

Abstract
Background
Polycystic kidney disease (PKD) is the most common hereditary kidney disease. The main mutational genes causing autosomal dominant polycystic kidney disease (ADPKD) are PKD1 and PKD2 as well as some rare pathogenic genes. Unilateral PKD is rare in clinics, and its association with gene mutations is unclear.
Methods
Targeted next‐generation sequencing (NGS) was performed to detect the renal ciliopathy‐associated genes (targeted NGS panel including 63 genes) in PKD patients.
Results
Forty‐eight PKD1 and PKD2 mutation sites were detected in 44 bilateral PKD patients, of which 48 were PKD1 mutation sites (87.5%) and six were PKD2 mutation sites (12.5%). All of which exhibited typical ADPKD. Furthermore, we detected HNF1B heterozygous mutations in three families. Although these three patients showed HNF1B heterozygous mutations, their clinical characteristics differed and showed phenotypic heterogeneity.
Conclusions
Targeted NGS panel was helpful in detecting typical ADPKD patients and even in non‐typical PKD patients. Macromutation in HNF1B may lead to bilateral PKD. The 16 novel PKD gene mutation sites and two novel PKD2 gene mutation sites discovered in this study have some significance in genetic counseling for ADPKD patients, and increase the number of studied families and expand the mutation database of ADPKD.
Keywords: gene mutations, next‐generation sequencing, PKD, targeted NGS panel
1. INTRODUCTION
Polycystic kidney disease (PKD) is a group of monogenic disorders that result in renal cyst development. Based on genetic patterns, PKD can be classified into autosomal dominant polycystic kidney disease (ADPKD) and autosomal recessive polycystic kidney disease (Harris & Torres, 2009). ADPKD is the most prevalent monogenic hereditary kidney disorder and the most common monogenic disorder that causes end‐stage renal disease (ESRD) (Torres, Harris, & Pirson, 2007). Its major presentations are multiple renal cysts that result in enlarged and irregular shaped kidneys. Depending on geographical location, its incidence is approximately 1:400–4000 (Torres et al., 2007). ADPKD develops primarily from PKD1 gene [MIM#601313] mutations on chromosome 16 and/or the PKD2 gene [MIM#173910] on chromosome 4 (Listed, 1994; Mochizuki et al., 1996). Clinical data indicate that PKD1 and PKD2 mutations account for 85% and 15% of ADPKD patients, respectively (Rossetti et al., 2007). ADPKD is heterogeneous with regard to locus and allele heterogeneity and phenotypic variability. PKD is usually bilateral, while unilateral PKD is rare. Unilateral PKD typically refers to segmental cystic abnormalities in 1 kidney. At present, few studies exist on unilateral PKD, and its pathological examination results are the same as those of ADPKD (Gouldesbrough & Fleming, 1998). Over time, unilateral PKD can change to bilateral PKD and further cause ESRD (Punia, Mohan, Bal, & Bansal, 2005). The pathogenesis of unilateral PKD and whether it is associated with gene mutations are unclear.
Currently, most ADPKD patients have PKD1 and/or PKD2 mutations; however, these mutations remain undetected in some patients. To better understand whether other genes cause ADPKD, as well as their resulting phenotypes, and to understand whether unilateral PKD is associated with gene mutations, we performed gene sequencing on PKD patients. Targeted next‐generation sequencing (NGS) was performed to screen 63 renal ciliopathy‐associated gene mutations in these patients.
2. MATERIALS AND METHODS
2.1. Study design
Gene sequencing was performed on 47 patients using our developed kidney disease panel. Possible pathogenic mutation sites were detected by Sanger sequencing. For some patients who did not detect suspected pathogenic genes, we further sequenced all exons and screened for suspected mutant genes. In addition, blood samples provided by family members of patients were used for segregation analyses. The detected mutation sites were carefully compared with the ADPKD Mutation Database (http://pkdb.mayo.edu), HGMD Professional (https://www.qiagenbioinformatics.com/products/human‐gene‐mutation‐database/) and relevant literature, and the mutation site pathogenicity were analyzed.
2.2. Patients
Forty‐seven unrelated PKD patients treated at the Chinese People's Liberation Army General Hospital between 2016 and 2017 were enrolled. All patients were confirmed to have unilateral or bilateral PKD by abdominal computed tomography or color Doppler ultrasound and volunteered for gene detection. The ages at PKD confirmation in these patients were 3–58 years. Forty‐four patients had bilateral PKD, and three had unilateral PKD. Among 44 bilateral PKD patients, 30 had clear family histories of dominant inheritance; therefore, validation was performed on immediate family members of 34 patients (a total of 65 family members). The three unilateral PKD patients were all isolated cases with no family history of this disease, and their parents did not have consanguineous marriages. All subjects or their legal guardians signed informed consent for genetic testing. The genetic analysis was approved by the Ethics Committee of the People's Liberation Army General Hospital (China).
2.3. Targeted NGS panel
Target region capture and next‐generation human gene analysis technology were performed for ciliopathy‐associated gene region and bioinformatics analyses. A kidney disease‐associated gene analysis panel including 63 genes was developed (Table S1). Combined with data including disease history and imaging examination, patients with kidney diseases and urinary system abnormalities could be screened at an early stage to reduce the damage of chronic kidney disease and provide gene diagnosis basis for personalized drugs in patients. This panel included many disease‐associated gene analyses including PKD, renal tuberculosis, Joubert syndrome, Meckel syndrome, short rib‐polydactyly syndrome (asphyxiating thoracic dysplasia/Jeune's syndrome), Bardet‐Biedl syndrome, and cranioectodermal dysplasia.
Targeted NGS panel was used for genetic analyses. NGS was performed on the NextSeq 500 apparatus (Illumina). This program included five main steps. (a) Nucleic acid extraction was performed on sample DNA using a genomic DNA extraction reagent kit, and the level of DNA quality was identified per the standard in Table S2. If the sample grade was level D, the sample was disqualified, and blood samples were collected again for DNA extraction. (b) The genomic library was constructed. Sample DNA was sheared to the range of 100–700 bp using the Covaris shearing method. The terminus was repaired, and an “A” was added. The product was purified for PCR amplification of the gene library, and the quality was detected per the standard in Table S3. If the sample grade was level D, the sample was disqualified and blood samples were collected again per the above procedures. (c) The target genes were captured. GenCap® Kidney disease gene capture probe (MyGenostics, China) and library DNA were hybridized under set conditions. Streptavidin‐modified magnetic beads were used to covalently bind to biotin‐labeled probes to capture target genes. Finally, a magnetic separator rack was used to adsorb magnetic beads that carried target genes. Target genes were then eluted, purified, and enriched. (d) The NextSeq 500 tabletop sequencer was used for large‐scale sequencing. (e) Data analyses were performed using BWA software (http://bio-bwa.sourceforge.net/), GATK software (http://www.broadinstitute. org/gsa/wiki/index.php/GATK_resource_bundle), and ANNOVAR software (http://www.broadinstitute.org/gsa/wiki/index.php/GATK_resource_bundle). After comparison with Homo sapiens (hg19), a mutation site that satisfied the following conditions was screened: (a) it was present in target regions; (b) it caused amino acid changes; (c) its mutation frequency in the local population was lower than 15%; and (d) if known in the databases, its minor allele frequency was below 1% for autosomal recessive inheritance and 0.2% for autosomal dominant transmission. All filtered variants were further analyzed using Alamut v.2.9.0 software (Interactive Biosoftware, La Rochelle, France) for predicting functional effects with SpliceSiteFinder, MaxEntScan, NNSPLICE, GeneSplicer, Human Splicing finder, Polyphen‐2, SIFT, MutationTaster, Align GVGD and UMD‐Predictor (Morais et al., 2017).
2.4. Whole exome sequencing
Genomic DNA was isolated from lymphocytes and subjected to exome capture using the SureSelect Human All ExonV6 human exome capture arrays (Agilent) followed by next generation sequencing on the NextSeq 500 tabletop sequencer. Data analyses were performed following the procedure of targeted NGS panel‐based analysis.
2.5. Sanger sequencing
After gene mutation sites were detected using NGS, they were validated using Sanger sequencing. In addition, blood samples provided by the patients’ family members were also validated by Sanger sequencing. Primers were designed using Primer software for PCR amplification. Next, capillary electrophoresis sequencing was performed using a 3130XL sequencer. When reference sequences were found, the reference sequences and raw data were analyzed using Mutation Surveyor software (https://softgenetics.com/mutationSurveyor.php).
2.6. Multiplex ligation‐dependent probe amplification
In order to confirm the presence of large gene rearrangements in the HNF1B gene we performed multiplex ligation‐dependent probe amplification (MLPA) using the MLPA kit P241 25R (HRC‐Holland, Amsterdam, Netherlands).
3. RESULTS
3.1. Genetic characterization
Among 44 bilateral PKD patients (Table 1), PKD1 heterozygous mutations were detected in 35 patients, PKD2 heterozygous mutations were detected in three patients, both PKD1 and PKD2 mutations were detected in three patients, one PKD1 heterozygous mutation and one PKHD1 (2‐point mutation sites) compound heterozygous mutation were detected in one patient, and HNF1B heterozygous mutations were detected in two patients. The MLPA results of P43 further confirmed the heterozygous deletion of exon 1‐9, that is, the complete HNF1B heterozygous deletion.(Figure S1) Among three unilateral PKD patients (Table 2), one HNF1B heterozygous mutation was detected in one patient, and no clear pathogenic gene mutations were detected in two patients. Whole exome sequencing (WES) was performed on patient P47, and the results showed that this patient had three heterozygous mutations in LYZ, FGA, and GLI3. Sanger sequencing validation was performed on the patient's father, who had a heterozygous mutation at the LYZ site but normal FGA and GLI3. Because a blood sample could not be collected from the patient's mother, Sanger validation was not performed. We do not think the heterozygous mutations detected in the autosomal recessive genes were significant in the disease's development and progression.
Table 1.
The mutation sites in 44 bilateral PKD patients
| Family No. | Mutated gene | Inheritance | Exon | Nucleotide change | Amino acid change | Status | Segregation tested | Reference | Clinical significance |
|---|---|---|---|---|---|---|---|---|---|
| (a) PKD1 mutations | |||||||||
| P1 | PKD1 | Dominant | exon45 | c.12310A>C | p.Ile4104Leu | Het | Yes | This study | Likely neutral |
| PKD1 | Dominant | exon15 | c.4340C>T | p.Ala1447Val | Het | Yes | Yu et al. (2011) | Likely neutral | |
| P2 | PKD1 | Dominant | exon40 |
c.11372_11373insGA TTACGACGTTGGCTGGGAG AGTCCTCACAATGG |
p.Gly3791fs | Het | Yes | PKDB | Definitely pathogenic |
| P3 | PKD1 | Dominant | exon23 | c.A8471C | p.Gln2824Pro | Het | Yes | This study | Likely neutral |
| P4 | PKD1 | Dominant | exon18 | c.7288C>T | p.Arg2430X | Het | Yes | Phakdeekitcharoen et al. (2000) | Definitely pathogenic |
| P5 | PKD1 | Dominant | exon13 | c.3140C>A | p.Ser1047X | Het | Yes | This study | Likely pathogenic |
| P6 | PKD1 | Dominant | exon15 | c.4306C>T | p.Arg1436X | Het | Yes | Garcia‐Gonzalez et al. (2007) | Definitely pathogenic |
| P7 | PKD1 | Dominant | exon36 | c.10678G>A | p.Gly3560Arg | Het | No | Tsuchiya et al. (2001) | Likely neutral |
| P8 | PKD1 | Dominant | exon31 | c.10081G>A | p.Gly3361Arg | Het | Yes | This study | Likely neutral |
| P9 | PKD1 | Dominant | exon6 | c.1291C>T | p.Gln431X | Het | Yes | This study | Likely pathogenic |
| P10 | PKD1 | Dominant | exon21 | c.7984C>T | p.Gln2662X | Het | Yes | PKDB | Definitely pathogenic |
| P11 | PKD1 | Dominant | exon16 | c.6935C>T | p.Ala2312Val | Het | Yes | PKDB | Likely neutral |
| P12 | PKD1 | Dominant | exon28 | c.9637T>G | p.Phe3213Val | Het | Yes | This study | Likely neutral |
| PKHD1 | Recessive | exon38 | c.6245C>T | p.Thr2082Ile | Het | Yes | Likely neutral | ||
| PKHD2 | Recessive | exon32 | c.4844C>T | p.Thr1615Met | Het | Yes | Likely neutral | ||
| P13 | PKD1 | Dominant | exon1 | c.108dupC | p.Cys37Leufs*77 | Het | No | Rossetti et al. (2012) | Definitely pathogenic |
| P14 | PKD1 | Dominant | exon23 | c.8311G>A | p.Glu2771Lys | Het | Yes | Rossetti et al. (2000) | Likely pathogenic |
| P15 | PKD1 | Dominant | exon10 | c.2039A>T | p.Tyr680Phe | Het | Yes | Liu et al. (2014) | Likely neutral |
| P16 | PKD1 | Dominant | exon15 | c.4810G>A | p.Val1604Met | Het | Yes | Yu et al. (2011) | Likely neutral |
| P17 | PKD1 | Dominant | exon36 | c.10678G>A | p.Gly3560Arg | Het | No | Tsuchiya et al. (2001) | Likely neutral |
| P18 | PKD1 | Dominant | exon23 | c.8426_8428del | p.2809_2810del | Het | Yes | This study | Likely neutral |
| P19 | PKD1 | Dominant | exon15 | c.5014_5015del | p.Arg1672Glyfs*9 | Het | Yes | Watnick et al. (1999) | Definitely pathogenic |
| P20 | PKD1 | Dominant | exon15 | c.6544C>T | p.Gln2182X | Het | Yes | This study | Likely pathogenic |
| P21 | PKD1 | Dominant | exon25 | c.9136C>T | p.Arg3046Cys | Het | Yes | Liu et al. (2015) | Likely pathogenic |
| PKD1 | Dominant | exon15 | c.6915+2T>G | splicing | Het | Yes | Perrichot et al. (2000) | Definitely pathogenic | |
| P22 | PKD1 | Dominant | exon23 | c.8464G>A | p.Val2822Met | Het | Yes | Hwang et al. (2016) | Likely neutral |
| P23 | PKD1 | Dominant | exon45 | c.12142G>T | p.Val4048Leu | Het | Yes | This study | Likely neutral |
| PKD1 | Dominant | exon25 | c.9157G>A | p.Ala3053Thr | Het | Yes | Chang et al. (2013) | Likely pathogenic | |
| PKD1 | Dominant | exon10 | c.2039A>T | p.Tyr680Phe | Het | Yes | Liu et al. (2014) | Likely neutral | |
| P24 | PKD1 | Dominant | exon23 | c.8464G>A | p.Val2822Met | Het | Yes | Hwang et al. (2016) | Likely neutral |
| PKD1 | Dominant | exon39 | c.11258G>C | p.Arg3753Pro | Het | Yes | This study | Likely pathogenic | |
| P25 | PKD1 | Dominant | exon26 | c.9272T>G | p.Met3091Arg | Het | Yes | This study | Likely neutral |
| PKD1 | Dominant | exon11 | c.2527T>C | p.Ser843Pro | Het | Yes | Yu et al. (2011) | Likely neutral | |
| P26 | PKD1 | Dominant | exon44 | c.12138+5G>A | splicing | Het | Yes | This study | Likely neutral |
| PKD1 | Dominant | exon36 | c.10678G>A | p.Gly3560Arg | Het | Yes | Tsuchiya et al. (2001) | Likely neutral | |
| P27 | PKD1 | Dominant | exon23 | c.8295_8296insATCCTCATGCGC | p.Ser2766delinsILMRS | Het | Yes | Rossetti et al. (2000) | Definitely pathogenic |
| P28 | PKD1 | Dominant | exon40 | c.11314delG | p.Ala3772Profs*54 | Het | Yes | Liu et al. (2015) | Definitely pathogenic |
| P29 | PKD1 | Dominant | exon33 | c.10321C>T | p.Gln3441X | Het | Yes | Obeidova et al. (2014) | Definitely pathogenic |
| P30 | PKD1 | Dominant | exon36 | c.10678G>A | p.Gly3560Arg | Het | Yes | Tsuchiya et al. (2001) | Likely neutral |
| PKD1 | Dominant | exon43 | c.11944C>T | p.Gln3982X | Het | Yes | Rossetti et al. (2007) | Definitely pathogenic | |
| P31 | PKD1 | Dominant | exon7 | c.1591G>A | p.Glu531K | Het | Yes | Hwang et al. (2016) | Likely neutral |
| P32 | PKD1 | Dominant | exon21 | c.7985dupA | p.Gln2663Alafs*159 | Het | Yes | This study | Likely pathogenic |
| P33 | PKD1 | Dominant | exon5 | c.862C>T | p.Gln288X | Het | Yes | Bataille, Berland, Fontes, and Burtey (2011) | Definitely pathogenic |
| P34 | PKD1 | Dominant | exon15 | c.3792C>A | p.Tyr1264X | Het | No | PKDB | Definitely pathogenic |
| P35 | PKD1 | Dominant | exon8 | c.1722+1G>C | splicing | Het | No | Audrézet et al. (2012) | Definitely pathogenic |
| P36 | PKD1 | Dominant | exon18 | c.7300C>T | p.Arg2434Trp | Het | Yes | Hoefele, Mayer, Scholz, and Klein (2011) | Likely pathogenic |
| (b) PKD2 mutations | |||||||||
| P37 | PKD2 | Dominant | exon4 | c.1094+1G>A | splicing | Het | Yes | Chung et al. (2010) | Definitely pathogenic |
| P38 | PKD2 | Dominant | exon4 | c.964C>G | p.Arg322Gly | Het | Yes | Audrézet et al. (2015) | Likely pathogenic |
| P39 | PKD2 | Dominant | exon5 | c.1249C>T | p.Arg417X | Het | Yes | Pei et al. (1998) | Definitely pathogenic |
| (c) PKD1&PKD2 mutations | |||||||||
| P40 | PKD1 | Dominant | exon23 | c.8444C>T | p.Ala2815Val | Het | No | Yu et al. (2011) | Likely neutral |
| PKD1 | Dominant | exon18 | c.7480G>A | p.Glu2494Lys | Het | No | This study | Likely neutral | |
| PKD2 | Dominant | exon6 | c.1546G>T | p.Val516Leu | Het | No | Yu et al. (2011) | Likely neutral | |
| P41 | PKD2 | Dominant | exon10 | c.2051dupA | p.Tyr684_S685delinsX | Het | Yes | This study | Likely pathogenic |
| PKD1 | Dominant | exon37 | c.10973A>G | p.Lys3658Arg | Het | Yes | This study | Likely neutral | |
| P42 | PKD2 | Dominant | exon10 | c.2083dupA | p.Ala696Sfs*2 | Het | Yes | This study | Likely pathogenic |
| PKD1 | Dominant | exon36 | c.10678G>A | p.Gly3560Arg | Het | Yes | Tsuchiya et al. (2001) | Likely neutral | |
| (d) HNF1B mutations | |||||||||
| P43 | HNF1B | Dominant | exon1−9 | Complete deletion | Complete deletion | Het | No | ||
| P44 | HNF1B | Dominant | exon4 | c.894_895delCT | p.Asn298Lysfs*21 | Het | No | ||
Table 2.
The mutation sites in three unilateral PKD patients
| Family No. | Mutated gene | Inheritance | Exon | Nucleotide change | Amino acid change | Status | Mutation frequency in the local population | Clinical significance |
|---|---|---|---|---|---|---|---|---|
| P45 | HNF1B | Dominant | exon1 | c.313G>A | p.Glu105Lys | het | 0.00217 | Uncertain |
| ZNF423 | Recessive | exon4 | c.2237A>G | p.Lys746Arg | het | 0.0001 | VUS | |
| P46 | ALMS1 | Recessive | exon8 | c.2351A>G | p.Flu784Gly | het | 0.0068 | VUS |
| BBS2 | Recessive | exon8 | c.865A>G | p.Ile289Val | het | 0.0174 | VUS | |
| BBS9 | Recessive | exon19 | c.2086G>A | p.Asp696Asn | het | 0.0068 | VUS | |
| CSPP1 | Recessive | exon27 | c.3298T>C | p.Trp1100Arg | het | 0.0308 | VUS | |
| IFT122 | Recessive | exon30 | c.3686G>A | p.Arg1229His | het | 0.0016 | VUS | |
| TTC8 | Recessive | exon14 | c.1328G>A | p.Arg443Gln | het | — | VUS | |
| P47 | CEP290 | Recessive | exon38 | c.5127G>T | p.Gln1709His | het | — | VUS |
| NPHP4 | Recessive | exon10 | c.1196A>G | p.Glu399Gly | het | 0.0026 | VUS | |
| PKHD1 | Recessive | exon28 | c.3179A>G | p.Asn1060Ser | het | — | VUS |
Forty‐eight mutation sites were detected in the PKD1 and PKD2 genes (Table 1). Compared with PKDB, HGMD Professional and literature reports, we found a total of 18 novel variants (16 in PKD1 and two in PKD2). All mutation sites were analyzed for pathogenicity in strict accordance with the American College of Medical Genetics and Genomics guidelines. Sixteen definite pathogenic mutation types (14 PKD1 gene sites and two PKD2 gene sites) were detected in 16 probands. These mutations included eight nonsense, four frameshift, three splicing, and one insertion mutation. We speculated that the splicing mutation (c.1722+1G>C, splicing) in PKD1 was a novel pathogenic mutation. There were 12 likely pathogenic mutations (nine PKD1 and three PKD2 gene sites) in 12 probands. These mutations included four nonsense, two frameshift, and six missense mutations. We predicted seven novel, likely pathogenic mutations (five PKD1 and two PKD2 gene sites) (Table 1). There were 20 likely neutral mutations (19 PKD1 and 1 PKD2 gene site) in 20 probands. These mutations included 18 missense, one splicing, and one frameshift mutation. We predicted 10 novel, likely neutral mutations (10 PKD1 gene sites) (Table 1).
3.2. Clinical manifestation
Among 42 patients with bilateral PKD caused by PKD1 and/or PKD2 mutations, 11 (26.2%) had combined hepatic cysts or polycystic liver. Some had already progressed to ESRD and required renal replacement therapy. Two with HNF1B mutations had no combined hepatic cysts, and their kidney functions were within the normal range. P44 had type 2 diabetes mellitus, while P43 had no history of diabetes mellitus but had increased uric acid (blood uric acid: 553.5 µmol/L). The three patients with unilateral PKD had a left polycystic kidney with normal morphology of the right kidney. P45 had no history of diabetes mellitus but had increased blood creatinine and uric acid (blood creatinine: 122.6 µmol/L; blood uric acid: 613 µmol/L). This patient's father also had a heterozygous mutation at the same HNF1B gene site (Table 2); however, the father had normal bilateral kidney morphology with no history of diabetes mellitus. The other two unilateral PKD patients (P46 and P47) with no detected pathogenic genes had normal creatinine and no family history of cystic diseases.
4. DISCUSSION
NGS can target the whole genome for detection. It has the advantages of high resolution, high throughput, high efficiency, and high sensitivity. It can increase gene detection efficiency and reduce gene detection costs (Mardis, 2008). After mutation sites are detected by NGS, Sanger sequencing validation can further increase detection accuracy; however, NGS cannot detect deleted or repeated fragments in nucleic acid sequences. Therefore, for typical ADPKD patients whose gene mutations cannot be detected by NGS, MLPA should be used to detect whether PKD1 or PKD2 have deleted or repeated fragments to avoid missed diagnoses. Synonymous mutations are generally considered not to affect amino acid changes in proteins, However, if a silent mutation in exonic splicing enhancer sequences may affect the splicing of MRNA (Ramser et al., 2005). Therefore, we need to make relevant analysis and prediction.
At present, diagnosis of ADPKD is based on family history and ultrasound imaging. In families of unknown genotype, the presence of three or more (unilateral or bilateral) renal cysts is sufficient for establishing the diagnosis in individuals aged 15–39 years, two or more cysts in each kidney is sufficient for individuals aged 40–59 years, and four or more cysts in each kidney is required for individuals 60 years (Pei et al., 2009). Which lead to a delay in or lack of diagnosis of ADPKD patients with no cysts in the kidneys and no apparent family history, which may result in inappropriate management. With the development of genetic testing technology, it is possible to make a definitive diagnosis before the onset age of a patient. Genotypes can also provide the basis for disease progression and prognosis (Jin et al., 2016). Based on our research and clinical experience, we designed the diagnosis process for patients with a positive family history (Figure 1).
Figure 1.
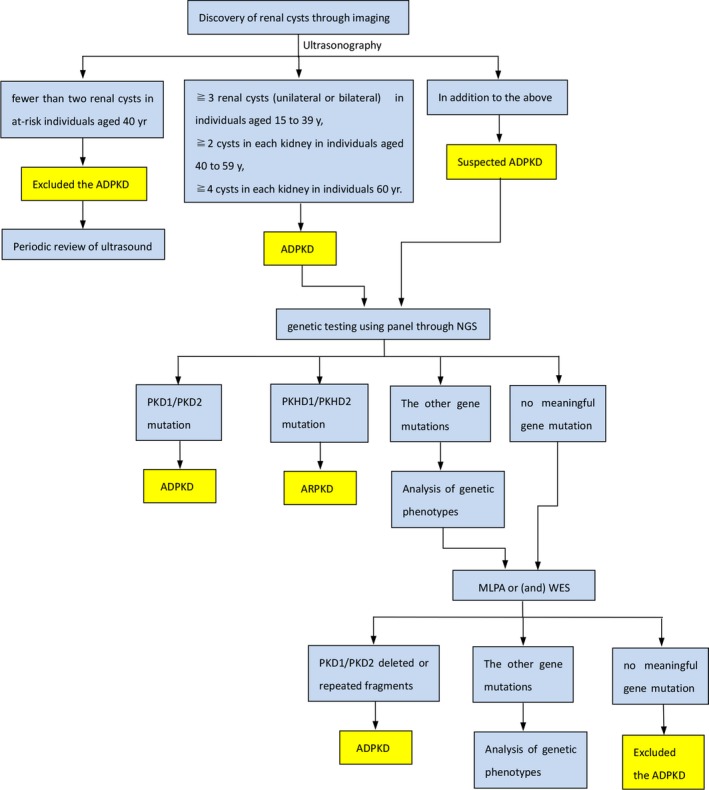
The diagnosis process for patients with a positive family history. ADPKD, autosomal dominant polycystic kidney disease; ARPKD, autosomal recessive polycystic kidney disease; MLPA, multiplex ligation‐dependent probe amplification; NGS, next‐generation sequencing; WES, whole exome sequencing
Applying our developed targeted NGS panel detected a pathogenic gene that caused this disease in 44 bilateral PKD patients. Forty‐two patients had detected PKD1 and/or PKD2 gene mutations, and two patients had detected HNF1B heterozygous mutations. Therefore, performing mutation analyses on only PKD1 and PKD2 in ADPKD patients who require gene diagnosis is insufficient, and application of our gene detection panel or even WES and MLPA are necessary (MLPA is a novel diagnostic tool for genetic screening, which is gradually becoming the principal method for the detection of exon deletion and duplication (Schouten et al., 2002), as they can detect HNF1B, gene repetition and deletion, or other possible pathogenic gene mutations. Currently, PKDB describes 2323 PKD1 mutations and 278 PKD2 mutations. We detected 42 PKD1 mutation sites (87.5%) and six PKD2 mutation sites (12.5%), which were similar to percentages previously reported in the literature. Among 44 bilateral PKD patients, the PKD1 (p.G3560R) missense mutation was detected in five families. This site might be a neutral site. We speculated that this mutation site had greater prevalence in the Chinese population. The pathogenicity of the novel mutation sites discovered in this study must be confirmed in other families in future studies. We detected no large gene deletions or repetitions in this patient group, which might be due to the limitations of NGS. We discovered 16 novel PKD1 gene mutation sites and two novel PKD2 gene mutation sites in the Chinese population, which could enrich the ADPKD Mutation Database.
The onset of ADPKD caused by PKD1 and/or PKD2 gene mutations and the severity of their phenotypes are not only associated with gene mutations (germ cell mutations) but are also associated with somatic cell mutations or deletions in normal alleles caused by environmental factors such as toxins and infection (Feng, Watnick, Onuchic, & Germino, 1996). ADPKD caused by PKD1 and/or PKD2 mutations is usually bilateral. Its phenotypes are associated with patient gender, whether the patient has hypertension or had a urologic event (gross hematuria, flank pain, or cyst infection) before age 35, and gene mutation characteristics (genotypes)(Cornec‐Le et al., 2016; Jin et al., 2016). The average age of ERSD onset caused by PKD1 mutations is 54.3 years, while the average age of ERSD onset caused by PKD2 mutations is 74 years (Kurashige et al., 2015). Because the average age of this patient group was 39.4 years and most patients had not reached the above ages, most patients had normal kidney functions. We have not performed genotype‐phenotype analyses on these patients; however, we will continue to monitor them and further perform genotype‐phenotype analyses.
The HNF1B gene [MIM 189907] that causes the phenotype similar to polycystic kidneys is transcription factor 2 located on chromosome 17q12. HNF1B can directly regulate PKHD1 transcription. Inhibiting PKHD1 gene expression may result in human renal cyst formation (Hiesberger et al., 2004). HNF1B's effects on the kidneys may include renal cysts, solitary kidney, horseshoe kidney, renal dysplasia, and hydronephrosis (Clissold, Hamilton, Hattersley, Ellard, & Bingham, 2015). Renal cysts caused by HNF1B mutations are more heterogeneous; they can present as multiple, few, or no cysts, and some patients will enter into ESRD (Faguer et al., 2011). The severity of the HNF1B mutation‐associated kidney disease phenotype had no clear association with the genotype (Heidet et al., 2010). We detected HNF1B heterozygous mutations in three patients. Two patients had bilateral PKD, and their HNF1B mutations were both large mutations (P43 had a complete deletion and P44 had a frameshift mutation). We speculated that these 2 patients’ bilateral PKD diseases were caused by HNF1B mutations. P45 had unilateral PKD with a point mutation in HNF1B. We found that the gene mutation levels of these three patients might be related to the kidney phenotype, which was more severe in patients with large mutations and was inconsistent with previous study results. This difference may have been due to our fewer patients. After a larger number of patients with PKD caused by HNF1B mutations is increased, whether kidney phenotype is associated with genotype can be further analyzed. The association between HNF1B mutations and unilateral PKD remains unclear. Mutations in the HNF1B gene usually cause diabetes maturity‐onset diabetes of the young type 5 (MODY5) (Roehlen et al., 2018). Of the three patients with the HNF1B mutation we detected, P44 (32 years old) had type 2 diabetes, while P43 (56 years old) and P45 (39 years old) had no diabetes. The incidence of diabetes in these three patients did not show age‐related. The phenotype caused by HNF1B mutations is diverse and does not necessarily lead to the onset of diabetes (Chen et al., 2010). Even mutations at the same site show multiple phenotypes (Yorifuji et al., 2004). The father of P45 also had an HNF1B mutation, however, he did not have diabetes mellitus or renal structural abnormalities, and we have not functionally validated this mutation site. Therefore, the significance of this mutation site on renal cyst development is unclear. Further larger studies would be required to confirm whether HNF1B mutations are associated with unilateral PKD.
Unilateral PKD is rare, as are studies of it. Among the three cases of unilateral PKD discovered in this study, P45’s case may have been caused by an HNF1B mutation, while P46 and P47 had no gene mutations on the kidney disease panel and P47 had LYZ, FGA, and GLI3 heterozygous mutations on the WES. These three genes have not been reported to be associated with PKD in the past, and this patient had no clinical presentations associated with these three genotypes. Whether unilateral PKD is associated with the above genes requires further confirmation using further larger studies.
In conclusion, using our developed targeted NGS panel for gene detection is necessary for PKD patients. It can be used to confirm patient genotypes (with/without mutations, mutation numbers, and mutation types) and has important significance in confirming molecular diagnoses and predicting patient prognosis. Targeted NGS panel and WES on unilateral PKD patients are significant. Macromutation in HNF1B may lead to bilateral PKD. While the relationship between HNF1B and unilateral PKD needs further studies to confirm. We discovered 16 novel PKD1 gene mutation sites and two novel PKD2 gene mutation sites that can enrich the PKDB and are significant in genetic counseling for ADPKD patients, and the use of effective targeted NGS method in the molecular diagnosis of ADPKD will increase the number of studied families and expand the mutation database of ADPKD.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
DATA AVAILABILITY STATEMENT
The datasets used and analyzed during the current study available from the corresponding author on reasonable request.
Supporting information
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (No. 81570597, and No. 81330019) and the Ministry of Science Technology of China (grant No. 2011CB944000).
Wang T, Li Q, Shang S, et al. Identifying gene mutations of Chinese patients with polycystic kidney disease through targeted next‐generation sequencing technology. Mol Genet Genomic Med. 2019;7:e720 10.1002/mgg3.720
Data Availability Statement: The datasets used and analyzed during the current study available from the corresponding author on reasonable request.
REFERENCES
- Audrézet, M. P. , Corbiere, C. , Lebbah, S. , Morinière, V. , Broux, F. , Louillet, F. , … Heidet, L. (2015). Comprehensive PKD1 and PKD2 mutation analysis in prenatal autosomal dominant polycystic kidney disease. Journal of the American Society of Nephrology, 27(3), 722 10.1681/ASN.2014101051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audrézet, M.‐P. , Gall, E.‐C.‐L. , Chen, J.‐M. , Redon, S. , Quéré, I. , Creff, J. , … Férec, C. (2012). Autosomal dominant polycystic kidney disease: Comprehensive mutation analysis of PKD1 and PKD2 in 700 unrelated patients. Human Mutation, 33(8), 1239–1250. 10.1002/humu.22103 [DOI] [PubMed] [Google Scholar]
- Bataille, S. , Berland, Y. , Fontes, M. , & Burtey, S. (2011). High resolution melt analysis for mutation screening in PKD1 and PKD2. BMC Nephrology, 12(1), 57 10.1186/1471-2369-12-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, M. Y. , Chen, H. M. , Jenq, C. C. , Lee, S. Y. , Chen, Y. M. , Tian, Y. C. , … Wu‐Chou, Y. H. (2013). Novel PKD1 and PKD2 mutations in Taiwanese patients with autosomal dominant polycystic kidney disease. Journal of Human Genetics, 58(11), 720–727. 10.1038/jhg.2013.91 [DOI] [PubMed] [Google Scholar]
- Chen, Y. Z. , Gao, Q. , Zhao, X. Z. , Chen, Y. Z. , Bennett, C. L. , Xiong, X. S. , … Chen, X. M. (2010). Systematic review of TCF2 anomalies in renal cysts and diabetes syndrome/maturity onset diabetes of the young type 5. Chinese Medical Journal (England), 123(22), 3326–3333. [PubMed] [Google Scholar]
- Chung, W. , Kim, H. , Hwang, Y. H. , Kim, S. Y. , Ko, A. R. , Ro, H. , … Ahn, C. (2010). PKD2 gene mutation analysis in Korean autosomal dominant polycystic kidney disease patients using two‐dimensional gene scanning. Clinical Genetics, 70(6), 502–508. [DOI] [PubMed] [Google Scholar]
- Clissold, R. L. , Hamilton, A. J. , Hattersley, A. T. , Ellard, S. , & Bingham, C. (2015). HNF1B‐associated renal and extra‐renal disease—an expanding clinical spectrum. Nature Reviews Nephrology, 11(2), 102 10.1038/nrneph.2014.232 [DOI] [PubMed] [Google Scholar]
- Cornec‐Le, G. E. , Audrézet, M. P. , Rousseau, A. , Hourmant, M. , Renaudineau, E. , Charasse, C. , … LeMeur, Y. (2016). The PROPKD score: A new algorithm to predict renal survival in autosomal dominant polycystic kidney disease. Journal of the American Society of Nephrology, 27(3), 942 10.1681/ASN.2015010016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faguer, S. , Decramer, S. , Chassaing, N. , Bellannéchantelot, C. , Calvas, P. , Beaufils, S. , … Chauveau, D. (2011). Diagnosis, management, and prognosis of HNF1B nephropathy in adulthood. Kidney International, 80(7), 768 10.1038/ki.2011.225 [DOI] [PubMed] [Google Scholar]
- Feng, Q. , Watnick, T. J. , Onuchic, L. F. , & Germino, G. G. (1996). The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell, 87(6), 979. [DOI] [PubMed] [Google Scholar]
- Garcia‐Gonzalez, M. A. , Jones, J. G. , Allen, S. K. , Palatucci, C. M. , Batish, S. D. , Seltzer, W. K. , … Watnick, T. J. (2007). Evaluating the clinical utility of a molecular genetic test for polycystic kidney disease. Molecular Genetics & Metabolism, 92(1–2), 160–167. 10.1016/j.ymgme.2007.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouldesbrough, D. R. , & Fleming, S. (1998). Unilateral and segmental localised polycystic kidney disease. Journal of Clinical Pathology, 51(9), 703–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, P. C. , & Torres, V. E. (2009). Polycystic kidney disease. Annual Review of Medicine, 60(60), 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidet, L. , Decramer, S. , Pawtowski, A. , Morinière, V. , Bandin, F. , Knebelmann, B. , … Salomon, R. (2010). Spectrum of HNF1B mutations in a large cohort of patients who harbor renal diseases. Clinical Journal of the American Society of Nephrology, 5(5), 1079–1090. 10.2215/CJN.06810909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiesberger, T. , Bai, Y. , Shao, X. , Mcnally, B. T. , Sinclair, A. M. , Tian, X. , … Igarashi, P. (2004). Mutation of hepatocyte nuclear factor‐1beta inhibits Pkhd1 gene expression and produces renal cysts in mice. Journal of Clinical Investigation, 113(6), 814 10.1172/jci20083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoefele, J. , Mayer, K. , Scholz, M. , & Klein, H. G. (2011). Novel PKD1 and PKD2 mutations in autosomal dominant polycystic kidney disease (ADPKD). Nephrology, Dialysis, Transplantation, 26(7), 2181–2188. 10.1093/ndt/gfq720 [DOI] [PubMed] [Google Scholar]
- Hwang, Y. H. , Conklin, J. , Chan, W. , Roslin, N. M. , Liu, J. , He, N. , … Pei, Y. (2016). Refining genotype‐phenotype correlation in autosomal dominant polycystic kidney disease. Journal of the American Society of Nephrology, 27(6), 1861 10.1681/ASN.2015060648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, M. , Xie, Y. , Chen, Z. , Liao, Y. , Li, Z. , Hu, P. , … Chen, X. (2016). System analysis of gene mutations and clinical phenotype in Chinese patients with autosomal‐dominant polycystic kidney disease. Scientific Reports, 6, 35945 10.1038/srep35945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurashige, M. , Hanaoka, K. , Imamura, M. , Udagawa, T. , Kawaguchi, Y. , Hasegawa, T. , … Maeda, S. (2015). A comprehensive search for mutations in the PKD1 and PKD2 in Japanese subjects with autosomal dominant polycystic kidney disease. Clinical Genetics, 87(3), 266–272. 10.1111/cge.12372 [DOI] [PubMed] [Google Scholar]
- Listed, N. (1994). The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. Cell, 8(5), 554–554. [DOI] [PubMed] [Google Scholar]
- Liu, B. , Chen, S. C. , Yang, Y. M. , Yan, K. , Qian, Y. Q. , Zhang, J. Y. , … Huang, H. F. (2015). Corrigendum: Identification of novel PKD1 and PKD2 mutations in a Chinese population with autosomal dominant polycystic kidney disease. Scientific Reports, 5, 17468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, W. , Chen, M. , Wei, J. , He, W. , Li, Z. , Sun, X. , & Shi, Y. (2014). Modification of PCR conditions and design of exon‐specific primers for the efficient molecular diagnosis of PKD1 mutations. Kidney and Blood Pressure Research, 39(6), 536–545. 10.1159/000368464 [DOI] [PubMed] [Google Scholar]
- Mardis, E. R. (2008). The impact of next‐generation sequencing technology on genetics. Trends in Genetics, 24(3), 133–141. 10.1016/j.tig.2007.12.007 [DOI] [PubMed] [Google Scholar]
- Mochizuki, T. , Wu, G. , Hayashi, T. , Xenophontos, S. L. , Veldhuisen, B. , Saris, J. J. , … Somlo, S. (1996). PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science, 272(5266), 1339–1342. [DOI] [PubMed] [Google Scholar]
- Morais, S. , Raymond, L. , Mairey, M. , Coutinho, P. , Brandão, E. , Ribeiro, P. , … Stevanin, G. (2017). Massive sequencing of 70 genes reveals a myriad of missing genes or mechanisms to be uncovered in hereditary spastic paraplegias. European Journal of Human Genetics, 25(11), 1217 10.1038/ejhg.2017.124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeidova, L. , Elisakova, V. , Stekrova, J. , Reiterova, J. , Merta, M. , Tesar, V. , … Kohoutova, M. (2014). Novel mutations of PKD genes in the Czech population with autosomal dominant polycystic kidney disease. BMC Medical Genetics, 15(1), 1–12. 10.1186/1471-2350-15-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei, Y. , He, N. , Wang, K. , Kasenda, M. , Paterson, A. D. , Chan, G. , … St George‐Hyslop, P. (1998). A spectrum of mutations in the polycystic kidney disease‐2 (PKD2) gene from eight Canadian kindreds. Journal of the American Society of Nephrology, 9(10), 1853. [DOI] [PubMed] [Google Scholar]
- Pei, Y. , Obaji, J. , Dupuis, A. , Paterson, A. D. , Magistroni, R. , Dicks, E. , … Ravine, D. (2009). Unified criteria for ultrasonographic diagnosis of ADPKD. Journal of the American Society of Nephrology, 20(1), 205 10.1681/ASN.2008050507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrichot, R. , Mercier, B. , Quere, I. , Carre, A. , Simon, P. , Whebe, B. , … Ferec, C. (2000). Novel mutations in the duplicated region of PKD1 gene. European Journal of Human Genetics, 8(5), 353–359. 10.1038/sj.ejhg.5200459 [DOI] [PubMed] [Google Scholar]
- Phakdeekitcharoen, B. , Watnick, T. J. , Ahn, C. , Whang, D. Y. , Burkhart, B. , & Germino, G. G. (2000). Thirteen novel mutations of the replicated region of PKD1 in an Asian population. Kidney International, 58(4), 1400 10.1046/j.1523-1755.2000.00302.x [DOI] [PubMed] [Google Scholar]
- Punia, R. P. , Mohan, H. , Bal, A. , & Bansal, V. K. (2005). Unilateral and segmental cystic disease of the kidney. International Journal of Urology Official Journal of the Japanese Urological Association, 12(3), 308–310. 10.1111/j.1442-2042.2005.01022.x [DOI] [PubMed] [Google Scholar]
- Ramser, J. , Abidi, F. E. , Burckle, C. A. , Lenski, C. , Toriello, H. , Wen, G. , … Nguyen, G. (2005). A unique exonic splice enhancer mutation in a family with X‐linked mental retardation and epilepsy points to a novel role of the renin receptor. Human Molecular Genetics, 14(8), 1019–1027. 10.1093/hmg/ddi094 [DOI] [PubMed] [Google Scholar]
- Roehlen, N. , Hilger, H. , Stock, F. , Glaser, B. , Guhl, J. , Schmitt‐Graeff, A. , … Laubner, K. (2018). 17q12 deletion syndrome as a rare cause for diabetes mellitus type MODY5. The Journal of Clinical Endocrinology and Metabolism, 103(10), 3601–3610. 10.1210/jc.2018-00955 [DOI] [PubMed] [Google Scholar]
- Rossetti, S. , Consugar, M. B. , Chapman, A. B. , Torres, V. E. , Guay‐Woodford, L. M. , Grantham, J. J. , … Harris, P. C. ; CRISP Consortium . (2007). Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. Journal of the American Society of Nephrology, 18(7), 2143. [DOI] [PubMed] [Google Scholar]
- Rossetti, S. , Hopp, K. , Sikkink, R. A. , Sundsbak, J. L. , Lee, Y. K. , Kubly, V. , … Harris, P. C. (2012). Identification of gene mutations in autosomal dominant polycystic kidney disease through targeted resequencing. Journal of the American Society of Nephrology, 23(5), 915–933. 10.1681/asn.2011101032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossetti, S. , Strmecki, L. , Gamble, V. , Burton, S. , Sneddon, V. , Peral, B. , … Harris, P. C. (2000). Mutation analysis of the entire PKD1 gene: Genetic and diagnostic implications. The American Journal of Human Genetics, 68(1), 46–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schouten, J. P. , Mcelgunn, A. C. J. , Waaijer, R. , Zwijnenburg, D. , Diepvens, F. , & Pals, G. (2002). Relative quantification of 40 nucleic acid sequences by multiplex ligation‐dependent probe amplification. Nucleic Acids Research, 30(12), e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres, V. E. , Harris, P. C. , & Pirson, Y. (2007). Autosomal dominant polycystic kidney disease. The Lancet, 369(9569), 1287–1301. [DOI] [PubMed] [Google Scholar]
- Tsuchiya, K. , Komeda, M. , Takahashi, M. , Yamashita, N. , Cigira, M. , Suzuki, T. , … Mochizuki, T. (2001). Mutational analysis within the 3' region of the PKD1 gene in Japanese families. Mutation Research/mutation Research Genomics, 458(3–4), 77–84. 10.1016/s0027-5107(01)00226-3 [DOI] [PubMed] [Google Scholar]
- Watnick, T. , Phakdeekitcharoen, B. , Johnson, A. , Gandolph, M. , Wang, M. , Briefel, G. , … Germino, G. G. (1999). Mutation detection of PKD1 identifies a novel mutation common to three families with aneurysms and/or very‐early‐onset disease. The American Journal of Human Genetics, 65(6), 1561–1571. 10.1086/302657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorifuji, T. , Kurokawa, K. , Mamada, M. , Imai, T. , Kawai, M. , Nishi, Y. , … Nakahata, T. (2004). Neonatal diabetes mellitus and neonatal polycystic, dysplastic kidneys: Phenotypically discordant recurrence of a mutation in the hepatocyte nuclear factor‐1β gene due to germline mosaicism. The Journal of Clinical Endocrinology & Metabolism, 89(6), 2905–2908. 10.1210/jc.2003-031828 [DOI] [PubMed] [Google Scholar]
- Yu, C. , Yuan, Y. , Lin, Z. , Hu, Z. , Jing, L. , Liu, Y. , … Zhang, S. (2011). Identification of novel mutations in Chinese Hans with autosomal dominant polycystic kidney disease. BMC Medical Genetics, 12(1), 164 10.1186/1471-2350-12-164 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and analyzed during the current study available from the corresponding author on reasonable request.