

Abstract
Background
Truncation FAM83H mutations cause human autosomal dominant hypocalcified amelogenesis imperfecta (ADHCAI), an inherited disorder characterized by severe hardness defects in dental enamel. No enamel defects were observed in Fam83h null mice suggesting that Fam83h truncation mice would better replicate human mutations.
Methods
We generated and characterized a mouse model (Fam83h Tr/Tr) expressing a truncated FAM83H protein (amino acids 1–296), which recapitulated the ADHCAI‐causing human FAM83H p.Tyr297* mutation.
Results
Day 14 and 7‐week Fam83h Tr/Tr molars exhibited rough enamel surfaces and slender cusps resulting from hypoplastic enamel defects. The lateral third of the Fam83h Tr/Tr incisor enamel layer was thinner, with surface roughness and altered enamel rod orientation, suggesting disturbed enamel matrix secretion. Regular electron density in mandibular incisor enamel indicated normal enamel maturation. Only mildly increased posteruption attrition of Fam83h Tr/Tr molar enamel was observed at 7‐weeks. Histologically, the Fam83h Tr/Tr enamel organ, including ameloblasts, and enamel matrices at sequential stages of amelogenesis exhibited comparable morphology without overt abnormalities, except irregular and less evident ameloblast Tomes' processes in specific areas.
Conclusions
Considering Fam83h−/− mice showed no enamel phenotype, while Fam83h Tr/Tr (p.Tyr297*) mice displayed obvious enamel malformations, we conclude that FAM83H truncation mutations causing ADHCAI in humans disturb amelogenesis through a neomorphic mechanism, rather than haploinsufficiency.
Keywords: amelogenesis imperfecta, hair defects, knockout mouse, neomorphic mechanism, skin defects, truncation mutation
1. INTRODUCTION
FAM83H (family with sequence similarity 83 member H on chromosome 8q24.3; OMIM *611,927) is a gene that was originally identified by gene prediction of the human genome through computational biology (Marchler‐Bauer et al., 2005). Based upon the predicted 1,179 amino acids of human FAM83H protein, the only functional domain motif was the N‐terminal domain (DUF1669) of unknown function that characterizes all eight members of the FAM83 protein family (Finn et al., 2016). Little attention was paid to FAM83H until it was reported to cause autosomal dominant hypocalcified amelogenesis imperfecta (ADHCAI, OMIM #130900) (Kim et al., 2008). ADHCAI is a specific type of inherited dental enamel malformation in humans. The defective enamel is hypomineralized with a marked decrease in hardness and is lost following tooth eruption, which imposes esthetic and functional burdens on affected individuals (Wright, Deaton, Hall, & Yamauchi, 1995). However, in spite of the high prevalence of ADHCAI in North America and the many FAM83H mutations that have been identified in ADHCAI patients, the pathogenesis of this disease is largely unknown.
To date, 27 different FAM83H autosomal dominant truncation mutations have been associated with ADHCAI (Chan et al., 2011; Ding et al., 2009; El‐Sayed, Shore, Parry, Inglehearn, & Mighell, 2010; Hart et al., 2009; Haubek et al., 2011; Hyun et al., 2009; Kantaputra, Intachai, & Auychai, 2016; Kim et al., 2008; Lee et al., 2008, 2011; Prasad et al., 2016; Song, Wang, Zhang, Yang, & Bian, 2012; Wang, Hu, Yang, Smith, Richardson, et al., 2015; Wright et al., 2009, 2011; Xin, Wenjun, Man, & Yuming, 2017; Zhang, Song, & Bian, 2015). Seven of these mutations have been found in multiple families. A contribution to the genetic etiology of ADHCAI has also been proposed for two heterozygous missense mutations, p.Ser342Thr (Pourhashemi et al., 2014) and p.Gly557Cys (Urzua et al., 2015), but their role in ADHCAI causality should be considered with caution. According to the ExAC database, the p.Gly557Cys mutation has a minor allele frequency of 3.57% in the non‐Finnish European population, suggesting that this mutation is most likely a neutral sequence variant, considering the rarity of ADHCAI. The p.Ser342Thr mutation has not been documented in the database, but the substituted amino acid is not highly conserved evolutionarily, and serine to threonine is usually not structurally significant and is predicted to be “benign” using the PolyPhen‐2 prediction algorithm of functionality, suggesting that this mutation is also not likely to be disease‐causing (Adzhubei, Jordan, & Sunyaev, 2013). Disregarding these two missense mutations of questionable causality, all of the other disease‐causing FAM83H mutations are either nonsense or frameshift mutations that generate a premature termination codon in the most 3‐prime exon of FAM83H. The resulting FAM83H transcripts apparently do not undergo nonsense mediated decay and are translated into truncated FAM83H proteins that include only the N‐terminal region of FAM83H, extending no less than 287 amino acids and no longer than 694 amino acids (out of 1,179 amino acids in the full‐length FAM83H protein). Neither truncation mutations outside of this specific region (287–694) nor other types of loss‐of‐function mutations seem to be disease‐causing. For this evident mutational homogeneity, it has been suspected that a dominant negative effect or a gain of function, rather than haploinsufficiency, underlies the pathological mechanism of ADHCAI. We previously generated and characterized Fam83h null mice, further demonstrating this hypothesis (Wang, Hu, Yang, Smith, Richardson, et al., 2015). The lack of evident enamel defects in Fam83h heterozygous or null mice suggested that FAM83H is not required for normal enamel development in mice. A simple decrease of FAM83H function, including haploinsufficiency and dominant negativity, is not the cause of defective enamel in human ADHCAI. Instead, the truncated FAM83H from the mutant allele appears to impair normal enamel development by executing certain “toxic” effects (neomorphism) in ameloblasts, which cause the observed dominant enamel malformations.
In order to discern the “toxic” effects of truncated FAM83H in ameloblasts and unravel the pathogenesis of human ADHCAI, we generated a mouse model (Fam83h Tr/Tr) by knocking in a truncation mutant Fam83h allele expressing only the N‐terminal 296 amino acids of the FAM83H protein to recapitulate the human FAM83H p.Tyr297* mutation in mice. By characterizing the enamel phenotypes of Fam83h Tr/Tr mice, we hypothesized potential disease mechanisms of ADHCAI at the cellular level and discussed difficulties and limitations of establishing a disease animal model to recapitulate a human disorder caused by neomorphic mutations.
2. MATERIALS AND METHODS
2.1. Generation of knockin mice (Fam83h Tr)
The knockin construct was designed and the mice (species: Mus musculus, strain C57BL/6) were generated by Ozgene (Perth, Australia). The Fam83h Tr mouse was generated by replacing Exon 4 of the endogenous Fam83h with a mini‐cDNA that introduced an early translation termination codon. The mini‐cDNA was composed of the whole of Exon 4 of Fam83h and the first 154 bp of Exon 5 followed by multiple polyadenylation signals. The 154th nucleotide of Exon 5 was mutated from a T to a G to introduce a stop codon (TAG) in the codon for Tyrosine 297 (TAT) of wild‐type Fam83h. The targeted Fam83h allele (Fam83hTr) is transcribed using its native promoter and translated from its native initiation codon, but generates a transcript encoding an open reading frame containing only the first 296 codons of wild‐type Fam83h, followed by multiple polyadenylation signals. This mutant transcript is predicted to produce a truncated mouse FAM83H protein (normally 1,209 amino acids) with only the N‐terminal 296 amino acids (FAM83H1‐296) within the natural physiological context of Fam83h expression in vivo. This mouse model mimics a specific human FAM83H disease‐causing mutation, p.Tyr297*. The deleted Fam83h and inserted NLS‐lacZ sequences are provided in Figures S3–S4.
2.2. Physical assessment and photography
Heterozygous mutant (Fam83h +/Tr) mice were mated to generate three mouse Fam83h genotypes (+/+, +/Tr, and Tr/Tr), which were then evaluated for their physical appearance, activity, growth rate, size difference, food intake, and fertility. The homozygous (Fam83h Tr/Tr) mice were viable and fertile. For gross evaluation of the dental enamel, mice were put under anesthesia using isofluorane, and their incisors were inspected under a dissection microscope. Seven‐week‐old mice were sacrificed and fixative‐perfused with 4% paraformaldehyde (PFA). The mandibles were removed and sliced through the mental symphysis with a razor blade to generate hemimandibles. These were carefully dissected free of soft tissues under a stereoscopic microscope using tissue forceps and a spoon excavator. The hemimandibles were submerged in 1% NaClO for 5 min, rinsed, air dried, and photographed using a Nikon SMZ1000 dissection microscope equipped with a Nikon digital camera DXM1200 (Melville, NY).
2.3. Molar histology
For histology of developing molars, Day 5, 11, and 14 mouse heads were quickly dissected free of skin, cut in half, and immersed in 4% PFA fixative overnight at 4°C and decalcified at 4°C by immersion in 1 L of 4.13% disodium EDTA (pH 7.3) with agitation. The EDTA solution was changed every other day for 19–21 days for D11 mice, and 30 days for D14 mice. The samples were washed in PBS at 4°C 4–5 times (every 0.5–1 hr) followed by one overnight wash. The samples were dehydrated using a graded ethanol series followed by xylene, embedded in paraffin, and sectioned at 5 μm thickness. For staining, the sections were rehydrated and stained with hematoxylin and eosin (H&E) stain.
2.4. Incisor histology
Fam83h +/+, Fam83h +/Tr, and Fam83h Tr/Tr mice at 7 weeks were deeply anesthetized with isoflurane, fixed by cardiac perfusion with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.2–7.4) containing 0.05% calcium chloride, postfixed for 2 hr at 4°C, and rinsed three times for 15 min each with 0.1 M sodium cacodylate buffer. The samples were decalcified at 4°C by immersion in 4 L of 4.13% disodium ethylenediaminetetraacetic acid (EDTA, pH 7.3) with agitation. The EDTA solution was changed every other day for 30 days. The samples were washed in PBS at 4°C 4–5 times every 0.5–1 hr, washed overnight, postfixed for 2 hr in 1% osmium tetroxide/1.5% potassium ferrocyanide, dehydrated using an acetone gradient, embedded in Epon812 substitute, semi thin‐sectioned and stained with 0.1% toluidine blue as described elsewhere (Smith, Hu, et al., 2011). Three maxillary (one from each genotype), two Fam83h +/+, two Fam83h +/Tr, and five Fam83h Tr/Tr mandibular incisors were processed for longitudinal sectioning. Three Fam83h +/+ and four Fam83h Tr/Tr mandibular incisors were processed for cross sectioning at 1 mm increments as described previously (Hu et al., 2016).
2.5. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE)
The detailed protocol of protein extraction from mouse molars and the analyses were previously described (Wang, Hu, Yang, Smith, Nunez, et al., 2015; Yamakoshi et al., 2011). Briefly, mouse maxillary and mandibular first Fam83h +/+; Fam83h +/Tr; Fam83h Tr/Tr; Fam83h− /Tr molars were extracted from mouse pups at D5, D11, and D14 using a dissecting microscope, stripped of soft tissue, and the mineralized portion of the teeth was incubated in 1 ml of 0.17 N HCl/0.95% formic acid for 2 hr at 4°C. After the undissolved materials were removed, the crude protein extract was buffer exchanged with 0.01% formic acid. The concentrate containing proteins extracted separately from the four molar genotypes was raised back to 250 μl of 0.01% formic acid and used for subsequent analysis by SDS‐PAGE stained with Coomassie Brilliant Blue (CBB). The amount of protein applied per lane for SDS‐PAGE was ~1/6 of the extract from a single molar. Recombinant mouse amelogenin (rM179) was used as a size control (Simmer et al., 1994).
2.6. Backscattered scanning electron microscopy
The procedures of backscattered scanning electron microscopy (bSEM) were described previously (Smith, Richardson, et al., 2011). Soft tissue was removed from the left and right hemimandibles of 7‐week‐old mice, and cross‐sectioned at 1 mm increments from the basal (growing) end of the incisors and imaged by bSEM using a JOEL 7,800 SEM at 20 kV (JOEL USA, Inc; Peabody, MA, USA). For whole incisor surface imaging, the bony soft tissue and bony caps covering the mandibular incisors were carefully removed and examined at 50X magnification with a Hitachi S‐3000N variable pressure SEM using the backscatter mode at 25 kV and 20 pascal pressure.
For molar surface SEM, the molars were prepared as follows: The D14 hemimandibles were submerged in 4% PFA overnight, carefully dissected of soft tissues under a stereoscopic microscope, submerged in 1% NaClO for 20 min, rinsed, and acetone dehydrated (30%, 50%, 70%, 80%, 90%, 100%). The hemimandibles were mounted on metallic stubs using conductive carbon cement and carbon coated to increase conductivity and examined using a Hitachi (Century City, Los Angeles, CA) S‐3000N variable pressure SEM using the backscatter mode.
2.7. Hardness evaluations
The procedures for tissue preparation and microhardness testing were previously described (Wang, Hu, Yang, Smith, Nunez, et al., 2015). In brief, hemimandibles from 7‐week‐old mice were cleaned free of soft tissue and embedded in Epon resin following graded acetone dehydration. After polymerization at 60°C, the incisors were cross‐sectioned 8 mm from the apical end of the incisor, approximately at the level of the crest of the alveolar bone close before the incisor erupts into the mouth. The cross‐sectioned incisor was reembedded in Castolite AC (Eager Polymers, Chicago, IL) using 25‐mm SteriForm molds (Struers Inc., Westlake, OH), allowed to harden overnight, and polished.
Microhardness testing was performed using a LM247AT microhardness tester (Leco Corp., St. Joseph, MI) with a load of 25 g for 10 s with a Knoop tip to obtain a Knoop hardness number (KHN). Measurements were made at 500X magnification. Indentations were placed in the outer, middle, and inner enamel as well as the dentin as a control reading for a total of four indentations per row. This series was performed three times in each animal, for a total of twelve points per animal. Hardness Data points were treated as independent, unweighted numbers and subjected to one‐way ANOVA than Tukey's HSD test using calculation spreadsheet at http://vassarstats.net/anova1u.html.Statistical significance was determined atp < 0.01.
Nano hardness test was done on (Hysitron TI 950 Triboindenter, Hysitron‐Bruker, Minneapolis, MN, USA). Using the nano DMA indenter, the loading force was 8,000 µN, 15 indents on each incisor spread among dentin, inner enamel, middle enamel and outer enamel. The data was analyzed by using Hysitron Troboscan.
2.8. Microcomputed tomography (µCT)
Sample preparation and imaging process for microcomputed tomography (µCT) were conducted as previously described (Yang et al., 2015). Briefly, the agarose‐embedded hemimandibles were scanned at 8 μm and analyzed with the SCANCO μCT‐100 series microcomputed tomography system. The data were analyzed using the MiiL program (Medical Image Illustrator; National Applied Research Laboratories, Taipei, Taiwan). For calculating the hard tissue volumes from the 3D molar images, the contour of each first molar was outlined by marking the border of the tooth on each scanning section. Three dimensional images were generated within the contour defined area. Based upon the distinct radiodensity of each tissue, the enamel, dentin, and pulp of the molars were isolated using density threshold ranges to measure the tissue volumes. The threshold range for measuring the pulp was 0 to 136, for dentin was 137 to 225, and for enamel was 226 to 255. Pulp, dentin, and enamel volumes as defined by these threshold ranges were calculated using the MiiL Program. The total volume of the whole tooth was the sum of volumes from its enamel, dentin, and pulp. The proportion of each tissue within a tooth was then calculated. The volume ratios of enamel to dentin (E/D) and enamel to dentin‐pulp complex (E/D + P) were also evaluated.
3. RESULTS
3.1. Generation of truncated Fam83h‐knockin mice
In order to recapitulate human FAM83H disease‐causing mutations and investigate the pathogenesis of human ADHCAI, we generated a mouse model expressing a truncated (Tr) FAM83H protein in the C57BL/6‐background. The structures of wild‐typeFam83h, the Fam83h targeting construct, the Cre‐inverted intermediate, and the final Fam83h Tr gene and strategy for PCR genotyping are provided in Figure S1. We also provide relevant DNA sequences, such as the wild‐type mouse Fam83h gene sequence and structure (Figure S2), the knockin intermediate following removal of the selection genes using Flp‐FRT recombination (Figure S3), and the final Fam83h Tr knockin sequence (Figure S4).
Intercrosses of heterozygous (Fam83h+/ Tr) mice produced pups of three genotypes at the expected Mendelian ratio. The homozygous (Fam83h Tr/Tr) mice were viable and fertile. At birth, they were indistinguishable from their wild‐type (Fam83h +/+) and heterozygous (Fam83h+/ Tr) littermates. However, when inspected at 7‐weeks, the homozygous mutants (Fam83h Tr/Tr) appeared normal in size but exhibited a sparse and scruffy coat, which allowed them to be readily discerned from their heterozygous and wild‐type littermates. Despite the coat distinctness of 7‐week Fam83h Tr/Tr mice, their incisors, when inspected under a dissection microscope (Figure 1), appeared normal with glossy enamel surfaces and sharp incisal tips, comparable to those of their Fam83h+/ Tr and Fam83h +/+ littermates. However, when carefully viewed from the lateral perspective (Figure 1b), the incisor enamel did not show a normal color of glazy yellow and a continuous curved line of light reflection, but instead exhibited a dull‐white appearance with disrupted patchy areas of light reflection. Basic tooth morphology looked comparable among the three genotypes, although the molar enamel surface textures were not as smooth as those of the wild‐type mice (Figure 1c). Notably the extensive posteruption attrition of enamel that is pathognomonic of the ADHCAI phenotype caused by heterozygous FAM83H truncation mutations in humans was absent from the mouse heterozygous molars and mild even in the Fam83h Tr/Tr molars.
Figure 1.
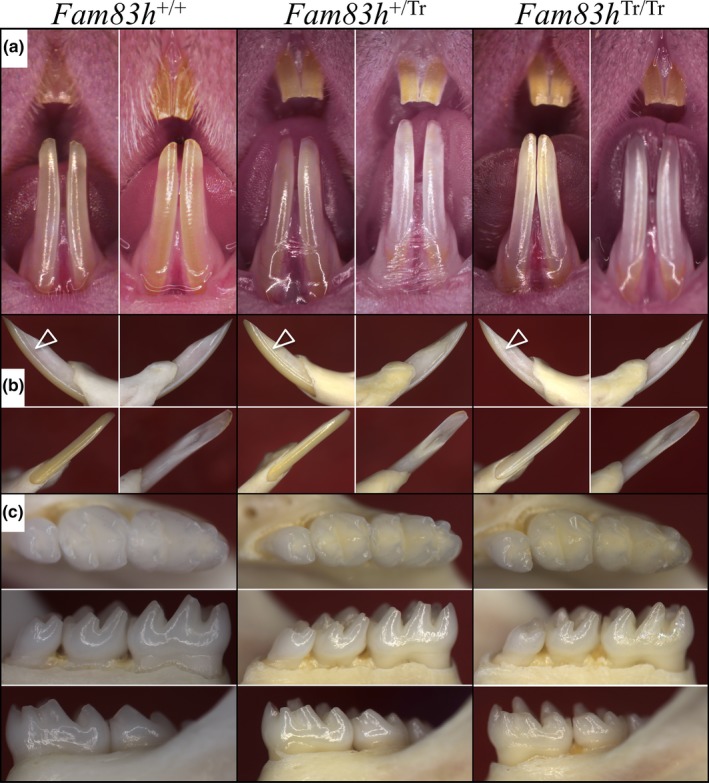
Photographs of Mouse Dentitions at 7‐weeks. (a) Frontal views of incisors. The teeth from all three genotypes look similar, with no signs of enamel chipping. (b) Going clockwise from upper left for each genotype: lateral, mesial, lingual, labial views of a mandibular incisor. Arrowheads indicate the lateral surface of the incisor enamel where the Fam83h Tr/Tr mice show a dull‐white surface rather than the reflective yellow surface of the Fam83h +/+ and Fam83h +/Tr mice. (c) Occlusal, lingual, and buccal views of the mandibular incisors. All molars show normal crown morphology without evidence of attrition, although the Fam83h Tr/Tr molars exhibited a subtle roughness that reduced its luster
3.2. Enamel defects in Fam83h Tr/Tr molars
To assess the potential enamel phenotypes in detail, we examined 7‐week molars using backscattered scanning electron microscopy (bSEM; Figure 2; Figure S5). The basic morphology of the molars was comparable in the three genotypes, although the Fam83h Tr/Tr molars sometimes appeared to be mildly smaller than the Fam83h+/ Tr and Fam83h +/+ molars, and their cusps seemed slender compared to those of the Fam83h+/ Tr and Fam83h +/+ mice, suggesting that their enamel layer might be thinner. The Fam83h Tr/Tr molar surfaces were less smooth than those of the Fam83h+/ Tr and Fam83h +/+ molars, confirming the impression made under dissection microscope. When inspected from the occlusal view, the cusp tips of Fam83h Tr/Tr first molars exhibited only slightly larger wear facets, suggesting that the Fam83h Tr/Tr molars had undergone similar mild attrition relative to the Fam83h+/ Tr and Fam83h +/+ molars.
Figure 2.
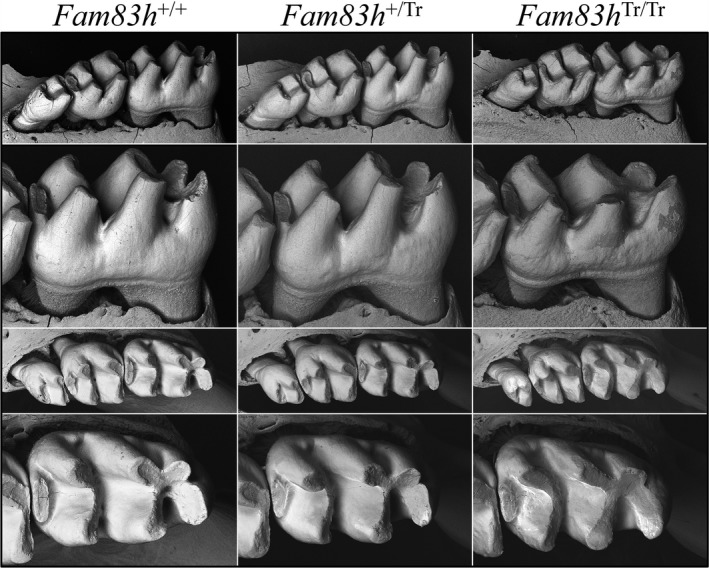
bSEM of Mandibular Molars at 7‐weeks. The homozygous Fam83h Tr/Tr mice show a mildly rougher surface texture than the Fam83h +/+ and Fam83h +/Tr mice. The first molars erupt into function after 2 weeks. By 7‐weeks (shown here) signs of abnormal attrition are suggested by shorter, broader cusps. The Fam83h Tr/Tr first molar (on the right) shows mildly reduced cusp height in the lateral view and broadened cusp width in the occlusal view, suggesting mildly increased attrition relative to the Fam83h +/+ and Fam83h +/Tr mice. Similar bSEM views of other 7‐week mouse molars from these genotypes are provided in Figure S5
We removed the overlying soft tissue covering the unerupted (but nearly erupted) Day 14 molars and characterized them using bSEM (Figure 3). The unerupted Fam83h Tr/Tr molars were similar to the 7‐week Fam83h Tr/Tr molars that had functioned in occlusion for 5 weeks by also exhibiting enamel surface roughness and cusp slenderness, indicating that these phenotypes resulted from developmental defects rather than posteruption changes.
Figure 3.
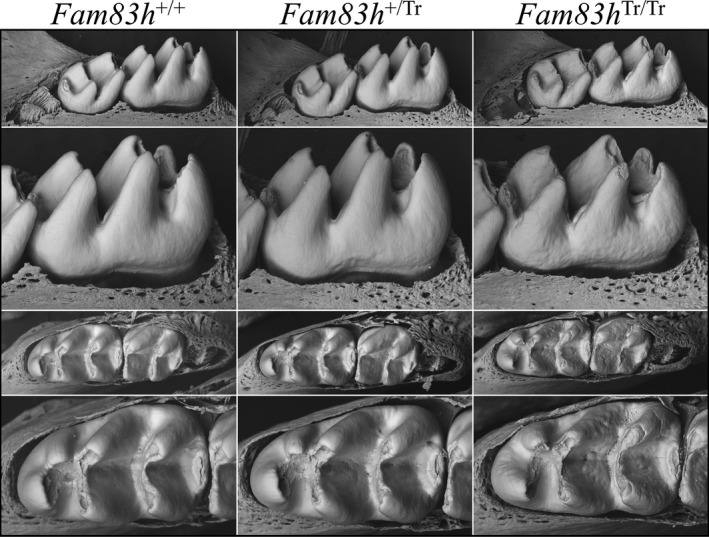
bSEM of D14 Mandibular Molars. Day 14 is the day before the first molar erupts into the oral cavity and is often the last day that the developmental form of the molars can be observed prior to potential attrition from function. Lingual views are shown on the upper two panels, and occlusal views on the lower two panels. The crowns from all three genotypes are similar, but the homozygous Fam83h Tr/Tr molars display a rougher surface texture. Note in the occlusal views that the molar cusp tips are thin and pointed on all teeth, as they have not yet undergone occlusal wear
To better assess the possibilities of smaller molar size and thinner enamel suggested by the dissection and bSEM microscopy, we performed µCT analyses to measure the volumes of enamel, dentin, and pulp in mandibular first molars at Day 14. Specific ranges of µCT threshold values were set to isolate each tissue (pulp, dentin and enamel): 0–136 for pulp, 137–225 for dentin, and 226–255 for enamel. The means of the measurements were calculated from six wild‐type, five heterozygous, and six homozygous D14 mice. There were no statistically significant differences in the volumes of the three dental tissues between the wild‐type and heterozygous mice. In contrast, the total molar volume, and the volumes of dentin, enamel were reduced between the Fam83h +/+ and the Fam83h Tr/Tr mice, and these differences were statistically significant (Figure 4). The total tooth, dentin, and enamel volumes decreased to 84%, 87%, 62% of those of the wild‐type, respectively. Overall, the mandibular first molars of the Fam83h Tr/Tr mice were smaller than those of the wild‐type. As the proportion of dentin increased in the homozygous (77%) relative to the wild‐type (75%), while the proportion of enamel fell in the homozygous (9.7%) relative to the wild‐type (13.2%), it is evident that the Fam83h Tr/Tr molars are smaller than those of the wild‐type principally because of a reduction in enamel volume, confirming a hypoplastic enamel defect in Fam83h Tr/Tr molars.
Figure 4.
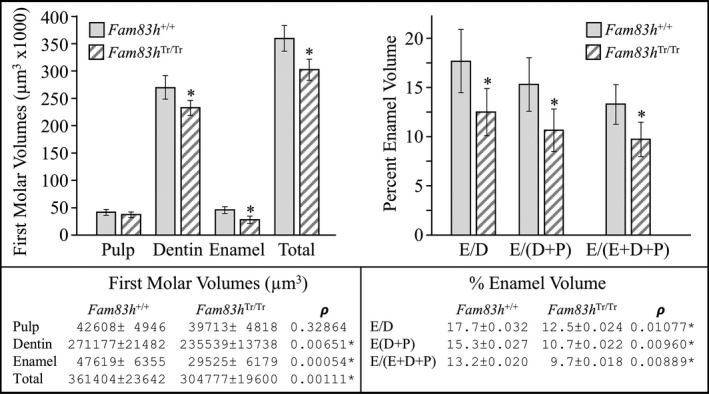
MicroCT Analyses of D14 Mandibular Molars. Day 14 precedes slightly the eruption of the first molar into function, before the potential effects of attrition on the volumes of enamel and dentin can occur. No statistically significant molar volumes were observed between the Fam83h +/+ and Fam83h +/Tr and are not displayed. Total molar volume was reduced in the Fam83h Tr/Tr to 84% that of the wild‐type. The volume of dentin was reduced by a lesser amount (to 87%), indicating that part of the total volume reduction was due to an overall reduction in tooth size. The volume of enamel was reduced in the Fam83h Tr/Tr to 62% that of the wild‐type indicating that most of the reduction in molar size was due to a reduction in enamel volume
We analyzed the histology of developing maxillary first molars at postnatal D5, D11, and D14 for three genotypes (Figure 5; Figure S6). All D5 first molars showed typical tall‐columnar secretory stage ameloblasts with accumulation of eosinophilic extracellular matrices in the enamel. Similarly, in D11 first molars of all genotypes, the maturation stage ameloblasts appeared comparable adjacent to an enamel space with some residual matrix. At D14, when the molar was about to erupt into the oral cavity, all molars of the three genotypes exhibited a thin layer of reduced enamel epithelium outlining the enamel space containing little residual matrix. Odontoblasts, developing dentin‐pulp complexes, developing roots, and surrounding periodontal tissues of D5, D11, and D14 first molars seemed to be comparatively normal in Fam83h+/ Tr, and Fam83h Tr/Tr mice. Light microscope histology of developing first molars did not provide evidence of pathology that could explain the enamel hypoplasia or surface roughness.
Figure 5.
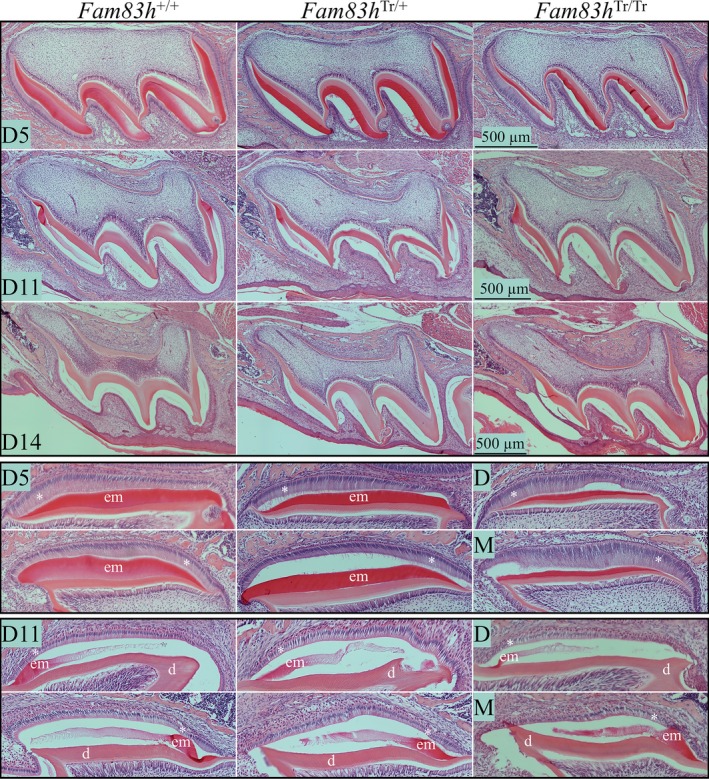
Histology of Fam83h +/+ (WT), Fam83h Tr/+, and Fam83h Tr/Tr Molars at Days 5, 11, and 14. Key: d, dentin; em, enamel matrix; *, ameloblasts
We also utilized SDS‐PAGE to analyze crude protein extracts from D5, D11, and D14 first molars of Fam83h+/+, Fam83h+/ Tr, and Fam83h Tr/Tr mice (Figure S7). No clear differences in the amount of matrix proteins were observed among the first molars of the different genotypes at D5, D11, and D14, which was consistent with the molar histology findings that ameloblast form and enamel architecture were not grossly altered in the Fam83h+/ Tr and Fam83h Tr/Tr mice.
3.3. Enamel defects in Fam83h Tr/Tr mandibular incisors
The mineralization of mandibular incisors was analyzed by comparing backscattered scanning electron microscopy (bSEM) images of 1‐mm incremental cross sections (from level 2 to 8) progressing from early (apical) to late (incisal) stage of development along the continuously growing mandibular incisors from Fam83h+/+, Fam83h+/ Tr, and Fam83h Tr/Tr mice. The incisor cross sections from all levels were imaged at three magnifications and the three genotypes compared at each magnification. These magnifications (from low to high) show the entire incisor cross sections (Figure S8), the enamel layer and underlying dentin (Figure S9), and the enamel layer at the height of contour (Figure S10), where the enamel is thickest and its rod pattern most evident. These comparisons, along with the study of bSEM image panels showing six Fam83h +/Tr (Figures S11–S16), six homozygous (Fam83h Tr/Tr) (Figures S17–S22), and one wild‐type (Fam83h +/+) (Figure S23) incisors at 1 mm cross sections at all three magnifications, demonstrated virtually normal progression of dentin and enamel mineralization in all three genotypes. The gist of these findings is evident in Figure 6, which shows a panel of bSEM images of incisor cross sections at level 8 that show the enamel layer after it had completed maturation and was about to erupt into the oral cavity. The enamel layer at level 8 in the WT, heterozygous (Fam83h +/Tr) and homozygous (Fam83h Tr/Tr) mice exhibited comparable thickness (from the DEJ to the enamel surface at the height of contour), electron density (reflecting degree of mineralization) and overall enamel rod patterns at sequential sections among the three genotypes, indicating that expression of the truncated FAM83H protein did not interfere with enamel and dentin mineralization, making it unlikely that enamel matrix secretion was disturbed. However, when inspected carefully, the lateral (lateral) 1/3 of the enamel layer of Fam83h Tr/Tr incisors was thinner relative to the Fam83h+/ Tr and Fam83h +/+ incisors (Figure 7). While the labial enamel surface of a normal incisor exhibited a smooth line gradually approaching the lateral cervical margin on a cross‐section, the contour of Fam83h Tr/Tr enamel abruptly steepened down at the lateral 1/3. This was evident at incremental sections starting from level 4, which represented the postsecretory (maturation) stage of enamel formation. In contrast, the dentin and surrounding alveolar bone of mandibular incisors in all three genotypes appeared to be comparable on bSEM cross sections.
Figure 6.
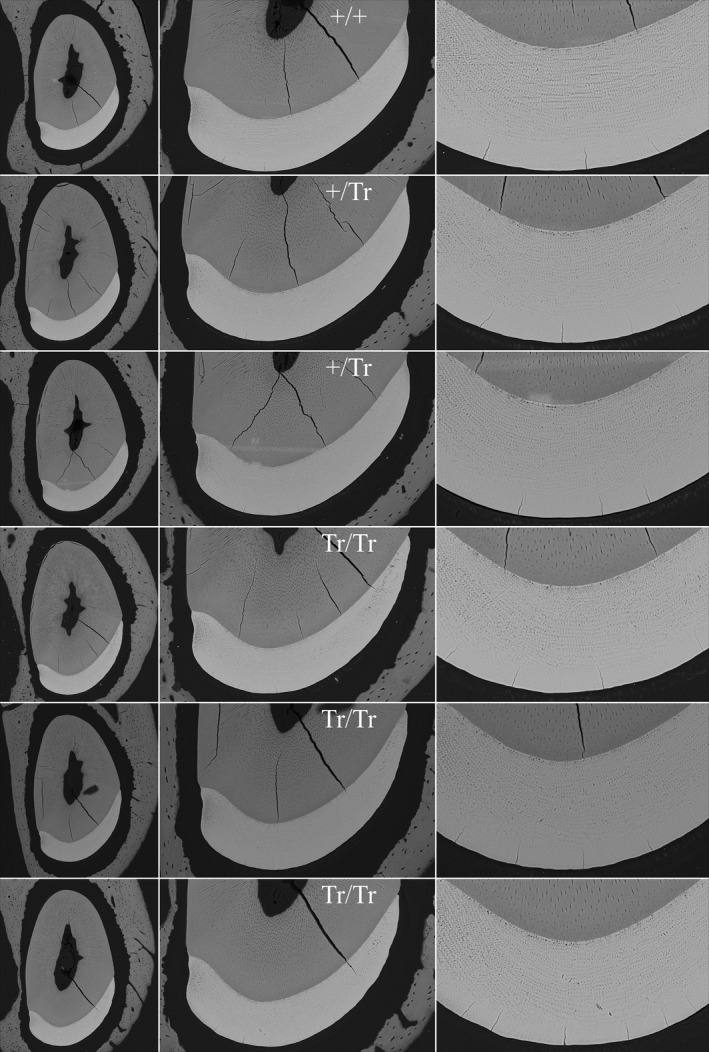
bSEM of 7‐week mandibular incisor cross sections. These incisor cross sections show the enamel layer at a level near the crest of the alveolar bone (level 8), which is prior to eruption into the oral cavity but after the enamel has undergone extensive maturation. The enamel looks similar in the WT (+/+), heterozygous (+/Tr) and homozygous (Tr/Tr) mice. The enamel of the Fam83h Tr/Tr incisors appears to have normal centro‐labial thickness, rod patterns and degree of mineralization. The only difference is in the contour of the enamel as it approaches the lateral side, so that the enamel layer is thinner than normal as it approaches the lateral cervical margin
Figure 7.
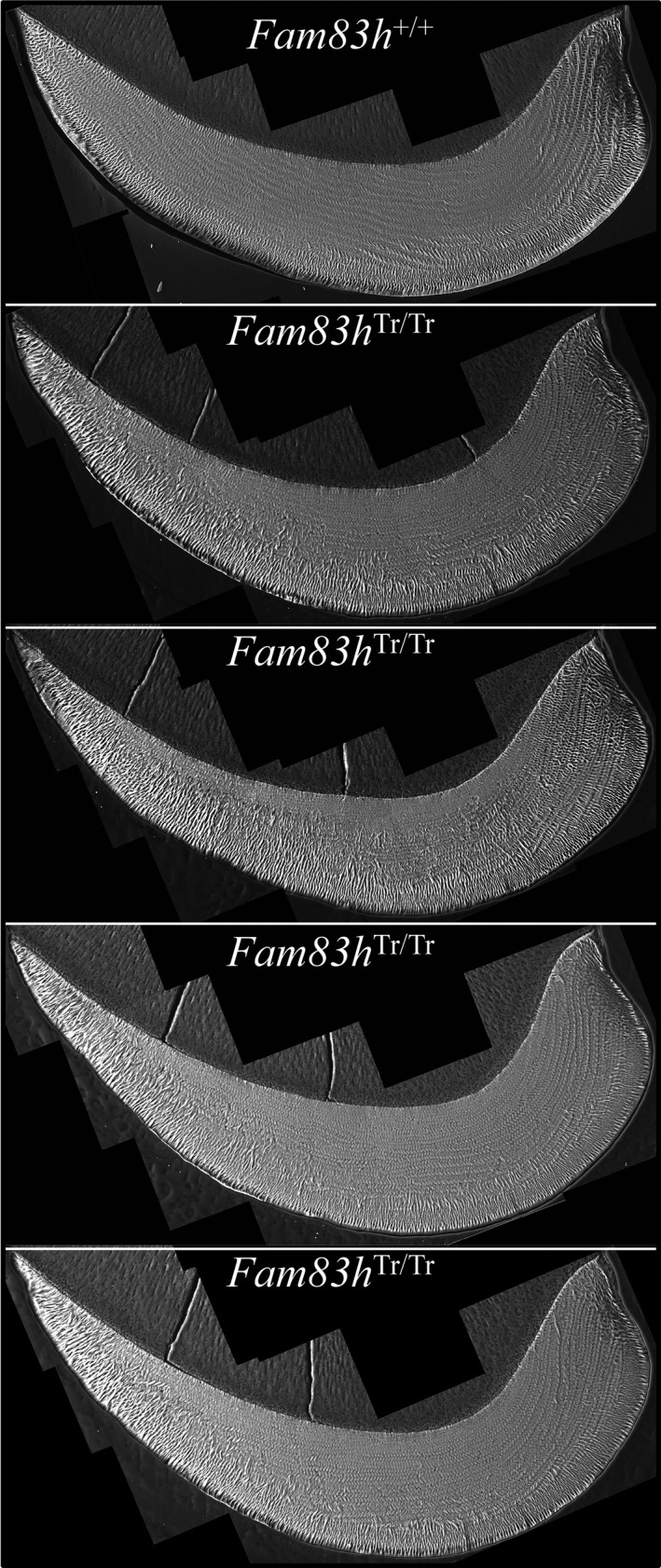
bSEM of 7‐week Mandibular Incisor Cross Sections. These incisor images are mosaics of cross sections imaged at high resolution. Note how the enamel layer thins prematurely as it approaches the lateral cervical margin and many of the enamel rods appear to be in the plane of section
To quantify this hypoplastic enamel defect, we analyzed the mesial and lateral enamel areas of level 8 cross sections from 10 Fam83h+/+ and eight Fam83h Tr/Tr 7‐week mandibular incisors (Figure 8; Figure S24). A bisecting line perpendicular to a line segment connecting the mesial cervical margin and the lateral cervical margin was used to divide the incisor enamel into mesial and lateral parts, and the areas of each part and the total enamel were calculated. In the wild‐type incisor, the average area of total enamel was around 73,700 µm2, roughly three‐fifths (45,600 µm2) of which was mesial enamel and two‐fifths (28,100 µm2) was lateral enamel. In contrast, the Fam83h Tr/Tr incisor exhibited significantly smaller areas of total and lateral enamel (66,800 and 22,300 µm2 respectively), but not mesial enamel (44,500 µm2). While the mesial enamel area appeared to be comparable between the wild‐type and Fam83h Tr/Tr incisors, the lateral enamel area of the Fam83h Tr/Tr incisor was less than 80% that of the wild‐type incisor. This uneven reduction of enamel area changed the proportion of lateral to mesial enamel from 2:3 to 1:2, confirming a thickness defect in enamel on the lateral aspect of a Fam83h Tr/Tr mandibular incisor.
Figure 8.
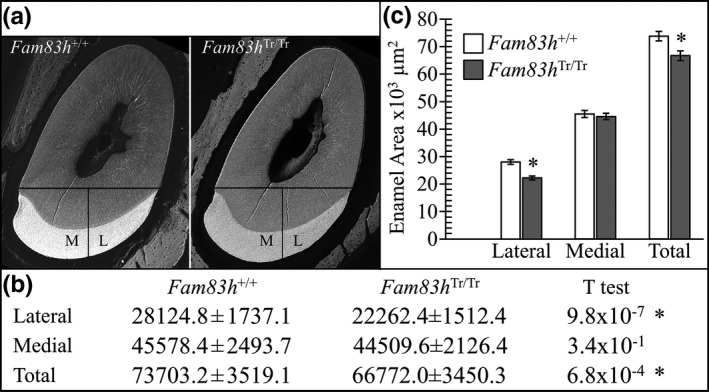
Enamel area on 7‐week mandibular incisor level 8 (alveolar crest) cross sections. (a) bSEM images showing how the enamel was divided into mesial and lateral regions by drawing a line from the mesial cervical margin to the lateral cervical margin and drawing a line perpendicular from its midpoint. The mesial, lateral, and total cross‐sectional areas (mm2) were determined by outlining the enamel on Photoshop (Fam83h +/+ n = 8; Fam83h Tr/Tr n = 10; see Figure S24). (b) Table showing the average areas and ρ values (T test). All ρ values 0.01 or below were considered to be significant (*). (c) Plot of the average lateral, mesial, and total surface areas of Fam83h+/+ (n = 8) and Fam83h Tr/Tr (n = 10) mice. Significant differences were observed between the wild‐type and the Fam83h homozygous truncation (Fam83h Tr/Tr) incisors in their lateral and total cross‐sectional areas, but not in the mesial enamel
To assess potential alteration of enamel functionality in Fam83h Tr/Tr mice, we conducted Knoop hardness testing (KHT) and Nano hardness testing (NHT) of dentin, the inner enamel (near dentin), the middle enamel, and outer enamel (near the surface) on 7‐week incisors of the three genotypes at level 8 (Figure 9; Figures S25–S27). However, neither Knoop hardness values nor Nano hardness values in respective areas of enamel among Fam83h+/+, Fam83h+/ Tr, and Fam83h Tr/Tr mice showed a significant difference. All samples demonstrated a rising trend of hardness values from dentin to the enamel surface, with dentin being the lowest and outer enamel the highest. Noticeably, for NHT, we included two indentation locations (location N and location O; Figure S26) at the lateral aspect of the incisor where we found reduced enamel thickness and altered rod–interrod structure. Nonetheless, the three genotypes exhibited comparable hardness values at both locations, indicating no apparent hardness defect at this area.
Figure 9.
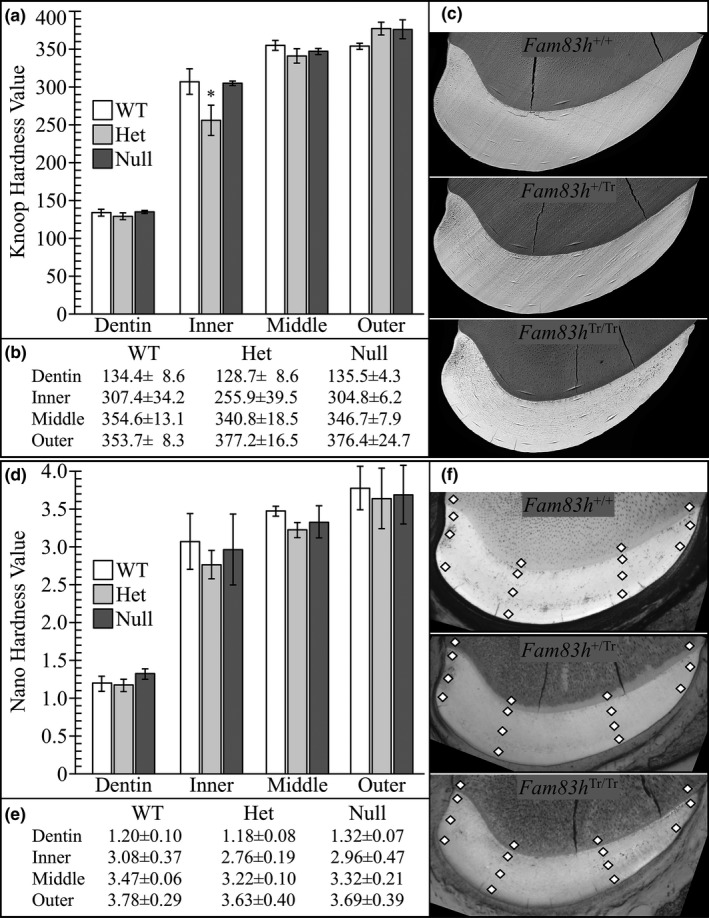
Micro and Nano Hardness Plots. Indents in dentin, inner enamel, middle enamel, and outer enamel on 7‐week mandibular incisor cross sections at the level of the alveolar crest (level 8). (a) Plot of the average calculated Knoop Hardness Value for Fam83h+/+, Fam83h+/ Tr and Fam83h Tr/Tr mice for dentin and enamel. Knoop hardness = 14.229p/d2; p = force (kgf) applied; d = length (mm) of the long diagonal of the indent. (b) Average calculated Knoop hardness values with standard deviations (Microhardness values of individual indents see Figure S25). (c) SEM images of selected microindented samples to show indent locations. Images of the entire set of samples are provided in Figure S25. (d) Plot of the average calculated nanohardness values in gigapascals: Gpa = 14.229p/d2; p = force (N) applied; d = length (mm) of the long diagonal of the indent (Figure S26). A ρ < 0.01 was considered significant. All ρ values were above 0.01 and did not support a hypothesis of significant hardness differences among the three genotypes. (e) Average nanohardness values with standard deviations. (f) SEM images of selected nano‐indented samples to show indent locations (indicated by diamond shapes). Images of the entire set of samples are provided in Figure S27
3.4. Histological analyses of Fam83h incisors at 7‐weeks
We analyzed the histology of longitudinal sections of continuously growing maxillary (Figures S28–S30) and mandibular (Figures S31–S48) incisors of 7‐week Fam83h+/+, Fam83h+/ Tr, and Fam83h Tr/Tr mice, which exhibited all stages of enamel formation. The basic morphology of the enamel organ, including ameloblasts, and enamel matrices at differentiation, secretory, transition, and maturation stages appeared comparable in the three genotypes, although the secretory stage ameloblasts of Fam83h Tr/Tr incisor gave the impression of being shorter, with a likely increased nucleus‐to‐cell height ratio, compared to those of Fam83h+/+ and Fam83h+/ Tr incisors (Figure 10). The Fam83h Tr/Tr incisor also exhibited normal onset of ameloblast differentiation and transition through all stages of amelogenesis without apparent delay, advance, or interruption. Furthermore, a clear enamel space with no residual staining was consistently observed beneath late maturation stage ameloblasts (Figures S28, S32, S34, S36, S38 and S40), which indicated that the process of enamel maturation in the Fam83h Tr/Tr incisor had proceeded normally.
Figure 10.

Histology of Mandibular Incisor Longitudinal Sections at 7‐weeks. Top: Apical region showing the secretory stage starting at the onset of mineralization. Bottom: Central region showing the end of the secretory stage, the transition stage (delineated by arrowheads), and early maturation stage. Ameloblast histology appears similar in the Fam83h +/+, Fam83h +/Tr, and Fam83h +/Tr mice, although the ameloblasts give the impression of being shorter in the Fam83h +/Tr and Fam83h Tr/Tr mice
Being aware of a hypoplastic defect at the lateral aspect of Fam83h Tr/Tr incisor, we examined the histology of amelogenesis in 1‐mm incremental cross sections (from level 1 to 10; Figures S49–S56) analogous to the previously‐mentioned bSEM analyses, including high magnification montages of the incisor cross sections (Figures S57–S61). Ameloblasts reverse their secretory polarity during differentiation to allow for extracellular release of large amounts of proteins from plasma membrane surfaces that were originally the embryonic bases of the cells (Smith & Nanci, 1995). In general, level 1 showed the onset of secretory stage with elongated ameloblasts being reversely‐polarized and developing Tomes' processes, while the midsecretory stage could be observed at level 2 showing tall‐columnar ameloblasts with well‐established Tomes' processes tilting mesially or laterally. The transition stage usually appeared around level 3, followed by the maturation stage extending from levels 3 through 8. With gradual removal of enamel matrix proteins and increased mineralization throughout the maturation stage, staining of the enamel matrix was barely observed after level 5. Consistent with the findings from longitudinal sections, the Fam83h Tr/Tr incisors exhibited comparable histology without overt morphological abnormalities at all section levels, relative to the Fam83h+/+incisors. However, at level 2 (mid‐secretory stage), the ameloblasts over the lateral aspect of Fam83h Tr/Tr incisor showed less evident Tomes' processes than those of wild‐type at the comparable area (Figure 11). Also, in some Fam83h Tr/Tr samples, the Tomes' processes of ameloblasts at the central area of level 2 were irregular in the enamel layer, which was rarely observed in the wild‐type samples (Compare the wild‐type Level 2 ameloblasts near the lateral cervical margin of Figure S57 to the Fam83h Tr/Tr ameloblasts in the same position in Figures S58–S61).
Figure 11.
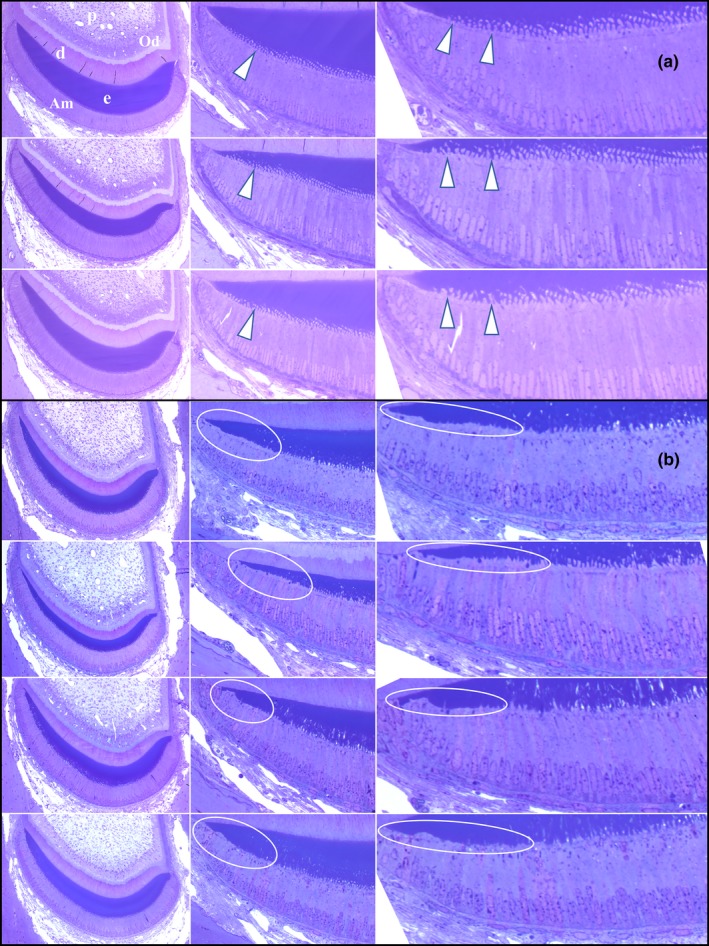
Histology of Secretory Stage Ameloblasts on the Lateral Aspect of 7‐weeks Mandibular Mouse Incisors. (a) Cross sections of three wild‐type incisors at Level 2. Arrowheads indicate Tomes' Processes extending into the enamel. Key: Am, ameloblasts; d, dentin; e, enamel; od, odontoblasts; p, pulp. Ameloblasts (Am) covering the lateral aspect of wild‐type mandibular incisors show prominent Tomes' processes (arrowheads). (b) Comparable images from four Fam83h Tr/Tr incisors showing ameloblast lateral membranes apparently lacking Tomes Processes (circled)
4. DISCUSSION
We recently demonstrated that Fam83h null mice exhibited no overt enamel defects, which suggested a nonessential role for FAM83H in enamel formation and supported a neomorphic disease mechanism of human autosomal dominant hypocalcified amelogenesis imperfecta (ADHCAI) (Wang, Hu, Yang, Smith, Richardson, et al., 2015). An amorphic (loss of function) disease mechanism for ADHCAI is ruled out in humans because only truncation mutations in a specific region of the last exon cause ADHCAI, whereas missense mutations in conserved positions and nonsense mutations in other parts of the gene that could cause a loss of function are not observed in ADHCAI patients. In mice, an amorphic mechanism for ADHCAI is ruled out because the Fam83h heterozygous and null mice do not exhibit an enamel phenotype (Wang, Hu, Yang, Smith, Richardson, et al., 2015). In this study we generated knockin (Fam83h Tr/Tr) mice with a truncation defect (p.Tyr297*) homologous to those that cause human ADHCAI. The (Fam83h Tr/Tr) mouse molars showed rough enamel surfaces and slender cusps while mandibular incisors showed enamel hypoplasia in their lateral enamel, indicating disturbed matrix secretion during enamel formation.
The mainly hypoplastic phenotype of Fam83h Tr/Tr enamel indicated that the secretory stage was more affected by the truncated FAM83H protein than was the maturation stage. We previously showed that in mouse molars, Fam83h expression in ameloblasts was generally weak and barely detectable until the transition to the maturation stage (Wang, Hu, Yang, Smith, Richardson, et al., 2015). This inconsistency suggests that mouse secretory stage ameloblasts are more highly vulnerable to the “toxic” effect of truncated FAM83H, given that their expression of the protein is low. Maturation stage ameloblasts were more resistant, since the molar enamel hardness of Fam83h Tr/Tr mice was only slightly reduced. Although Fam83h Tr/Tr enamel was malformed, the defects were considerably different from those observed in ADHCAI patients. In human ADHCAI, the enamel of affected individuals is of normal thickness in unerupted or newly erupted teeth, but is hypomineralized, soft, and undergoes rapid attrition following eruption. It is unknown if human ameloblasts express FAM83H at higher levels than mouse ameloblasts, which might account for the more severe enamel defects in ADHCAI patients. Alternatively, human ameloblasts, particularly the maturation stage ones, might be more sensitive to the “toxic” effect of the mutant FAM83H. This potential difference in FAM83H expression level and ameloblast susceptibility to the mutant protein might explain why enamel defects could be seen in homozygous (Fam83h Tr/Tr), but not heterozygous (Fam83h +/Tr) mutant mice, while a single copy of mutant FAM83H can cause ADHCAI in humans.
An interesting feature of Fam83h Tr/Tr incisors was that enamel defects were only evident on the lateral aspect. The mutant FAM83H protein in mice more significantly impacts appositional growth of enamel over the lateral aspect (of the incisor) than the mesial, causing a localized hypoplastic defect. In that area the enamel was thinner, but also the normal pattern of enamel rod rows was lost (Figure 7). Interestingly, the altered Fam83h Tr/Tr incisor did not seem to affect enamel functionality, namely hardness, since we did not detect significant differences with either KHT or NHT.
Tomes' processes are cell projections of secretory stage ameloblasts into the enamel matrix. They are considered to be critical for establishment of enamel rod‐interrod structure. Aberration of Tomes' processes on the lateral aspect of incisors might explain the hypoplastic enamel in this area of Fam83h Tr/Tr incisors. Kuga et al. recently showed that FAM83H and its associated protein CK‐1ε colocalized with keratin filaments in mouse ameloblasts, and demonstrated that FAM83H and CK‐1 regulate the organization of the keratin cytoskeleton, and maintain the formation of desmosomes in human ameloblastoma cell lines (Kuga et al., 2016). Formation of Tomes' processes by ameloblasts reflects a significant reorganization of the cytoskeleton, so perhaps truncated FAM83H disturbs the organization of the cytoskeleton, although the normal FAM83H protein is not critical for proper organization of the keratin cytoskeleton in ameloblasts, given the fact that there are no evident enamel defects in Fam83h null mice (Wang, Hu, Yang, Smith, Richardson, et al., 2015).
Another interesting finding of Fam83h Tr/Tr enamel defects was that the molars appeared more affected and showed more prominent hypoplastic defects than the incisors did. This discrepancy might result from a fundamental difference in enamel structure between molars and incisors. The spatial distribution and three‐dimensional arrangement of enamel types, namely schmelzmuster, in rodents have been well characterized and documented by paleontologists for phylogenetic purposes (von Koenigswald, 2004a, 2004b; von Koenigswald & Clemens, 1992). In most myomorph rodents, including mice (Mus musculus), the enamel microstructure of incisors comprises an inner layer of uniserial lamellar enamel (inner enamel) and an outer layer of radial enamel (outer enamel), as described above (von Koenigswald & Clemens, 1992; Moinichen, Lyngstadaas, & Risnes, 1996). The inner layer exhibits a decussation structure on cross sections and an architecture of Hunter‐Schreger Bands (HSB) on sagittal (longitudinal) sections. On the other hand, the molars showed a more complicated enamel microstructure, named C‐type schmelzmuster (von Koenigswald, 2004b). A basal ring of lamellar enamel (BRLE) surrounds the cervical part of the crown, covered by an outer layer of radial enamel, while the upper part of the enamel cap is formed by mostly radial enamel with rods perpendicular to the enamel surface. The uniserial HSBs in incisors are identical to the BRLE in molars. The truncated FAM83H protein disturbed the ameloblast Tomes' processes and altered the enamel rod profiles they produced. These effects were more significant on radial enamel with rods perpendicular to the enamel surface, which comprise the greater part of a molar crown, contributing to the surface roughness of Fam83h Tr/Tr molars. In contrast, the incisor outer enamel with rods running parallel to the enamel surface might be less susceptible to the altered rod–interrod profile caused by truncated FAM83H and therefore exhibit a minor hypoplastic defect. Interestingly, mice with ablation of serotonin 2B receptor (Htr2b null mice) have a similar molar phenotype of generally thinner enamel with surface roughness, and the enamel microstructure and rod profiles of Htr2b−/− molars were also abnormal (Dimitrova‐Nakov et al., 2014; Harichane et al., 2011). Although the alteration of the rod profiles of molar enamel was not well detailed, primarily due to the high complexity of molar enamel microstructure and a lack of standardized methodology for molar characterization, it was clearly indicated that enamel analysis of mutant genotypes based exclusively on incisor defects can be biased (Goldberg, Kellermann, Dimitrova‐Nakov, Harichane, & Baudry, 2014). Correspondingly, the Fam83h Tr/Tr mice in this study also demonstrated distinct enamel phenotypes between incisors and molars, indicating a necessity of comprehensive characterization of both tooth types in a given mouse model and an imperativeness of developing standardized methods to investigate molar enamel microstructure.
We previously showed that Fam83h null mice died at around 2 weeks, and the few older survivors were generally weak, suggesting an essential role of FAM83H in certain critical physiological functions (Wang, Hu, Yang, Smith, Richardson, et al., 2015). Nonetheless, in this study, Fam83h Tr/Tr mice appeared to be viable and fertile. This discrepancy in viability suggested that the N‐terminal which is composed of 296 amino acids of FAM83H is sufficient for its critical functions. In other words, the truncated part of the protein in the C‐terminus, composed of about 900 amino acids, is not necessary for viability functions. The protein family of FAM83 is characterized by a highly conserved N‐terminal domain of unknown function, DUF1669, comprising ~ 300 amino acids (Cipriano et al., 2014). Except for this domain, the eight members of FAM83 family have vastly different C‐terminal regions with variable lengths. The FAM83 proteins were recently identified as novel transforming oncogenes that function as intermediaries in EGFR/RAS signaling, and the oncogenic properties were dependent upon the conserved DUF1669 domain (Cipriano et al., 2012, 2014). It was also demonstrated that these proteins, including FAM83H, were overexpressed in many human tumors (Cipriano et al., 2014; Snijders et al., 2017). Therefore, FAM83H might play a role in cell proliferation or survival mediated by EGFR/RAS signaling, presumably through its DUF1669 domain, which is essential for certain critical physiological functions. In this regard, complete loss of FAM83H in Fam83h null mice would be fatal. In contrast, the Fam83h Tr/Tr mice, although only expressing the first 296 amino acids of FAM83H containing the DUF1669 domain, would be able to survive. In other words, for certain cells other than ameloblasts, the mutant (truncated) FAM83H protein is not only not “toxic”, but can serve critical functions. Nevertheless, despite the viability, Fam83h Tr/Tr mice exhibited sparse and scruffy coat as did Fam83h null mice (Wang, Hu, Yang, Smith, Richardson, et al., 2015), which suggested that the DUF1669 domain of FAM83H was not sufficient to serve its functions in hair follicle cells. Since FAM83H is believed to interact with keratin through its C‐terminus (Kuga et al., 2013), FAM83H might be critical for maintaining the homeostasis of hair follicles via this interaction. Therefore, loss of FAM83H C‐terminus, in both Fam83h null and Fam83h Tr/Tr mice, leads to hair pathology.
CONFLICTS OF INTEREST
The authors have no conflict of interest to declare regarding this manuscript.
Supporting information
ACKNOWLEGEMNTS
This work was supported by NTUH (National Taiwan University Hospital) Grant 106‐N3424, the MOST (Ministry of Science and Technology in Taiwan) Grant 107–2314‐B‐002–014, NIDCR/NIH grant 1R01DE015846 and 1R01DE27675.
Wang S‐K, Hu Y, Smith CE, et al. The Enamel Phenotype in Homozygous Fam83h Truncation Mice. Mol Genet Genomic Med. 2019;7:e724 10.1002/mgg3.724
Contributor Information
Shih‐Kai Wang, Email: shihkaiw@ntu.edu.tw.
James P. Simmer, Email: jsimmer@umich.edu.
REFERENCES
- Adzhubei, I. , Jordan, D. M. , & Sunyaev, S. R. (2013). Predicting functional effect of human missense mutations using PolyPhen‐2. Current Protocols in Human Genetics, Chapter, 7(Unit7), 20 10.1002/0471142905.hg0720s76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, H. C. , Estrella, N. M. , Milkovich, R. N. , Kim, J. W. , Simmer, J. P. , & Hu, J. C. (2011). Target gene analyses of 39 amelogenesis imperfecta kindreds. European Journal of Oral Sciences, 119(Suppl 1), 311–323. 10.1111/j.1600-0722.2011.00857.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipriano, R. , Graham, J. , Miskimen, K. L. , Bryson, B. L. , Bruntz, R. C. , Scott, S. A. , … Jackson, M. W. (2012). FAM83B mediates EGFR‐ and RAS‐driven oncogenic transformation. Journal of Clinical Investigation, 122(9), 3197–3210. 10.1172/JCI60517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipriano, R. , Miskimen, K. L. , Bryson, B. L. , Foy, C. R. , Bartel, C. A. , & Jackson, M. W. (2014). Conserved oncogenic behavior of the FAM83 family regulates MAPK signaling in human cancer. Molecular Cancer Research, 12(8), 1156–1165. 10.1158/1541-7786.MCR-13-0289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova‐Nakov, S. , Baudry, A. , Harichane, Y. , Collet, C. , Marchadier, A. , Kellermann, O. , & Goldberg, M. (2014). Deletion of serotonin 2B receptor provokes structural alterations of mouse dental tissues. Calcified Tissue International, 94(3), 293–300. 10.1007/s00223-013-9810-y [DOI] [PubMed] [Google Scholar]
- Ding, Y. , Estrella, M. R. , Hu, Y. Y. , Chan, H. L. , Zhang, H. D. , Kim, J. W. , … Hu, J. C. (2009). Fam83h is associated with intracellular vesicles and ADHCAI. Journal of Dental Research, 88(11), 991–996. 10.1177/0022034509349454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Sayed, W. , Shore, R. C. , Parry, D. A. , Inglehearn, C. F. , & Mighell, A. J. (2010). Ultrastructural analyses of deciduous teeth affected by hypocalcified amelogenesis imperfecta from a family with a novel Y458X FAM83H nonsense mutation. Cells Tissues Organs, 191(3), 235–239. 10.1159/000252801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn, R. D. , Coggill, P. , Eberhardt, R. Y. , Eddy, S. R. , Mistry, J. , Mitchell, A. L. , … Bateman, A. (2016). The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Research, 44(D1), D279–D285. 10.1093/nar/gkv1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg, M. , Kellermann, O. , Dimitrova‐Nakov, S. , Harichane, Y. , & Baudry, A. . (2014). Comparative studies between mice molars and incisors are required to draw an overview of enamel structural complexity. Frontiers in Physiology, 5, 359 10.3389/fphys.2014.00359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harichane, Y. , Dimitrova‐Nakov, S. , Marchadier, A. , Collet, C. , Baudry, A. , Vidal, C. , … Goldberg, M. (2011). Enamel alterations in serotonin 2B receptor knockout mice. European Journal of Oral Sciences, 119(Suppl 1), 177–184. 10.1111/j.1600-0722.2011.00908.x [DOI] [PubMed] [Google Scholar]
- Hart, P. S. , Becerik, S. , Cogulu, D. , Emingil, G. , Ozdemir‐Ozenen, D. , Han, S. T. , … Hart, T. C. (2009). Novel FAM83H mutations in Turkish families with autosomal dominant hypocalcified amelogenesis imperfecta. Clinical Genetics, 75(4), 401–404. 10.1111/j.1399-0004.2008.01112.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubek, D. , Gjorup, H. , Jensen, L. G. , Juncker, I. , Nyegaard, M. , Borglum, A. D. , … Hertz, J. M. (2011). Limited phenotypic variation of hypocalcified amelogenesis imperfecta in a Danish five‐generation family with a novel FAM83H nonsense mutation. International Journal of Paediatric Dentistry, 21(6), 407–412. 10.1111/j.1365-263X.2011.01142.x [DOI] [PubMed] [Google Scholar]
- Hu, Y. , Smith, C. E. , Richardson, A. S. , Bartlett, J. D. , Hu, J. C. , & Simmer, J. P. (2016). MMP20, KLK4, and MMP20/KLK4 double null mice define roles for matrix proteases during dental enamel formation. Molecular Genetics & Genomic Medicine, 4(2), 178–196. 10.1002/mgg3.194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun, H. K. , Lee, S. K. , Lee, K. E. , Kang, H. Y. , Kim, E. J. , Choung, P. H. , & Kim, J. W. (2009). Identification of a novel FAM83H mutation and microhardness of an affected molar in autosomal dominant hypocalcified amelogenesis imperfecta. International Endodontic Journal, 42(11), 1039–1043. 10.1111/j.1365-2591.2009.01617.x [DOI] [PubMed] [Google Scholar]
- Kantaputra, P. N. , Intachai, W. , & Auychai, P. (2016). All enamel is not created equal: Supports from a novel FAM83H mutation. American Journal of Medical Genetics Part A, 170A(1), 273–276. 10.1002/ajmg.a.37406 [DOI] [PubMed] [Google Scholar]
- Kim, J. W. , Lee, S. K. , Lee, Z. H. , Park, J. C. , Lee, K. E. , Lee, M. H. , … Simmer, J. P. (2008). FAM83H mutations in families with autosomal‐dominant hypocalcified amelogenesis imperfecta. The American Journal of Human Genetics, 82(2), 489–494. 10.1016/j.ajhg.2007.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuga, T. , Kume, H. , Adachi, J. , Kawasaki, N. , Shimizu, M. , Hoshino, I. , … Tomonaga, T. (2016). Casein kinase 1 is recruited to nuclear speckles by FAM83H and SON. Scientific Reports, 6, 34472 10.1038/srep34472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuga, T. , Kume, H. , Kawasaki, N. , Sato, M. , Adachi, J. , Shiromizu, T. , … Tomonaga, T. (2013). A novel mechanism of keratin cytoskeleton organization through casein kinase Ialpha and FAM83H in colorectal cancer. Journal of Cell Science, 126(Pt 20), 4721–4731. 10.1242/jcs.129684 [DOI] [PubMed] [Google Scholar]
- Lee, S. K. , Hu, J. C. , Bartlett, J. D. , Lee, K. E. , Lin, B. P. , Simmer, J. P. , & Kim, J. W. (2008). Mutational spectrum of FAM83H: The C‐terminal portion is required for tooth enamel calcification. Human Mutation, 29, E95–E99. 10.1002/humu.20789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S. K. , Lee, K. E. , Jeong, T. S. , Hwang, Y. H. , Kim, S. , Hu, J. C. , … Kim, J. W. (2011). FAM83H mutations cause ADHCAI and alter intracellular protein localization. Journal of Dental Research, 90(3), 377–381. 10.1177/0022034510389177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler‐Bauer, A. , Anderson, J. B. , Cherukuri, P. F. , DeWeese‐Scott, C. , Geer, L. Y. , Gwadz, M. , … Bryant, S. H. (2005). CDD: A Conserved Domain Database for protein classification. Nucleic Acids Research, 33(Database, issue), D192–D196. 10.1093/nar/gki069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moinichen, C. B. , Lyngstadaas, S. P. , & Risnes, S. (1996). Morphological characteristics of mouse incisor enamel. Journal of Anatomy, 189(Pt 2), 325–333. [PMC free article] [PubMed] [Google Scholar]
- Pourhashemi, S. J. , Ghandehari Motlagh, M. , Meighani, G. , Ebrahimi Takaloo, A. , Mansouri, M. , Mohandes, F. , … Heidari, M. (2014). Missense mutation in Fam83H gene in Iranian patients with amelogenesis imperfecta. Iranian Journal of Public Health, 43(12), 1680–1687. [PMC free article] [PubMed] [Google Scholar]
- Prasad, M. K. , Geoffroy, V. , Vicaire, S. , Jost, B. , Dumas, M. , Le Gras, S. , … Bloch‐Zupan, A. (2016). A targeted next‐generation sequencing assay for the molecular diagnosis of genetic disorders with orodental involvement. Journal of Medical Genetics, 53(2), 98–110. 10.1136/jmedgenet-2015-103302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmer, J. P. , Lau, E. C. , Hu, C. C. , Aoba, T. , Lacey, M. , Nelson, D. , … Fincham, A. G. (1994). Isolation and characterization of a mouse amelogenin expressed in Escherichia coli . Calcified Tissue International, 54(4), 312–319. [DOI] [PubMed] [Google Scholar]
- Smith, C. E. , Hu, Y. , Richardson, A. S. , Bartlett, J. D. , Hu, J. C. , & Simmer, J. P. (2011). Relationships between protein and mineral during enamel development in normal and genetically altered mice. European Journal of Oral Sciences, 119(Suppl 1), 125–135. 10.1111/j.1600-0722.2011.00871.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, C. E. , & Nanci, A. (1995). Overview of morphological changes in enamel organ cells associated with major events in amelogenesis. The International Journal of Developmental Biology, 39(1), 153–161. [PubMed] [Google Scholar]
- Smith, C. E. , Richardson, A. S. , Hu, Y. , Bartlett, J. D. , Hu, J. C. , & Simmer, J. P. (2011). Effect of kallikrein 4 loss on enamel mineralization: Comparison with mice lacking matrix metalloproteinase 20. The Journal of Biological Chemistry, 286(20), 18149–18160. 10.1074/jbc.M110.194258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijders, A. M. , Lee, S. Y. , Hang, B. , Hao, W. , Bissell, M. J. , & Mao, J. H. (2017). FAM83 family oncogenes are broadly involved in human cancers: An integrative multi‐omics approach. Molecular Oncology, 11(2), 167–179. 10.1002/1878-0261.12016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, Y. L. , Wang, C. N. , Zhang, C. Z. , Yang, K. , & Bian, Z. (2012). Molecular characterization of amelogenesis imperfecta in Chinese patients. Cells Tissues Organs, 196(3), 271–279. 10.1159/000334210 [DOI] [PubMed] [Google Scholar]
- Urzua, B. , Martinez, C. , Ortega‐Pinto, A. , Adorno, D. , Morales‐Bozo, I. , Riadi, G. , … Reyes, M. (2015). Novel missense mutation of the FAM83H gene causes retention of amelogenin and a mild clinical phenotype of hypocalcified enamel. Archives of Oral Biology, 60(9), 1356–1367. 10.1016/j.archoralbio.2015.06.016 [DOI] [PubMed] [Google Scholar]
- von Koenigswald, W. (2004a). Enamel microstructure of rodent molars, classification, and parallelisms, with a note on the systematic affiliation of the enigmatic eocene rodent protoptychus. Journal of Mammalian Evolution, 11(2), 127–142. 10.1023/B:JOMM.0000041192.79808.52 [DOI] [Google Scholar]
- von Koenigswald, W. (2004b). The three basic types of schmelzmuster in fossil and extant rodent molars and their distribution among rodent clades. Palaeontographica, Abteilung A, 270, 95–132. [Google Scholar]
- von Koenigswald, W. , & Clemens, W. A. (1992). Levels of complexity in the microstructure of mammalian enamel and their application in studies of systematics. [Review]. Scanning Microscopy, 6(1), 195–217. [PubMed] [Google Scholar]
- Wang, S.‐K. , Hu, Y. , Yang, J. , Smith, C. E. , Nunez, S. M. , Richardson, A. S. , … Simmer, J. P. (2015). Critical roles for WDR72 in calcium transport and matrix protein removal during enamel maturation. Molecular Genetics & Genomic Medicine, 3(4), 302–319. 10.1002/mgg3.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S.‐K. , Hu, Y. , Yang, J. , Smith, C. E. , Richardson, A. S. , Yamakoshi, Y. , … Simmer, J. P. (2015). Fam83h null mice support a neomorphic mechanism for human ADHCAI. Molecular Genetics & Genomic Medicine, 4(1), 46–67. 10.1002/mgg3.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, J. T. , Deaton, T. G. , Hall, K. I. , & Yamauchi, M. (1995). The mineral and protein content of enamel in amelogenesis imperfecta. Connective Tissue Research, 32(1–4), 247–252. [DOI] [PubMed] [Google Scholar]
- Wright, J. T. , Frazier‐Bowers, S. , Simmons, D. , Alexander, K. , Crawford, P. , Han, S. T. , … Hart, T. C. (2009). Phenotypic variation in FAM83H‐associated amelogenesis imperfecta. Journal of Dental Research, 88(4), 356–360. 10.1177/0022034509333822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, J. T. , Torain, M. , Long, K. , Seow, K. , Crawford, P. , Aldred, M. J. , … Hart, T. C. (2011). Amelogenesis imperfecta: Genotype‐phenotype studies in 71 families. Cells Tissues Organs, 194(2–4), 279–283. 10.1159/000324339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin, W. , Wenjun, W. , Man, Q. , & Yuming, Z. (2017). Novel FAM83H mutations in patients with amelogenesis imperfecta. Scientific Reports, 7(1), 6075 10.1038/s41598-017-05208-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakoshi, Y. , Richardson, A. S. , Nunez, S. M. , Yamakoshi, F. , Milkovich, R. N. , Hu, J. C. , … Simmer, J. P. (2011). Enamel proteins and proteases in Mmp20 and Klk4 null and double‐null mice. European Journal of Oral Sciences, 119(Suppl 1), 206–216. 10.1111/j.1600-0722.2011.00866.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Wang, S. K. , Choi, M. , Reid, B. M. , Hu, Y. , Lee, Y. L. , … Hu, J. C. (2015). Taurodontism, variations in tooth number, and misshapened crowns in Wnt10a null mice and human kindreds. Mol Genet Genomic Med, 3(1), 40–58. 10.1002/mgg3.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, C. , Song, Y. , & Bian, Z. (2015). Ultrastructural analysis of the teeth affected by amelogenesis imperfecta. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontology, 119(2), e69–e76. 10.1016/j.oooo.2014.09.002 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials