

Abstract
Background
Lynch syndrome (LS) is an autosomal dominant cancer predisposition condition caused by germline heterozygous mutations in mismatch repair (MMR) genes. However, as one of the MMR genes, PMS2 mutation‐induced LS‐associated ovarian cancer (LSAOC) has rarely been reported.
Methods
Next‐generation sequencing (NGS) or Sanger sequencing was used to detect the genetic status of one family including four generations with 16 members. Then, quantitative real‐time PCR (qPCR), western blotting, immunohistochemistry (IHC) staining, and Swiss‐Model software were used to identify the function of the PMS2 mutation.
Results
Five individuals [I‐1, II‐1, II‐2, II‐4, and III‐2 (proband)] suffered from LS‐associated cancers, for example, colon cancer, gastric cancer, and ovarian cancer, with the age of onset ranging from 39 to 70 years old. A PMS2 germline heterozygous mutation (c.943C>T) was confirmed in three members [II‐9, III‐2, and IV‐1] by gene sequencing. In addition, this PMS2 mutation was verified by qPCR, western blotting, and IHC, and a dramatic change with partial loss of the C‐terminal domain in an α‐helix might be exhibited.
Conclusion
Carrying PMS2 germline mutations (c.943C>T) confers an extremely high susceptibility of suffering from LS‐associated cancers. Thus, close clinical monitoring and prophylactic surgery is highly recommended to help reduce the morbidity and mortality of LS‐associated cancers.
Keywords: Lynch syndrome, nonsense mutation, ovarian cancer, PMS2 gene
1. INTRODUCTION
Ovarian cancer (OC) is a common gynecologic malignancy, and more women die from OC than from cervical cancer and uterine cancer combined (Barrett et al., 2014; Siegel, Miller, & Jemal, 2017). Recently, studies have shown that the lifetime risk of Lynch syndrome (LS)‐associated ovarian cancer (LSAOC) is approximately 4%–24% (Barrow, Hill, & Evans, 2013; Bonadona et al., 2011), which is about 2% of all OC (Malander et al., 2006).
Lynch syndrome was first described in 1913 by Warthin and was further delineated by Lynch in 1966 (Lynch, Shaw, Magnuson, Larsen, & Krush, 1966). As an autosomal dominant tumor predisposition condition, LS is caused by germline heterozygous mutations in one of four mismatch repair (MMR) genes including MLH1, MSH2, MSH6, and PMS2 (Bhattacharya, 2018). Individuals found to have deleterious LS mutations are at an increased risk of cancer, primarily colorectal cancer (CRC) and endometrial cancer, followed by gastric cancer (GC) and OC (Bhattacharya, 2018).
In January 2017, a patient of our gynecologic oncology group underwent a laparoscopic comprehensive staging surgery and was pathologically diagnosed with ovarian clear cell carcinoma, FIGO Stage IC. This patient was only 39 years old and had a family history of aggregation malignancies (her biological grandfather and two uncles suffered from CRC and another uncle suffered from GC), and was further confirmed to carry the PMS2 (OMIM: *600,259) germline heterozygous mutation c.943C>T (p.Arg315*) by next‐generation sequencing (NGS).
PMS2 germline heterozygous mutations account for fewer than 5% of LS cases (Bhattacharya, 2018). Correspondingly, the malignancies caused by PMS2 mutations are also relatively rare compared with other LS‐associated MMR genes. Even the largest single institution cohort study of LSAOC did not report a single case of a PMS2 heterozygous mutation (Ryan, Evans, Green, & Crosbie, 2017). In view of the infrequent incidence of LSAOC with PMS2 mutations, it is necessary to share the vital clinicopathological features of such cases to formulate definitive conclusions and improve the prognosis of this subgroup of patients.
2. MATERIALS AND METHODS
2.1. Ethical compliance
All procedures performed in this study involving human participants were in accordance with the ethical standards of the Medical Ethics Committee at Shanghai First Maternity and Infant Hospital and the 2014 Declaration of Helsinki and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study.
2.2. Patient and pedigree
The proband (III‐2) with ovarian clear cell carcinoma was diagnosed and treated at Shanghai First Maternity and Infant Hospital, and this four‐generation Chinese pedigree with 16 members was enrolled in our study. The diagnostic criteria for patients with LS are based on the Amsterdam II criteria.
2.3. Histochemistry and immunohistochemistry
Ovarian cancer tissues from the proband and control tissues from benign ovarian cysts and OC patients with wild‐type PMS2 (NM_000535.6) were fixed in 10% formalin for 24 hr at 4°C and embedded in paraffin. Then, the tissues were sectioned into 4‐µm‐thick slices and placed on glass slides.
For the histochemistry, the tissue slides were dewaxed in xylene, hydrated with graded ethanol, and stained with hematoxylin and eosin for 5 min at room temperature; then, they were viewed and photographed under a Nikon eclipse TE2000 fluorescence microscope.
For the immunohistochemical staining (IHC), ovarian tissue sections were incubated with primary mouse monoclonal antibodies against PMS2 (Cat. 66075‐1, Proteintech, USA), MLH1 (Maxim Biotechnologies, Cat. MAB‐0642, China), MSH2 (Maxim Biotechnologies, MAB‐0291, China), and MSH6 (Maxim Biotechnologies, MAB‐0642, China) overnight at 4°C, diluted in antibody diluent (Cat. 8112 L, Cell Signaling Technology, USA). The slides were then incubated with secondary antibody, and finally counterstained with 3,3‐diaminobenzidine and hematoxylin. The stained sections were assessed by two independent pathology experts. The extent of PMS2 staining in the tissue cores was quantified using a four‐tier grading system as previously described (Liu et al., 2017).
2.4. NGS‐based clinical cancer gene test
Next‐generation sequencing with a multiple gene panel of 7,708 exons from 508 cancer‐related genes and 78 introns from 19 genes recurrently rearranged in solid tumors (Table S1) were carried out by BGI‐Shenzhen (Thomson, 1966; West, McAdams, & Northway, 1968; Yang et al., 2017). Briefly, tumor DNA and genomic DNA were isolated from formalin‐fixed paraffin‐embedded (FFPE) tumor specimens and peripheral blood, respectively. NGS was performed with an Illumina HiSeq 2500 platform (Illumina, San Diego, CA, USA). In this study, NGS was conducted in the proband.
2.5. Sanger sequencing
To validate putative mutations, Sanger sequencing was performed by BGI‐Shenzhen. Briefly, primers were designed based on the reference genomic sequences of the human genome downloaded from NCBI's GenBank and synthesized by Invitrogen (Shanghai, China). Polymerase chain reaction (PCR) amplification was carried out with an ABI 9700 Thermal Cycler and sequenced on an ABI PRISM 3730 automated sequencer (Applied Biosystems) (Banerjee et al., 2016).
2.6. Microsatellite instability assay
DNA from both tumor and normal tissues was extracted from FFPE blocks for each patient. Microsatellite instability (MSI) was performed by multiplex PCR amplification of the five microsatellite loci (BAT25, BAT26, D5S346, D2S123, and D17S250) recommended by the National Cancer Institute (NCI). The fluorescence‐labeled products were analyzed by capillary electrophoresis. A difference in the length of a microsatellite marker in tumor tissue compared to that of normal tissue was interpreted as an unstable microsatellite. MSI‐high (MSI‐H) was defined as two or more markers being affected, and the involvement of one marker was interpreted as MSI‐low. Microsatellite stable (MSS) was reported if all five microsatellites showed stability.
2.7. Tumor mutation burden
The tumor mutation burden (TMB) was calculated based on the number of somatic mutations in the sequenced genes, and the value was extrapolated to the genome as a whole using a validated algorithm. The TMB was reported as the number of mutations per megabase (mb) of the genome. Based on the FoundationOne™ Heme reports, TMB results were also categorized into three groups: low (1–5 mutations/mb), intermediate (6–19 mutations/mb), and high (≥ 20 mutations/mb). Values were rounded to the nearest integer.
2.8. Quantitative real‐time PCR
To validate the targeted NGS results and to further quantify the PMS2 mutation gene, quantitative real‐time PCR (qPCR) was performed. Total RNA was extracted from ovarian tissues using the TRIzol Reagent (Invitrogen), and cDNA was synthesized by a reverse transcription kit (TaKaRa). PMS2 mRNA levels were measured using a Super Real PreMix Plus (SYBR Green) Kit (Tiangen Biotech, China) and an Applied Biosystems Step One Plus™ Real‐Time PCR System according to the manufacturer's instructions. The PMS2 mRNA relative level was calculated using the 2−ΔΔCt method, and GAPDH was used as a positive control. The qRT‐PCR results were as follows:
GAPDH, forward: 5′‐ACAACTTTGGTATCGTGGAAGG‐3′
and reverse: 5′‐GCCATCACGCCACAGTTTC‐3′,
PMS2 exon 8–9, forward: 5′‐TGCACGCATGGAGTTGGAAG‐3′
and reverse: 5′‐CTGCAGACCTTTGCTGGGTC‐3′, and
PMS2 exon 9–10, forward: 5′‐TTCTGTTGATTCAGAATGCGTTGAT‐3′
and reverse: 5′‐CTTCAACATCCAGCAGTGGCT‐3′.
2.9. Western blot analysis
Total protein from tissues was lysed in a whole‐cell lysis assay (Cat. KGP250, KeyGen BioTECH, China) containing protease inhibitors (Cat. KGP603, KeyGen BioTECH, China) and phosphatase inhibitors (Cat. KGP602, KeyGen BioTECH, China). Then, 10 µg of protein per sample was resolved by 10% SDS‐PAGE and transferred onto PVDF membranes. The membranes were first incubated overnight at 4°C in BSA in TBS containing 0.05% Tween 20 with primary antibodies against β‐actin (1:5,000, Cat. M20010, Abmart, USA) and PMS2 (1:1,000, Cat. 66075‐1, Proteintech, USA), followed by incubation with secondary antibodies (Cat. KGAA3d5, KeyGen BioTECH, China) conjugated with horseradish peroxidase at room temperature for 1 hr. The protein bands were detected using an enhanced chemiluminescence plus kit (Millipore) according to the manufacturer's instructions.
2.10. Structure prediction of the mutant protein
The amino acid (aa) sequences of the PMS2 protein (GenBank accession number NP_000526.1, Data S2) were obtained from the GenBank database. The homology modeling program, Swiss‐Model (http://swissmodel.expasy.org), was used to create an appropriate model to mimic the structure of the mutated region (Biasini et al., 2014).
2.11. Statistical analysis
All experiments were repeated at least three times in duplicate. The data are presented as the mean ± SD. Differences between the treated and control groups were analyzed using Student's t test and one‐way ANOVA. The level of significance was set at p < 0.05. All statistical analyses were performed with SPSS 24.0 (IBM, USA).
3. RESULTS
3.1. Clinical findings
One Chinese family was found to have significant characteristics of tumor aggregation, including three members (I‐1, II‐1, and II‐2) who suffered from CRC, one member (II‐4) with GC, and one member (III‐2) with OC. The onset age of CRC and GC were approximately 60–70 years old, but that of OC was merely 39 years old. The proband (III‐2) was pathologically diagnosed with ovarian clear cell carcinoma (Type 1, FIGO Stage IC). No tumor metastasis was detected in the omentum, peritoneum, diaphragmatic dome, pelvic or para‐aortic lymph nodes. After a comprehensive staging operation, the proband was treated with six courses of adjuvant paclitaxel combined with carboplatin chemotherapy, and subsequently monitored under routine clinical follow‐up every 3 months; no tumor recurrence and metastasis was observed until now (24 months). In addition, the expression of MMR genes was measured in the OC tissues of the proband by IHC staining, and the results showed that PMS2 presented only foci cytoplasm positivity, while MLH1, MSH2, and MSH6 showed diffuse strong positivity (Figure 1).
Figure 1.
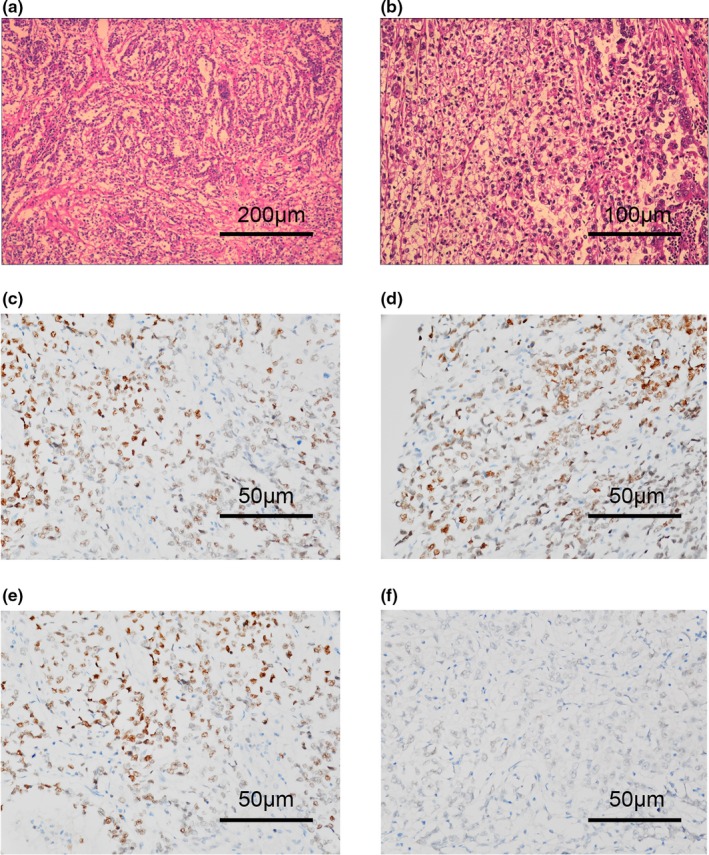
Clinical characteristics of the proband. (a and b) Representative pictures of HE staining in nodular lesions confirmed by pathology experts. Representative IHC staining of (c) MSH1, (d) MSH2, (e) MSH6, and (f) PMS2 (NM_000535.6) in the proband's tumor tissues. IHC, immunohistochemistry
3.2. Identification of PMS2 germline heterozygous mutations in the family
A PMS2 germline heterozygous mutation was confirmed in the proband (III‐2) by NGS, and this mutation is a nonsense mutation (c.943C>T) in exon 9 (mutation accession number CM_102797), which was predicted to lead to the early termination of translation at p.Arg315*, while the wild‐type PMS2 gene encodes a protein with a total length of 862 aa. Moreover, MSI‐H status and high TMB were detected in this proband. In addition, in this family, the biological father (II‐9) and son (IV‐1) of the proband also were validated to carry the same PMS2 heterozygous mutation by Sanger sequencing; samples from the CRC and GC patients (I‐1, II‐1, II‐2, and II‐4) were unavailable because of prior death (Figure 2). Therefore, according to the Amsterdam II criteria, this family was diagnosed with LS, and the proband as diagnosed with LSAOC.
Figure 2.
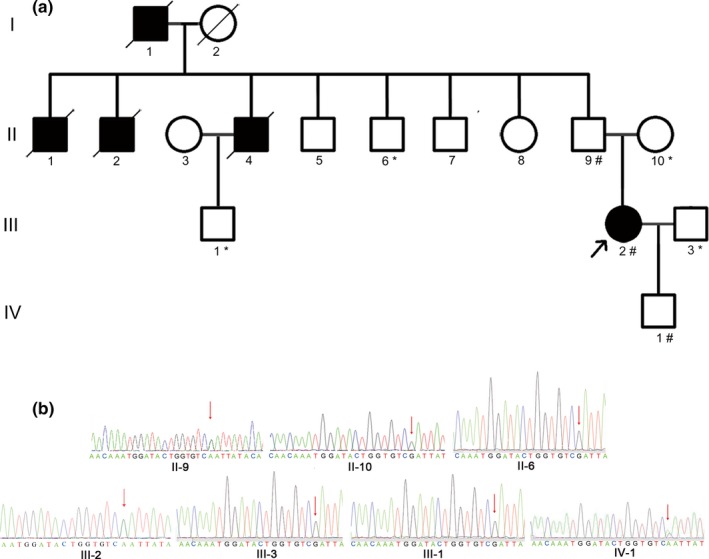
Pedigree structure of this Chinese family and validation of the PMS2 (NM_000535.6) mutation by Sanger sequencing. (a) Pedigree structure of the Chinese family with LS. The family members that suffered from tumors are indicated with shading. Squares and circles denote males and females, respectively. Roman numerals indicate generations. The arrow indicates the proband (III‐2). The sign “#” indicates which family members were tested for mutations and found to carry the mutation in the pedigree; the sign “*” indicates which family members were tested and found not to carry the mutation. (b) Validation of the heterozygous germline PMS2 mutation by Sanger sequencing. The red arrows show the heterozygous mutation c.943C>T (p.Arg315*). LS, Lynch syndrome
3.3. Identification of the function of the PMS2 mutation at the mRNA and protein levels
First, cross exon primers were designed to detect the identical mutations in PMS2 (c.943C>T) in exon 9. As the qPCR results showed, the amplification level of the PMS2‐mutated transcript was significantly lower in the proband, compared to OC and benign ovarian cysts without PMS2 mutations, as validated by Sanger sequencing (p < 0.05) (Figure 3a). Next, translation of the PMS2 mutation was further validated by western blot analysis and IHC staining. The immunogenic synthetic peptide used in this study was specific to human PMS2 aa 513–862, while the PMS2 mutation (c.943C>T) induced aa 315 termination; thus, this specific antibody could not detect the PMS2‐mutated protein. As the results showed, the protein level of PMS2 in the mutation carrier was remarkably decreased compared to the wild‐type, as confirmed by western blot analysis (Figure 3b) and IHC staining (Figure 3c).
Figure 3.
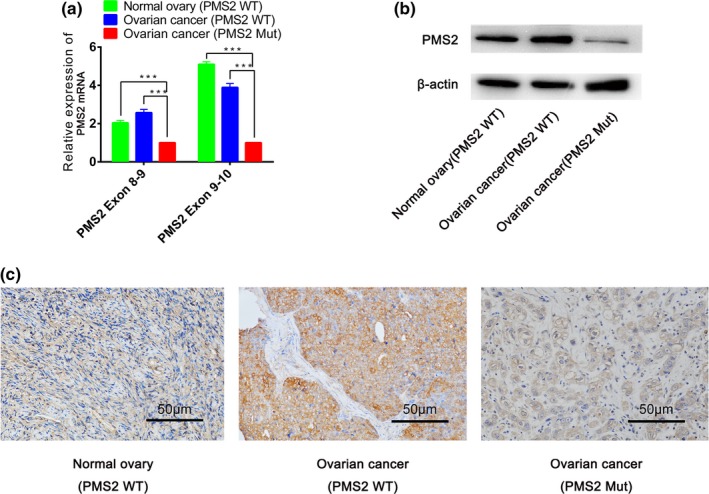
Functional verification of the large heterozygous deletion in PMS2 Mut (mutant‐type) and PMS2 WT (wild‐type). (a) The relative mRNA expression level of PMS2 was detected by qPCR. (b) The relative protein expression level of PMS2 was measured by western blot analysis, and (c) IHC (***p < 0.001, Scale bar = 50 µm). IHC, immunohistochemistry; qPCR, quantitative real‐time PCR
3.4. Prediction of PMS2‐mutated protein structure
In order to explore the spatial configuration of the PMS2 mutation, Swiss‐Model online software was utilized. As the results showed, a partial loss of the C‐terminal domain in the α‐helix of the mutated PMS2 protein was observed, compared with the wild‐type, as marked by red arrows in the figure (Figure 4).
Figure 4.
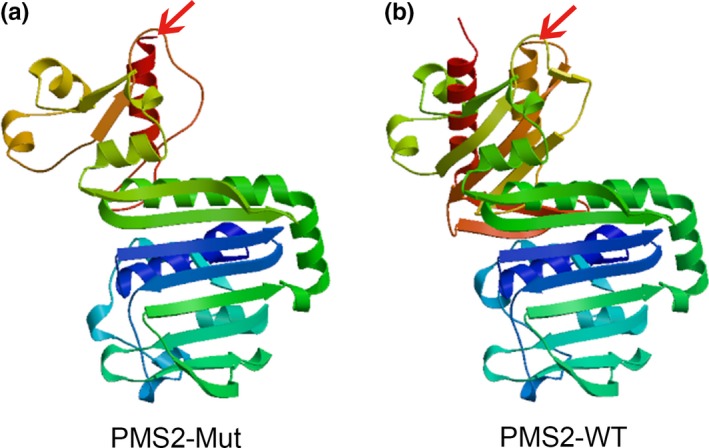
Structure prediction of the mutant protein. (a) The mutant protein structure of PMS2 (PMS2‐Mut) was predicted to result in the partial loss of the C‐terminal domain of the α‐helix by Swiss‐Model online software (marked by the red arrows). (b) The wild‐type PMS2 protein structure (PMS2‐WT)
4. DISCUSSION
LS is well known for its close association with CRC in familial aggregated hereditary tumors, and it accounts for 2%–5% of all CRC. Recently, studies have found that the LS spectrum also includes tumors of the endometrium, ovaries, small bowel, urothelium, biliary tract, and stomach (Cohen & Leininger, 2014). LS is inherited in an autosomal dominant manner: thus, first‐degree relatives (parents, siblings, children) have a 50% chance of being affected. At present, the detection of MMR (MLH1, MSH2, MSH6, and PMS2) protein expression in tumor tissues by IHC is traditionally used for preliminary screening for LS. If at least one MMR protein staining result is negative, referred to as MMR‐deficient (dMMR), then further germline genetic testing should be offered. If all of the MMR proteins are present, the probability of MMR gene mutation is small.
In this study, first, IHC was used to validate negative PMS2 expression, while that of the other three MMR genes MLH1, MSH2, and MSH6 were all positive in the proband's OC tissues. Second, a germline heterozygous mutation, c.943C>T (p.Arg315*), in PMS2 was confirmed in the proband by NGS. In addition, the biological father and son of the proband were also proved to carry the same PMS2 mutation by Sanger sequencing, while the proband's husband and biological mother did not. Considering the family history of gastrointestinal tumor aggregation, according to the Amsterdam II criteria, the family was diagnosed with LS.
Previous studies have found that the clinicopathological features of LSAOC are obviously distinct from those of sporadic OC. First, the age of onset of LSAOC is relatively young, with a mean age of diagnosis of 45 years (Ketabi et al., 2011), which is 15–20 years earlier than the mean age in sporadic OC (Buys et al., 2011). Some scholars have proposed that the onset age distribution of PMS2 mutation‐associated CRC and endometrium carcinoma is very broad, including both those that are very young or very old (ten Broeke et al., 2015). Second, the degree of malignancy is relatively low in LSAOC. Low‐grade type I OC with endometrioid morphology accounts for the vast majority of LSAOC with a proportion of more than 50% (Ryan et al., 2017), which is significantly different from the non‐LS and BRCA mutation types of OC, in which high‐grade type 2 with serous tumors are more common (Lu, 2008). Third, the prognosis of LSAOC is relatively better. The 10‐year overall survival of patients with LSAOC is approximately 75% compared to 35% for non‐LS associated OC (Niskakoski et al., 2013). Fourth, early FIGO‐stage cases are more common in LSAOC. Approximately 65% of LSAOC present at FIGO Stage I–II (Helder‐Woolderink et al., 2016; Ryan et al., 2017), whereas most cases of sporadic OC are diagnosed at advanced stages (FIGO Stage III–IV) (Sopik, Iqbal, Rosen, & Narod, 2015); this may help to explain the better survival rates of LSAOC, similar to the LSAOC case reported here.
It is still controversial as to whether prophylactic hysterectomy and oophorectomy should be recommended in LS‐associated MMR gene mutation carriers. Some scholars believe that prophylactic surgery will bring benefits to the high‐risk group of MMR gene mutation carriers who have no desire to bear children (Schmeler et al., 2006), while some other scholars suggest that tight clinical monitoring is enough among the relatively lower risk MMR gene carriers, such as those with mutations in PMS2 (ten Broeke et al., 2015; Pylvanainen, Lehtinen, Kellokumpu, Jarvinen, & Mecklin, 2012); alternatively, some researchers have recommended that annual screening from the age of 30, followed by prophylactic surgery at the age 40 is the most cost‐effective gynecologic cancer prevention strategy in women with LS (Chen, Yang, Little, Cheung, & Caughey, 2007). In view of the fact that, the onset age of LSAOC carrying this specific PMS2 mutation (c.943C>T) is significantly younger than that of gastrointestinal malignancies, we recommended prophylactic hysterectomy and bilateral salpingo‐oophorectomy for women >35 years old with no desire to bear children.
Previous researchers have confirmed that alone or combined use of transvaginal ultrasonography and serum tumor markers (such as CA 125) cannot effectively improve the early detection rate of OC and reduce mortality in average risk women, but will increase invasive diagnostic testing (e.g., surgery) and the related complications that result from false‐positive test results. According to the latest ACOG Committee Opinion, obtaining a detailed personal and family history of breast, gynecologic, and colon cancer is recommended to facilitate categorizing women by their risk of developing epithelial OC (average risk or high risk) (Committee on Gynecologic Practice, 2017). However, the present situation is that the majority of LSAOC cases is diagnosed after OC diagnosis. Perhaps more LS and BRCA mutations will be diagnosed early in the future. Most importantly, these high‐risk groups should receive more adaptive monitoring, thereby improving the early diagnosis of OC and other related malignancies.
During normal DNA replication, proficient MMR genes play vital roles in detecting DNA mismatch errors and correcting DNA strands. Being dMMR leads to the formation of diffuse MSI, which subsequently results in additive mutations throughout the genome leading to MSI tumors (Lee, Murphy, Le, & Diaz, 2016). Compared with MSS tumors, MSI tumors present with higher mutation loads and neoantigens, which leads to dense tumor infiltrating lymphocytes (TILs) and a Th1‐associated cytokine‐rich environment. However, the combination of programmed death 1 (PD‐1) in TILs and ligand PD‐L1 (PD‐L1) on MSI tumor cells will continuously inhibit TIL activity and result in tumor immune escape. We know that PD‐1/PD‐L1 inhibitors can specifically block the immunosuppressive response mediated by PD‐1/PD‐L1 (Diaz & Le, 2015; Le et al., 2017; Topalian, Drake, & Pardoll, 2015) and subsequently enhance T‐cell immunotherapy responsiveness and antitumor function. Therefore, PD‐1/PD‐L1 inhibitors may benefit for the patients with dMMR MSI tumors (Lee & Le, 2016; Poliak & Indeikin, 1969), such as this proband.
In conclusion, the specific PMS2 germline mutation (c.943C>T) has obvious susceptibility to LS‐associated tumors, and the onset age of OC is significantly younger than that of gastrointestinal tumors. Thus prophylactic hysterectomy and bilateral salpingo‐oophorectomy are recommended for women over 35 years old with no desire of childbirth. In addition, the PMS2 mutation (c.943C>T) associated cancers with MSI‐H and high load of TMB status may benefit from immunological checkpoints blocking PD‐1/PD‐L1 inhibitors individual treatment. However, a large sample of prospective studies and vital clinicopathological features are needed to further define the cancer risk faced by PMS2 germline mutation carriers and improve strategies for the prevention, early diagnosis, treatment, and prognosis supervision of this subgroup of LS.
CONFLICT OF INTERESTS
The authors declare that they have no conflict of interests.
Supporting information
Guo X, Wu W, Gao H, et al. PMS2 germline mutation c.943C>T (p.Arg315*)‐induced Lynch syndrome‐associated ovarian cancer. Mol Genet Genomic Med. 2019;7:e721 10.1002/mgg3.721
Funding information
This work was supported by the National Natural Science Foundation of China (grant number 81372305), the Shanghai Municipal Medical and Health Discipline Construction Projects (grant number 2017ZZ02015), and the Fundamental Research Funds for the Central Universities (grant numbers 22120170047 and 22120170104).
REFERENCES
- Banerjee, S. , Chen, H. , Huang, H. , Wu, J. , Yang, Z. , Deng, W. , … Li, X. (2016). Novel mutations c.28G>T (p.Ala10Ser) and c.189G>T (p.Glu63Asp) in WDR62 associated with early onset acanthosis and hyperkeratosis in a patient with autosomal recessive microcephaly type 2. Oncotarget, 7(48), 78363–78371. 10.18632/oncotarget.13279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett, J. , Jenkins, V. , Farewell, V. , Menon, U. , Jacobs, I. , Kilkerr, J. , … Fallowfield, L. ; UKCTOCS trialists . (2014). Psychological morbidity associated with ovarian cancer screening: Results from more than 23,000 women in the randomised trial of ovarian cancer screening (UKCTOCS). BJOG, 121(9), 1071–1079. 10.1111/1471-0528.12870 [DOI] [PubMed] [Google Scholar]
- Barrow, E. , Hill, J. , & Evans, D. G. (2013). Cancer risk in Lynch syndrome. Familial Cancer, 12(2), 229–240. 10.1007/s10689-013-9615-1 [DOI] [PubMed] [Google Scholar]
- Bhattacharya, P. (2018). Lynch syndrome. In McHugh T. W. (Ed.). Treasure Island, FL: StatPearls. [Google Scholar]
- Biasini, M. , Bienert, S. , Waterhouse, A. , Arnold, K. , Studer, G. , Schmidt, T. , … Schwede, T. (2014). SWISS‐MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Research, 42(W1), W252–W258. 10.1093/nar/gku340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonadona, V. , Bonaiti, B. , Olschwang, S. , Grandjouan, S. , Huiart, L. , Longy, M. , … Bonaïti‐Pellié, C. ; French Cancer Genetics Network . (2011). Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA, 305(22), 2304–2310. 10.1001/jama.2011.743 [DOI] [PubMed] [Google Scholar]
- Buys, S. S. , Partridge, E. , Black, A. , Johnson, C. C. , Lamerato, L. , Isaacs, C. , … Berg, C. D. ; PLCO Project Team . (2011). Effect of screening on ovarian cancer mortality: The prostate, lung, colorectal and ovarian (PLCO) cancer screening randomized controlled trial. JAMA, 305(22), 2295–2303. 10.1001/jama.2011.766 [DOI] [PubMed] [Google Scholar]
- Chen, L. M. , Yang, K. Y. , Little, S. E. , Cheung, M. K. , & Caughey, A. B. (2007). Gynecologic cancer prevention in Lynch syndrome/hereditary nonpolyposis colorectal cancer families. Obstetrics and Gynecology, 110(1), 18–25. 10.1097/01.AOG.0000267500.27329.85 [DOI] [PubMed] [Google Scholar]
- Cohen, S. A. , & Leininger, A. (2014). The genetic basis of Lynch syndrome and its implications for clinical practice and risk management. The Application of Clinical Genetics, 7, 147–158. 10.2147/TACG.S51483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Committee on Gynecologic Practice , S. o. G. O. (2017). Committee opinion No. 716: The role of the obstetrician‐gynecologist in the early detection of epithelial ovarian cancer in women at average risk. Obstetrics & Gynecology, 130(3), e146–e149. 10.1097/AOG.0000000000002299 [DOI] [PubMed] [Google Scholar]
- Diaz, L. A. Jr. , & Le, D. T. (2015). PD‐1 blockade in tumors with mismatch‐repair deficiency. New England Journal of Medicine, 373(20), 1979 10.1056/NEJMc1510353 [DOI] [PubMed] [Google Scholar]
- Helder‐Woolderink, J. M. , Blok, E. A. , Vasen, H. F. , Hollema, H. , Mourits, M. J. , & De Bock, G. H. (2016). Ovarian cancer in Lynch syndrome; a systematic review. European Journal of Cancer, 55, 65–73. 10.1016/j.ejca.2015.12.005 [DOI] [PubMed] [Google Scholar]
- Ketabi, Z. , Bartuma, K. , Bernstein, I. , Malander, S. , Grönberg, H. , Björck, E. , … Nilbert, M. (2011). Ovarian cancer linked to Lynch syndrome typically presents as early‐onset, non‐serous epithelial tumors. Gynecologic Oncology, 121(3), 462–465. 10.1016/j.ygyno.2011.02.010 [DOI] [PubMed] [Google Scholar]
- Le, D. T. , Durham, J. N. , Smith, K. N. , Wang, H. , Bartlett, B. R. , Aulakh, L. K. , … Diaz, L. A. Jr. (2017). Mismatch repair deficiency predicts response of solid tumors to PD‐1 blockade. Science, 357(6349), 409–413. 10.1126/science.aan6733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, V. , & Le, D. T. (2016). Efficacy of PD‐1 blockade in tumors with MMR deficiency. Immunotherapy, 8(1), 1–3. 10.2217/imt.15.97 [DOI] [PubMed] [Google Scholar]
- Lee, V. , Murphy, A. , Le, D. T. , & Diaz, L. A. Jr. (2016). Mismatch repair deficiency and response to immune checkpoint blockade. The Oncologist, 21(10), 1200–1211. 10.1634/theoncologist.2016-0046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, N. , Peng, S. M. , Zhan, G. X. , Yu, J. , Wu, W. M. , Gao, H. , … Guo, X. Q. (2017). Human chorionic gonadotropin beta regulates epithelial‐mesenchymal transition and metastasis in human ovarian cancer. Oncology Reports, 38(3), 1464–1472. 10.3892/or.2017.5818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, K. H. (2008). Hereditary gynecologic cancers: Differential diagnosis, surveillance, management and surgical prophylaxis. Familial Cancer, 7(1), 53–58. 10.1007/s10689-007-9144-x [DOI] [PubMed] [Google Scholar]
- Lynch, H. T. , Shaw, M. W. , Magnuson, C. W. , Larsen, A. L. , & Krush, A. J. (1966). Hereditary factors in cancer. Study of two large midwestern kindreds. Archives of Internal Medicine, 117(2), 206–212. 10.1001/archinte.1966.03870080050009 [DOI] [PubMed] [Google Scholar]
- Malander, S. , Rambech, E. , Kristoffersson, U. , Halvarsson, B. , Ridderheim, M. , Borg, A. , & Nilbert, M. (2006). The contribution of the hereditary nonpolyposis colorectal cancer syndrome to the development of ovarian cancer. Gynecologic Oncology, 101(2), 238–243. 10.1016/j.ygyno.2005.10.029 [DOI] [PubMed] [Google Scholar]
- Niskakoski, A. , Kaur, S. , Renkonen‐Sinisalo, L. , Lassus, H. , Järvinen, H. J. , Mecklin, J.‐P. , … Peltomäki, P. (2013). Distinct molecular profiles in Lynch syndrome‐associated and sporadic ovarian carcinomas. International Journal of Cancer, 133(11), 2596–2608. 10.1002/ijc.28287 [DOI] [PubMed] [Google Scholar]
- Poliak, B. L. , & Indeikin, E. N. (1969). Acute paroxysmal attack (decompensation) in the initial stage of primary glaucoma. Vestnik Oftalmologii, 6, 35–40. [PubMed] [Google Scholar]
- Pylvanainen, K. , Lehtinen, T. , Kellokumpu, I. , Jarvinen, H. , & Mecklin, J. P. (2012). Causes of death of mutation carriers in Finnish Lynch syndrome families. Familial Cancer, 11(3), 467–471. 10.1007/s10689-012-9537-3 [DOI] [PubMed] [Google Scholar]
- Ryan, N. A. J. , Evans, D. G. , Green, K. , & Crosbie, E. J. (2017). Pathological features and clinical behavior of Lynch syndrome‐associated ovarian cancer. Gynecologic Oncology, 144(3), 491–495. 10.1016/j.ygyno.2017.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeler, K. M. , Lynch, H. T. , Chen, L.‐M. , Munsell, M. F. , Soliman, P. T. , Clark, M. B. , … Lu, K. H. (2006). Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. New England Journal of Medicine, 354(3), 261–269. 10.1056/NEJMoa052627 [DOI] [PubMed] [Google Scholar]
- Siegel, R. L. , Miller, K. D. , & Jemal, A. (2017). Cancer statistics, 2017. CA: A Cancer Journal for Clinicians, 67(1), 7–30. 10.3322/caac.21387 [DOI] [PubMed] [Google Scholar]
- Sopik, V. , Iqbal, J. , Rosen, B. , & Narod, S. A. (2015). Why have ovarian cancer mortality rates declined? Part I. Incidence. Gynecologic Oncology, 138(3), 741–749. 10.1016/j.ygyno.2015.06.017 [DOI] [PubMed] [Google Scholar]
- ten Broeke, S. W. , Brohet, R. M. , Tops, C. M. , van der Klift, H. M. , Velthuizen, M. E. , Bernstein, I. , … Wijnen, J. T. (2015). Lynch syndrome caused by germline PMS2 mutations: Delineating the cancer risk. Journal of Clinical Oncology, 33(4), 319–325. 10.1200/JCO.2014.57.8088 [DOI] [PubMed] [Google Scholar]
- Thomson, D. S. (1966). Blurring of vision on exercise. Transactions of the Ophthalmological Societies of the United Kingdom, 86, 479–492. [PubMed] [Google Scholar]
- Topalian, S. L. , Drake, C. G. , & Pardoll, D. M. (2015). Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell, 27(4), 450–461. 10.1016/j.ccell.2015.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West, C. D. , McAdams, A. J. , & Northway, J. D. (1968). Focal glomerulonephritis in children. Histopathology and clinical observations. The Journal of Pediatrics, 73(2), 184–194. 10.1016/S0022-3476(68)80067-8 [DOI] [PubMed] [Google Scholar]
- Yang, S. , Wang, X. , Jiang, H. , Wang, Y. , Li, Z. , & Lu, H. (2017). Effective treatment of aggressive fibromatosis with celecoxib guided by genetic testing. Cancer Biology & Therapy, 18(10), 757–760. 10.1080/15384047.2017.1373215 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials