

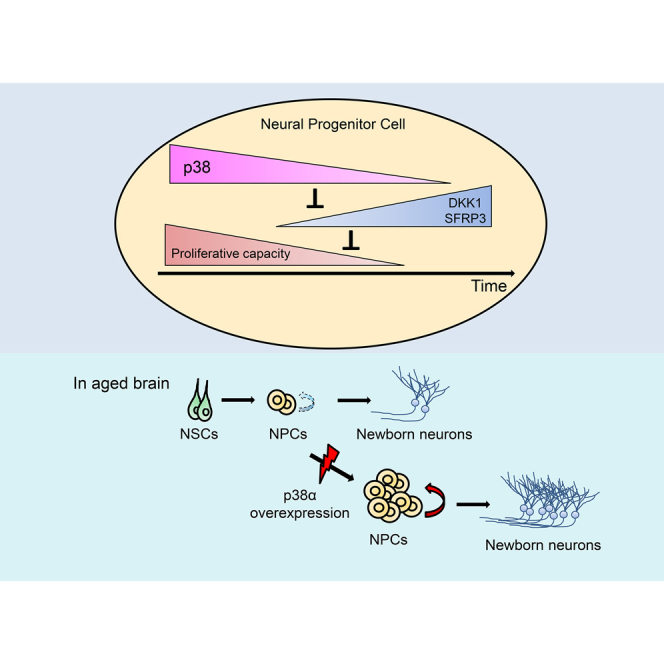
Summary
Neurogenesis in specific brain regions in adult mammals decreases with age. Progressive reduction in the proliferation of neural stem and progenitor cells (NS/PCs) is a primary cause of this age-associated decline. However, the mechanism responsible for this reduction is poorly understood. We identify p38 MAPK as a key factor in the proliferation of neural progenitor cells (NPCs) in adult neurogenic niches. p38 expression in adult NS/PCs is downregulated during aging. Deletion of p38α in NS/PCs specifically reduces the proliferation of NPCs but not stem cells. Conversely, forced expression of p38α in NS/PCs in the aged mouse subventricular zone (SVZ) restores NPC proliferation and neurogenesis, and prevents age-dependent SVZ atrophy. We also found that p38 is necessary for suppressing the expression of Wnt antagonists DKK1 and SFRP3, which inhibit the proliferation of NPCs. Age-related reduction in p38 thus leads to decreased adult neurogenesis via downregulation of Wnt signaling.
Key words: adult neurogenesis, aging, DKK1, neural progenitor cell, neural stem cell, p38, SFRP3, transit-amplifying cell, Wnt signaling
Graphical Abstract
Highlights
-
•
p38 expression in adult neural stem and progenitor cells declines during aging
-
•
p38 regulates the proliferation of neural progenitor cells in the adult brain
-
•
Reduction of p38 is a major cause of age-related decline of adult neurogenesis
-
•
Forced expression of p38α restores adult neurogenesis without exhaustion of NSCs
Kase et al. show that p38 expression in neural stem and progenitor cells (NS/PCs) in the adult brain decreases with aging. This reduction specifically causes proliferation defect in neural progenitor cells (NPCs), leading to the age-dependent decline of adult neurogenesis. Conversely, overexpression of p38α in NS/PCs in the aged brain restores NPC proliferation without exhaustion of neural stem cells.
Introduction
Neurogenesis persists mainly in two brain regions, the subventricular zone (SVZ) of the lateral ventricle and the subgranular zone (SGZ) of the dentate gyrus, throughout adulthood in mammals (Ming and Song, 2011). In these regions, slowly dividing quiescent neural stem cells (NSCs) give rise to activated NSCs, which rapidly differentiate into transit-amplifying cells (TACs) and subsequently into immature neurons (Encinas et al., 2011, Ming and Song, 2011, Obernier et al., 2018). Adult neurogenesis in these neurogenic niches declines with aging (Capilla-Gonzalez et al., 2015, Encinas et al., 2011), largely due to the reduced proliferation of neural stem and progenitor cells (NS/PCs). Several studies have reported that this reduction is likely to be caused by declines in extrinsic signals that support the proliferation of NS/PCs, including mitotic signals such as epidermal growth factor (EGF) and fibroblast growth factor 2 (FGF-2) (Conover and Todd, 2017, Seib and Martin-Villalba, 2015) and increases in systemic pro-aging factors (Bond et al., 2015). Moreover, it has recently been suggested that mitochondrial dysfunction is a cause of the age-related decline in neurogenesis in the SGZ (Beckervordersandforth et al., 2017). In these studies, exposure of the SGZ or SVZ to mitogens or enhancement of mitochondrial function restored neurogenesis; however, the specific cell types targeted by these interventions were not clearly identified, and the underlying age-associated processes have not been studied in detail.
The p38 MAPKs (mitogen-activated protein kinases) include four family members: p38α (MAPK14), p38β (MAPK11), p38γ (MAPK12), and p38δ (MAPK13). p38α was first identified as a stress-activated protein kinase associated with inflammation (Ashwell, 2006). The p38 MAPK signaling pathway mediates a range of extrinsic signals, including environmental stressors, growth factors, and cytokines, to ensure appropriate cellular outputs (Cuadrado and Nebreda, 2010). Although many studies have reported the involvement of p38 MAPK signaling in the proliferation, differentiation, migration, and apoptosis of NS/PCs, the results reported to date have been disparate (Cheng et al., 2001, Faigle et al., 2004, Wang et al., 2017). Specifically, different groups using various experimental systems have reported both positive and negative effects of p38 MAPK signaling on the proliferation and apoptosis of NS/PCs (Kim et al., 2008, Kim and Wong, 2009, Sato et al., 2008, Yoshioka et al., 2015).
In the present study, we report that p38 MAPK signaling is required for the sustained proliferation of neural progenitor cells (NPCs) in the adult neurogenic niche via modulation of Wnt signaling and that decreased p38 MAPK signaling is responsible for the age-related decline in adult neurogenesis. Moreover, we found that forced expression of p38α in NS/PCs in the SVZ of aged mice prevented the decline in adult neurogenesis and inhibited age-related SVZ atrophy without exhaustion of NSCs. Our findings provide new mechanistic insights into the aging of neurogenic niches and a new molecular target for inducing regeneration of the aged brain via the mobilization of NPCs.
Results
p38 Expression in Adult Neurogenic Niches Decreases with Aging
We previously reported that p38 is essential for the generation of large numbers of glial cells during late embryonic development (Naka-Kaneda et al., 2014). In contrast, NS/PCs mainly generate neurons in adult neurogenic niches (Ming and Song, 2011). This neurogenesis declines with aging (Figure S1), which prompted us to ask whether p38 facilitates the age-dependent reduction in adult neurogenesis by increasing gliogenesis. We immunohistochemically analyzed the expression of p38 and its activated form, phosphorylated p38 (P-p38), in the SVZ and SGZ of postnatal mice at various ages. Unexpectedly, the expression levels of both p38 and P-p38 decreased with aging (Figures S2A–S2D). Further immunohistochemical analyses using markers for NSCs (SOX2+/GFAP+ cells) and for TACs (ASCL1+ cells) (Ming and Song, 2011) revealed that P-p38 is expressed in most NSCs and TACs in young animals. However, the proportion of P-p38+ cells in these cells decreased with age (Figures 1A–1D and 1I; Table S1). This age-related decrease in P-p38+ NSCs was more pronounced in the SGZ than in the SVZ. Decreases in the P-p38+ population of NSCs in the SVZ in the maturing mouse brain were significant but slight, and the P-p38 levels in NSCs of the SVZ did not show dramatic declines, even in aged mice. In TACs, on the other hand, the P-p38+ population dramatically decreased with aging in both the SVZ and SGZ (Figures 1E–1H and 1J; Table S1). The spatiotemporal pattern of changes in P-p38 expression appears to be correlated with the age-dependent reduction of NSCs and TACs in the SVZ and SGZ (Figure S1).
Figure 1.

Age-Dependent Reduction in the Phospho-p38 and p38 Expression Levels in the SVZ and SGZ
(A–D) Representative confocal images of the SVZ (A and B) and SGZ (C and D) from 3- and 77-week-old mice, immunostained with antibodies raised against markers for NSCs (SOX2+ [magenta]/GFAP+ [cyan] cells) and P-p38 (green).
(E–H) Representative confocal images of the SVZ (E and F) and SGZ (G and H) from 8- and 28-week-old mice, immunostained with antibodies raised against markers for TACs (ASCL1+ cells [magenta] and P-p38 [green]).
(I) Percentages of phospho-p38 (P-p38)-positive cells in SOX2+/GFAP+ NSCs in the SVZ (red line) and SGZ (blue line), which decreased with aging.
(J) Percentages of phospho-p38 (P-p38)-positive cells in ASCL1+ TACs in the SVZ (red line) and SGZ (blue line), which also decreased with aging.
Values in the line graphs represent the mean ± SD (n = 3 mice). Nuclei were stained with Hoechst (gray). LV, lateral ventricle. Scale bars, 50 μm.
Adult Neurogenesis Is Impaired in p38α Conditional Knockout Mice
To determine the role of p38 in adult neurogenesis, we analyzed p38α (Mapk14) conditional knockout mice (p38α CKO mice) because p38α is the major isozyme of the p38 MAPK family (Ashwell, 2006). To generate the p38α CKO mice, in which an exon of p38α is selectively deleted in adult NS/PCs at specific time points, we crossed p38αflox/flox mice (Nishida et al., 2004) with transgenic mice expressing CreERT2 recombinase under the control of the Nestin gene enhancer/promoter (Nes-creER mice) (Imayoshi et al., 2006). Selective deletion was induced by intraperitoneal (i.p.) injection of tamoxifen (TAM) (75 mg/kg) for 5 consecutive days, followed 4 weeks later by the injection of 5-ethynyl-2′-deoxyuridine (EdU) (50 mg/kg, i.p.) 2 h prior to brain fixation to assess NS/PC proliferation (Figure 2A). To confirm the conditional deletion, we crossed p38αflox/flox;Nes-creER mice with a reporter mouse line that expresses monomeric teal fluorescent protein 1 (mTFP1) in a Cre-dependent manner from the ROSA26 locus (Imayoshi et al., 2012). As expected, we detected the expression of mTFP1 in SOX2+/GFAP+ NSCs in the SVZ and SGZ (Figures S3A and S3B). We also confirmed that TAM has no detrimental effect on the proliferation of NS/PCs in the SVZ and SGZ of either wild-type or Nes-creER mice (Figure S3C).
Figure 2.

Reduction of TACs and Neurogenesis in the SVZ and SGZ of p38α CKO Mice
(A) Strategy for inducible conditional deletion of p38α in p38αflox/flox;Nes-creER mouse and EdU labeling of cycling NS/PCs. Tamoxifen (TAM) was injected intraperitoneally (i.p.) for five consecutive days. EdU was injected i.p. 2 h before perfusion.
(B and C) There was no significant difference in the number of SOX2+/GFAP+ NSCs in the SVZ (B) and SGZ (C) between control floxed p38α and p38α CKO mice at 10 or 65 weeks (n = 5 mice).
(D and E) ASCL1+ TACs in the SVZ (D) and SGZ (E) were significantly lower in p38α CKO mice than in control mice at 10 weeks but not at 65 weeks (n = 5 mice).
(F) EdU+ cycling cells in the SVZ were also significantly lower in p38α CKO mice compared with control mice at both 10 and 65 weeks (n = 5 mice).
(G) EdU+ cycling cells in the SGZ were also significantly lower in p38α CKO mice than in control mice at 10 weeks but not at 65 weeks (n = 5 mice).
(H and I) Representative confocal images of EdU+ cells (magenta) in the SVZ (H) and SGZ (I) of control and p38α CKO mice at 10 weeks. Scale bars, 100 μm.
(J and K) TBR2+ intermediate progenitor cells in SGZ were significantly reduced in p38α CKO mice compared with control mice at 10 weeks (n = 3 mice). Scale bars in (K), 50 μm.
(L and M) DCX+ immature newborn neurons (red) in the SVZ were significantly lower in p38α CKO mice compared with control mice at 10 weeks (n = 5 mice). Lower panels of (M) are the higher-magnification images corresponding to dashed-line squares in the upper panels. Scale bars, 200 μm.
(N and O) DCX+ immature newborn neurons (red) in the SGZ were also significantly lower in p38α CKO mice compared with control mice at 10 weeks (n = 5 mice). Scale bars in (P), 100 μm.
Statistical analysis was performed with an unpaired two-tailed Student's t test. Values in the bar graphs represent the mean ± SD. ∗∗p < 0.01, ∗∗∗p < 0.001. NS, not significant; LV, lateral ventricle.
There was no significant difference between control (without TAM) and p38α CKO mice regarding the number of NSCs in the SVZ and SGZ at 10 weeks and 65 weeks (Figures 2B, 2C, and S3D). In contrast, the number of ASCL1+ TACs was significantly lower in both SVZ and SGZ in p38α CKO mice at 10 weeks compared with that in control mice; however, no significant difference was observed at 65 weeks (Figures 2D, 2E, and S3E). We observed similar reductions of EdU incorporation in p38α CKO mice, even at 65 weeks, in the SVZ (Figures 2F–2I). Moreover, the number of TBR2 (T-box brain gene 2) expressing intermediate progenitor cells, a type of TAC in SGZ (Hodge et al., 2008), were also significantly reduced in p38α CKO mice compared with control mice at 10 weeks (Figures 2J and 2K). There was no significant difference in the number of apoptotic cells detected by TUNEL staining between control and p38α CKO mice, and this number increased with age in both groups (Figures S3J–S3M). These results suggest that p38α is required for the amplification of TACs. The number of DOUBLECORTIN (DCX)-expressing immature neurons, which are progeny cells of TACs (Ming and Song, 2011), was also significantly reduced in both SVZ and SGZ in 10-week-old mice through p38α deficiency (Figures 2L–2O). The reduction in the number of TACs should cause the subsequent reduction in the number of newborn neurons. Indeed, EdU pulse-chase experiments revealed that p38α deletion results in significant reductions of EdU+/NeuN+ newborn neurons in the olfactory bulb (OB) and granule cell layer in the dentate gyrus (DG) after a 4-week chase at 14 weeks (Figures S3F–S3I). The reduction rates of the EdU+ newborn neurons by p38α deletion in both regions were not significantly different from that of DCX+ immature neurons in SVZ and SGZ (DCX+ cells: 0.546 ± 0.011 in SVZ, 0.720 ± 0.074 in SGZ; EdU+/NeuN+ cells: 0.486 ± 0.106 in OB, 0.626 ± 0.151 in DG, relative to control mice), indicating that p38α is not involved either in differentiation or survival of adult-born neurons in these regions.
Specific Requirement of p38 for Proliferation of TACs
To determine how p38α functions in NS/PCs, we assessed the proliferation of TACs by quantification of ASCL1+EdU+/ASCL1+ cells and the activation of NSCs as defined by Ki-67 expression within the GFAP+/SOX2+ population (Kawaguchi et al., 2013) in the SVZ and SGZ at 6 and 10 weeks. Conditional disruption of p38α and EdU labeling was carried out as described above (Figure 2A). The proliferation of TACs was significantly reduced by the disruption of p38α in both the SVZ and SGZ (Figure 3), whereas there was no change in the activation of NSCs (Figure S4).
Figure 3.

Specifically Impaired Proliferation of TACs in p38α CKO Mice
(A) The EdU+ population (magenta) within ASCL1+ TACs (green) in the SVZ was significantly lower in p38α CKO mice than in control mice at 6 and 10 weeks (n = 3 mice).
(B) Representative confocal images of ASCL1+ TACs (green) and EdU+ cells (magenta) in the SVZ of control and p38α CKO mice (10 weeks old). Arrows indicate ASCL1+/EdU+ cells.
(C) The EdU+ population (magenta) within ASCL1+ TACs (green) in the SGZ was significantly lower in p38α CKO mice than in control mice at 6 and 10 weeks (n = 3 mice).
(D) Representative confocal images of ASCL1+ TACs (green) and EdU+ cells (magenta) in the SGZ of control and p38α CKO mice (10 weeks old). Arrows indicate ASCL1+/EdU+ cells.
Statistical analysis was performed with an unpaired two-tailed Student's t test. Values in the bar graphs represent the mean ± SD. ∗∗p < 0.01, ∗∗∗p < 0.001. LV, lateral ventricle. Scale bars, 100 μm.
To examine whether the altered proliferation of TACs by p38α deletion was due to an environmental change within the neurogenic niche, we conducted a neurosphere assay (Figure 4A) in which TACs were specifically amplified to form clonally derived cell clusters composed mainly of proliferating NPCs in the presence of FGF-2 and EGF in vitro (Mich et al., 2014). Owing to difficulty in obtaining a sufficient volume of neurospheres from SGZ of aged mice to enable a detailed analysis, we analyzed neurospheres derived from adult SVZs in the following in vitro studies. Pan-p38, p38α, and P-p38 immunoreactivities detected in control neurospheres were barely detectable in neurospheres derived from p38α CKO mice (Figures 4B and 4C), which strongly indicated that p38α is a major isozyme in NPCs. To assess NPC self-renewal and proliferation, we measured the number, size, and EdU incorporation of secondary neurospheres after a passage of primary neurospheres. EdU (10 μM) was administered 30 min before fixation of the neurospheres. The disruption of p38α resulted in a significant reduction in the total number of neurospheres, a significant population shift to smaller sizes of neurospheres, and a significant reduction in EdU incorporation (Figures 4D–4F). These results suggest that p38α plays a crucial role in the self-renewing expansion of NPCs and that p38 is required to support the full proliferation of TACs in the adult brain.
Figure 4.

Impaired Proliferation of the NS/PCs of p38α CKO Mice and Aged Mice In Vitro
(A) Strategy for inducible conditional deletion of p38α in p38αflox/flox;Tg (Nes-cre/ERT2) mice for an in vitro NPC proliferation assay via neurosphere formation. Four-week-old mice were injected i.p. with TAM for 5 consecutive days and processed for neurosphere formation 7 days after injection.
(B and C) Representative confocal images of pan-p38, p38α, P-p38 (all green), and NESTIN (red) reactivities in neurospheres from control (B) and p38α CKO mice (C) (6 weeks old). Nuclei were counterstained with Hoechst (blue). Scale bars, 100 μm.
(D) Distribution of the different sizes of secondary neurospheres and their total numbers between control and p38α CKO mice (n = 3 independent cultures). The p38α CKO neurospheres were relatively smaller and less expandable.
(E) Representative confocal images of EdU+ cells (red) in control and p38α CKO neurospheres. EdU (10 μM) was administered 30 min before fixation of neurospheres. Nuclei were stained with Hoechst (blue). Scale bars, 100 μm.
(F) EdU incorporation into the neurospheres was significantly reduced by p38α deletion (n = 6 independent cultures; at least 1,500 cells were analyzed).
(G) SA-β-gal staining of control and p38α CKO neurospheres in blue. The color is darker in the p38α CKO neurosphere than in the control neurosphere. Scale bars, 100 μm.
(H) Quantitative analysis of SA-β-gal activity. SA-β-gal activity was significantly increased in p38α CKO neurospheres compared with that in control neurospheres (n = 3 independent cultures). RFU, relative fluorescence units.
(I) The number of secondary neurospheres from aged mice (6 months old) was significantly lower than that of young mice (6 weeks old) (n = 6 independent cultures).
(J) Representative confocal images of EdU+ cells (red) in neurospheres from young and aged mice. EdU labeling was performed as described. Nuclei were counterstained with Hoechst (blue). Scale bars, 100 μm.
(K) EdU incorporation in the neurospheres was significantly reduced in aged mice (n = 6 independent cultures; at least 600 cells were analyzed).
(L) Quantitative analysis of the p38 mRNA expression in secondary neurospheres. The relative expression of p38 mRNA decreased with age (n = 6 independent experiments).
(M and N) Significant reduction of p38 expression in the neurospheres from aged mice. Pan-p38, p38α, and P-p38 immunoreactivities (green) were barely detected in neurospheres from aged mice (N), whereas all of these immunoreactivities were clearly detected in the neurospheres from young mice (M). There was no significant difference in NESTIN expression (red) between neurospheres from young and aged mice. Nuclei were stained with Hoechst (blue). Scale bars, 100 μm.
Statistical analysis was performed with an unpaired two-tailed Student's t test (D, F, H, I, and K) or one-way ANOVA, Tukey-Kramer post hoc test (L). Values in the bar graphs represent the mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. NS, not significant; 6W, 6 weeks old; 6M, 6 months old, 8M, 8 months old; TAM, tamoxifen.
The age-dependent decline in p38 expression by TACs in the SVZ and SGZ (Figure 1) is strongly correlated with similar decreases in TACs (Figure S1), raising the possibility that the reduction in p38α induces cellular senescence. To test this possibility, we measured senescence-associated β-galactosidase (SA-β-gal) activity (Dimri et al., 1995). Although it has been shown that SA-β-gal activity is not specific to cellular senescence (Yang and Hu, 2005), such activity remains useful as a marker of cell-cycle arrest. Compared with neurospheres derived from control mice, SA-β-gal activity was significantly increased in those from p38α CKO mice (Figures 4G and 4H), which indicated a role of p38α in supporting the proliferation of NPCs and raising the possibility that p38 regulates the timing of cellular senescence in TACs. We also examined whether p38α deletion alters the expressions of two senescence-associated genes, p16INK4a, whose expression in NS/PC in the SVZ increases with age (Daynac et al., 2016), and p19ARF, which regulates NSC self-renewal in the SVZ (Nishino et al., 2008), in neurospheres from the SVZ. However, no significant change was observed in their expressions (Figure S6A). This suggests that these factors are not involved in the regulation of TAC proliferation downstream of p38.
Reduction in p38 Is a Major Cause of Age-Related Decline in Adult Neurogenesis
Given its activity and expression pattern, p38 may regulate the aging of NS/PCs in adult neurogenic niches. To test this hypothesis, we first examined self-renewing expansion and p38 expression in NPCs derived from the SVZ of young (6-week-old) and aged (6-month-old) mice by using a neurosphere assay in vitro. In comparison with young mice, in aged mice the numbers of secondary neurospheres or degree of EdU incorporation were significantly lower (Figures 4I–4K). Moreover, the p38 mRNA expression in neurospheres was significantly decreased in an age-dependent manner (Figure 4L). Pan-p38, p38α, and P-p38 immunoreactivity was barely detectable in neurospheres from aged mice, whereas such immunoreactivity was clearly detectable in neurospheres from young mice (Figures 4M and 4N). These age-dependent differences in the characteristics of NPCs in vitro resemble those in vivo (Figures 1 and S1), suggesting that the reduction of p38 expression in NS/PCs are largely responsible for the decline in adult neurogenesis with age.
Forced Expression of p38α Restores the Proliferative Capacity of NPCs and Prevents Age-Related SVZ Atrophy without Exhaustion of NSCs
We next asked whether forced expression of p38 would rescue the age-dependent decline in NPC proliferation. Forced expression of p38α in secondary neurospheres derived from aged (6-month-old) mice via a lentiviral vector (Naka-Kaneda et al., 2014) resulted in an approximately 2-fold increase in EdU incorporation compared with that via a control VENUS-expressing vector (Figures 5A–5C). Similarly, infusion of the p38α-expressing lentiviral vector in the lateral ventricles of the 6-month-old mice significantly increased the number of ASCL1+ TACs and the EdU incorporation in the SVZ at 7 days post infection compared with the corresponding levels in the control brain infected with the VENUS-expressing lentiviral vector (Figures 5D–5G), whereas no significant changes were observed in EdU incorporation in SOX2+/GFAP+ NSCs (Figures 5H and 5I). Moreover, forced expression of p38α in SVZ restored the number of DCX+ immature neurons to the level of 10-week-old mice (Figures 5J and 5K). These findings indicated that the reduction of p38 in NPCs, but not in NSCs, is largely responsible for the age-dependent decline in adult neurogenesis.
Figure 5.

Forced Expression of p38α Restores the Proliferative Capacity of NPCs and Neurogenesis in Aged Mice without Exhaustion of NSCs
(A) EdU incorporation in the neurospheres was significantly increased by p38α overexpression (OE) (n = 3 independent cultures; at least 700 cells were analyzed).
(B and C) Representative confocal images of EdU+ cells (magenta) in neurospheres from aged mice (6 months old) transduced with lentiviral vectors expressing VENUS or p38α. VENUS-expressing (green) control vector. HA staining (green) for detecting the exogenous expression of p38α (B). P-p38 staining (green) for confirmation of p38 activation (C). Nuclei were stained with Hoechst (blue). Scale bars, 100 μm.
(D and E) p38α OE via infusion of a lentiviral vector significantly increased the number of ASCL1+ cells in the SVZ of aged mice (n = 6 mice) (D). Representative confocal images of ASCL1+ cells (magenta) in the SVZ of aged (6-month-old) mice infected with lentiviruses (E). VENUS-expressing (green) control vector. p38α OE was detected by HA staining (green). Only VENUS+ or HA+ cells are counted. Scale bars, 50 μm.
(F and G) p38α OE via infusion of a lentiviral vector significantly increased EdU incorporation into the SVZ of aged (6-month-old) mice (F). EdU labeling was performed as described in Figure 2A (n = 6 mice). Representative confocal images of EdU+ cells (magenta) in the SVZ of aged (6-month-old) mice infected with lentiviruses (G). VENUS-expressing control vector (green). p38α OE was detected by HA staining (green). Only VENUS+ or HA+ cells are counted. Scale bars, 100 μm.
(H and I) p38α OE did not alter the number of SOX2+/EdU+/GFAP+ cells in the SVZ of aged mice (H). EdU labeling was performed as described in Figure 2A (n = 6 mice). Representative confocal images of activated NSCs defined as SOX2+ (pseudocolored green)/EdU+ (magenta)/GFAP+ (cyan) cells in the SVZ of aged mice infected with control and p38α OE lentiviruses (I). Only VENUS+ or HA+ cells are counted. Scale bars, 50 μm.
(J and K) p38α OE restored neurogenesis in the SVZ of aged mice. The number of DCX+ immature neurons in the SVZ of the aged mice was increased to the level of that in young mice by p38α OE (n = 6 mice) (J). Representative confocal images of DCX+ cells (red) in the SVZ of aged mice infected with lentiviruses and in the SVZ of 10-week-old wild-type mice (K). Only VENUS+ or HA+ cells are counted. Scale bars, 200 μm.
(L and M) A significant increase in the number of ASCL1+ TACs in the SVZ of 18-month-old mice by p38α OE (n = 4 mice) (L). Representative confocal images of ASCL1+ (magenta) cells in the SVZ of 18-month-old mice (M). Control VENUS-expressing lentiviruses or p38α OE lentiviruses were infused into the lateral ventricles of 6-month-old mice, followed by analysis at 18 months of age. Scale bars, 50 μm.
(N and O) A significant increase in EdU incorporation into the SVZ of 18-month-old mice by p38α OE (n = 4 mice) (N). Representative confocal images of EdU+ (red) cells in the SVZ of 18-month-old mice (O). Control VENUS-expressing lentiviruses or p38α OE lentiviruses were infused into the lateral ventricles of 6-month-old mice, followed by analysis at 18 months of age. Scale bars, 100 μm.
(P and Q) A significant increase in the number of DCX+ immature neurons in the SVZ of 18-month-old mice by p38α OE (n = 4 mice) (P). Representative confocal images of DCX+ (green) cells in the SVZ of 18-month-old mice (Q). Control VENUS-expressing lentiviruses or p38α OE lentiviruses were infused into the lateral ventricles of 6-month-old mice, followed by analysis at 18 months of age. Scale bars: 100 μm.
Statistical analysis was performed with an unpaired two-tailed Student's t test (A, D, F, H, L, N, and P) or one-way ANOVA, Tukey-Kramer post hoc test (J). Values in the bar graphs represent the mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. NS, not significant; LV, lateral ventricle.
We further analyzed 18-month-old mice infused with the p38α-expressing lentiviral vector in the lateral ventricles at the age of 6 months to determine whether the increase in NPC proliferation driven by the forced expression of p38α induces exhaustion of NSCs. Compared with VENUS-expressing control mice, mice with the forced expression of p38α exhibited significant increases in ASCL1+ TACs, EdU incorporation, and DCX+ immature neurons in the SVZ (Figures 5L–5Q and S5A), while the transgene expression decreased significantly, which necessitated an enhancement of the signal to detect hemagglutinin (HA) immunoreactivity (Figure S5B). Moreover, the size of the lateral ventricle in the p38α-overexpressing (OE) mice was significantly smaller than that in the control mice (Figures S5C and S5D), and the SVZ defined by SOX2+ cell layers was significantly thicker in the p38α OE mice compared with the control mice (Figures S5E and S5F). Thus, sustained expression of p38 prevented the age-related decline in neurogenesis and the progression of atrophy in SVZ without exhaustion of NSCs.
p38 Regulates NPC Proliferation via the Modulation of Wnt Signaling in the Adult SVZ
We next sought to determine how the p38 MAPK pathway facilitates NPC proliferation. We first screened 28 genes that are reportedly involved in NS/PC proliferation in the adult brain as candidate downstream effectors of the p38 MAPK pathway. We analyzed the expression levels of these genes in neurospheres (derived from the SVZ of 10-week-old mice) transduced with a lentiviral vector expressing small hairpin RNA (shRNA) targeting p38α (p38α KD) or control shRNA (sh Control) (Naka-Kaneda et al., 2014) by using qRT-PCR (Figure S6A). We found that the expression levels of Dickkopf-1 (Dkk1) and secreted frizzled-related protein 3 (Sfrp3), which are antagonists of canonical Wnt signaling (Niehrs, 2006, Jones and Jomary, 2002), were significantly increased in the p38α KD neurospheres compared with the corresponding levels in the sh Control neurospheres (Figure S6B). DKK1 and SFRP3 have been shown to attenuate NS/PC proliferation and adult neurogenesis (Jang et al., 2013, Seib et al., 2013). We also confirmed significant increases in the expression levels of these genes in p38α CKO neurospheres derived from the SVZ of 6-week-old mice compared with those of control neurospheres (Figure 6A). Additionally, expression levels of DKK1 and SFRP3 in SGZ at 14 weeks also significantly increased in p38α CKO mice compared with the control mice (Figure S6C).
Figure 6.

DKK1 and SFRP3 Regulate NPC Proliferation Downstream of p38
(A) Expression levels of DKK1 and SFRP3 increased in p38α CKO neurospheres. Neurospheres were obtained as described in Figure 4A (n = 3 independent experiments).
(B) Decreased EdU incorporation into neurospheres by deletion of p38α was restored by knockdown (KD) of Dkk1 and Sfrp3 via a lentiviral vector expressing specific shRNAs targeting each mRNA (Dkk1 KD and Sfrp3 KD). EdU labeling was performed as described in Figure 4 (n = 3 independent cultures; at least 1,000 cells were analyzed).
(C) Representative images of EdU (red) incorporation into each neurospheres. Nuclei were counterstained with Hoechst (blue). Scale bars, 100 μm.
(D) Distribution of the different sizes of secondary neurospheres and their total numbers. Decreased neurosphere formation by deletion of p38α was canceled either by Dkk1 KD or Sfrp3 KD (n = 6 independent cultures).
Statistical analysis was performed with an unpaired two-tailed Student's t test (A), one-way ANOVA and Tukey-Kramer post hoc test (B and D). Values in the bar graphs represent the mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Next, we examined whether the impaired NPC proliferation caused by the loss of p38 is due to increases in DKK1 and SFRP3 expression. Individual knockdown of Dkk1 and Sfrp3 by the lentiviral vector expressing shRNA targeting each of these genes (Dkk1 KD and Sfrp3 KD) (Figure S7A) in the p38α CKO neurospheres resulted in marked recoveries of NPC proliferation assessed by EdU incorporation (Figures 6B and 6C) and neurosphere growth and self-renewal ability assessed by the neurosphere formation efficiency (Figure 6D) to the level of control neurospheres. These results indicated that the repression of DKK1 and SFRP3 expression downstream of p38 supports the proliferation of NPCs.
We further examined whether DKK1 and SFRP3 are responsible for the age-related decline in NPC proliferation caused by the decrease in p38 expression in the adult SVZ. The analyses of both DKK1 and SFRP3 expression in neurospheres derived from mouse SVZs at various ages (3, 8, and 15 months old) by qRT-PCR revealed robust increases in NS/PCs in an age-dependent manner (Figure 7A). Similar age-dependent increases in DKK1 and SFRP3 expressions were observed in the SGZ (Figure S6D). Moreover, both Dkk1 KD and Sfrp3 KD in neurospheres derived from the SVZ of 14-week-old mice caused significant increases in EdU incorporation and neurosphere formation, while no synergistic or additive effect was observed upon double knockdown of Dkk1 and Sfrp3 (Figures 7B, S7B, and S7C). The Dkk1 KD and Sfrp3 KD in the SVZ of aged (6-month-old) mice also caused a significant increase in the EdU-incorporated dividing cells and DCX+ immature neurons compared with those in the control mice (Figures 7C, 7D, and S7D–S7F).
Figure 7.

Age-Dependent Increases in DKK1 and SFRP3 Cause a Decline in NPC Proliferation
(A) DKK1 and Sfrp3 mRNA expression levels in the secondary neurospheres from adult SVZs increased in an age-dependent manner (n = 3 independent experiment). 3M, 3 month old; 8M, 8 months old; 15M, 15 months old.
(B) EdU incorporation into the secondary neurospheres from the SVZs of 14-week-old mice was significantly increased by Dkk1 KD and Sfrp3 KD compared with control shRNA (sh Control) (n = 3 independent cultures; at least 600 cells were analyzed), while neither an additive effect nor a synergistic effect was observed upon double knockdown of these genes (n = 3 independent cultures).
(C) EdU incorporation into the SVZ of 6-month-old mice was significantly increased by Dkk1 KD and Sfrp3 KD compared with the sh Control. Neither an additive effect nor a synergistic effect was observed by their double knockdown (n = 5 mice).
(D) Lentiviral vectors were infused into the lateral ventricles of 6-month-old mice, and sections were processed for immunohistochemical analyses 1 week after injection. Nuclei were counterstained with Hoechst (blue). Scale bars, 100 μm.
(E) Canonical Wnt signaling recognized by the accumulation of non-phospho β-CATENIN in the secondary neurospheres from the SVZ of 6-week-old mice was reduced by p38α deletion. Western blot analysis of three independent cultures from three individual mice is shown.
(F) Accumulation of non-phospho β-CATENIN was significantly reduced in the p38α CKO (10-week-old) SVZ. Representative immunohistochemical micrographs of non-phospho β-CATENIN are shown. Nuclei were counterstained with Hoechst (blue). Scale bars, 50 μm.
Lentiviral infections for the knockdown of Dkk1 and Sfrp3 and EdU labeling were performed as described in Figures 2, 5, and 6. Statistical analysis was performed with one-way ANOVA, and Tukey-Kramer post hoc test (A– C), or an unpaired two-tailed Student's t test (B and E). Values in the bar graphs represent the mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. NS, not significant; LV, lateral ventricle.
Finally, we examined whether canonical Wnt signaling in NS/PCs is modulated by the p38-DKK1/SFRP3 axis in the adult SVZ. Western blotting and immunohistochemical analyses of non-phosphorylated β-CATENIN, which is induced to accumulate by canonical Wnt signaling (Clevers and Nusse, 2012), showed that the level of non-phosphorylated β-CATENIN was significantly decreased in p38α CKO neurospheres (6-week-old) and SVZ cells of p38α CKO (10-week-old) mice compared with those of control mice (Figures 7E and 7F).
These results collectively suggested that p38 MAPK promotes NPC proliferation in the adult SVZ, primarily by preventing the antagonism of canonical Wnt signaling by DKK1 and SFRP3, which locally act via autocrine and/or paracrine mechanisms. Age-dependent decreases in p38 cause increases in DKK1 and SFRP3, which is a major cause of the age-dependent decrease in NPC proliferation at least in the SVZ.
Discussion
In this study, we provide evidence that downregulation of p38 MAPK expression in NPCs during aging causes an age-dependent decline in neurogenesis in neurogenic niches in the adult brain. We found that the NS/PC-specific deletion of p38α in the adult mouse brain induces a specific defect in the proliferation of NPCs in neurogenic niches, resembling the age-dependent decline in NPC proliferation in these regions, in which p38 expression also decreases with age. We also showed that the age-dependent decline in adult neurogenesis and incidental SVZ atrophy can be rescued by forced expression of p38α. Moreover, we were able to recapitulate these in vivo phenotypes in vitro, indicating that p38 is a major factor that modulates the aging of NS/PCs in adult neurogenic niches. The age-dependent decline in adult neurogenesis is caused largely by a reduction of NS/PCs. The reduction in NPC proliferation is particularly robust (Conover and Todd, 2017, Encinas et al., 2011). Several extrinsic and intrinsic factors have been shown to be involved in this age-dependent decline (Conover and Todd, 2017, Gontier et al., 2018, Seib and Martin-Villalba, 2015, Wyss-Coray, 2016). Although it has been suggested that combinatorial effects of both intrinsic and extrinsic factors may give rise to the aging-related phenotype, the underlying mechanisms remain poorly understood.
In the present study, we found that the signal transducer p38 MAPK modulates canonical Wnt signaling via the repression of Wnt antagonists. Wnt signaling has been shown to positively regulate adult neurogenesis in the SGZ and SVZ at multiple levels of differentiation status: from activation of stem cells to neuronal differentiation (Adachi et al., 2007, Kuwabara et al., 2009). Moreover, a decline in Wnt signaling caused by a decrease in Wnt3/3a in astrocytes and an increase in DKK1 in granule neurons has been shown to be associated with respect to the age-dependent decline in neurogenesis in the SGZ (Lie et al., 2005, Seib et al., 2013). Here, we also provide evidence indicating that the age-dependent decrease in p38 leads to increases in DKK1 and SFRP3 expressions in NS/PCs and their differentiated progenies, which downregulate canonical Wnt signaling via autocrine and/or paracrine mechanisms, and that this effect is a major cause of age-related decline in NPC proliferation in the adult SVZ. This may be the same in the SGZ because the similar expression patterns of DKK1 and SFRP3 in response to p38α deficiency and aging were observed.
The roles of the p38 MAPK pathway in the age-related decline in stem cell function have been studied in several tissues (Schultz and Sinclair, 2016). In most reports to date, this pathway has been shown to transmit extrinsic cues that induce age-related phenotypes, including cellular senescence and reduced capacity for self-renewal. The functions of p38 MAPK in NS/PCs reported to date, however, have been more controversial. Groups using different experimental systems have reported both positive and negative roles for p38 MAPK signaling in the proliferation of NS/PCs (Kim et al., 2008, Kim and Wong, 2009, Sato et al., 2008, Yoshioka et al., 2015). However, the positive function of p38 MAPK signaling has been observed only in cell-culture conditions that facilitate the proliferation of NS/PCs (Kim et al., 2008, Kim et al., 2015, Kumar et al., 2016). Other than these studies, the inhibitory role of p38 MAPK signaling on NS/PC proliferation has been described, which is contrary to our results. In the adult mouse SVZ, p38 mediates the proliferation defect of ATM (ataxia telangiectasia mutated)-deficient NS/PCs (Kim and Wong, 2009). In the adult mouse SGZ, reduction or pharmacological inhibition of p38α facilitates the proliferation of NS/PCs in vitro (Yoshioka et al., 2015). The role of p38 in NS/PC proliferation may differ depending on the cellular environment, which changes the activation levels of p38 and of other signaling pathways.
Since the forced mobilization of NSCs may result in their exhaustion (Kippin et al., 2005, Porlan et al., 2013), new insights into how NPCs regulate their proliferative capacity will be necessary to enable the development of novel therapies for neurodegenerative and cerebrovascular diseases, which affect increasing numbers of individuals. Targeted manipulation of p38 MAPK signaling may provide unique opportunities for regenerative medicine approaches in diseases of the CNS. Although there are intensive ongoing debates regarding the persistence of postnatal neurogenesis in the human brain (Kempermann et al., 2018, Sorrells et al., 2018), our findings that forced expression of p38α prevents age-related decline in the proliferative capacity of NPCs and SVZ atrophy without exhaustion of NSCs may specifically contribute to the development of strategies for brain regeneration via the mobilization of adult NS/PCs by supporting more efficient mobilization of NS/PCs in the aged brain. The mobilization of endogenous NS/PCs by the infusion of mitogens, neurotrophic factors, and/or cytokines has been shown to ameliorate cerebral ischemia-induced behavioral defects to some extent in rodent models (Bacigaluppi et al., 2016), which suggests that cerebral infarction may represent an ideal first target. Alzheimer's disease and psychiatric disorders, including schizophrenia and depression, may also be viable therapeutic candidates, as a significant decrease in hippocampal neurogenesis is observed in these disorders (Hamilton et al., 2015). Stress and depression have been shown to reduce hippocampal neurogenesis, and the recovery of neurogenesis by antidepressant treatment is required for behavioral improvements in animal models (Morais et al., 2017), suggesting that targeting of the p38 MAPK pathway may show utility in the treatment of depression, especially in aged patients.
For the discovery of new drug candidates that facilitate adult neurogenesis in the aged brain, identifying direct downstream targets of the p38 MAPK pathway in the regulation of NPC proliferation is a key process. Elucidation of the mechanisms of age-dependent decreases in p38 expression in adult NS/PCs is also important for a better understanding of NS/PC aging and to accomplish the restoration of neurogenesis in the aged brain without genetic manipulation.
Experimental Procedures
Mice
All experimental procedures were approved by the ethics committee of Keio University (Assurance No. 09091) and were in accordance with the Guide for the Care and Use of Laboratory Animals (U.S. National Institutes of Health). C57BL/6JJmsSlc mice were obtained from Japan SLC. B6.129-Mapk14 <tm1.2Otsu> (Riken, RBRC02192) (p38αflox/flox), C57BL/6-Tg(Nes-cre/ERT2)5-1Imayo, and B6.129S6-GtROSA26Sor <tm1.1(CAG-mTFP1)> (Riken, RBRC05147) mice were maintained C57BL/6JJmsSlc genetic background and were intercrossed to generate an NS/PC-specific p38α inducible CKO mouse line (Imayoshi et al., 2006, Imayoshi et al., 2012, Nishida et al., 2004). The mice were housed in groups under a 12-h light/dark cycle with free access to food and water.
Conditional Deletion of p38α
Genotyping to obtain the p38α CKO mouse line was performed as described previously (Nishida et al., 2004). Males were used for the experiments. The selective deletion was induced by an intraperitoneal injection of tamoxifen (Sigma-Aldrich, T5648-1G) (75 mg/kg) dissolved in corn oil (Sigma-Aldrich, C8267-500ML) for 5 consecutive days for in vivo experiments. For in vitro studies, tamoxifen (37.5 mg/kg) was injected into the 4-week-old mice for 5 consecutive days and subjected to neurosphere cultures 7 days after the final injection. The control mice were injected with corn oil only.
Statistical Information
For each statistical analysis, at least three independent experiments were carried out. Statistical significance was determined by an unpaired two-tailed Student's t test (two-sided) for comparison of two sets of data and one-way ANOVA with Tukey-Kramer post hoc test. Throughout the study, p values of <0.05 were considered statistically significant. The following significance thresholds were used throughout: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; NS, not significant. Values in the bar and line graphs represent the mean ± SD in all figures.
Author Contributions
All experiments were designed by Y.K. and T.S. T.S. guided the experimental processes. K.O. generated the p38αflox/flox mice. Experiments and data analyses were carried out by Y.K. The project was supervised by T.S. and H.O.
Acknowledgments
We are grateful to Dr. Shengpu Chou (University of Tokyo) for advice on the injection of lentivirus into the mouse lateral ventricle, Katsuyuki Nakanishi (Keio University) for assistance with lentiviral vector administration into the mouse lateral ventricles, Dr. Hiroyuki Miyoshi (Keio University) for the lentivirus constructs, Dr. Ryoichiro Kageyama (Kyoto University) for providing Nes-creER mice, and Douglas Sipp for critical reading of the manuscript. This study was supported by JSPS KAKENHI grant numbers JP18K06469 (T.S.) and JP26117007 (H.O.).
Published: May 9, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stemcr.2019.04.010.
Contributor Information
Takuya Shimazaki, Email: shimazaki@keio.jp.
Hideyuki Okano, Email: hidokano@keio.jp.
Supplemental Information
References
- Adachi K., Mirzadeh Z., Sakaguchi M., Yamashita T., Nikolcheva T., Gotoh Y., Peltz G., Gong L., Kawase T., Alvarez-Buylla A. Beta-catenin signaling promotes proliferation of progenitor cells in the adult mouse subventricular zone. Stem Cells. 2007;25:2827–2836. doi: 10.1634/stemcells.2007-0177. [DOI] [PubMed] [Google Scholar]
- Ashwell J.D. The many paths to p38 mitogen-activated protein kinase activation in the immune system. Nat. Rev. Immunol. 2006;6:532–540. doi: 10.1038/nri1865. [DOI] [PubMed] [Google Scholar]
- Bacigaluppi M., Russo G.L., Peruzzotti-Jametti L., Rossi S., Sandrone S., Butti E., De Ceglia R., Bergamaschi A., Motta C., Gallizioli M. Neural stem cell transplantation induces stroke recovery by upregulating glutamate transporter GLT-1 in astrocytes. J. Neurosci. 2016;36:10529–10544. doi: 10.1523/JNEUROSCI.1643-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckervordersandforth R., Ebert B., Schaffner I., Moss J., Fiebig C., Shin J., Moore D.L., Ghosh L., Trinchero M.F., Stockburger C. Role of mitochondrial metabolism in the control of early lineage progression and aging phenotypes in adult hippocampal neurogenesis. Neuron. 2017;93:560–573. doi: 10.1016/j.neuron.2016.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond A.M., Ming G.L., Song H. Adult mammalian neural stem cells and neurogenesis: five decades later. Cell Stem Cell. 2015;17:385–395. doi: 10.1016/j.stem.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capilla-Gonzalez V., Herranz-Perez V., Garcia-Verdugo J.M. The aged brain: genesis and fate of residual progenitor cells in the subventricular zone. Front. Cell. Neurosci. 2015;9:365. doi: 10.3389/fncel.2015.00365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A., Chan S.L., Milhavet O., Wang S., Mattson M.P. p38 MAP kinase mediates nitric oxide-induced apoptosis of neural progenitor cells. J. Biol. Chem. 2001;276:43320–43327. doi: 10.1074/jbc.M107698200. [DOI] [PubMed] [Google Scholar]
- Clevers H., Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Conover J.C., Todd K.L. Development and aging of a brain neural stem cell niche. Exp. Gerontol. 2017;94:9–13. doi: 10.1016/j.exger.2016.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado A., Nebreda A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010;429:403–417. doi: 10.1042/BJ20100323. [DOI] [PubMed] [Google Scholar]
- Daynac M., Morizur L., Chicheportiche A., Mouthon M.A., Boussin F.D. Age-related neurogenesis decline in the subventricular zone is associated with specific cell cycle regulation changes in activated neural stem cells. Sci. Rep. 2016;6:21505. doi: 10.1038/srep21505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri G.P., Lee X., Basile G., Acosta M., Scott G., Roskelley C., Medrano E.E., Linskens M., Rubelj I., Pereira-Smith O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. U S A. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encinas J.M., Michurina T.V., Peunova N., Park J.H., Tordo J., Peterson D.A., Fishell G., Koulakov A., Enikolopov G. Division-coupled astrocytic differentiation and age-related depletion of neural stem cells in the adult hippocampus. Cell Stem Cell. 2011;8:566–579. doi: 10.1016/j.stem.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faigle R., Brederlau A., Elmi M., Arvidsson Y., Hamazaki T.S., Uramoto H., Funa K. ASK1 inhibits astroglial development via p38 mitogen-activated protein kinase and promotes neuronal differentiation in adult hippocampus-derived progenitor cells. Mol. Cell. Biol. 2004;24:280–293. doi: 10.1128/MCB.24.1.280-293.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gontier G., Iyer M., Shea J.M., Bieri G., Wheatley E.G., Ramalho-Santos M., Villeda S.A. Tet2 rescues age-related regenerative decline and enhances cognitive function in the adult mouse brain. Cell Rep. 2018;22:2094–2106. doi: 10.1016/j.celrep.2018.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton L.K., Dufresne M., Joppe S.E., Petryszyn S., Aumont A., Calon F., Barnabe-Heider F., Furtos A., Parent M., Chaurand P. Aberrant lipid metabolism in the forebrain niche suppresses adult neural stem cell proliferation in an animal model of Alzheimer's disease. Cell Stem Cell. 2015;17:397–411. doi: 10.1016/j.stem.2015.08.001. [DOI] [PubMed] [Google Scholar]
- Hodge R.D., Kowalczyk T.D., Wolf S.A., Encinas J.M., Rippey C., Enikolopov G., Kempermann G., Hevner R.F. Intermediate progenitors in adult hippocampal neurogenesis: TBR2 expression and coordinate regulation of neuronal output. J. Neurosci. 2008;28:3707–3717. doi: 10.1523/JNEUROSCI.4280-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imayoshi I., Hirano K., Sakamoto M., Miyoshi G., Imura T., Kitano S., Miyachi H., Kageyama R. A multifunctional teal-fluorescent Rosa26 reporter mouse line for Cre- and Flp-mediated recombination. Neurosci. Res. 2012;73:85–91. doi: 10.1016/j.neures.2012.02.003. [DOI] [PubMed] [Google Scholar]
- Imayoshi I., Ohtsuka T., Metzger D., Chambon P., Kageyama R. Temporal regulation of Cre recombinase activity in neural stem cells. Genesis. 2006;44:233–238. doi: 10.1002/dvg.20212. [DOI] [PubMed] [Google Scholar]
- Jang M.-H., Bonaguidi M.A., Kitabatake Y., Sun J., Kang E., Jun H., Zhong C., Su Y., Guo J.U., Wang M.X. Secreted frizzled-related protein 3 regulates activity-dependent adult hippocampal neurogenesis. Cell Stem Cell. 2013;12:215–223. doi: 10.1016/j.stem.2012.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S.E., Jomary C. Secreted Frizzled-related proteins: searching for relationships and patterns. Bioessays. 2002;24:811–820. doi: 10.1002/bies.10136. [DOI] [PubMed] [Google Scholar]
- Kawaguchi D., Furutachi S., Kawai H., Hozumi K., Gotoh Y. Dll1 maintains quiescence of adult neural stem cells and segregates asymmetrically during mitosis. Nat. Commun. 2013;4:1880. doi: 10.1038/ncomms2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempermann G., Gage F.H., Aigner L., Song H., Curtis M.A., Thuret S., Kuhn H.G., Jessberger S., Frankland P.W., Cameron H.A. Human adult neurogenesis: evidence and remaining questions. Cell Stem Cell. 2018;23:25–30. doi: 10.1016/j.stem.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Wong P.K. Loss of ATM impairs proliferation of neural stem cells through oxidative stress-mediated p38 MAPK signaling. Stem Cells. 2009;27:1987–1998. doi: 10.1002/stem.125. [DOI] [PubMed] [Google Scholar]
- Kim K.T., Kim H.J., Cho D.C., Bae J.S., Park S.W. Substance P stimulates proliferation of spinal neural stem cells in spinal cord injury via the mitogen-activated protein kinase signaling pathway. Spine J. 2015;15:2055–2065. doi: 10.1016/j.spinee.2015.04.032. [DOI] [PubMed] [Google Scholar]
- Kim S.J., Son T.G., Park H.R., Park M., Kim M.S., Kim H.S., Chung H.Y., Mattson M.P., Lee J. Curcumin stimulates proliferation of embryonic neural progenitor cells and neurogenesis in the adult hippocampus. J. Biol. Chem. 2008;283:14497–14505. doi: 10.1074/jbc.M708373200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kippin T.E., Martens D.J., van der Kooy D. p21 loss compromises the relative quiescence of forebrain stem cell proliferation leading to exhaustion of their proliferation capacity. Genes Dev. 2005;19:756–767. doi: 10.1101/gad.1272305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V., Pandey A., Jahan S., Shukla R.K., Kumar D., Srivastava A., Singh S., Rajpurohit C.S., Yadav S., Khanna V.K. Differential responses of Trans-Resveratrol on proliferation of neural progenitor cells and aged rat hippocampal neurogenesis. Sci. Rep. 2016;6:28142. doi: 10.1038/srep28142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwabara T., Hsieh J., Muotri A., Yeo G., Warashina M., Lie D.C., Moore L., Nakashima K., Asashima M., Gage F.H. Wnt-mediated activation of NeuroD1 and retro-elements during adult neurogenesis. Nat. Neurosci. 2009;12:1097–1105. doi: 10.1038/nn.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie D.C., Colamarino S.A., Song H.J., De' sire L., Mira H., Consiglio A., Lein E.S., Jessberger S., Lansford H., Dearie A.R., Gage F.H. Wnt signalling regulates adult hippocampal neurogenesis. Nature. 2005;437:1370–1375. doi: 10.1038/nature04108. [DOI] [PubMed] [Google Scholar]
- Mich J.K., Signer R.A., Nakada D., Pineda A., Burgess R.J., Vue T.Y., Johnson J.E., Morrison S.J. Prospective identification of functionally distinct stem cells and neurosphere-initiating cells in adult mouse forebrain. Elife. 2014;3:e02669. doi: 10.7554/eLife.02669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming G.L., Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70:687–702. doi: 10.1016/j.neuron.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais M., Patricio P., Mateus-Pinheiro A., Alves N.D., Machado-Santos A.R., Correia J.S., Pereira J., Pinto L., Sousa N., Bessa J.M. The modulation of adult neuroplasticity is involved in the mood-improving actions of atypical antipsychotics in an animal model of depression. Transl. Psychiatry. 2017;7:e1146. doi: 10.1038/tp.2017.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naka-Kaneda H., Nakamura S., Igarashi M., Aoi H., Kanki H., Tsuyama J., Tsutsumi S., Aburatani H., Shimazaki T., Okano H. The miR-17/106-p38 axis is a key regulator of the neurogenic-to-gliogenic transition in developing neural stem/progenitor cells. Proc. Natl. Acad. Sci. U S A. 2014;111:1604–1609. doi: 10.1073/pnas.1315567111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehrs C. Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene. 2006;25:7469–7481. doi: 10.1038/sj.onc.1210054. [DOI] [PubMed] [Google Scholar]
- Nishida K., Yamaguchi O., Hirotani S., Hikoso S., Higuchi Y., Watanabe T., Takeda T., Osuka S., Morita T., Kondoh G. p38α mitogen-activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Mol. Cell. Biol. 2004;24:10611–10620. doi: 10.1128/MCB.24.24.10611-10620.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino J., Kim I., Chada K., Morrison S.J. Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf expression. Cell. 2008;135:227–239. doi: 10.1016/j.cell.2008.09.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obernier K., Cebrian-Silla A., Thomson M., Parraguez J.I., Anderson R., Guinto C., Rodas Rodriguez J., Garcia-Verdugo J.M., Alvarez-Buylla A. Adult neurogenesis is sustained by symmetric self-renewal and differentiation. Cell Stem Cell. 2018;22:221–234. doi: 10.1016/j.stem.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porlan E., Morante-Redolat J.M., Marques-Torrejon M.A., Andreu-Agullo C., Carneiro C., Gomez-Ibarlucea E., Soto A., Vidal A., Ferron S.R., Farinas I. Transcriptional repression of Bmp2 by p21(Waf1/Cip1) links quiescence to neural stem cell maintenance. Nat. Neurosci. 2013;16:1567–1575. doi: 10.1038/nn.3545. [DOI] [PubMed] [Google Scholar]
- Sato K., Hamanoue M., Takamatsu K. Inhibitors of p38 mitogen-activated protein kinase enhance proliferation of mouse neural stem cells. J. Neurosci. Res. 2008;86:2179–2189. doi: 10.1002/jnr.21668. [DOI] [PubMed] [Google Scholar]
- Schultz M.B., Sinclair D.A. When stem cells grow old: phenotypes and mechanisms of stem cell aging. Development. 2016;143:3–14. doi: 10.1242/dev.130633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seib D.R., Martin-Villalba A. Neurogenesis in the normal ageing hippocampus: a mini-review. Gerontology. 2015;61:327–335. doi: 10.1159/000368575. [DOI] [PubMed] [Google Scholar]
- Seib D.R.M., Corsini N.S., Ellwanger K., Plaas C., Mateos A., Pitzer C., Niehrs C., Celikel T., Martin-Villalba A. Loss of Dickkopf-1 restores neurogenesis in old age and counteracts cognitive decline. Cell Stem Cell. 2013;12:204–214. doi: 10.1016/j.stem.2012.11.010. [DOI] [PubMed] [Google Scholar]
- Sorrells S.F., Paredes M.F., Cebrian-Silla A., Sandoval K., Qi D., Kelley K.W., James D., Mayer S., Chang J., Auguste K.I. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature. 2018;555:377–381. doi: 10.1038/nature25975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Engstrom A.K., Xia Z. Cadmium impairs the survival and proliferation of cultured adult subventricular neural stem cells through activation of the JNK and p38 MAP kinases. Toxicology. 2017;380:30–37. doi: 10.1016/j.tox.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T. Ageing, neurodegeneration and brain rejuvenation. Nature. 2016;539:180–186. doi: 10.1038/nature20411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N.C., Hu M.L. The limitations and validities of senescence associated-β-galactosidase activity as an aging marker for human foreskin fibroblast Hs68 cells. Exp. Gerontol. 2005;40:813–819. doi: 10.1016/j.exger.2005.07.011. [DOI] [PubMed] [Google Scholar]
- Yoshioka K., Namiki K., Sudo T., Kasuya Y. p38α controls self-renewal and fate decision of neurosphere-forming cells in adult hippocampus. FEBS Open Biol. 2015;5:437–444. doi: 10.1016/j.fob.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.