

Abstract
Background
Major depressive disorder (MDD) is a serious mental health problem in modern society, which is difficult to identify and diagnose in the early stages. Despite strong evidence supporting the heritability of MDD, progresses in large‐scale and individual genetic studies remain preliminary.
Methods
In this study, a multi‐data source‐based prioritization (MDSP) method was proposed, and an appropriate threshold was determined for the optimization of depression‐related genes (DEPgenes). Analyses on Gene Ontology biological processes, KEGG pathway and the specific pathway crosstalk network were further proposed.
Results
A total of 143 DEPgenes were identified and the MDD‐specific network was constructed for the pathogenesis investigation and therapeutic methods development of MDD. Comparing with existing research strategies, the genetic optimization and analysis results were confirmed to be reliable. Finally, the pathway enrichment and crosstalk analyses revealed two unique pathway interaction modules that were significantly enriched with MDD genes. The related core pathways of neuroactive ligand‐receptor interaction and dopaminergic synapse supported the neuropathology hypothesis of MDD. And the pathways of serotonergic synapse and morphine addiction indicated the mechanism of drug addiction caused by serotonin used in the treatment.
Conclusions
This work provided a reference for the study of MDD, although future validation by extensive experimentation is still required.
Keywords: gene ontology, KEGG pathway, major depressive disorder, multi‐data‐source based prioritization
1. INTRODUCTION
Major depressive disorder (MDD) is a severe psychiatric disease with high morbidity and mortality worldwide (Culpepper, Lam, & McIntyre, 2017). This growing recognition of the public health burden has led to the development of depression detection and treatment. However, novel interventions of depression are still hindered by a limited understanding of the neurobiological mechanisms (Bayes & Parker, 2018). The efforts to clarify this biology through common or rare variant association studies seemed to be unsuccessful with the lack of distinct understanding of heterogeneity and absence of a biological gold‐standard diagnosis (Krystal & State, 2014). Nowadays, strong shreds of heritability evidence of mental diseases have been revealed (Alnaes et al., 2018; Pain et al., 2018), which attracted the studies on the generation of numerous genetic and genomic datasets in MDD studies.
During the past decade, rapid advances in high throughput technologies have helped investigators, aiming to uncover disease causal genes and their actions in complex diseases. Specifically, in psychiatric genetics, there have been numerous datasets from different platforms or sources such as association studies, including genome‐wide association studies, genome‐wide linkage scans, microarray gene expression, and copy number variation (Michaelson, 2017). Large‐scale and individual genetic studies revealed various polymorphisms and overexpression of certain genes in patients presenting with depressive symptoms (Lacerda‐Pinheiro et al., 2014; Milanesi et al., 2015). Zhang's group has found that, increased 5‐HT1A expression inversely correlated with 5‐HT activity via a negative feedback mechanism (Zhang et al., 2014). Moreover, HPA axis hyperactivity was reported as a trigger of MDD due to findings of GR and mineralocorticoid receptor dysfunction in depressed patients (Pariante & Lightman, 2008). However, a pervasive limitation in the existing research is the inherent heterogeneity in MDD studies, which impacts the validity of biomarker data (Young et al., 2016). Thus it is still necessary to simplify these depression‐related candidate genes to an optimal set for the subsequent biological experiments. Moreover, the incompletion of information resources used in existing calculation and the fixed screening threshold of corresponding online tools also result in arbitrarily preferred results and lower reliability.
In this study, gene information from multiple sources (including OMIM, Phenolyzer, GeneCards and GLAD4U) were integrated and analyzed for MDD. A multi‐data‐source based prioritization (MDSP) was proposed and an appropriate threshold was determined for the optimization of depression‐related genes (DEPgenes). Finally, the acquired genes which were significantly related to depression (DEPgenes) were verified by the receiver operating characteristic (ROC) curve and functional and pathway enrichment analysis. Our work demonstrated a practical framework for complex disease candidate gene analysis, which is of great significance for the comprehensive functional assessment of optimized pathogenic genes.
2. MATERIALS AND METHODS
2.1. MDD candidate genes and optimizing process
OMIM (www.omim.org), which provides vast repositories of rich clinical and genetic knowledge, was considered as a core gene database in this study. For association studies, the susceptibility genes were retrieved by searching all human genetic association studies deposited in Phenolyzer (phenolyzer.usc.edu), GeneCards (www.genecards.org) and GLAD4U (bioinfo.vanderbilt.edu/glad4u), which used as training gene categories. However, the background information of the dataset‐related patients is not provided in the database. For all the genes collected, genes presented in a certain training category were assigned a score of 1 point; otherwise, 0 was assigned. Thus, a gene could be represented by a vector of three elements, with each element being 1 or 0. When a gene showed up in all the training categories, all the elements in the vector would be 1's; on the other hand, a gene had at least one element being 1. For each training category, a weight was assigned to measure the category's reliability. A combined score derived from the category‐specific weight and gene score in the corresponding category was adopted to measure the correlation between a gene and the phenotype. All the candidate genes were ranked by their combined scores computed from their scores corresponding to the categories and the optimal weights. The combined scores were calculated by equation 1:
| (1) |
where i was the training category index, N = 3, Wi was the corresponding weight of categoryi, and Scorei and was equal to 1 when a gene showed up in categoryi; otherwise, Scorei=0.
The combined score of a gene depends on its score from each training category and the corresponding weight value. In order to prioritize the genes collected so that the genes more likely correlated with MDD can be ranked higher in the list, a suitable weight for each training category needs to be determined. In this study, the following procedure was adopted:
Randomly selecting weight value between 0 and 1.0 for each training category and normalizing the weight matrix (consisted of the three weights) to have a sum of 1;
Calculating the combined score S for all genes by equation 1 and ranking all genes according to their combined scores;
Calculating ratio R: calculating the proportion k of core genes known to be related to MDD selected from OMIM in the top 3% of all candidate genes and R = k/23;
Reallocate values into the weight matrix and keeping the weight matrix to have a sum of 1.
Calculating ratio R after obtaining the new score S and ranking of all candidate genes;
Repeating steps 2–5 until no larger R can be found, and then the weight matrix obtained is the optimal weight matrix.
2.2. Evaluation of genetic optimizing results
The ROC curve was employed to assess the discrimination capability of the classifiers proposed in this study. ROC curves represent the performance of a classifier without taking into consideration class distribution or error overheads. And the classification success is then calculated by area under ROC curve (AUC) (Wray, Yang, Goddard, & Visscher, 2010). When the ROC curve deviated from the diagonal, i.e. the AUC value was close to 1, the verified method was evaluated as better reliability.
2.3. Functional and pathway enrichment tests
The relation of the prioritized genes with MDD was evaluated by analyzing the Gene Ontology (GO) biological processes or biochemical pathways enriched in these genes. The Database for Annotation, Visualization, Integration and Discovery (DAVID, david-d.ncifcrf.gov) was used for GO term enrichment analysis, followed by the correction of multiple testing using the Benjamini & Hochberg (BH) method. And the biological processes (BP) term was considered as significantly enriched with a cutoff of PBH < 0.01. In addition, KEGG pathway analysis was performed by WebGestalt online tool (www.webgestalt.org) (Wang, Vasaikar, Shi, Greer, & Zhang, 2017) and PBH < 0.05 was set as the cutoff criterion.
2.4. Pathway crosstalk
The pathway crosstalk analysis was performed to further investigate the interactions of significantly enriched pathways of optimized MDD‐related genes. Two pathways are considered to crosstalk if they share a proportion of DEPgenes. Two measurements were introduced to computationally indicate the overlap of a pair of pathways: Overlap coefficient (OC) = and Jaccard coefficient (JC) = , where A and B denote the number of DEPgenes in the two pathways, respectively. The averages of OC and JC were calculated to reflect the overlap degree between pairs of pathways. And the crosstalk results were visualized by Cytoscape (Uzoma et al., 2018).
2.5. Depression‐specific network and cluster analysis by Cytoscape
To construct a depression‐specific network, the DEPgenes were imported into the STRING (string-db.org). The information on gene interaction was extracted and used to form a specific network. Module cluster analysis of the depression‐specific network was performed using the MCODE plug‐in in Cytoscape. Besides, to verify the nonrandomness of the obtained depression‐specific network, the following verification steps were performed:
Random network generation: generating 1,000 random networks which had the same node and interaction numbers as the depression‐specific network using Erdos‐Renyi model in an igraph package of R software;
Calculating the average shortest path distance (SPD) and average clustering coefficient (CC) of all the random networks, respectively.
Statistics: Calculating the number of the random networks that have shorter SPD than MDD‐specific network and the number of random network that have higher CC than MDD‐specific network, which denoted as ND and NC, respectively.
Calculating the experience p‐value: PD = ND/1,000 and PC = NC/1,000, which should reflect the significance of nonrandomness of MDD‐specific network.
3. RESULTS
3.1. Collection of MDD candidate and core genes
A total of 23 genes were collected from OMIM (Table 1), which were regarded as core genes. Besides, 14,144 genes from Phenolyzer, 5,358 genes from GeneCards and 149 genes from GLAD4U were collected regarded as MDD candidate genes. These genes were collected from multi‐source, and each gene is showed up in a certain source in Figure 1a. MDSP was proposed and an appropriate threshold was determined for the optimization of MDD candidate genes. As the optimization algorithm flow chart of MDD candidate genes shown in Figure 1b, when a gene shows up in a certain training category, a score of 1 point is assigned; otherwise, 0 is assigned. Each of the four categories has a weight value, which is determined by the optimization algorithm as described in the "Material and Methods" section. The genes are ranked by their combined scores computed from scores of three training categories and their weights. Genes are ranked and prioritized by their combined scores, and further analysis is performed for the selected genes.
Table 1.
Major depressive disorder core genes collected from OMIM
| Gene symbol | MIM ID | Gene symbol | MIM ID |
|---|---|---|---|
| MDD1 | 608516 | DRD4 | 608516 |
| MDD2 | 608516 | TPH1 | 608516 |
| FKBP5 | 608516 | HTR2C | 608516 |
| TPH2 | 608516 | HTR1D | 608516 |
| HTR2A | 608516 | HTR1B | 608516 |
| CALCA | 608516 | MAOB | 608516 |
| DUSP1 | 608516 | SLC6A4 | 608516 |
| MTHFR | 608516 | BCR | 608516 |
| CREB1 | 608516 | PER3 | 608516 |
| HSP90AA1 | 608516 | APAF1 | 608520 |
| CHRM2 | 608516 | SLC6A15 | 608520 |
| TOR1A | 608516 |
Figure 1.
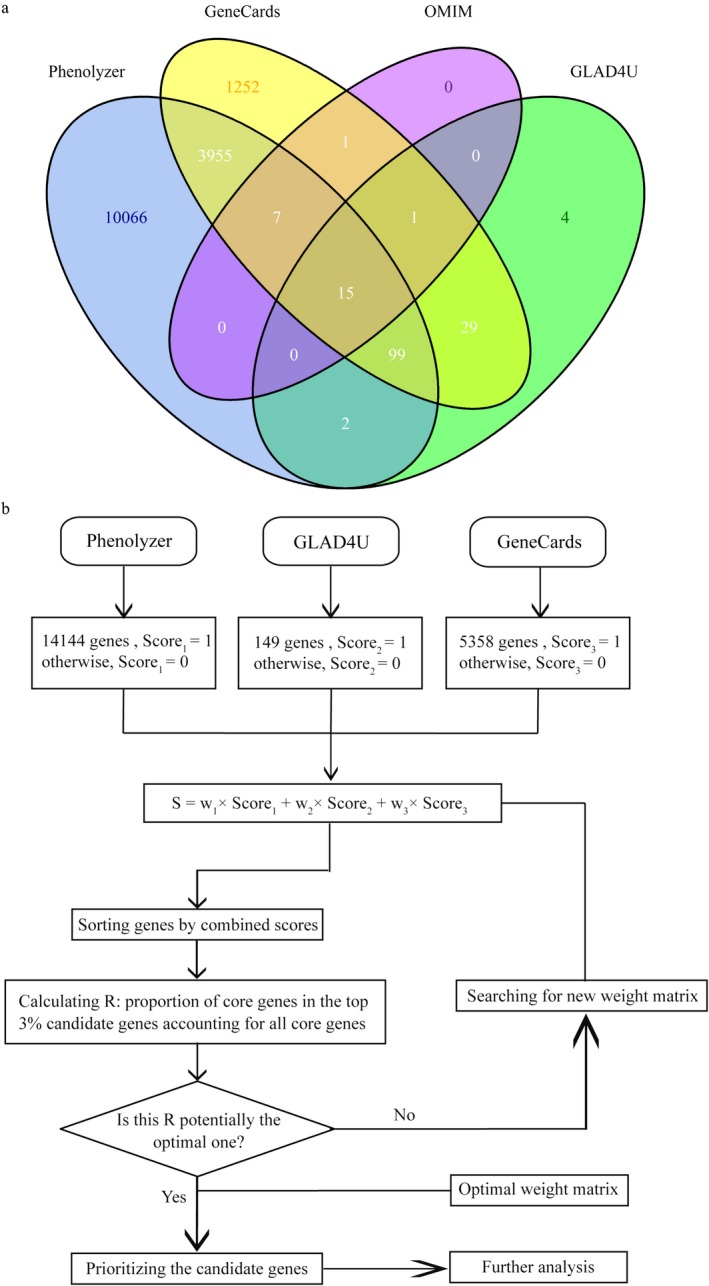
Overview of gene prioritization method. (a) Venn diagram of major depressive disorder (MDD)‐related candidate genes collected from different sources; (b) The flow chart for MDD‐related genes prioritization
3.2. Optimization and evaluation of MDD candidate genes
The combined scores of all candidate genes were calculated based on the optimal weight matrix and the candidate gene score in each source. The MDD candidate genes were ranked according to the combined scores. The gene list and the combined scores distribution of core genes and all candidate genes optimized by our process are shown in Figure 2a. Most of the core genes with higher combined scores appeared in front of the sorted list, and only several appeared in the posterior position, indicating that the distribution of the candidate genes' combined scores was in line with our expectations.
Figure 2.
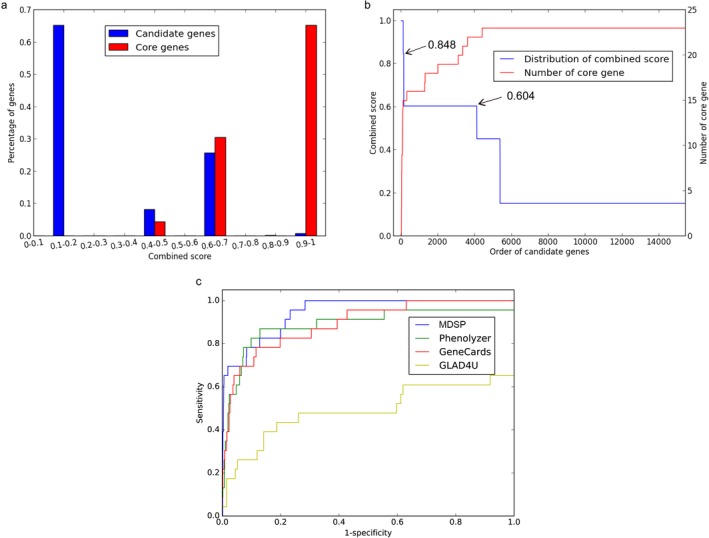
Optimization and evaluation of MDD candidate genes. (a) Distribution of the combined scores of all candidate genes and the core genes. The percentage of each histogram bin is measured by the genes with scores falling in the bin divided by the total number of candidate genes or the number of the core genes; (b) The distribution of the combined scores of the candidate genes. The genes are ranked by their combined scores. The x‐axis is the order of the candidate genes. The y‐axis on the left side is the combined score of the candidate genes, and the y‐axis on the right side is the number of core genes with higher combined score. (c) ROC curve of different prioritization tools. MDD: major depressive disorder; ROC: receiver operating characteristic
From Figure 2b, it was inferred that, the score drops quickly from 1.0 to about 0.848 and then drops to about 0.604; after that, the combined scores decrease slowly. Such a distribution indicated that a relatively small number of genes have higher combined scores, while the majority of genes has moderate or small scores. With a threshold of 0.848, 65.2% of the core genes (15/23) were contained. Although with a threshold of 0.604, 95.7% of the core genes (22/23) could be contained, the number of selected candidate genes would also dramatically increase to 4,105. As the smaller the comprehensive score was, the higher the false positive rate of the prioritized gene was, 143 DEPgenes were identified with a threshold of 0.848 (Table S1).
Finally, the reliability of our method for prioritizing MDD candidate genes was compared with Phenolyzer, GeneCards and GLAD4U through ROC curve. As a result, AUC of MDSP (0.944) is the largest followed by GeneCards (0.893) and Phenolyzer (0.888), and GLAD4U had the smallest AUC value (0.490), which indicated that the results of the MDSP optimization were the best.
3.3. GO enrichment analysis
To explore specific functional features of the 143 DEPgenes, GO enrichment analysis was performed using DAVID. Seventy‐two biological processes (BP terms) which related to synaptic transmission, neurodevelopment and drug reaction were significantly enriched in DEPgenes (Table 2). The GO terms related to synaptic transmission included synaptic transmission, regulation of synaptic transmission, positive regulation of synaptic transmission and negative regulation of synaptic transmission. The GO terms related to nerve signal transduction included second‐messenger‐mediated signaling, regulation of transmission of nerve impulse, cell surface receptor linked signal transduction, G‐protein coupled receptor protein signaling pathway and glutamate signaling pathway. The GO terms related to neurotransmitter, such as regulation of neurotransmitter levels, regulation of neurotransmitter transport, regulation of neurotransmitter uptake, regulation of catecholamine secretion, regulation of dopamine secretion and regulation of glutamate secretion, while that related to drug reaction (response to tropane, response to cocaine, response to amphetamine and response to histamine) and learning or memory were also significantly enriched.
Table 2.
Significantly enriched BP terms of the 143 DEPgenes
| GO terms | Biological process | No. of genes | p‐value | PBH |
|---|---|---|---|---|
| GO:0007268 | Synaptic transmission | 36 | 1.24E‐32 | 1.02E‐29 |
| GO:0019932 | Second‐messenger‐mediated signaling | 22 | 5.46E‐17 | 2.25E‐14 |
| GO:0030808 | Regulation of nucleotide biosynthetic process | 16 | 4.37E‐15 | 1.19E‐12 |
| GO:0050804 | Regulation of synaptic transmission | 17 | 5.37E‐15 | 1.10E‐12 |
| GO:0006140 | Regulation of nucleotide metabolic process | 16 | 9.79E‐15 | 1.61E‐12 |
| GO:0051969 | Regulation of transmission of nerve impulse | 17 | 1.89E‐14 | 2.60E‐12 |
| GO:0031644 | Regulation of neurological system process | 17 | 3.56E‐14 | 4.21E‐12 |
| GO:0007166 | Cell surface receptor linked signal transduction | 46 | 8.23E‐14 | 8.49E‐12 |
| GO:0045761 | Regulation of adenylate cyclase activity | 14 | 3.60E‐13 | 3.30E‐11 |
| GO:0007186 | G‐protein coupled receptor protein signaling pathway | 33 | 2.50E‐11 | 2.06E‐09 |
| GO:0051046 | Regulation of secretion | 16 | 3.56E‐11 | 2.67E‐09 |
| GO:0001505 | Regulation of neurotransmitter levels | 11 | 8.09E‐11 | 5.57E‐09 |
| GO:0051952 | Regulation of amine transport | 9 | 1.12E‐10 | 7.10E‐09 |
| GO:0031280 | Negative regulation of cyclase activity | 10 | 3.17E‐10 | 1.87E‐08 |
| GO:0051350 | Negative regulation of lyase activity | 10 | 3.17E‐10 | 1.87E‐08 |
| GO:0007611 | Learning or memory | 12 | 8.15E‐10 | 4.49E‐08 |
| GO:0051050 | Positive regulation of transport | 15 | 1.55E‐09 | 8.01E‐08 |
| GO:0014073 | Response to tropane | 7 | 4.54E‐09 | 2.21E‐07 |
| GO:0042220 | Response to cocaine | 7 | 4.54E‐09 | 2.21E‐07 |
| GO:0051940 | Regulation of catecholamine uptake during transmission of nerve impulse | 5 | 1.66E‐08 | 7.64E‐07 |
| GO:0051588 | Regulation of neurotransmitter transport | 7 | 3.69E‐08 | 1.61E‐06 |
| GO:0051580 | Regulation of neurotransmitter uptake | 5 | 4.96E‐08 | 2.05E‐06 |
| GO:0007242 | Intracellular signaling cascade | 29 | 1.45E‐07 | 5.70E‐06 |
| GO:0009712 | Catechol metabolic process | 7 | 2.05E‐07 | 7.70E‐06 |
| GO:0006584 | Catecholamine metabolic process | 7 | 2.05E‐07 | 7.70E‐06 |
| GO:0006576 | Biogenic amine metabolic process | 9 | 7.77E‐07 | 2.79E‐05 |
| GO:0014059 | Regulation of dopamine secretion | 5 | 1.06E‐06 | 3.65E‐05 |
| GO:0051047 | Positive regulation of secretion | 9 | 1.89E‐06 | 6.25E‐05 |
| GO:0051954 | Positive regulation of amine transport | 5 | 3.16E‐06 | 1.00E‐04 |
| GO:0030003 | Cellular cation homeostasis | 12 | 3.99E‐06 | 1.22E‐04 |
| GO:0001662 | Behavioral fear response | 5 | 4.28E‐06 | 1.26E‐04 |
| GO:0031281 | Positive regulation of cyclase activity | 7 | 4.80E‐06 | 1.37E‐04 |
| GO:0006939 | Smooth muscle contraction | 6 | 4.96E‐06 | 1.37E‐04 |
| GO:0001964 | Startle response | 5 | 5.68E‐06 | 1.51E‐04 |
| GO:0050806 | Positive regulation of synaptic transmission | 6 | 5.78E‐06 | 1.49E‐04 |
| GO:0051349 | Positive regulation of lyase activity | 7 | 5.89E‐06 | 1.47E‐04 |
| GO:0008306 | Associative learning | 5 | 7.38E‐06 | 1.79E‐04 |
| GO:0015844 | Monoamine transport | 5 | 7.38E‐06 | 1.79E‐04 |
| GO:0051971 | Positive regulation of transmission of nerve impulse | 6 | 8.89E‐06 | 2.10E‐04 |
| GO:0043269 | Regulation of ion transport | 8 | 1.10E‐05 | 2.52E‐04 |
| GO:0014075 | Response to amine stimulus | 6 | 1.16E‐05 | 2.59E‐04 |
| GO:0031646 | Positive regulation of neurological system process | 6 | 1.16E‐05 | 2.59E‐04 |
| GO:0008217 | Regulation of blood pressure | 8 | 1.17E‐05 | 2.55E‐04 |
| GO:0050433 | Regulation of catecholamine secretion | 5 | 1.19E‐05 | 2.51E‐04 |
| GO:0001975 | Response to amphetamine | 5 | 1.19E‐05 | 2.51E‐04 |
| GO:0050805 | Negative regulation of synaptic transmission | 5 | 1.81E‐05 | 3.74E‐04 |
| GO:0044106 | Cellular amine metabolic process | 12 | 2.24E‐05 | 4.52E‐04 |
| GO:0042053 | Regulation of dopamine metabolic process | 4 | 2.44E‐05 | 4.79E‐04 |
| GO:0055082 | Cellular chemical homeostasis | 13 | 3.46E‐05 | 6.65E‐04 |
| GO:0042069 | Regulation of catecholamine metabolic process | 4 | 3.63E‐05 | 6.82E‐04 |
| GO:0010959 | Regulation of metal ion transport | 7 | 3.70E‐05 | 6.79E‐04 |
| GO:0007215 | Glutamate signaling pathway | 5 | 3.74E‐05 | 6.71E‐04 |
| GO:0051970 | Negative regulation of transmission of nerve impulse | 5 | 3.74E‐05 | 6.71E‐04 |
| GO:0060134 | Prepulse inhibition | 4 | 5.16E‐05 | 9.06E‐04 |
| GO:0060191 | Regulation of lipase activity | 7 | 5.55E‐05 | 9.54E‐04 |
| GO:0031645 | Negative regulation of neurological system process | 5 | 5.95E‐05 | 1.00E‐03 |
| GO:0050801 | Ion homeostasis | 13 | 7.05E‐05 | 1.16E‐03 |
| GO:0032309 | Icosanoid secretion | 4 | 7.06E‐05 | 1.14E‐03 |
| GO:0050482 | Arachidonic acid secretion | 4 | 7.06E‐05 | 1.14E‐03 |
| GO:0007632 | Visual behavior | 5 | 8.98E‐05 | 1.43E‐03 |
| GO:0014048 | Regulation of glutamate secretion | 4 | 1.21E‐04 | 1.88E‐03 |
| GO:0033238 | Regulation of cellular amine metabolic process | 4 | 1.21E‐04 | 1.88E‐03 |
| GO:0034776 | Response to histamine | 3 | 1.76E‐04 | 2.70E‐03 |
| GO:0046717 | Acid secretion | 4 | 3.35E‐04 | 5.03E‐03 |
| GO:0048699 | Generation of neurons | 14 | 3.51E‐04 | 5.17E‐03 |
| GO:0015909 | Long‐chain fatty acid transport | 4 | 5.38E‐04 | 7.76E‐03 |
| GO:0019614 | Catechol catabolic process | 3 | 5.82E‐04 | 8.26E‐03 |
| GO:0015718 | Monocarboxylic acid transport | 5 | 5.87E‐04 | 8.19E‐03 |
| GO:0032102 | Negative regulation of response to external stimulus | 5 | 5.87E‐04 | 8.19E‐03 |
| GO:0010648 | Negative regulation of cell communication | 9 | 6.57E‐04 | 9.01E‐03 |
| GO:0022008 | Neurogenesis | 14 | 6.97E‐04 | 9.39E‐03 |
| GO:0043271 | Negative regulation of ion transport | 4 | 7.08E‐04 | 9.39E‐03 |
DEPgenes: depression‐related genes; GO: gene ontology.
3.4. Crosstalk among significantly enriched pathways
Since abundant genes and pathways seemed to be involved in MDD, a pathway crosstalk analysis was performed to deeply investigate the relationship between the pathways. As shown in Figure 3a, 16 significantly enriched pathways were identified, including nervous system pathways, such as Dopaminergic synapse, serotonergic synapse, glutamatergic synapse, retrograde endocannabinoid signaling and GABAergic synapse. Besides, the pathways related to drug addiction (cocaine addiction, amphetamine addiction, nicotine addiction, alcoholism and morphine addiction), signal transduction (cAMP signaling pathway, taste transduction and calcium signaling pathway) were enriched. Interestingly, the environmental adaptation processes (circadian entrainment and circadian rhythm) were also involved in the DEPgenes' pathways. In Figure 3b, it was clear that the significantly enriched pathways were clustered into a module which was relevant to the pathogenesis of neurological diseases.
Figure 3.
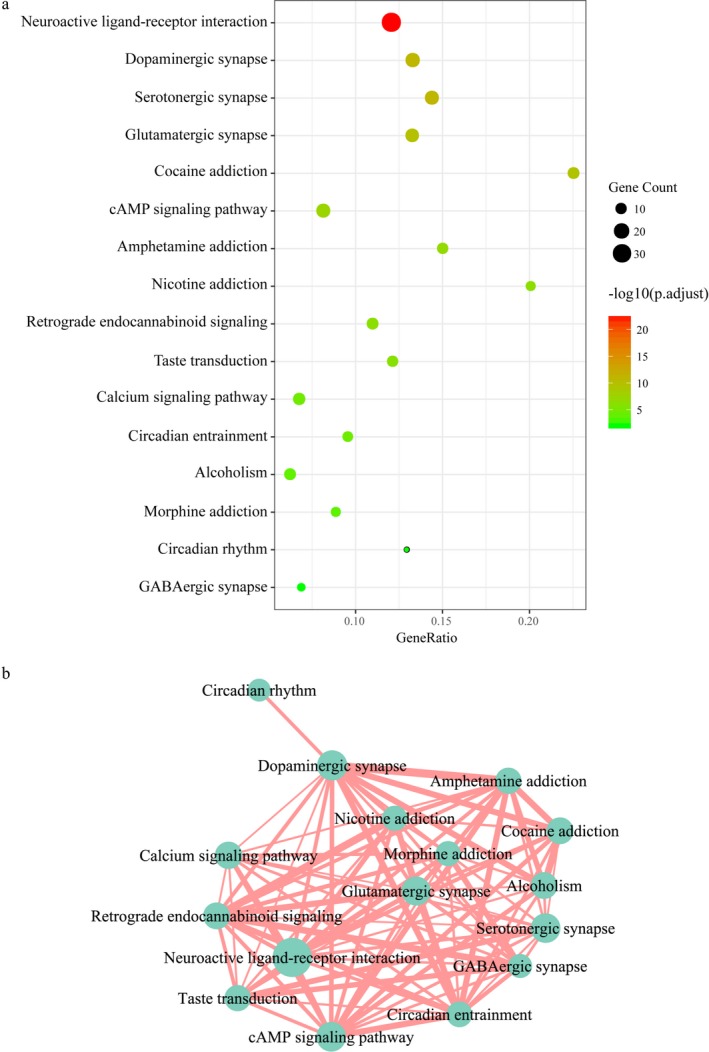
KEGG pathway enrichment analysis of DEPgenes. (a) Significantly enriched KEGG pathways of DEPgenes. The abscissa GeneRatio was the ratio of DEPgenes mapped to a KEGG pathway to the total number of genes in the pathway; (b) Visual crosstalk of KEGG pathways. The nodes size represented the number of DEPgenes contained in the pathway. The larger the node was, the more DEPgenes were included. The width of the edge indicated the overlapping degree of genes contained in two pathways. DEPgenes: depression‐related genes
3.5. MDD‐specific networks
The information on gene interaction was extracted from the STRING database and used to form a specific network (Figure 4a). To test nonrandomness of the MDD‐specific network, we generated 1,000 random networks with same node and edge number with MDD‐specific network and compared their SPD and CC. As a result, the average SPD of these random networks was 3.4, which was significantly larger than that of the MDD‐specific network with an SPD of 2.5, PD < 0.001. Meanwhile, the CC of random networks was 0.1, which was significantly smaller than that of the MDD‐specific networks with a CC of 0.5 (PC < 0.001). So, the nonrandomness of the MDD‐specific network could be inferred. Furthermore, two modules were identified by the modular cluster analysis of MDD‐specific networks (Figure 4b,c). KEGG pathway analysis of genes contained in Figure 4b indicated significantly enriched pathways of neuroactive ligand‐receptor interaction, dopaminergic synapse and morphine addiction. For genes contained in Figure 4c, the serotonergic synapse was the most significantly enriched pathway.
Figure 4.
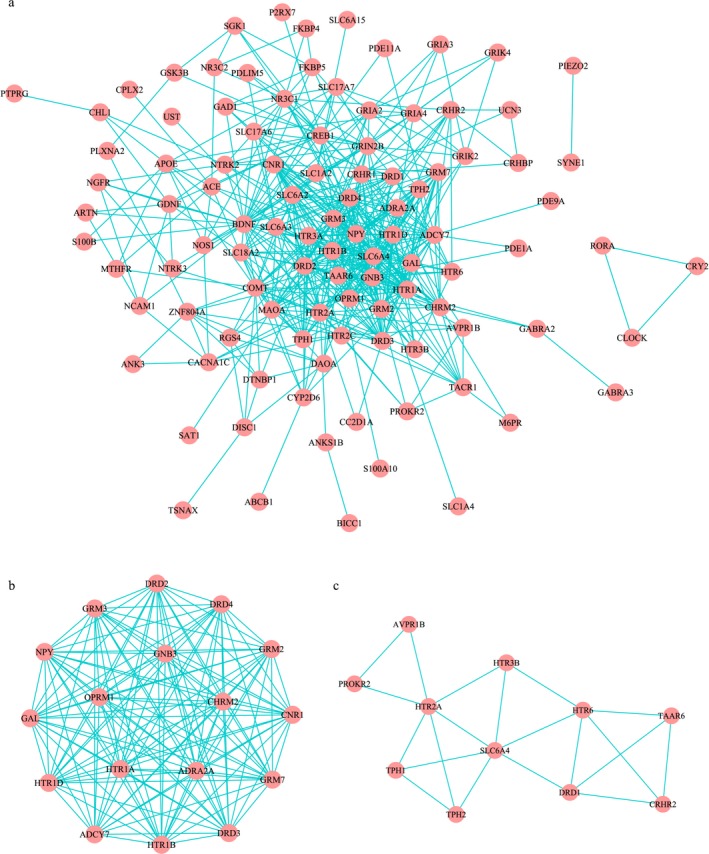
MDD‐specific network analysis. (a) The specific network of MDD; (b and c) Module Cluster analyses by MCODE. MDD: major depressive disorder
4. DISCUSSION
Drug therapy is still the preferred current clinical treatment for MDD. The most widely used antidepressant drugs are selective serotonin reuptake inhibitors (SSRIs), including fluoxetine, citalopram, and sertraline, which can significantly improve cognitive function of MDD patients (Jakubovski, Varigonda, Freemantle, Taylor, & Bloch, 2016). However, current antidepressant drugs used clinically bring lots of adverse reactions, such as xerostomia, constipation, drowsiness, obesity, cardiotoxicity, and drug withdrawal (Fava, Gatti, Belaise, Guidi, & Offidani, 2015; Hieronymus, Emilsson, Nilsson, & Eriksson, 2016). The lack of approaches on early identification and intervention of MDD patients limits the establishment of safe and effective individualized treatment (Duman, Aghajanian, Sanacora, & Krystal, 2016). Although numerous reports of susceptibility genes or loci to MDD have been reported previously, no disease causal genes and therapeutic target genes were confirmed (Rao et al., 2016). Thus, it is important to reduce the data noise and prioritize candidate genes from multiple datasets and then explore their functional relationships for further validation (Jia, Kao, Kuo, & Zhao, 2011).
In this study, we presented a complete process to collect large‐scale genotypic data on MDD from different sources, and provided optimization and comprehensive analyses for the exploration of the pathogenesis and treatment of depression. Twenty‐three DEPgenes from OMIM, 14,144 DEPgenes from Phenolyzer, 5,358 DEPgenes from GeneCards and 149 DEPgenes from GLAD4U were collected and optimized for further analyzation. MDSP was proposed and an appropriate threshold was determined for the optimization of MDD‐related genes. One hundred and forty‐three DEPgenes were identified and used for additional functional and pathway enrichment analyses. Most of these genes, such as PCDH9, MDD1, MDD2, CREB1 and DISC1, have been identified to be associated with MDD (Cacabelos, Torrellas, & Fernandez‐Novoa, 2016; Xiao et al., 2018), and some of them (e.g. TPH1, GRIN2B and MAOA) were also related to other mental disorders (van Donkelaar et al., 2017; Perlis, 2016; Tovilla‐Zarate et al., 2014). This indicated that our preferred solution designed was able to be utilized to get the expected data.
So far, the study of the pathogenesis of depression mainly focuses on the biological mechanisms, such as autophagy and apoptosis of nerve cells, neurotransmitter secretion disorders, immune inflammatory reactions, dysfunction of hypothalamus pituitary adrenal axis, and other biological mechanisms (Cattaneo et al., 2015; Menard, Hodes, & Russo, 2016; Smith, 2015). With functional enrichment analysis, a more specific functional pattern implicated in these DEPgenes was revealed. In this study, 72 GO BP terms and 16 KEGG pathways were identified to be significantly enriched. The terms related to synaptic transmission, nerve signal transduction, neurotransmitter and learning or memory reflected the pathogenesis of MDD, which was consistent with the literature reports. Interestingly, the BP term of drug reaction and the KEGG pathway of drug addiction were both enriched, indicating that the key requirement of avoiding drug dependence in MDD drug development and clinical treatment.
The occurrence and development of MDD involve complex biological processes, which is the result of a combination of multiple genes and environmental factors. Therefore, the study of the interactions between DEPgenes from the perspective of networks can provide insights into the pathogenesis of depression and contribute to the discovery of new drug targets. Thus, the network information on MDD was mined from the STRING database which contains experimental data, the PubMed abstract text database and results predicted by bioinformatics methods for specific analysis. Besides, applied bioinformatics methods in this process included gene adjacency, gene fusion, phylogenetic profiles, and gene co‐expression based on chip data. A comprehensive score was calculated with the weight matrix of these different methods determined by a scoring mechanism demonstrated above. Finally, the core pathways involved in MDD were shown in the module. The pathways of neuroactive ligand‐receptor interaction, dopaminergic synapse and morphine addiction are presented in Figure 4b. And as shown in Figure 4c, the serotonergic synapse seemed to be higher specificity than other pathways. From these results, we inferred that the drug addiction caused by serotonin used in the treatment of MDD might relate to the mechanism of morphine addiction.
The main problems that limit the development of a reliably viable MDD biomarker are the heterogeneity of depressive disorder pathophysiology, etiology, and study designs, which may bring in conflicting data. In this study, a systems biology framework for the genetic information collection, advanced function and pathway analyses for MDD was developed. A total of 143 DEPgenes were identified and the MDD‐specific network was constructed for the pathogenesis investigation and therapeutic methods development of MDD. Comparing with existing research strategies, the genetic optimization and analysis results were confirmed to be reliable. As most studies collected data from small samples sizes often consisting of fewer than 100 subjects, this study would contribute to improving the precision and generalizability of MDD‐related genes in these three databases. However, although this computational framework applied quantity of valuable information that required future validation by extensive experimental, it still provided a reference for the study of other complex disease.
5. ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHORS' CONTRIBUTIONS
Yi Liu and Shiyuan Zhang conceived and designed the project, Pengfei Fan acquired the data, Yi Liu, Pengfei Fan and Yidan Wang analyzed and interpreted the data, Yidan Wang and Dan Liu wrote the paper. Shiyuan Zhang approved the final version.
Supporting information
Liu Y, Fan P, Zhang S, Wang Y, Liu D. Prioritization and comprehensive analysis of genes related to major depressive disorder. Mol Genet Genomic Med. 2019;7:e659 10.1002/mgg3.659
Funding information
Not applicable.
REFERENCES
- Alnæs, D. , Kaufmann, T. , Doan, N. T. , Córdova‐Palomera, A. , Wang, Y. , Bettella, F. , … Westlye, L. T. (2018). Association of heritable cognitive ability and psychopathology with white matter properties in children and adolescents. JAMA Psychiatry, 75(3), 287–295. 10.1001/jamapsychiatry.2017.4277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayes, A. J. , & Parker, G. B. (2018). Comparison of guidelines for the treatment of unipolar depression: A focus on pharmacotherapy and neurostimulation Acta Psychiatrica Scandinavica, 137(6), 459–471. 10.1111/acps.12878 [DOI] [PubMed] [Google Scholar]
- Cacabelos, R. , Torrellas, C. , & Fernandez‐Novoa, L. (2016). Pharmacogenomics of antidepressant drugs In Holzer P. (Ed.), Melatonin, neuroprotective agents and antidepressant therapy (pp. 545–609). New Delhi: Springer. [Google Scholar]
- Cattaneo, A. , Macchi, F. , Plazzotta, G. , Veronica, B. , Bocchio‐Chiavetto, L. , Riva, M. A. , & Pariante, C. M. (2015). Inflammation and neuronal plasticity: A link between childhood trauma and depression pathogenesis. Frontiers in Cellular Neuroscience, 9, 40 10.3389/fncel.2015.00040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culpepper, L. , Lam, R. W. , & McIntyre, R. S. (2017). Cognitive impairment in patients with depression: Awareness, assessment, and management. The Journal of Clinical Psychiatry, 78(9), 1383–1394. 10.4088/JCP.tk16043ah5c [DOI] [PubMed] [Google Scholar]
- Duman, R. S. , Aghajanian, G. K. , Sanacora, G. , & Krystal, J. H. (2016). Synaptic plasticity and depression: New insights from stress and rapid‐acting antidepressants. Nature Medicine, 22(3), 238–249. 10.1038/nm.4050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fava, G. A. , Gatti, A. , Belaise, C. , Guidi, J. , & Offidani, E. (2015). Withdrawal symptoms after selective serotonin reuptake inhibitor discontinuation: A systematic review. Psychotherapy and Psychosomatics, 84(2), 72–81. 10.1159/000370338 [DOI] [PubMed] [Google Scholar]
- Hieronymus, F. , Emilsson, J. F. , Nilsson, S. , & Eriksson, E. (2016). Consistent superiority of selective serotonin reuptake inhibitors over placebo in reducing depressed mood in patients with major depression. Molecular Psychiatry, 21(4), 523–530. 10.1038/mp.2015.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubovski, E. , Varigonda, A. L. , Freemantle, N. , Taylor, M. J. , & Bloch, M. H. (2016). Systematic review and meta‐analysis: Dose‐response relationship of selective serotonin reuptake inhibitors in major depressive disorder. American Journal of Psychiatry, 173(2), 174–183. 10.1176/appi.ajp.2015.15030331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia, P. , Kao, C. F. , Kuo, P. H. , & Zhao, Z. (2011). A comprehensive network and pathway analysis of candidate genes in major depressive disorder. BMC Systems Biology, 5(Suppl 3), S12 10.1186/1752-0509-5-S3-S12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal, J. H. , & State, M. W. (2014). Psychiatric disorders: Diagnosis to therapy. Cell, 157(1), 201–214. 10.1016/j.cell.2014.02.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacerda‐Pinheiro, S. F. , Pinheiro Junior, R. F. F. , Lima, M. A. P. d. , Silva, C. G. L. d. , Santos, M. d. S. V. D. , Teixeira Júnior, A. G. , … Bianco, B. A. V. (2014). Are there depression and anxiety genetic markers and mutations? A systematic review. Journal of Affective Disorders, 168, 387–398. 10.1016/j.jad.2014.07.016 [DOI] [PubMed] [Google Scholar]
- Menard, C. , Hodes, G. E. , & Russo, S. J. (2016). Pathogenesis of depression: Insights from human and rodent studies. Neuroscience, 321, 138–162. 10.1016/j.neuroscience.2015.05.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson, J. J. (2017). Genetic approaches to understanding psychiatric disease. Neurotherapeutics, 14(3), 564–581. 10.1007/s13311-017-0551-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milanesi, E. , Bonvicini, C. , Congiu, C. , Bortolomasi, M. , Gainelli, G. , Gennarelli, M. , & Minelli, A. (2015). The role of GRIK4 gene in treatment‐resistant depression. Genetics Research, 97, e14 10.1017/S0016672315000142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pain, O. , Dudbridge, F. , Cardno, A. G. , Freeman, D. , Lu, Y. i. , Lundstrom, S. , … Ronald, A. (2018). Genome‐wide analysis of adolescent psychotic‐like experiences shows genetic overlap with psychiatric disorders. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics, 177(4), 416–425. 10.1002/ajmg.b.32630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pariante, C. M. , & Lightman, S. L. (2008). The HPA axis in major depression: Classical theories and new developments. Trends in Neurosciences, 31(9), 464–468. 10.1016/j.tins.2008.06.006 [DOI] [PubMed] [Google Scholar]
- Perlis, R. H. (2016). Genomic studies of treatment resistance in major depressive disorder In Rybakowski J. K. & Serretti A. (Eds.), Genetic influences on response to drug treatment for major psychiatric disorders (pp. 55–65). Cham, Switzerland: Adis; [Google Scholar]
- Rao, S. , Yao, Y. , Ryan, J. , Li, T. , Wang, D. , Zheng, C. , … Xu, Q. i. (2016). Common variants in FKBP5 gene and major depressive disorder (MDD) susceptibility: A comprehensive meta‐analysis. Scientific Reports, 6, 32687 10.1038/srep32687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, H. R. (2015). Depression in cancer patients: Pathogenesis, implications and treatment (Review). Oncology Letters, 9(4), 1509–1514. 10.3892/ol.2015.2944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovilla‐Zárate, C. A. , González‐Castro, T. B. , Juárez‐Rojop, I. , Pool García, S. , Velázquez‐Sánchez, M. P. , Villar‐Soto, M. , … Jiménez‐Santos, M. A. (2014). Study on genes of the serotonergic system and suicidal behavior: Protocol for a case‐control study in Mexican population. BMC Psychiatry, 14, 29 10.1186/1471-244X-14-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzoma, I. , Hu, J. , Cox, E. , Xia, S. , Zhou, J. , Rho, H.‐S. , … Zhu, H. (2018). Global identification of small ubiquitin‐related modifier (SUMO) substrates reveals crosstalk between SUMOylation and phosphorylation promotes cell migration. Molecular & Cellular Proteomics, 17(5), 871–888. 10.1074/mcp.RA117.000014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Donkelaar, M. M. J. , Hoogman, M. , Shumskaya, E. , Buitelaar, J. K. , Bralten, J. , & Franke, B. (2017). Monoamine and neuroendocrine gene‐sets associate with frustration‐based aggression in a gender‐specific manner. European Neuropsychopharmacology, 27, 1–12. 10.1016/j.euroneuro.2017.11.016 [DOI] [PubMed] [Google Scholar]
- Wang, J. , Vasaikar, S. , Shi, Z. , Greer, M. , & Zhang, B. (2017). WebGestalt 2017: A more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Research, 45(W1), W130–W137. 10.1093/nar/gkx356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray, N. R. , Yang, J. , Goddard, M. E. , & Visscher, P. M. (2010). The genetic interpretation of area under the ROC curve in genomic profiling. PLoS Genetics, 6(2), e1000864 10.1371/journal.pgen.1000864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, X. , Zheng, F. , Chang, H. , Ma, Y. , Yao, Y. G. , Luo, X. J. , & Li, M. (2018). The gene encoding protocadherin 9 (PCDH9), a novel risk factor for major depressive disorder. Neuropsychopharmacology, 43(5), 1128–1137. 10.1038/npp.2017.241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, J. J. , Silber, T. , Bruno, D. , Galatzer‐Levy, I. R. , Pomara, N. , & Marmar, C. R. (2016). Is there progress? An overview of selecting biomarker candidates for major depressive disorder. Frontiers in Psychiatry, 7, 72 10.3389/fpsyt.2016.00072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z.‐J. , Wang, D. i. , Man, S. C. , Ng, R. , McAlonan, G. M. , Wong, H. K. , … Tan, Q.‐R. (2014). Platelet 5‐HT(1A) receptor correlates with major depressive disorder in drug‐free patients. Progress in Neuro‐Psychopharmacology and Biological Psychiatry, 53, 74–79. 10.1016/j.pnpbp.2014.03.004 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials