (P < 0.001), WHO grade (P = 0.001), ALT positivity (P < 0.001), and DAXX loss (P = 0.008). On multivariate analysis, only Ki-67 index (OR 1.369; 95% CI 1.121–1.673; P = 0.002), N stage (OR 4.568; 95% CI 1.458–14.312; P = 0.009), and ALT positivity (OR 3.486; 95% CI 1.093–11.115; P = 0.035) were confirmed to be independent risk factors for liver metastases (Table 2).
TABLE 2.
Multivariate Analysis of Clinical, Pathological, and Biomarker Data Associated With Liver Metastases
| Variables | OR | IC 95% | P |
|---|---|---|---|
|
| |||
| Ki-67 | 1.369 | 1.121 to 1.673 | 0.002 |
| N stage (N1) | 4.568 | 1.458 to 14.312 | 0.009 |
| ALT positivity | 3.486 | 1.093 to 11.115 | 0.035 |
Mutational and Copy Number Analysis
Genetic analyses were performed on a subset of these cases, which included 48 primary PanNETs, of which 24 from the metastatic group, from which the extracted DNA passed the quality controls, and 24 from the control group matched for tumor size and WHO grade (Table 1, Supplemental material, http://links.lww.com/SLA/B503).
Targeted next-generation sequencing detected a total of 38 somatic mutations in 7 genes among the 48 tumors, with a range of 0 to 3 mutations per tumor (Fig. 3). MEN1 was mutated in 16 (33%) cases and mTOR pathway genes were mutated in 10 cases, more specifically TSC2 in 9 cases (19%) and PTEN in 1 case (2%). DAXX was mutated in 9 cases (19%) and ATRX in 1 (2%). TP53 and CDKN2A were mutated in 1 case (2%) each. Comparison of mutations with metastatic status showed that MEN1 was mutated in 9 metastatic and 7 nonmetastatic cases, TSC2 in 6 metastatic and 3 nonmetastatic, ATRX and CDKN2A were mutated in 1 nonmetastatic case each, and PTEN and TP53 were mutated in 1 metastatic case each. Strikingly, DAXX was mutated in 8 metastatic cases and 1 nonmetastatic case (P = 0.023).
FIGURE 3.
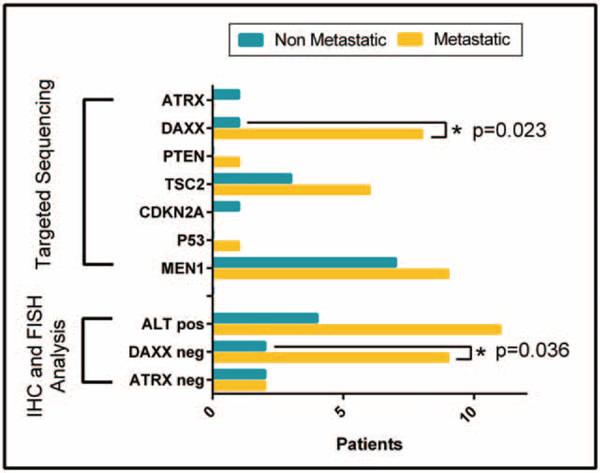
Somatic mutations, ATRX/DAXX protein expression, and ALT status in metastatic and nonmetastatic PanNETs.
Genome-wide SNP array analysis revealed large chromosomal events including recurrent regions of gain and loss. All chromosomal alterations, along with associated genes and noncoding regions, are listed in Supplementary Dataset S1, http://links.lww.com/SLA/B502. Tumors that metastasized were characterized by a higher prevalence of chromosomal gains (1046 regions of gain vs. 661), while nonmetastatic tumors had more chromosomal losses (756 regions of loss vs. 701). Copy number alterations consisted in broad genomic regions generally spanning entire chromosomes (Fig. 4). The most frequent chromosomal gains involved chromosomes 4, 5, 7, 9, 14, 18, and 19 with frequencies ranging from 25% to 50%, while recurrent chromosomal losses were detected in chromosomes 11, 16, and 22 with frequencies ranging from 25% to 75% (Fig. 4). PanNETs with frequent chromosomal gains also exhibited recurrent CN-LOH events, involving the other chromosomes (Supplementary Dataset S2, http://links.lww.com/SLA/B502).
FIGURE 4.
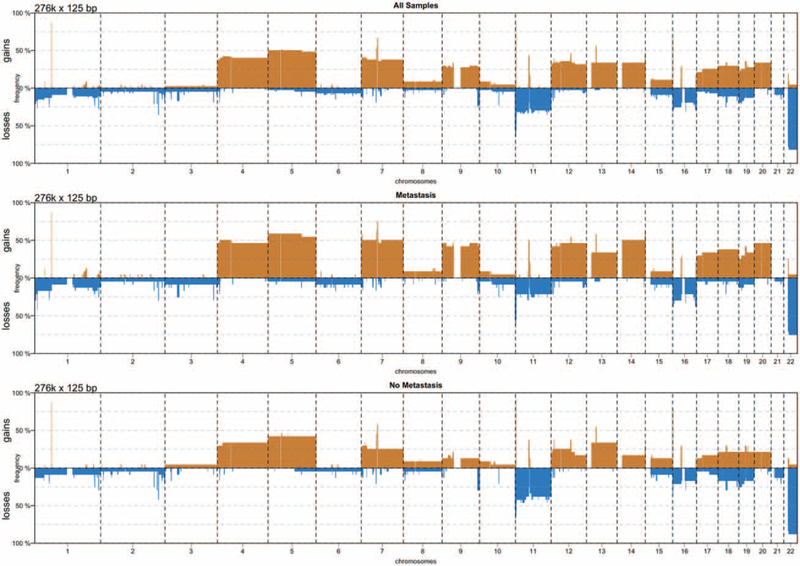
Rate of chromosomal gains (red) and losses (blue) per chromosome in metastatic and nonmetastatic PanNETs.
Chromosome 11q13.1 containing the MEN1 locus was lost in 16 cases (33%), while several regions previously described to be gained in PanNETs were also identified in our study: ERBB2 (chr 17q12) in 12 (25%) cases, PSPN (chr 19p13.3) in 10 (21%), and ULK1 (chr 12q24.33) in 15 (31%). AKT2 (chr 19q13.2), a serine/threonine-protein kinase that inactivates TSC2 and therefore upregulates the mTORC1 complex, was amplified in 12 cases (25%), none of which harboring a TSC2 mutation.
Chromothripsis was detected at chromosome 11 in 4 cases (PanNET2, 11, 27, and 37); at chr 7 in 7 cases (PanNET4, 11, 15, 23, 26, 30, 46); and at chr 9 in PanNET46.
Genomic Subtypes
Cluster analysis of copy number status stratified the tumors into 3 different molecular subtypes on the basis of recurrent chromosomal alterations (Fig. 5): Group 1, characterized by chromosomal gains; Group 2, with limited number of chromosomal events; Group 3, presenting a recurrent pattern of chromosome loss, mainly affecting chromosomes 11 and 22. Integrating copy number alterations, single nucleotide variants, and ALT status revealed distinct molecular and clinical patterns for each subtype. Overall, Group 1 was strongly associated with metastases compared with the other groups combined (chi-squared test, P < 0.03).
FIGURE 5.
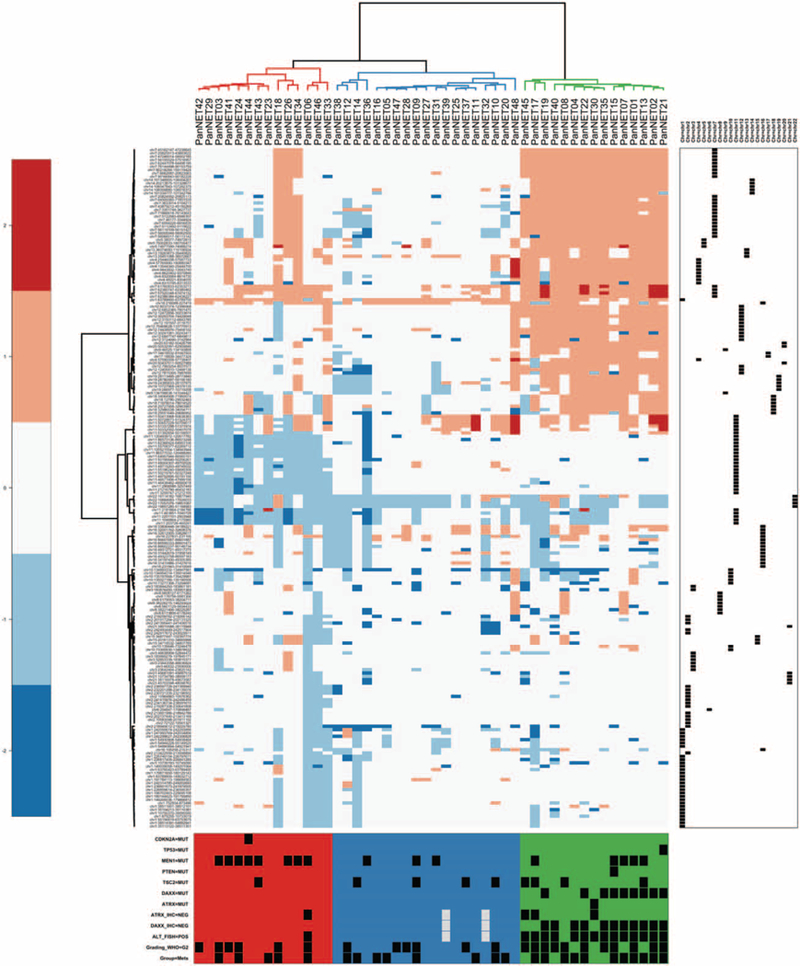
Cluster analysis of the copy number state stratified the tumors into 3 different subtypes characterized by different mutational and clinical patterns (as shown at the bottom). Hierarchical clustering was based on copy number alterations, using the Manhattan distance and the Ward clustering method.
Group 1 included 15 PanNETs that, all except one (PanNET8), were ALT-positive. Strikingly, 14 cases lacked DAXX or ATRX protein expression (respectively in 11 and 3 cases), which was due to mutations in 10 cases (DAXX in 9, ATRX in 1). As expected, all cases that were wild type at the genetic level for ATRX and DAXX retained nuclear protein expression. MEN1 was mutated in 5 cases and mTOR pathway genes in 5 cases (TSC2 in 4, PTEN in 1). The majority of Group 1 tumors were G2(11/15) and 73% (11/15) were metastatic.
Group 2 included 19 cases characterized by limited copy number and mutational events. TSC2 and MEN1 were mutated in 4 (21%) and 3 (16%) cases, respectively. All cases were ALT-negative, wild-type for the ATRX/DAXX genes, and preserved nuclear ATRX/DAXX protein expression. In this group, 42% (8/19) of tumors developed metastases, including 5 G1 and 3 G2 tumors.
Group 3 included 14 cases, all exhibiting recurrent loss of chromosome 11q MEN1 locus and associated with MEN1somatic mutations in 8 (57%) cases. TSC2 and CDKN2A were mutated in 1 case each. Only 1 G2 case that metastasized (PanNET6) was ALT-positive and displayed loss of nuclear ATRX protein expression. In this group, 35% (5/14) of the tumors were metastatic (4 G2 cases and 1G1).
DISCUSSION
PanNETs are characterized by a variable spectrum of clinical behavior.20 Several lines of evidence suggest that most small primary PanNETs are indolent tumors, supporting the idea that surveillance, rather than surgical resection, could be indicated.4,8,29,30 However, a limited subset of small primary PanNETs are aggressive and will metastasize, predominantly to the liver. The discrimination of small PanNETs with a high risk of metastasis from those with low risk has important prognostic and therapeutic implications. First, we evaluated clinicopathological features and several tissue-based biomarkers in a cohort of 87 primary well-differentiated PanNETs that were all < 3cm in size. We observed that high Ki-67 (OR 1.369), N-stage (OR 4.568), and ALT-positivity (OR 3.486) were independently associated with liver metastases. While our cohort was limited to small PanNETs, these findings are similar to other recent studies assessing cohorts encompassing the entire range of tumor sizes.15,16,22
Next, vailing of a recent whole genome sequencing study,10 we investigated a series of small primary PanNETs (<3 cm) using whole genome copy number analysis and targeted deep sequencing of 18 relevant genes to assess the genetic alterations associated with metastatic spread. To this end, we compared the genetic alterations in small primary PanNETs that developed liver metastases with those that were metastases-free after a long postresection follow-up period. We identified 3 different molecular subtypes of small primary PanNETs on the basis of the copy number status, matching those recently described in Scarpa et al,10 which were also characterized by different mutational and clinical features.
In Group 1, a large proportion (11/15, 73%) of tumors were metastatic. The defining molecular features of these tumors were recurrent chromosomal gains, CN-LOH, and a high rate of somatic mutations in the driver genes in our targeted panel. In addition, 14 (of 15) cases were also ALT-positive and all cases displayed loss of either DAXX or ATRX nuclear protein expression and inactivating mutations in either DAXX or ATRX were observed in 10 of 15 cases, indicating the presence of other possible mechanisms of loss, such as complex karyotype rearrangements in ALT-positive PanNETs.10 We observed 3 ALT-positive cases, not included in the genetic analysis, which retained DAXX and ATRX nuclear protein expression. These observations may indicate potential additional biological mechanisms, independent of DAXX and ATRX, that promote ALT in a minority of cases, or by genetic alterations conferring a loss-of-function of either DAXX and ATRX (eg, missense mutations), but not loss of nuclear protein expression.13,15,16,22,31 In this study, we observed an almost perfect correlation between recurrent patterns of chromosomal gains and ALT, suggesting that chromosomal gains may represent an early stage of genomic instability associated with ALT. Previous studies that investigated ALT prevalence in large cohorts of resected PanNETs described a direct correlation between ALT and larger size or higher grade.16,21,22 In our series of small primary PanNETs that metastasized, most of the ALT-positive tumors were G2, suggesting that either ALT increases cell proliferation and metastatic potential or that a high proliferative index in PanNETs increases the probability of acquiring the requisite mutations that promote ALT. Interestingly, TSC2 was mutated in 4 cases (27%), however, all the remnant tumors in Group 1 with wild-type TSC2 showed copy number gains of chromosome 19q (11/15, 73%), suggesting that potential proto-oncogenes functioning in the mTOR pathway may be located in this region. One possible candidate is AKT2, which may upregulate the mTORC1 complex when TSC2 function is retained.32,33
In Group 2, a proportion (8/19, 42%) of tumors were metastatic, despite having a low proliferative index in the majority of cases. This group included the majority of G1 metastatic tumors (5 of 9 G1 metastatic PanNETs, 55%). Interestingly, these tumors were characterized by the presence of few mutations and limited chromosomal alterations.
In Group 3, tumors were characterized by frequent biallelic inactivation of MEN1 and the lowest metastatic potential. In total, PanNETs in our series harbored MEN1 locus alterations in 69% (33/ 48) of cases, including chromosomal loss, CN-LOH or chromotripsis, and MEN1 somatic mutations in 33% (16/48) of cases, which were distributed mainly between Groups 1 and 3. Our data do not provide strong evidence supporting an interaction between MEN1 and the ALT pathway. However, since menin has a critical role in the biology of telomeres by negatively regulating telomerase,34,35 it is tempting to speculate that tumors at an early disease stage with partial or complete loss of menin may use upregulated telomerase for telomere maintenance, and thus do not require the ALT pathway.
While ALT and ATRX/DAXX alterations play pivotal roles in the progression of a subset of PanNETs, metastatic tumors are present also in the other subtypes and the absence of recurrent genetic changes in Groups 2 and 3 suggests that additional biological mechanisms, or genetic alterations not included in our analysis, may contribute to neoplastic progression in these groups. Limitations of our approach were represented by the relatively limited size of the study cohort and by the absence of confirmation of our results in an independent dataset, which are direct consequences of designing our study around rigorous clinical case selection. Our cohort included patients with either synchronous or metachronous liver metastases; we therefore cannot exclude the possibility that additional genetic alterations occurred after the development of metastatic disease in PanNETs with synchronous metastases. However, in our cohort, no substantial genetic differences were observed between PanNETs with metachronous or synchronous metastases (see Supplementary material, http://links.lww.com/SLA/B503), while we observed a high allele frequency for each mutation indicating that these alterations are present in the majority of neoplastic clones of the primary tumor, regardless of the timing of metastasis. This is also consistent with previous reports evaluating ALT in the primary and metastatic lesions from the same patient indicating that molecular alterations that promote ALT occur prior to the development of metastases.16
Recent ENETS consensus guidelines for the management of PanNETs suggest that only asymptomatic G1 tumors < 2cmin size may be suitable for close observation, due to their very low risk of neoplastic progression.8,36 In our cohort, biased for the development of liver metastases, 9 metastatic cases were <2cm. While no specific genetic alterations were observed in these tumors (see Supplementary material, http://links.lww.com/SLA/B503), DAXX was mutated in 4 cases (3 with Ki67 < 5%) highlighting its importance as potential risk-stratification biomarker. We limited our genetic analysis to copy number changes and to targeting driver genes frequently mutated in PanNETs. We did not include analysis of other genes, such as those involved in chromatin remodeling (mutated in approximately 5% of PanNETs), or those containing pathogenic germline variants thought to have a role in the development of up to 17% of sporadic PanNETs.10 Thus, we cannot exclude that such infrequently altered genes also play some role in metastasis of small PanNETs.
In conclusion, we characterize the biological variability among morphologically similar well-differentiated PanNETs. We identified subtypes of small PanNETs characterized by distinct metastatic potential on the basis of integrated genetic and immuno-staining expression data. ALT-positive tumors have the highest prevalence of metastases suggesting that ALT should be considered a potential biomarker of an aggressive phenotype among small tumors and employed for risk stratification. Importantly, NGS with targeted panels can be performed on small samples37 and ALT can be accurately be detected on endoscopic ultrasound-guided fine needle aspiration.31 Thus, we propose that, the concomitant integrated evaluation of targeted sequencing, Ki-67, and ALT on endoscopic ultrasound-guided fine needle aspiration specimens could guide the choice of the surgical treatment, as well as the enrollment into surveillance programs for small and asymptomatic tumors.
Supplementary Material
Acknowledgments
This study was supported by the “Department of Surgery Pancreatic Tumor Research Fund—PI: C.L.W.” Part of the study was supported by the NIH grant P30CA006973, NIH/NCI P50 CA62924; NIH/NIDDK K08 DK107781; Sol Goldman Pancreatic Cancer Research Center; Buffone Family Gastrointestinal Cancer Research Fund; Kaya Tuncer Career Development Award in Gastrointestinal Cancer Prevention; AGA-Bernard Lee Schwartz Foundation Research Scholar Award in Pancreatic Cancer; Sidney Kimmel Foundation for Cancer Research Kimmel Scholar Award; AACR-Incyte Corporation Career Development Award for Pancreatic Cancer Research; Rolfe Pancreatic Cancer Foundation; Joseph C Monastra Foundation; The Gerald O Mann Charitable Foundation (Harriet and Allan Wulfstat, Trustees); Sigma Beta Sorority; the Neuroendocrine Tumor Research Foundation, the Margie & Robert E. Petersen and by the 2016 Basic/Translational Science Investigator Award to C. Heaphy from the North American Neuroendocrine Tumor Society; Fondazione Italiana Malattie Pancreas (FIMP-Ministero Salute, CUP_J33G13000210001); Associazione Italiana Ricerca Cancro (grant 12182); FP7 European Community Grant Cam-Pac (n: 602783); Italian Cancer Genome Project—Ministry of University (FIRB RBAP10AHJB). M.N. and L.A.B. were supported by the Dutch Digestive Foundation (CDG 14–02).
Conceived and designed the study: A.P., J.Y., L.D.W., and C.L.W.; performed the experiments: J.Y., A.P., C.M.H.; acquisition of data: A.P., J.Y., L.M., M.N., C.L., A.P., R.F.d.W., L.A.B., N.R., A.J., S.G., R.G., R.S., C.B., J.H., M.J.W., J.L.C., J.A.O., R.H.H., R.T.L., A.S., C.M.H., L.D.W., and C.L.W.; analysis andinterpretation ofdata: A.P., J.Y., L.M., C.L., A.S., C.M.H., L.D.W., and C.L.W.; statistical analysis: A.P., J.Y., L.M., A.P., and C.M.H.; drafted the manuscript: A.P., J.Y., L.M., A.S., C.M.H., L.D.W., and C.L.W.; revised the manuscript and agreed with the manuscript’s results and conclusions: all the authors; obtained funding: C.L.W., A.P., L.M., A.S., C.M.H., M.N., and L.A.B.; study supervision: C.L.W.
Footnotes
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s Web site (www.annalsofsurgery.com).
The authors report no conflicts of interest.
REFERENCES
- 1.Yao JC, Hassan M, Phan A, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26:3063–3072. [DOI] [PubMed] [Google Scholar]
- 2.Grimelius L, Hultquist GT, Stenkvist B. Cytological differentiation of asymptomatic pancreatic islet cell tumours in autopsy material. Virchows Arch A Pathol Anat Histol. 1975;365:275–288. [DOI] [PubMed] [Google Scholar]
- 3.Kimura W, Kuroda A, Morioka Y. Clinical pathology of endocrine tumors of the pancreas. Analysis of autopsy cases. Dig Dis Sci. 1991;36:933–942. [DOI] [PubMed] [Google Scholar]
- 4.Kuo EJ, Salem RR. Population-level analysis of pancreatic neuroendocrine tumors 2 cm or less in size. Ann Surg Oncol. 2013;20:2815–2821. [DOI] [PubMed] [Google Scholar]
- 5.Partelli S, Gaujoux S, Boninsegna L, et al. Pattern and clinical predictors of lymph node involvement in nonfunctioning pancreatic neuroendocrine tumors (NF-PanNETs). JAMA Surg. 2013;148:932–939. [DOI] [PubMed] [Google Scholar]
- 6.Bettini R, Partelli S, Boninsegna L, et al. Tumor size correlates with malignancy in nonfunctioning pancreatic endocrine tumor. Surgery. 2011;150:75–82. [DOI] [PubMed] [Google Scholar]
- 7.Vagefi PA, Razo O, Deshpande V, et al. Evolving patterns in the detection and outcomes of pancreatic neuroendocrine neoplasms: the Massachusetts General Hospital experience from 1977 to 2005. Arch Surg. 2007;142:347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Falconi M, Eriksson B, Kaltsas G, et al. ENETS consensus guidelines update for the management of patients with functional pancreatic neuroendocrine tumors and non-functional pancreatic neuroendocrine tumors. Neuroendocrinology. 2016;103:153–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haynes AB, Deshpande V, Ingkakul T, et al. Implications of incidentally discovered, nonfunctioning pancreatic endocrine tumors: short-term and long-term patient outcomes. Arch Surg. 2011;146:534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scarpa A, Chang DK, Nones K, et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature. 2017;543:65–71. [DOI] [PubMed] [Google Scholar]
- 11.Missiaglia E, Dalai I, Barbi S, et al. Pancreatic endocrine tumors: expression profiling evidences a role for AKT-mTOR pathway. J Clin Oncol. 2010;28:245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiao Y, Shi C, Edil BH, et al. DAXX/ATRX, MEN1, and mTORpathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331: 1199–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heaphy CM, de Wilde RF, Jiao Y, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011;333:425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heaphy CM, Subhawong AP, Hong SM, et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am J Pathol. 2011;179:1608–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marinoni I, Kurrer AS, Vassella E, et al. Loss of DAXX and ATRX are associated with chromosome instability and reduced survival of patients with pancreatic neuroendocrine tumors. Gastroenterology. 2014;146:453.e5–460.e5. [DOI] [PubMed] [Google Scholar]
- 16.Singhi AD, Liu TC, Roncaioli JL, et al. Alternative lengthening of telomeres and loss of DAXX/ATRX expression predicts metastatic disease and poor survival in patients with pancreatic neuroendocrine tumors. Clin Cancer Res. 2017;23:600–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laplante M Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yao JC, Pavel M, Lombard-Bohas C, et al. Everolimus for the treatment of advanced pancreatic neuroendocrine tumors: overall survival and circulating biomarkers from the randomized, Phase III RADIANT-3 Study. J Clin Oncol. 2016;34:3906–3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lloyd RVOR, Kloppel G, Rosai J. WHO Classification of Tumours of Endocrine Organs, 4th ed. Lyon: IARC Press; 2017. [Google Scholar]
- 21.de Wilde RF, Heaphy CM, Maitra A, et al. Loss of ATRX or DAXX expression and concomitant acquisition of the alternative lengthening of telomeres phenotype are late events in a small subset of MEN-1 syndrome pancreatic neuroendocrine tumors. Mod Pathol. 2012;25:1033–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim JY, Brosnan-Cashman JA, An S, et al. Alternative lengthening of telomeres in primary pancreatic neuroendocrine tumors is associated with aggressive clinical behavior and poor survival. Clin Cancer Res. 2017;23: 1598–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pea A, Yu J, Rezaee N, et al. Targeted DNA sequencing reveals patterns of local progression in the pancreatic remnant following resection of intraductal papillary mucinous neoplasm (IPMN) of the pancreas. Ann Surg. 2017;266: 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luchini C, Pea A, Lionheart G, et al. Pancreatic undifferentiated carcinoma with osteoclast-like giant cells is genetically similar to, but clinically distinct from, conventional ductal adenocarcinoma. J Pathol. 2017;243:148–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diskin SJ, Li M, Hou C, et al. Adjustment of genomic waves in signal intensities from whole-genome SNP genotyping platforms. Nucleic Acids Res. 2008;36:e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song S, Nones K, Miller D, et al. qpure: A tool to estimate tumor cellularity from genome-wide single-nucleotide polymorphism profiles. PLoS One. 2012;7:e45835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang J, Liu J, Ouyang L, et al. CTLPScanner: a web server for chromothripsis-like pattern detection. Nucleic Acids Res. 2016;44:W252–W258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amin MB, Edge SB, Greene FL, et al. , eds. AJCC Cancer Staging Manual, 8th ed. New York; Springer; 2017. [Google Scholar]
- 29.Sadot E, Reidy-Lagunes DL, Tang LH, et al. Observation versus resection for small asymptomatic pancreatic neuroendocrine tumors: a matched case- control study. Ann Surg Oncol. 2016;23:1361–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaujoux S, Partelli S, Maire F, et al. Observational study of natural history of small sporadic nonfunctioning pancreatic neuroendocrine tumors. J Clin Endocrinol Metab. 2013;98:4784–4789. [DOI] [PubMed] [Google Scholar]
- 31.VandenBussche CJ, Allison DB, Graham MK, et al. Alternative lengthening of telomeres and ATRX/DAXX loss can be reliably detected in FNAs of pancreatic neuroendocrine tumors. Cancer Cytopathol. 2017;125:544–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005;24:7455–7464. [DOI] [PubMed] [Google Scholar]
- 33.Cheng JQ, Ruggeri B, Klein WM, et al. Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc Natl Acad Sci USA. 1996;93:3636–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin SY, Elledge SJ. Multiple tumor suppressor pathways negatively regulate telomerase. Cell. 2003;113:881–889. [DOI] [PubMed] [Google Scholar]
- 35.Mirabello L, Yu K, Kraft P, et al. The association of telomere length and genetic variation in telomere biology genes. Hum Mutat. 2010;31:1050–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Falconi M, Bartsch DK, Eriksson B, et al. ENETS Consensus Guidelines for the management of patients with digestive neuroendocrine neoplasms of the digestive system: well-differentiated pancreatic non-functioning tumors. Neuroendocrinology. 2012;95:120–134. [DOI] [PubMed] [Google Scholar]
- 37.Gleeson FC, Voss JS, Kipp BR, et al. Assessment of pancreatic neuroendocrine tumor cytologic genotype diversity to guide personalized medicine using a custom gastroenteropancreatic next-generation sequencing panel. Oncotarget. 2017;8:93464–93475. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.