

Abstract
Background.
Despite established clinical associations among major depression (MD), alcohol dependence (AD), and alcohol consumption (AC), the nature of the causal relationship between them is not completely understood. We leveraged genome-wide data from the Psychiatric Genomics Consortium (PGC) and UK Biobank to test for the presence of shared genetic mechanisms and causal relationships among MD, AD, and AC.
Methods.
Linkage disequilibrium score regression and Mendelian randomization (MR) were performed using genome-wide data from the PGC (MD: 135 458 cases and 344 901 controls; AD: 10 206 cases and 28 480 controls) and UK Biobank (AC-frequency: 438 308 individuals; AC-quantity: 307 098 individuals).
Results.
Positive genetic correlation was observed between MD and AD (rgMD−AD = + 0.47, P = 6.6 × 10−10). AC-quantity showed positive genetic correlation with both AD (rgAD−AC quantity = + 0.75, P = 1.8 × 10−14) and MD (rgMD−AC quantity = + 0.14, P = 2.9 × 10−7), while there was negative correlation of AC-frequency with MD (rgMD−AC frequency = −0.17, P = 1.5 × 10−10) and a non-significant result with AD. MR analyses confirmed the presence of pleiotropy among these four traits. However, the MD-AD results reflect a mediated-pleiotropy mechanism (i.e. causal relationship) with an effect of MD on AD (beta = 0.28, P = 1.29 × 10−6). There was no evidence for reverse causation.
Conclusion.
This study supports a causal role for genetic liability of MD on AD based on genetic datasets including thousands of individuals. Understanding mechanisms underlying MD-AD comorbidity addresses important public health concerns and has the potential to facilitate prevention and intervention efforts.
Keywords: Alcohol consumption, alcohol dependence, genetic correlation, genome-wide association, major depression, Mendelian randomization
Introduction
Major depression (MD) and alcohol dependence (AD) are common psychiatric disorders, contribute substantially to global morbidity, and often co-occur (Ferrari et al., 2013; Shield et al., 2013). Epidemiological studies report that individuals with MD are at increased risk for AD and vice versa (Kessler et al., 1997; Swendsen and Merikangas, 2000; Boden and Fergusson, 2011). Leading hypotheses suggest these associations may be due to shared risk factors (genetic and environmental) or causal processes of one disorder leading to the other, such as the self-medication hypothesis of MD (Khantzian, 1997). However, the mechanisms underlying MD-AD dual diagnosis remain unclear.
Twin studies show genetic factors influence susceptibility to MD, AD, and alcohol consumption (AC) (Sullivan et al., 2000; Vrieze et al., 2013; Verhulst et al., 2015). Large-scale genome-wide association studies (GWAS) have identified risk variants for these disorders and have revealed polygenic architectures with multiple common variants (CONVERGE consortium, 2015; Schumann et al., 2016; Clarke et al., 2017; Walters et al., 2018; Wray et al., 2018). Twin studies report moderate shared genetic liability between MD and AD, estimating the genetic correlation from 0.3 to 0.6 (Kendler et al., 1993; Prescott et al., 2000). Although emerging molecular genetic studies have reported shared genetic risk between these disorders, they have not yet illuminated mechanisms of association underlying genetic correlations (Almeida et al., 2014; Bulik-Sullivan et al., 2015a; Wium-Andersen et al., 2015; Clarke et al., 2017; Walters et al., 2018; Wray et al., 2018). That is, questions remain whether these traits show genetic correlation because of shared genetic effects independently on each trait (i.e. horizontal pleiotropy) (Hemani et al., 2018a) or because of causal processes (e.g. mediated pleiotropy).
GWAS data can be used to assess causal mechanisms by applying Mendelian randomization (MR). MR is an instrumental variables technique that uses genetic variants to index if an observational association between a risk factor (e.g. MD) and an outcome (e.g. AD) is consistent with a causal effect (e.g. MD causes AD). MR relies on random assortment of genetic variants during meiosis which are typically unassociated with confounders since they are randomly distributed in the population at birth. The differences in outcome between those who carry genetic risk variants and those who do not can be attributed to the difference in the risk factor. The validity of the genetic instrument is dependent on meeting three core assumptions: (i) the genetic variant is associated with the risk factor/exposure; (ii) the genetic variant is not associated with confounders; and (iii) the genetic variant influences the outcome only through the risk factor. Although random controlled trials (RCTs) are considered the gold standard for establishing causality, MR is a viable alternative to provide support for causal mechanisms, especially when RCTs are not possible or ethical.
Two studies have previously evaluated the causal effect of AC on depression using MR. Almeida et al., investigated the impact of ADH1B rs1229984 in a sample of 3873 men and did not find evidence of a causal influence on depression (Almeida et al., 2014). In another study, the causal influence of two alcohol dehydrogenase (ADH) genes, ADH1B (rs1229984) and ADH1C (rs698) on depression was assessed in a sample of 68 486 participants from the general population and reported a lack of evidence for a causal influence on depression (Wium-Andersen et al., 2015). However, these studies did not investigate the causal influence of MD on AC or on AD risk. Furthermore, explorations of the causal nature were based on ADH candidate genetic variants only, which does not model the polygenic nature of these disorders.
Here, we leverage GWAS summary statistics generated by large datasets from the Psychiatric Genomics Consortium (PGC) and the UK Biobank to estimate genetic correlations between MD, AD, and two measures of AC (AC-quantity, AC-frequency) via linkage disequilibrium (LD) score regression (Bulik-Sullivan et al., 2015a, 2015b). Further, we investigated support for causal mechanisms linking these psychiatric disorders and AC via two-sample MR analyses, which use genetic variants to assess whether an exposure has a causal effect on an outcome in a non-experimental setting (Burgess et al., 2015).
Materials and methods
Samples
-
Major depression (Wray et al., 2018)
MD summary association data were obtained from the latest GWAS meta-analysis including 135 458 MD cases and 344 901 controls from the MD working group of the PGC (PGC-MDD2), which included seven cohorts. A detailed description of the cohorts is reported in the main GWAS analysis and a summary appears in the Supplemental Methods.
-
Alcohol Dependence (Walters et al., 2018)
AD summary association data from unrelated subjects of European descent (10 206 cases; 28 480 controls) were obtained from GWAS meta-analysis of 14 cohorts conducted by the PGC Substance Use Disorder Workgroup. Detailed descriptions of the AD samples have been previously reported and a summary appears in the Supplemental Methods.
-
UK Biobank – AC
The UK Biobank cohort consists of 502 000 middle-aged (40–69 years) individuals recruited from the UK. Information on alcohol intake was obtained through various self-report questionnaires. Frequency of consumption (AC-frequency) was assessed in 501 718 participants (UK Biobank field IDs: 1558) with the item ‘About how often do you drink alcohol?’. Frequency was originally assessed at a scale ranging from 1 (daily or almost daily) to 6 (never), but was reverse coded so that a lower score represented less frequent drinking. Online Supplemental Figure S1 shows the distribution in the UK Biobank population. In those who drink at least once or twice a week, information on the quantity of consumption (AC-quantity) was assessed (n = 348 039). Details can be found in the online Supplemental Methods.
Approximately 488 000 participants were genotyped and imputed using Haplotype Reference Consortium (HRC) and UK10 K haplotype resources (The UK10K Consortium et al., 2015; McCarthy et al., 2016; Bycroft et al., 2018). Due to the UK Biobank’s reported QC issues with non-HRC single nucleotide polymorphisms (SNPs), we retained only the ∼40 M HRC SNPs for analysis. In light of a large number of related individuals in the UK Biobank cohort, the GWAS was performed using BOLT-LMM (Loh et al., 2015). Using the criteria reported in the supplemental methods, we identified 438 870 individuals for this study who are genetically similar to those of white-British ancestry. After exclusion of ethnic outliers, we included 438 308 participants in the AC-frequency and 307 098 participants in the AC-quantity GWAS.
SNP-based heritability analysis
The proportion of variance in phenotypic liability that could be explained by the aggregated effect of all SNPs (h2-SNP) was estimated using LD-Score Regression analysis (online Supplemental Methods) (Bulik-Sullivan et al., 2015b). For this analysis, we included in the regression 1 217 311 SNPs that were present in the HapMap 3 reference panel. Analyses were performed using pre-computed LD scores based on 1000 Genomes Project reference data on individuals of European ancestry. The h2-SNP estimates for the two binary traits were converted to the liability scale, using sample prevalence of 0.159 for AD and 0.15 for MD.
Genetic correlations between MD, AD, AC-quantity, and AC-frequency
We used cross-trait LD-Score regression to estimate the bivariate genetic correlations between MD, AD, and AC using GWA summary statistics (Bulik-Sullivan et al., 2015a). For each pair of traits, the genetic covariance is estimated using the slope from the regression of the product of z-scores from two GWA studies on the LD score. The estimate represents the genetic covariation between the two traits based on all polygenic effects captured by SNPs. To correct for multiple testing, we adopted a Bonferroni corrected P-value threshold of significance of 0.05/ 6 = 0.0083.
Mendelian randomization
To assess causality among MD, AD, AC-quantity, and AC-frequency, we used GWAS summary association data to conduct two-sample MR analyses (Davey Smith and Hemani, 2014; Burgess et al., 2015). Since different MR methods have sensitivities to different potential issues, accommodate different scenarios, and vary in their statistical efficiency (Polimanti et al., 2017, 2018; Ravera et al., 2018; Wendt et al., 2018), we considered multiple MR methods (online Supplemental Table S1). These include methods based on median (Bowden et al., 2017), mean (Bowden et al., 2016), and mode (Hartwig et al., 2017), and various adjustments, such as fixed v. random effects (Bowden et al., 2017), Rucker framework (Rucker et al., 2011), and Steiger filtering (Hemani et al., 2017). We verified the stability of the results, comparing the effect directions across the different MR-variant filtering methods (online Supplemental Table S1). MR-Egger regression intercept was considered to verify the presence of pleiotropic effects of the SNPs on the outcome (i.e. to verify whether the instrumental variable is associated with the outcome independently from its association with the exposure) (Bowden et al., 2015). In total we performed 17 MR tests (online Supplemental Table S2). This number is due to the fact we were not able to test AD using a genetic instrument based on genome-wide significant (GWS) loci and, since we are conducting a two-sample MR analysis, we did not test causal relationship between AC-quantity and AC-frequency because they are based on UK Biobank cohort. For the variants included in the instrumental variable, we performed LD clumping by excluding alleles that have R2 ≥ 0.01 with another variant with a smaller association P-value considering a 1Mb window. Additionally, during the harmonization of exposure and outcome data, palindromic variants with an ambiguous allele frequency (i.e. minor allele frequency close to 50%) were removed from the analysis to avoid possible issues (Bowden et al., 2015; Hartwig et al., 2016). The variants included in each genetic instrument used in the present analysis are listed in online Supplemental Table S3. For each exposure, two instrumental variables were built considering GWS loci (P < 5× 10−8) and suggestive loci (P < 5× 10−5). We verified these MR estimates using the MR-RAPS approach, which is a method designed to identify and estimate confounded causal effects using weak genetic instrumental variables (Zhang et al., 2018). To ensure the reliability of the significant findings, we performed heterogeneity tests based on three different methods: inverse-variance weighted, MR-Egger regression, and maximum likelihood (online Supplemental Table S4). To further confirm the absence of possible distortions due to heterogeneity and pleiotropy, we tested the presence of horizontal pleiotropy among the variants included in the genetic instrument using MR-PRESSO (Verbanck et al., 2018). Finally, the funnel plot and leave-one-out analysis were conducted to identify potential outliers among the variants included in the genetic instruments tested. The MR analyses were conducted using the TwoSampleMR R package (Hemani et al., 2018b).
Data availability
MD GWAS:
The PGC’s policy is to make genome-wide summary results public. Summary statistics for a combined meta-analysis of PGC29 with five of the six expanded samples (deCODE, Generation Scotland, GERA, iPSYCH, and UK Biobank) are available on the PGC web site (https://www.med.unc.edu/pgc/results-and-downloads). Results for 10 000 SNPs for all seven cohorts are also available on the PGC web site. GWA summary statistics for the Hyde et al., cohort (23andMe, Inc.) must be obtained separately. These can be obtained by qualified researchers under an agreement with 23andMe that protects the privacy of the 23andMe participants. Contact David Hinds (dhinds@23andme.com) to apply for access to the data. Researchers who have the 23andMe summary statistics can readily recreate our results by meta-analyzing the six-cohort results file with the Hyde et al., results file from 23andMe.
AD GWAS:
The PGC’s policy is to make genome-wide summary results public. Summary statistics are available on the PGC web site is (https://www.med.unc.edu/pgc/results-and-downloads).
AC quantity and frequency GWAS: Summary statistics will be made publicly available through LD hub http://ldsc.broadinstitute.org/ldhub/ before publication of this paper or can be obtained upon request from the corresponding author.
Results
SNP-based heritabilities and genetic correlations
We confirmed previously reported heritability estimates of MD (h2-SNP = 8.5%, s.e. = 0.003, K = 0.15) and AD (h2-SNP = 9.0%, s.e. = 0.019, K = 0.16), with K defined as the disease prevalence in the population (Walters et al., 2018; Wray et al., 2018). The h2-SNP of AC-frequency, which has not been previously reported, was estimated at 8.0% (s.e. = 0.003). The h2-SNP of AC-quantity using LD-score regression was estimated at 6.9% (s.e. = 0.004), which is lower than the GCTA-estimate reported by Clarke et al. (13%) who analyzed a smaller subset (n = 112 117) (Clarke et al., 2017) from the current data (n = 307 098). The lower estimate may be explained by differential methodology (i.e. LD-score regression v. GCTA) and by the fact that the first release of UK Biobank included a subset of individuals that was selected based on smoking and may be less representative of the general population than the current sample (Wain et al., 2015). These h2-SNP estimates are capturing 17–23% of heritabilities reported by twin studies (Sullivan et al., 2000; Verhulst et al., 2015).
The high genetic correlation between AD and AC-quantity (rgAD−AC quantity = + 0.75, 95%CI = (0.56, 0.94), P = 1.8 × 10−14) (Fig. 1) suggests that these phenotypes capture overlapping constructs and that quantity of consumption is an indicator of problematic alcohol use. Of note, the genetic correlation between AD and AC-frequency is not significantly different from zero, indicating that it is not a reliable indicator of genetic risk for AD.
Fig. 1.
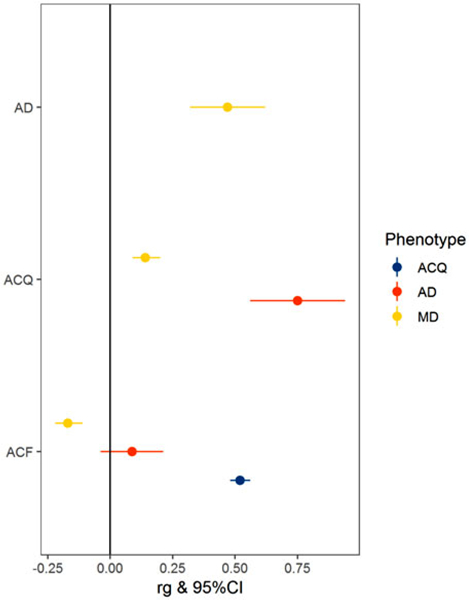
Genetic correlations of major depression (MD), alcohol dependence (AD), and alcohol consumption quantity (ACQ), and alcohol consumption frequency (ACF).
Consistent with twin studies, MD and AD show moderate overlap of genetic factors [rgMD−AD = + 0.47, 95% CI = (0.32, 0.62), P = 6.6 × 10−10] (Kendler et al., 1993; Prescott et al., 2000). A significant genetic correlation between AC-quantity and AC-frequency was observed [rgAC quantity−AC frequency = + 0.52, 95% CI = (0.48, 0.56), P = 1.3 × 10−149], but MD showed significant correlations with these traits in opposite directions [rgMD−AC quantity = + 0.14, 95% CI = (0.09, 0.20), P = 2.9 × 10−7; rgMD−AC frequency = −0.17, 95% CI = (−0.22, −0.11), P = 1.5 × 10−10].
Mendelian randomization
We investigated the presence of mediated pleiotropy via two-sample MR as this allows us to test for causative mechanisms linking MD, AD, AC-frequency, and AC-quantity. This is a strategy to investigate causal relationships in which evidence on the associations of genetic variants (i.e. instrumental variable) with the risk factor (i.e. exposure) and with the outcome are derived from two samples (Davey Smith and Hemani, 2014; Burgess et al., 2015). The instrumental variables were built considering GWS loci (P < 5× 10−8) and suggestive loci (P < 5× 10−5). Since different MR methods have sensitivities to different potential issues, we considered 28 MR/variant-filtering approaches (online Supplemental Table S1). A Bonferroni correction (P < 1.05 × 10−4) was applied to correct for the number of MR tests performed (n = 17; online Supplemental Table S2) and the number of methods/variant-filtering considered for each test (n = 28; online Supplemental Table S1). Of the 17 MR tests conducted, we observed that 14 survived multiple testing correction (Table 1). This outcome was expected due to the strong genetic correlations observed among the traits investigated. To verify that the significant results were not due to the presence of biases in the genetic instruments, we conducted three main sensitivity analyses: (i) inspected consistency of direction of effects across MR methods (online Supplemental Table S5); (ii) tests of horizontal pleiotropy between the exposure and the outcome (MR-Egger regression intercept P > 0.1; online Supplemental Table S6); (iii) assessed heterogeneity of effect sizes among the variants included in the genetic instrument (heterogeneity test P > 0.05; online Supplemental Table S3). Of 14 MR tests surviving Bonferroni multiple testing correction, only the causal relationship of MD on AD passed all three sensitivity analyses. We observed that the MD instrumental variable based on suggestive variants (259 SNPs) was associated with AD (fixed-effect inverse-variance weighted method: β = 0.28, P = 1.3 × 10−6; Fig. 2). Since this causal estimate was generated from a genetic instrument including suggestive variants, we confirmed this result using the MR-RAPS method: β = 0.28, P = 5.6 × 10−5. A similar effect size was also observed for the MD instrumental variable based on GWS loci (40 SNPs; fixed-effect inverse-variance weighted method: β = 0.27, P = 0.054). Results indicated that MD is associated with a 32% increase in the odds for AD risk per unit increase in the log(OR) for MD (95% CI 18–48%) and were consistent across multiple MR approaches (online Supplemental Table S5.17). As mentioned above, the MD genetic instrument did not show evidence of horizontal pleiotropic effects as demonstrated by MR-Egger regression intercept (P = 0.297, online Supplemental Table S6), confirming that the causal effect of MD on AD does not appear to be biased by horizontal pleiotropy. The heterogeneity tests indicated no evidence of heterogeneity in the MD-AD result (P > 0.13; online Supplemental Table S3). The MR-PRESSO global test (Verbanck et al., 2018) also supported the absence of horizontal pleiotropy (RSSobs = 285.6, P = 0.143). The MR-RAPS overdispersion test did not observe significant horizontal pleiotropy (estimated pleiotropy variance = 1 × 10−4; P = 0.249). Finally, the funnel plot and leave-one-out analyses provided additional support that the MD-AD result was not biased by outliers included in the genetic instrument (online Supplemental Figure S2). The same MD genetic instrument also showed significant effects on AC-quantity and AC-frequency (Table 1), but, in contrast to the AD outcome, these causal effects showed evidence of non-consistency across MR methods, heterogeneity, and horizontal pleiotropy (online Supplemental Table S3, S5, S6). No reverse causal effect was observed between AD genetic instrument and MD (fixed-effect inverse-variance weighted method: β = 0.01, P = 0.1), which also showed non-concordant direction of effects across MR methods (online Supplemental Figure S3). Conversely, the AD genetic instrument showed significant effects on AC-quantity and AC-frequency but was affected by heterogeneity and horizontal pleiotropy (Table 1; online Supplemental Table S3, S6).
Table 1.
Results of the most significant MR approach among those surviving Bonferroni multiple testing correction for each of the MR tests conducted
| Test | Method | SNP n | Estimate | P | Concordance | Het. (P > 0.05) | MR-Egger intercept (P > 0.1) |
|---|---|---|---|---|---|---|---|
| ACF→AD | Egger | 92 | −1.97 | 2.51×10−11 | pass | violated | violated |
| ACF×10−5→AD | Egger | 773 | −0.75 | 3.24×10−06 | pass | violated | violated |
| ACF×10−5→MD | IVW | 795 | 0.07 | 6.50×10−20 | violated | violated | violated |
| ACQ→AD | IVW | 31 | 0.12 | 3.81×10−11 | pass | violated | violated |
| ACQ→MD | IVW | 30 | 0.01 | 8.06×10−05 | violated | violated | pass |
| ACQ×10−5→AD | IVW | 385 | 0.06 | 7.34×10−20 | pass | violated | violated |
| ACQ×10−5→MD | IVW | 405 | 0.01 | 2.43×10−17 | violated | violated | violated |
| AD×10−5→ACF | Egger | 96 | −0.05 | 9.11×10−48 | pass | violated | violated |
| AD×10−5→ACQ | IVW | 95 | 0.26 | 3.26×10−36 | pass | violated | pass |
| MD→ACF | IVW | 36 | 0.05 | 4.06×10−08 | pass | violated | pass |
| MD→ACQ | Egger | 36 | −4.77 | 1.93×10−10 | pass | violated | violated |
| MD×10−5→ACF | IVW | 252 | 0.02 | 1.27×10−08 | violated | violated | pass |
| MD×10−5→ACQ | IVW | 251 | 0.31 | 1.46×10−06 | violated | violated | violated |
| MD×10−5→AD | IVW | 259 | 0.28 | 1.29×10−06 | pass | pass | pass |
ACF, alcohol consumption frequency; ACQ, alcohol consumption quantity; AD, alcohol dependence; MD, major depression; Het., heterogeneity test; suggestive loci (P < 5× 10−5). All top-results reported in the table were obtained using fixed effects and tophits adjustments (see online Supplemental Table S2)
Fig. 2.
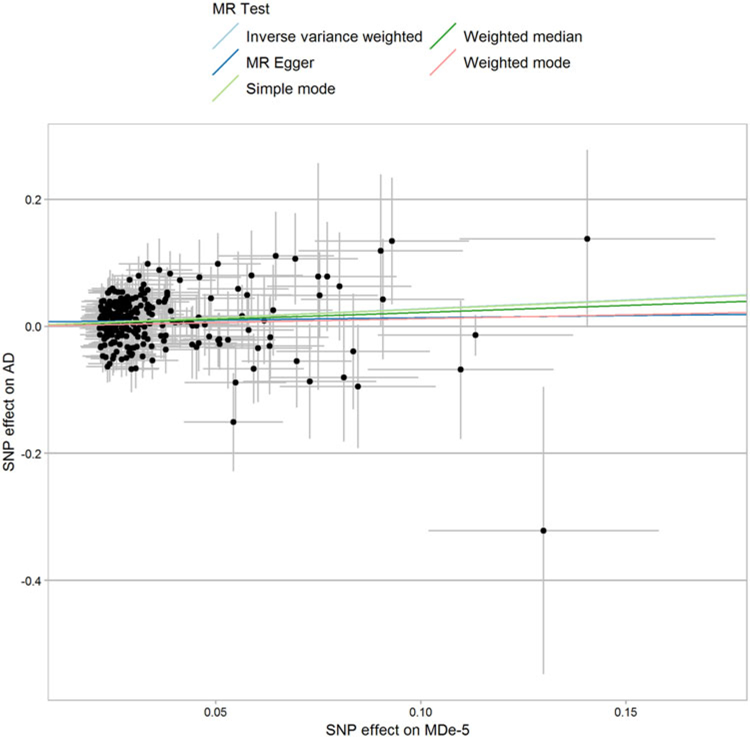
Single nucleotide polymorphism (SNP)-exposure (major depression (MD) associations, logOR) and SNP-outcome (alcohol dependence (AD) associations, logOR) coefficients used in the Mendelian randomization (MR) analysis. Error bars (95% CIs) are reported for each association.
Discussion
Data from large-scale GWAS are redefining the boundaries of psychiatric disorders, identifying the contribution of common risk alleles and pervasive genetic correlations. Here, leveraging the polygenic architecture of these complex traits and the large sample size of GWAS results from the PGC and UK Biobank, we observed genetic overlap between MD and AD and provide support for a causal effect of MD on AD, which does not appear to be affected by horizontal pleiotropy or other detectable biases. To our knowledge, this is the first report of causality between MD and AD based on molecular genetic information. Consistent with the two previously published MR studies (Almeida et al., 2014; Wium-Andersen et al., 2015), we did not find a robust causal influence of AC on depression.
We detected significant genetic correlations between all pairs of phenotypes, except for AD and AC-frequency, suggesting that frequency of AC is not a good proxy for AD. In contrast, the genetic correlation between AD and AC-quantity is high at rg = 0.75. Of note, our estimate is higher than previously reported by Walters et al. (rg = 0.37) who used the same GWAS results for AD (Walters et al., 2018) but the earlier, smaller subset of the UK Biobank data on consumption (Clarke et al., 2017). However, our estimate is comparable to the estimate reported by Walters et al. (rgAD−AC quantity = + 0.70) who calculated the genetic correlation between AD and AC from the Alcohol Genome-wide Association and Cohorts for Aging and Research in Genomic Epidemiology Plus consortium (Schumann et al., 2016), suggesting that the genetic correlation between these traits is indeed high (Walters et al., 2018).
The finding of a negative correlation between AC-frequency and MD may seem counterintuitive, but is supported by earlier studies that report opposing effects of quantity and frequency on health. For example, high drinking quantity is associated with increased all-cause mortality risk while the frequency of drinking does not show such an association (Breslow and Graubard, 2008). Additionally, in line with our findings, it was recently shown that MD is associated in opposite directions with two aspects of problematic drinking as assessed by the AUDIT: AUDIT-consumption (i.e. assessing frequency of consumption) (rg = −0.23) and AUDIT-problematic consequences (rg = + 0.26) (Sanchez-Roige et al., 2018). Our findings support the notion that MD is genetically positively correlated with measures of problematic drinking (i.e. AD and AC-quantity), but is negatively correlated with the frequency of consumption.
In contrast to some epidemiological reports (Boden and Fergusson, 2011), our results do not support evidence of reverse causation, that is, AD causing MD. One could posit that this is due to the relative power of the AD instrumental variable compared to those for MD and AC given the greater number of GWS variants detected for those traits. However, we would like to bring forward three arguments that support the notion that the null AD→MD result is due to the absence of a causal effect of AD on MD rather than a lack of power. First, our findings are in line with the results of an earlier MR study that explored the causal effect of ADH1B rs1229984 on depression and reported no significant association (Almeida et al., 2014). Since this variant is significantly associated with AD risk (Walters et al., 2018), this supports our premise that AD does not have a causal influence on MD. Second, the AD genetic instrument showed different associations between the traits tested: significant causal effect with respect to AC scales, while no effect on MD. Third, the genetic-correlation results indicated that AD is informative of the pleiotropy (mediated or horizontal) with ACQ and MD. Therefore, although the AD GWAS has a smaller sample size than the other GWAS used in the present analysis, it is informative of AD polygenic architecture as indicated by quantifiable and statistically significant SNP-based heritability and genetic correlation results. In particular, MD showed a much stronger genetic correlation with AD than that observed with AC scales. However, we note that larger AD and MD datasets will be required to confirm the current findings using genetic instruments based on genetic variants that reached the more conservative genome-wide significance threshold.
Conclusions
These results support the utility of using genetic approaches to advance the understanding of complex trait comorbidities. Given the significant morbidity and mortality associated with the comorbid conditions, AD and MD, understanding mechanisms underlying these associations not only address important public health concerns but also has the potential to facilitate prevention and intervention efforts. As discovery GWAS increase in sample size, future research will have the power to examine patterns of genetic correlation and causal mechanisms by important stratifications such as across diverse ancestries and sex.
Supplementary Material
Acknowledgements.
The Psychiatric Genomics Consortium (PGC): We are deeply indebted to the investigators who comprise the PGC, and to the hundreds of thousands of subjects who have shared their life experiences with PGC investigators. This work was conducted using the UK Biobank Resource (application number 25331). The PGC has received major funding from the National Institute of Mental Health and the National Institute on Drug Abuse (PGC3: U01 MH109528 and U01 MH109532, PGC2: U01 MH094421, PGC1:U01MH085520). The Substance Use Disorders Working Group of the Psychiatric Genomics Consortium (PGC-SUD) is supported by funds from NIDA and NIMH to MH109532 and, previously, with analyst support from NIAAA to U01AA008401 (COGA). We gratefully acknowledge the contributing studies and the participants in those studies without whom this effort would not be possible. For a full list of acknowledgements of all individual cohorts included in the PGC-SUD and PGC-MD groups, please see the original publications. Statistical analyses were carried out on the Genetic Cluster Computer (http://www.geneticcluster.org) hosted by SURFsara, which is financially supported by the Netherlands Scientific Organization (NWO 480-05-003) along with a supplement from the Dutch Brain Foundation and the VU University Amsterdam. Renato Polimanti was supported by a Young Investigator Grant from the American Foundation for Suicide Prevention. Roseann E. Peterson was supported by National Institutes of Health K01 grant MH113848. Nathan A. Gillespie was supported by National Institutes of Health R00 grant R00DA023549. This paper represents independent research part-funded by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care.
Footnotes
Supplementary material. The supplementary material for this article can be found at https://doi.org/10.1017/S0033291719000667
Ethical standards. Contributing studies provided individual-level genotype data or summary statistics consistent with their institutional review board-approved protocols. All procedures complied with the ethical standards of the relevant national and institutional committees on human experimentation and with the Helsinki Declaration of 1975, as revised in 2008.
Conflict of interest. 23andMe Research Team are employees of 23andMe, Inc. and hold stock or stock options in 23andMe. The other authors reported no biomedical financial interests or potential conflicts of interest.
References
- Almeida OP, Hankey GJ, Yeap BB, Golledge J and Flicker L (2014) The triangular association of ADH1B genetic polymorphism, alcohol consumption and the risk of depression in older men. Molecular Psychiatry 19, 995–1000. [DOI] [PubMed] [Google Scholar]
- Boden JM and Fergusson DM (2011) Alcohol and depression. Addiction 106, 906–914. [DOI] [PubMed] [Google Scholar]
- Bowden J, Davey Smith G and Burgess S (2015) Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. International Journal of Epidemiology 44, 512–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden J, Davey Smith G, Haycock PC and Burgess S (2016) Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genetic Epidemiology 40, 304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan N and Thompson J (2017) A framework for the investigation of pleiotropy in two-sample summary data Mendelian randomization. Statistics in Medicine 36, 1783–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow RA and Graubard BI (2008) Prospective study of alcohol consumption in the United States: quantity, frequency, and cause-specific mortality. Alcoholism, Clinical and Experimental Research 32, 513–521. [DOI] [PubMed] [Google Scholar]
- Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, ReproGen C, Psychiatric Genomics C, Genetic Consortium for Anorexia Nervosa of the Wellcome Trust Case Control C, Duncan L, Perry JR, Patterson N, Robinson EB, Daly MJ, Price AL and Neale BM (2015a) An atlas of genetic correlations across human diseases and traits. Nature Genetics 47, 1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulik-Sullivan BK, Loh PR, Finucane HK, Ripke S, Yang J, Schizophrenia Working Group of the Psychiatric Genomics C, Patterson N, Daly MJ, Price AL and Neale BM (2015b) LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nature Genetics 47, 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess S, Scott RA, Timpson NJ, Davey Smith G and Thompson SG and Consortium E-I (2015) Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. European Journal of Epidemiology 30, 543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O’Connell J, Cortes A, Welsh S, Young A, Effingham M, McVean G, Leslie S, Allen N, Donnelly P and Marchini J (2018) The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke TK, Adams MJ, Davies G, Howard DM, Hall LS, Padmanabhan S, Murray AD, Smith BH, Campbell A, Hayward C, Porteous DJ, Deary IJ and McIntosh AM (2017) Genome-wide association study of alcohol consumption and genetic overlap with other health-related traits in UK Biobank (N = 112 117). Molecular Psychiatry 22, 1376–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONVERGE consortium (2015) Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature 523, 588–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey Smith G and Hemani G (2014) Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Human Molecular Genetics 23, R89–R98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari AJ, Charlson FJ, Norman RE, Patten SB, Freedman G, Murray CJ, Vos T and Whiteford HA (2013) Burden of depressive disorders by country, sex, age, and year: findings from the global burden of disease study 2010. PLoS Medicine 10, e1001547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig FP, Davies NM, Hemani G and Davey Smith G (2016) Two-sample Mendelian randomization: avoiding the downsides of a powerful, widely applicable but potentially fallible technique. International Journal of Epidemiology 45, 1717–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig FP, Davey Smith G and Bowden J (2017) Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. International Journal of Epidemiology 46, 1985–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemani G, Tilling K and Davey Smith G (2017) Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genetics 13, e1007081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemani G, Bowden J and Davey Smith G (2018a) Evaluating the potential role of pleiotropy in Mendelian randomization studies. Human Molecular Genetics 27, R195–R208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, Laurin C, Burgess S, Bowden J, Langdon R, Tan VY, Yarmolinsky J, Shihab HA, Timpson NJ, Evans DM, Relton C, Martin RM, Davey Smith G, Gaunt TR and Haycock PC (2018b) The MR-base platform supports systematic causal inference across the human phenome. Elife 7, e34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendler KS, Heath AC, Neale MC, Kessler RC and Eaves LJ (1993) Alcoholism and major depression in women. A twin study of the causes of comorbidity. Archives of General Psychiatry 50, 690–698. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Crum RM, Warner LA, Nelson CB, Schulenberg J and Anthony JC (1997) Lifetime co-occurrence of DSM-III-R alcohol abuse and dependence with other psychiatric disorders in the National Comorbidity Survey. Archives of General Psychiatry 54, 313–321. [DOI] [PubMed] [Google Scholar]
- Khantzian EJ (1997) The self-medication hypothesis of substance use disorders: a reconsideration and recent applications. Harvard Review of Psychiatry 4, 231–244. [DOI] [PubMed] [Google Scholar]
- Loh PR, Tucker G, Bulik-Sullivan BK, Vilhjalmsson BJ, Finucane HK, Salem RM, Chasman DI, Ridker PM, Neale BM, Berger B, Patterson N and Price AL (2015) Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nature Genetics 47, 284–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, Kang HM, Fuchsberger C, Danecek P, Sharp K, Luo Y, Sidore C, Kwong A, Timpson N, Koskinen S, Vrieze S, Scott LJ, Zhang H, Mahajan A, Veldink J, Peters U, Pato C, van Duijn CM, Gillies CE, Gandin I, Mezzavilla M, Gilly A, Cocca M, Traglia M, Angius A, Barrett JC, Boomsma D, Branham K, Breen G, Brummett CM, Busonero F, Campbell H, Chan A, Chen S, Chew E, Collins FS, Corbin LJ, Smith GD, Dedoussis G, Dorr M, Farmaki AE, Ferrucci L, Forer L, Fraser RM, Gabriel S, Levy S, Groop L, Harrison T, Hattersley A, Holmen OL, Hveem K, Kretzler M, Lee JC, McGue M, Meitinger T, Melzer D, Min JL, Mohlke KL, Vincent JB, Nauck M, Nickerson D, Palotie A, Pato M, Pirastu N, McInnis M, Richards JB, Sala C, Salomaa V, Schlessinger D, Schoenherr S, Slagboom PE, Small K, Spector T, Stambolian D, Tuke M, Tuomilehto J, Van den Berg LH, Van Rheenen W, Volker U, Wijmenga C, Toniolo D, Zeggini E, Gasparini P, Sampson MG, Wilson JF, Frayling T, de Bakker PI, Swertz MA, McCarroll S, Kooperberg C, Dekker A, Altshuler D, Willer C, Iacono W, Ripatti S, Soranzo N, Walter K, Swaroop A, Cucca F, Anderson CA, Myers RM, Boehnke M, McCarthy MI, Durbin R and the Haplotype Reference Consortium (2016) A reference panel of 64976 haplotypes for genotype imputation. Nature Genetics 48, 1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polimanti R, Amstadter AB, Stein MB, Almli LM, Baker DG, Bierut LJ, Bradley B, Farrer LA, Johnson EO, King A, Kranzler HR, Maihofer AX, Rice JP, Roberts AL, Saccone NL, Zhao H, Liberzon I, Ressler KJ, Nievergelt CM, Koenen KC, Gelernter J and Psychiatric Genomics Consortium Posttraumatic Stress Disorder W (2017) A putative causal relationship between genetically determined female body shape and posttraumatic stress disorder. Genome Medicine 9, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polimanti R, Gelernter J and Stein DJ (2018) Genetically determined schizophrenia is not associated with impaired glucose homeostasis. Schizophrenia Research 195, 286–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott CA, Aggen SH and Kendler KS (2000) Sex-specific genetic influences on the comorbidity of alcoholism and major depression in a population-based sample of US twins. Archives of General Psychiatry 57, 803–811. [DOI] [PubMed] [Google Scholar]
- Ravera S, Carrasco N, Gelernter J and Polimanti R (2018) Phenomic impact of genetically-determined euthyroid function and molecular differences between thyroid disorders. Journal of Clinical Medicine 7, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rucker G, Schwarzer G, Carpenter JR, Binder H and Schumacher M (2011) Treatment-effect estimates adjusted for small-study effects via a limit meta-analysis. Biostatistics (Oxford, England) 12, 122–142. [DOI] [PubMed] [Google Scholar]
- Sanchez-Roige S, Palmer AA, Fontanillas P, Elson SL, and Me Research T, Substance Use Disorder Working Group of the Psychiatric Genomics C, Adams MJ, Howard DM, Edenberg HJ, Davies G, Crist RC, Deary IJ, McIntosh AM and Clarke TK (2018) Genome-wide association study meta-analysis of the Alcohol Use Disorders Identification Test (AUDIT) in two population-based cohorts. American Journal of Psychiatry 176, 107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann G, Liu C, O’Reilly P, Gao H, Song P, Xu B, Ruggeri B, Amin N, Jia T, Preis S, Segura Lepe M, Akira S, Barbieri C, Baumeister S, Cauchi S, Clarke TK, Enroth S, Fischer K, Hallfors J, Harris SE, Hieber S, Hofer E, Hottenga JJ, Johansson A, Joshi PK, Kaartinen N, Laitinen J, Lemaitre R, Loukola A, Luan J, Lyytikainen LP, Mangino M, Manichaikul A, Mbarek H, Milaneschi Y, Moayyeri A, Mukamal K, Nelson C, Nettleton J, Partinen E, Rawal R, Robino A, Rose L, Sala C, Satoh T, Schmidt R, Schraut K, Scott R, Smith AV, Starr JM, Teumer A, Trompet S, Uitterlinden AG, Venturini C, Vergnaud AC, Verweij N, Vitart V, Vuckovic D, Wedenoja J, Yengo L, Yu B, Zhang W, Zhao JH, Boomsma DI, Chambers J, Chasman DI, Daniela T, de Geus E, Deary I, Eriksson JG, Esko T, Eulenburg V, Franco OH, Froguel P, Gieger C, Grabe HJ, Gudnason V, Gyllensten U, Harris TB, Hartikainen AL, Heath AC, Hocking L, Hofman A, Huth C, Jarvelin MR, Jukema JW, Kaprio J, Kooner JS, Kutalik Z, Lahti J, Langenberg C, Lehtimaki T, Liu Y, Madden PA, Martin N, Morrison A, Penninx B, Pirastu N, Psaty B, Raitakari O, Ridker P, Rose R, Rotter JI, Samani NJ, Schmidt H, Spector TD, Stott D, Strachan D, Tzoulaki I, van der Harst P, van Duijn CM, Marques-Vidal P, Vollenweider P, Wareham NJ, Whitfield JB, Wilson J, Wolffenbuttel B, Bakalkin G, Evangelou E, Liu Y, Rice KM, Desrivières S, Kliewer SA, Mangelsdorf DJ, Müller CP, Levy D and Elliott P (2016) KLB is associated with alcohol drinking, and its gene product beta-Klotho is necessary for FGF21 regulation of alcohol preference. Proceedings of the National Academy of Sciences of the United States of America 113, 14372–14377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shield KD, Parry C and Rehm J (2013) Chronic diseases and conditions related to alcohol use. Alcohol Research 35, 155–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PF, Neale MC and Kendler KS (2000) Genetic epidemiology of major depression: review and meta-analysis. American Journal of Psychiatry 157, 1552–1562. [DOI] [PubMed] [Google Scholar]
- Swendsen JD and Merikangas KR (2000) The comorbidity of depression and substance use disorders. Clinical Psychology Review 20, 173–189. [DOI] [PubMed] [Google Scholar]
- The UK10K Consortium, Walter K, Min JL, Huang J, Crooks L, Memari Y, McCarthy S, Perry JR, Xu C, Futema M, Lawson D, Iotchkova V, Schiffels S, Hendricks AE, Danecek P, Li R, Floyd J, Wain LV, Barroso I, Humphries SE, Hurles ME, Zeggini E, Barrett JC, Plagnol V, Richards JB, Greenwood CM, Timpson NJ, Durbin R and Soranzo N (2015) The UK10 K project identifies rare variants in health and disease. Nature 526, 82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbanck M, Chen CY, Neale B and Do R (2018) Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nature Genetics 50, 693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhulst B, Neale MC and Kendler KS (2015) The heritability of alcohol use disorders: a meta-analysis of twin and adoption studies. Psychological Medicine 45, 1061–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrieze SI, McGue M, Miller MB, Hicks BM and Iacono WG (2013) Three mutually informative ways to understand the genetic relationships among behavioral disinhibition, alcohol use, drug use, nicotine use/dependence, and their co-occurrence: twin biometry, GCTA, and genome-wide scoring. Behavior Genetics 43, 97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wain LV, Shrine N, Miller S, Jackson VE, Ntalla I, Soler Artigas M, Billington CK, Kheirallah AK, Allen R, Cook JP, Probert K, Obeidat M, Bosse Y, Hao K, Postma DS, Pare PD, Ramasamy A, Consortium UKBE, Magi R, Mihailov E, Reinmaa E, Melen E, O’Connell J, Frangou E, Delaneau O, Ox GSKC, Freeman C, Petkova D, McCarthy M, Sayers I, Deloukas P, Hubbard R, Pavord I, Hansell AL, Thomson NC, Zeggini E, Morris AP, Marchini J, Strachan DP, Tobin MD and Hall IP (2015) Novel insights into the genetics of smoking behaviour, lung function, and chronic obstructive pulmonary disease (UK BiLEVE): a genetic association study in UK Biobank. The Lancet. Respiratory Medicine 3, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters RK, Adams MJ, Adkins AE, Aliev F, Bacanu S-A, Batzler A, Bertelsen S, Biernacka J, Bigdeli TB, Chen L-S, Clarke T-K, Chou Y-L, Degenhardt F, Docherty AR, Fontanillas P, Foo J, Fox L, Frank J, Giegling I, Gordon S, Hack L, Hartz SM, Heilmann-Heimbach S, Herms S, Hodgkinson C, Hoffmann P, Hottenga J-J, Kennedy MA, Alanne-Kinnunen M, Konte B, Lahti J, Lahti-Pulkkinen M, Ligthart L, Loukola A-M, Maher BS, Mbarek H, McIntosh AM, McQueen MB, Milaneschi Y, Palviainen T, Pearson JF, Peterson RE, Polimanti R, Ripatti S, Ryu E, Saccone NL, Salvatore JE, Sanchez-Roige S, Schwandt M, Sherva R, Streit F, Strohmaier J, Thomas N, Wang J-C, Webb BT, Wedow R, Wetherill L, Wills AG, Boardman JD, Chen D, Choi D-S, Copeland WE, Culverhouse RC, Dahmen N, Degenhardt L, Domingue BW, Elson SL, Frye M, Gäbel W, Ising M, Johnson EC, Keyes M, Kiefer F, Kramer J, Kuperman S, Lucae S, Lynskey MT, Maier W, Mann K, Männistö S, McClintick JN, Meyers JL, Müller-Myhsok B, Nurnberger JI, Palotie A, Preuss U, Räikkönen K, Reynolds MD, Ridinger M, Scherbaum N, Shuckit M, Soyka M, Treutlein J, Witt S, Wodarz N, Zill P, Adkins DE, Boden JM, Boomsma D, Bierut LJ, Brown SA, Bucholz KK, Cichon S, Costello EJ, de Wit H, Diazgranados N, Dick DM, Eriksson JG, Farrer LA, Foroud TM, Gillespie NA, Goate AM, Goldman D, Grucza RA, Hancock DB, Harris KM, Heath AC, Hesselbrock V, Hewitt JK, Hopfer CJ, Horwood J, Iacono W, Johnson EO, Kaprio JA, Karpyak VM, Kendler KS, Kranzler HR, Krauter K, Lichtenstein P, Lind PA, McGue M, MacKillop J, Madden PAF, Maes HH, Magnusson P, Martin NG, Medland SE, Montgomery GW, Nelson EC, Nöthen MM, Palmer AA, Pedersen NL, Penninx BWJH, Porjesz B, Rice J, Rietschel M, Riley BP, Rose R, Rujescu D, Shen PH, Silberg J, Stallings MC, Tarter RE, Vanyukov MM, Vrieze S, Wall TL, Whitfield JB, Zhao H, Neale BM, Gelernter J, Edenberg HJ and Agrawal A (2018) Trans-ancestral GWAS of alcohol dependence reveals common genetic underpinnings with psychiatric disorders. Nature Neuroscience 21, 1656–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt FR, Carvalho C, Gelernter J and Polimanti R (2018) DRD2 and FOXP2 are implicated in the associations between computerized device use and psychiatric disorders. bioRxiv, 497420.
- Wium-Andersen MK, Orsted DD, Tolstrup JS and Nordestgaard BG (2015) Increased alcohol consumption as a cause of alcoholism, without similar evidence for depression: a Mendelian randomization study. International Journal of Epidemiology 44, 526–539. [DOI] [PubMed] [Google Scholar]
- Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, Adams MJ, Agerbo E, Air TM, Andlauer TMF, Bacanu SA, Baekvad-Hansen M, Beekman AFT, Bigdeli TB, Binder EB, Blackwood DRH, Bryois J, Buttenschon HN, Bybjerg-Grauholm J, Cai N, Castelao E, Christensen JH, Clarke TK, Coleman JIR, Colodro-Conde L, Couvy-Duchesne B, Craddock N, Crawford GE, Crowley CA, Dashti HS, Davies G, Deary IJ, Degenhardt F, Derks EM, Direk N, Dolan CV, Dunn EC, Eley TC, Eriksson N, Escott-Price V, Kiadeh FHF, Finucane HK, Forstner AJ, Frank J, Gaspar HA, Gill M, Giusti-Rodriguez P, Goes FS, Gordon SD, Grove J, Hall LS, Hannon E, Hansen CS, Hansen TF, Herms S, Hickie IB, Hoffmann P, Homuth G, Horn C, Hottenga JJ, Hougaard DM, Hu M, Hyde CL, Ising M, Jansen R, Jin F, Jorgenson E, Knowles JA, Kohane IS, Kraft J, Kretzschmar WW, Krogh J, Kutalik Z, Lane JM, Li Y, Li Y, Lind PA, Liu X, Lu L, MacIntyre DJ, MacKinnon DF, Maier RM, Maier W, Marchini J, Mbarek H, McGrath P, McGuffin P, Medland SE, Mehta D, Middeldorp CM, Mihailov E, Milaneschi Y, Milani L, Mill J, Mondimore FM, Montgomery GW, Mostafavi S, Mullins N, Nauck M and Ng B, Nivard MG, Nyholt DR, O’Reilly PF, Oskarsson H, Owen MJ, Painter JN, Pedersen CB, Pedersen MG, Peterson RE, Pettersson E, Peyrot WJ, Pistis G, Posthuma D, Purcell SM, Quiroz JA, Qvist P, Rice JP, Riley BP, Rivera M, Saeed Mirza S, Saxena R, Schoevers R, Schulte EC, Shen L, Shi J, Shyn SI, Sigurdsson E, Sinnamon GBC, Smit JH, Smith DJ, Stefansson H, Steinberg S, Stockmeier CA, Streit F, Strohmaier J, Tansey KE, Teismann H, Teumer A, Thompson W, Thomson PA, Thorgeirsson TE, Tian C, Traylor M, Treutlein J, Trubetskoy V, Uitterlinden AG, Umbricht D, Van der Auwera S, van Hemert AM, Viktorin A, Visscher PM, Wang Y, Webb BT, Weinsheimer SM, Wellmann J, Willemsen G, Witt SH, Wu Y, Xi HS, Yang J, Zhang F; eQTLGen; 23andMe, Arolt V, Baune BT, Berger K, Boomsma DI, Cichon S, Dannlowski U, de Geus ECJ, DePaulo JR, Domenici E, Domschke K, Esko T, Grabe HJ, Hamilton SP, Hayward C, Heath AC, Hinds DA, Kendler KS, Kloiber S, Lewis G, Li QS, Lucae S, Madden PFA, Magnusson PK, Martin NG, McIntosh AM, Metspalu A, Mors O, Mortensen PB, Müller-Myhsok B, Nordentoft M, Nöthen MM, O’Donovan MC, Paciga SA, Pedersen NL, Penninx BWJH, Perlis RH, Porteous DJ, Potash JB, Preisig M, Rietschel M, Schaefer C, Schulze TG, Smoller JW, Stefansson K, Tiemeier H, Uher R, Völzke H, Weissman MM, Werge T, Winslow AR, Lewis CM, Levinson DF, Breen G, Børglum AD, Sullivan PF and Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium (2018) Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nature Genetics 50, 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Yang C, Wang J and Small DS (2018) Powerful genome-wide design and robust statistical inference in two-sample summary-data Mendelian randomization. arXiv:1804.07371v2 [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
MD GWAS:
The PGC’s policy is to make genome-wide summary results public. Summary statistics for a combined meta-analysis of PGC29 with five of the six expanded samples (deCODE, Generation Scotland, GERA, iPSYCH, and UK Biobank) are available on the PGC web site (https://www.med.unc.edu/pgc/results-and-downloads). Results for 10 000 SNPs for all seven cohorts are also available on the PGC web site. GWA summary statistics for the Hyde et al., cohort (23andMe, Inc.) must be obtained separately. These can be obtained by qualified researchers under an agreement with 23andMe that protects the privacy of the 23andMe participants. Contact David Hinds (dhinds@23andme.com) to apply for access to the data. Researchers who have the 23andMe summary statistics can readily recreate our results by meta-analyzing the six-cohort results file with the Hyde et al., results file from 23andMe.
AD GWAS:
The PGC’s policy is to make genome-wide summary results public. Summary statistics are available on the PGC web site is (https://www.med.unc.edu/pgc/results-and-downloads).
AC quantity and frequency GWAS: Summary statistics will be made publicly available through LD hub http://ldsc.broadinstitute.org/ldhub/ before publication of this paper or can be obtained upon request from the corresponding author.