

Abstract
Misfolded proteins in the endoplasmic reticulum (ER) are selected for ER‐associated degradation (ERAD). More than 60 disease‐associated proteins are substrates for the ERAD pathway due to the presence of missense or nonsense mutations. In yeast, the Hsp104 molecular chaperone disaggregates detergent‐insoluble ERAD substrates, but the spectrum of disease‐associated ERAD substrates that may be aggregation prone is unknown. To determine if Hsp104 recognizes aggregation‐prone ERAD substrates associated with human diseases, we developed yeast expression systems for a hydrophobic lipid‐binding protein, apolipoprotein B (ApoB), along with a chimeric protein harboring a nucleotide‐binding domain from the cystic fibrosis transmembrane conductance regulator (CFTR) into which disease‐causing mutations were introduced. We discovered that Hsp104 facilitates the degradation of ER‐associated ApoB as well as a truncated CFTR chimera in which a premature stop codon corresponds to a disease‐causing mutation. Chimeras containing a wild‐type version of the CFTR domain or a different mutation were stable and thus Hsp104 independent. We also discovered that the detergent solubility of the unstable chimera was lower than the stable chimeras, and Hsp104 helped retrotranslocate the unstable chimera from the ER, consistent with disaggregase activity. To determine why the truncated chimera was unstable, we next performed molecular dynamics simulations and noted significant unraveling of the CFTR nucleotide‐binding domain. Because human cells lack Hsp104, these data indicate that an alternate disaggregase or mechanism facilitates the removal of aggregation‐prone, disease‐causing ERAD substrates in their native environments.
Keywords: endoplasmic‐reticulum–associated degradation, molecular chaperone, Hsp104, AAA‐ATPase, proteasome, ubiquitin, apolipoprotein B, cystic fibrosis transmembrane conductance regulator, protein aggregation, yeast
Introduction
Approximately one‐third of all proteins in eukaryotes enter the endoplasmic reticulum (ER), which serves as the first destination for soluble and membrane proteins that traverse the secretory pathway. However, proteins targeted to the ER can misfold or fail to acquire critical post‐translational modifications, which in some cases lead to the generation of “protein conformational diseases”. In most of these instances, maturation of the disease‐causing protein is compromised due to genetic mutations. During or soon after synthesis, these proteins are recognized by molecular chaperones, ubiquitinated, and then extracted or “retrotranslocated” from the ER for degradation by the cytosolic proteasome.1, 2, 3, 4, 5 This pathway is known as ER‐associated degradation (ERAD), and to date greater than 60 proteins linked to specific human diseases are ERAD substrates.6
Some misfolded proteins that reside in the cytosol or nucleus aggregate or alternatively form amyloids, which also leads to protein conformational diseases.7 Emerging data indicate that the chemical and/or chaperone‐rich environment in the lumen of the ER prevents the formation of protein aggregates in this compartment.8, 9 However, disease‐associated membrane proteins that contain aberrant domains residing in the cytosol could also aggregate. Therefore, human cells might express a specialized chaperone or chaperone system to disassemble ER membrane protein aggregates, or the aggregates might be handled by an alternative degradation pathway, such as ER‐phagy.10, 11
Yeast and bacteria express a hexameric, cytosolic AAA+‐ATPase protein disaggregase that dissolves insoluble species and either refolds them with the assistance of other chaperones or delivers the solubilized species to cytosolic proteases.12, 13, 14 AAA+‐ATPases couple ATP hydrolysis to drive energy‐expensive processes, such as the unwinding of DNA, remodeling of the lipid bilayer, protein transport, chromatin condensation, transcription regulation, prion severing, as well as protein disaggregation and degradation.15, 16, 17, 18 In yeast, the disaggregase is known as Hsp104.19, 20 To investigate whether Hsp104 plays a role in the ERAD of a detergent‐insoluble membrane protein, we fused a transmembrane helical hairpin to an aggregation‐prone domain that resides in the cytosol.21, 22 We discovered that degradation of this unstable substrate in the ER was Hsp104 dependent and that Hsp104 function was required prior to substrate ubiquitination, which serves as a prerequisite for proteasome‐mediated degradation during ERAD.4, 23
Human cells lack Hsp104. Therefore, we wished to investigate if an ER membrane‐associated protein expressed in human cells with an appended aggregation‐prone cytosolic domain is targeted for ERAD. To begin to answer this question, we asked if human ERAD substrates expressed in yeast require Hsp104 function.
We first developed a new yeast expression system for a truncated form of apolipoprotein B (ApoB). ApoB is the major proteinaceous component in chylomicrons and low‐density lipoproteins (LDLs) and is a large (~540 kDa), amphipathic protein produced in two isoforms: full length ApoB (ApoB100) and an isoform that is ~48% of the full‐length protein (ApoB48). ApoB100 is synthesized in the liver, enters the ER cotranslationally, becomes incorporated into LDLs, and is secreted into the serum when excess hepatic lipids are available.24, 25 ApoB48 is instead synthesized in the small intestine and after entry into the ER is incorporated into chylomicrons when lipids are abundant, for example, after a triacylglycerol‐ and cholesterol‐rich meal. Both forms of ApoB contain multiple aggregation‐prone β‐sheets and shorter α‐helices,26 creating its amphipathic nature and allowing for cholesterol, triacylglycerol, and fatty acid binding and transport in the blood. Shorter forms of ApoB (e.g., ApoB29) have also been detected in humans, and although these individuals suffer from hypolipidemia because the protein binds lower levels of lipids/cholesterol, ApoB29 still undergoes proper regulation in the secretory pathway and has been expressed and examined in yeast.24, 27 Second, we fused the second nucleotide‐binding domain (NBD2) from the cystic fibrosis transmembrane conductance regulator (CFTR) to a transmembrane helical hairpin to tether this domain to the ER membrane. Cystic fibrosis is the most common, lethal inherited disease in Caucasians in North America. To date, ~200 disease‐causing mutations in this integral membrane ion channel have been identified, including some in NBD2.28 Among these mutations is a truncation in NBD2 (W1282X), which results in a poorly expressed yet potentially correctable mutant protein,29, 30 as well as a point mutation (N1303K) that appears to stabilize the protein.31 Based on the fact that the W1282X truncation resides in a central core of the NBD2—and because of the highly hydrophobic nature of ApoB, which is targeted for ERAD under lipid‐poor conditions24, 25—we hypothesized that these substrates would be aggregation prone and thus Hsp104‐dependent ERAD substrates in yeast. As outlined below, this hypothesis was correct, lending support to the view that ERAD substrates can also aggregate in human cells and that an Hsp104‐like activity must exist in the cytosol to ensure clearance of these toxic species.
Results
Characterization of a new ERAD substrate derived from the cystic fibrosis transmembrane conductance regulator
We previously reported on a model aggregation‐prone ERAD substrate in yeast, termed Chimera A [Fig. 1(A)].21 This chimera contained a truncated NBD2 (NBD2*) from a yeast transporter, Ste6, which is homologous to CFTR. NBD2 was tethered to the ER membrane by virtue of a synthetic transmembrane domain (tm1‐2). The resulting protein—Chimera A*—was unstable, exhibited lower detergent solubility and was targeted for ERAD in an Hsp104‐dependent manner.22 Thus, we reasoned that CFTR NBD2 might similarly be used to develop a series of new—and potentially aggregation‐prone—ERAD substrates. Indeed, CFTR NBD1 and NBD2 are aggregation prone, and resolution of their X‐ray crystal structures required the presence of numerous solubilizing mutations or deletion of a regulatory insertion motif.32, 33, 34 To test this hypothesis, we appended the CFTR NBD2 onto the same transmembrane helical hairpin (tm1‐2) that resides in Chimera A* [Fig. 1(A)]. Into this chimera, we then introduced the following disease‐causing mutations: (1) a missense mutation, N1303K, which is relatively common in European populations and exhibits greater stability than several other misfolded CFTR variants31, 35; and (2) a stop codon at position W1282 (W1282X), which results in premature termination, accounts for ~11% the population with cystic fibrosis, and leads to severe disease.36 Although Food and Drug Administration (FDA)‐approved drugs help correct the most common disease‐causing allele, F508del CFTR, as well as select other mutant alleles,37, 38 pharmaceuticals that rectify molecular defects associated with W1282X and N1303K are lacking. Nevertheless, some improvement was seen in a patient homozygous for the W1282X allele who was administered one of the FDA‐approved drugs.36
Figure 1.
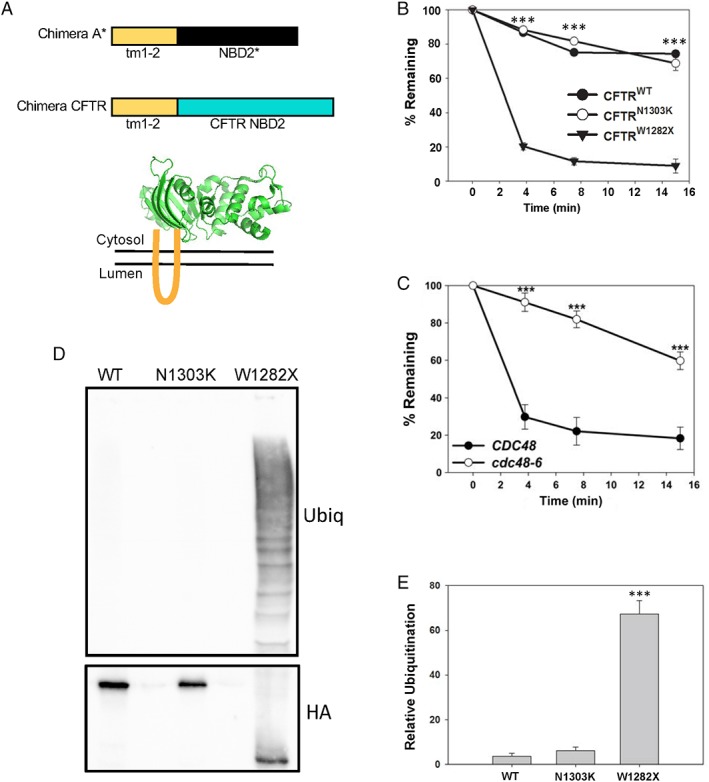
Truncation of a nucleotide‐binding domain destabilizes a CFTR‐derived chimeric protein. (A) Top, schematic of the new CFTR chimera described in this report in comparison to Chimera A.21 The “tm1‐2” sequence is derived from Chimera A, and the NBD2 of Ste6 and CFTR, depicted in black and blue, respectively, are ~20% identical. Bottom, tm1‐2 (in orange) links the chimera to the ER membrane, and NBD2 is displayed in the yeast cytosol. Representative structure of the NBD2 from CFTR is modified from the PDB (3GD7). (B) Metabolic stabilities of the WT and N1303K and W1282X mutant forms of the CFTR‐derived chimeras were determined by cycloheximide chase at 26°C. The results from three independent experiments, ±SEM, are shown. (C) Metabolic stability of the W1282X mutant form of the CFTR‐derived chimera was determined by cycloheximide chase in WT (CDC48) and cdc48‐6 yeast. Cells were grown at 26°C and then shifted to 39°C for 3 h prior to the start of the chase to induce the mutant phenotype. The results from eight independent experiments, ±SEM, are shown. (D) The level of chimera ubiquitination was examined in a WT yeast strain grown at 26°C. Cells were lysed and the designated proteins were isolated after immunoprecipitation using anti‐HA‐conjugated agarose beads to precipitate the chimera. Western blotting was then performed to detect the substrate (HA) and ubiquitin. (E) Quantification of results from 6 to 10 independent experiments, ±SEM, are shown. *** denotes P < 0.001.
We first examined the degradation rates of the wild‐type (WT), N1303K, and W1282X CFTR chimeras by cycloheximide chase and found that the WT protein was stable, as predicted based on the relative stability of Chimera A, which contains the full‐length NBD2 [Fig. 1(B)].21 In contrast, a chimera harboring the W1282X premature termination codon was unstable, but the N1303K mutant chimera exhibited an identical profile as the WT protein. This result is consistent with the relative stability of N1303K CFTR in mammalian cells31 and suggests that disease presentation arises from another phenomenon. As anticipated for an ERAD substrate, the degradation of W1282X was also Cdc48 dependent [Fig. 1(C)]. Next, we examined whether differences in stability reflected the extent of polyubiquitination, which is required for proteasome‐targeting during ERAD.4, 23 As shown in Figure 1(D,E), instability correlated with substrate ubiquitination, that is, the W1282X mutant was polyubiquitinated to a greater extent than the other, stable constructs. Together, the W1282X chimera is a new ERAD substrate, one that might help define the molecular basis of CF in patients who carry this allele.
Degradation of the W1282X chimera is Hsp104 dependent
To confirm that W1282X is an ERAD substrate and therefore is retrotranslocated from the ER in a ubiquitin‐dependent manner, we utilized an assay in which the ER extraction (i.e., retrotranslocation) of a ubiquitinated membrane protein can be recapitulated in vitro.39 To this end, yeast ER‐derived microsomes were prepared from cells expressing the W1282X chimera. The microsomes were then incubated with 125I‐radiolabeled ubiquitin, an ATP‐regenerating system, and yeast cytosol, which contains factors required for substrate recognition and ubiquitination. After centrifugation, the retrotranslocated fraction of W1282X should reside in the supernatant. As shown in Figure 2(A), ubiquitination was ATP dependent, as expected. We also found that ~25% of the ubiquitinated W1282X chimera was retrotranslocated (compare the “S” and “P” fractions), which is consistent with the extent of retrotranslocation reported for the truncated form of Chimera A, Chimera A*.21 Because Chimera A* was also degraded in an Hsp104‐dependent manner,22 we surmised that the unstable W1282X chimera might behave similarly. As predicted, W1282X turnover was significantly slowed in yeast lacking Hsp104 [Fig. 2(B)].
Figure 2.
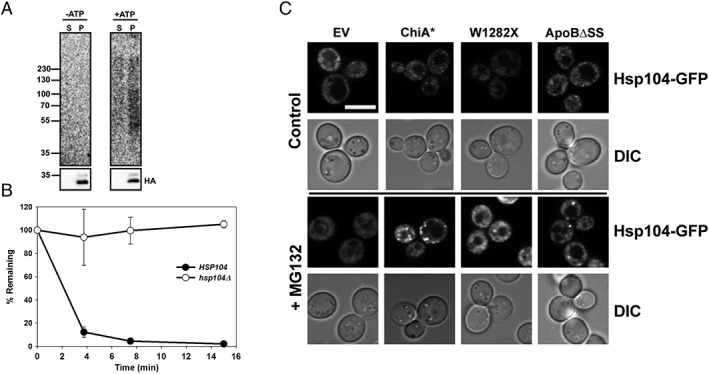
Hsp104 facilitates the ERAD of the W1282X CFTR chimera. (A) Microsomes were prepared from WT yeast expressing W1282X and were subjected to in vitro ubiquitination for 45 min at 26°C. Reactions were performed in the absence (−ATP) or presence (+ATP) of an ATP regenerating system. After centrifugation, W1282X was immunoprecipitated from the supernatant (S) and pellet (P) fractions. The supernatant represents the fraction (~25% of the total) of the W1282X chimera retrotranslocated during the assay. Representative autoradiograph (top) and corresponding anti‐HA‐HRP blot (bottom) is shown from two independent experiments performed with replicates. (B) The stability of the W1282X chimera was examined in WT and hsp104Δ yeast. Data represent the means of three independent experiments, ±SEM. The WT and N1303K mutant proteins were unaffected by deletion of the HSP104 gene, as they are already highly stable (data not shown). (C) Hsp104‐GFP‐expressing pdr5Δ yeast were transformed with an empty vector (EV), Chimera A* (Chi A*), the W1282X chimera, or ApoBΔSS. Early log‐phase cells were plated onto glass bottom microwell dishes for imaging using a Nikon A1 confocal microscope. Fields of cells were imaged before treatment and following a 1 h treatment with 100 μM MG132 at 37°C, as indicated. Representative images of Hsp104‐GFP fluorescence and cells (DIC) are shown from pretreatment (control) and post‐treatment (+MG132). White scale bar ~5 μM.
Because Hsp104 is required for the ERAD of detergent‐insoluble substrates,22 the data presented above strongly suggested that the W1282X protein is aggregation prone. We previously reported that Hsp104 is also recruited to inclusions in the ER to help dissolve these species.22 Other work showed that Hsp104 can be recruited to cytoplasmic inclusions.40 To address whether Hsp104 was recruited to the ER when W1282X was expressed—especially under conditions in which it was stabilized (i.e., in the presence of MG132)—we expressed Chimera A*, the W1282X chimera, and an ApoB variant (see below) in yeast containing an integrated form of Hsp104‐GFP. Yeast containing an empty vector served as a control. In this experiment, the presence of puncta in cells expressing these substrates would be consistent with the formation of cellular inclusions. As shown in Figure 2(C), puncta were only evident in cells expressing Chimera A*, as anticipated,22 as well as W1282X and the ApoB variant (see below). These data are consistent with cellular aggregation of W1282X and Hsp104‐mediated substrate solubilization.
Prior work linked the degradation of unstable Hsp104‐dependent ERAD substrates to reduced detergent solubility,22 which is consistent with Hsp104‐mediated disaggregation. Therefore, we hypothesized that the WT and N1303K chimeras would be more soluble in a nonionic detergent than the W1282X chimera. As anticipated, the W1282X chimera was solubilized ~50% less efficiently with 0.5 mM dodecyl maltoside (DDM) than the WT and N1303K chimeras [Fig. 3(A,B)]. We then examined whether the W1282X chimeric protein had formed denser inclusions in the ER membrane than the N1303K chimera. To this end, membrane lysates were fractionated to equilibrium in a sucrose gradient [Fig. 3(C)]. In our previous study, the Hsp104‐dependent substrate migrated to a fraction with greater density compared to a more soluble and stable substrate.22 Indeed, the new Hsp104‐dependent chimera with lower detergent solubility (i.e., W1282X) also resided in denser (i.e., 1.4M–1.6M) fractions compared to the more soluble and stable chimera (i.e., N1303K) or the WT protein [Fig. 3(D)]. These collective results suggest that protein disaggregation is required to facilitate the retrotranslocation of a ubiquitinated, aggregation‐prone CFTR mutant.
Figure 3.
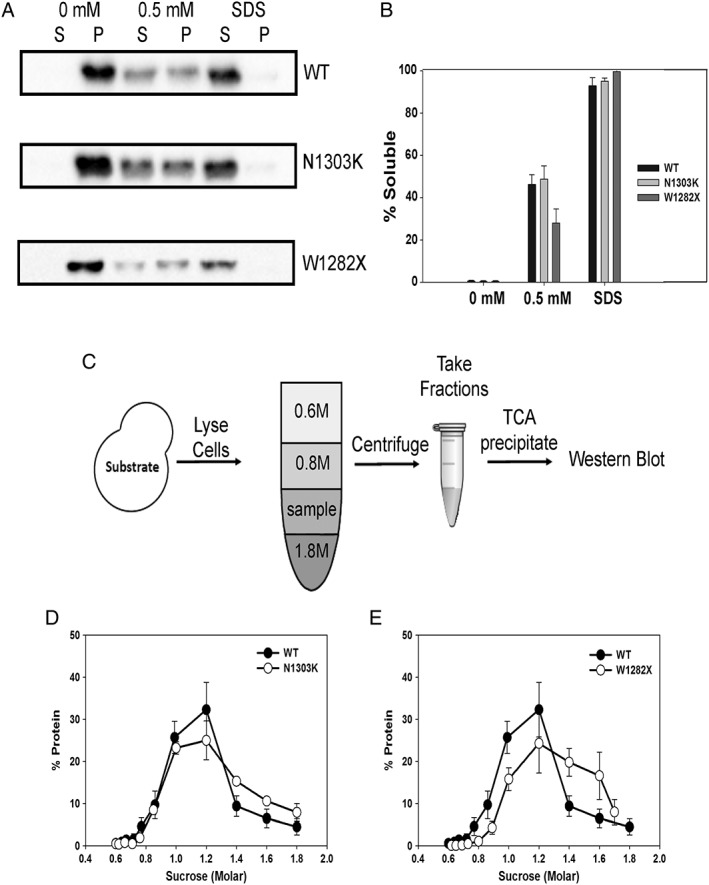
The W1282X CFTR chimera exhibits lower detergent solubility and oligomerizes in the ER membrane. (A) The solubility of the WT, N1303K, and W1282X CFTR chimeras in ER‐derived microsomes was examined in the presence of buffer (0 mM), 0.5 mM DDM, or 1% SDS. (B) Quantification of the results of the solubility assay, as performed in part (A), based on an analysis of four to six independent experiments, ±SEM. (C) A schematic of the flotation assay used to measure the oligomerization of membrane proteins.41 (D, E) LYSATES from WT yeast expressing the designated chimeras, and WT CFTR were prepared and analyzed by sucrose density centrifugation. Gradient samples were processed and immunoblotted against the HA tag to detect protein residence. The quantification of experiments with the (D) N1303K stable CFTR chimera and the (E) W1282X unstable chimera are shown. For comparison, the fractionation of the WT species (closed circles) is reproduced in both panels. Data were standardized to the amount of protein in each gradient fraction relative to the total from four independent experiments, ±SEM.
The W1282X mutation destabilizes an α‐helix in the NBD2
The W1282X premature stop codon truncates CFTR within a short α‐helix. Given the decreased detergent solubility and stability of the W1282X ER‐embedded chimera, we reasoned that the truncation either directly exposes a prominent hydrophobic region within NBD2 or unravels a portion of the domain upstream of position 1282, thereby exposing hydrophobic sequences. To test these hypotheses and to gain atomic‐level insight into the structure and dynamics of NBD2 constructs, molecular dynamics (MD) simulations were performed on WT NBD2, the N1303K mutant, and the W1282X truncated construct, and the resulting trajectories were analyzed (Fig. 4). First, we derived residue‐based root‐mean‐squared fluctuation (RMSF) profiles and found them to be identical for the WT and N1303K species, consistent with their identical properties in each of the assays performed above. RMSF values represent the mean fluctuation for each residue calculated over the entire MD trajectory, and large values mean larger fluctuations. In contrast, the RMSF values for the ~54 residues toward the N‐terminus from W1282X (1228–1281; part of the Cβ and the ATP binding core subdomains in NBD2) were significantly higher, indicating higher fluctuations and reduced stability in this region, which is consistent with the overall experimentally observed reduced stability of this construct. The increased fluctuations in the α‐helix part (residues 1249–1258) in the truncated mutant are also consistent with a partial loss of secondary structure as observed from a Defined second structure of proteins (DSSP) analysis (Supporting Information Fig. S1). DSSP analysis provides the secondary structure of the protein as determined from geometric features and hydrogen bond patterns.42 Next, we calculated the radius of gyration (Rg) as a function of simulation time for residues 1202–1282 (the common part) for all constructs. Rg is the measure of protein compactness and defined as the root‐mean‐square average of the distance of all atoms from the center of the mass of the protein. The results of this calculation are presented in Supporting Information Figure S2 and demonstrate identical behavior for the WT and the N1303K construct. In contrast, the truncation mutant exhibits a reduced Rg profile. Finally, we calculated the per‐residue solvent accessible surface area (SASA) for residues 1202–1281 for the WT and the truncation constructs, again, as a function of simulation time [Supporting Information Fig. S3(A,B)]. SASA is defined as the surface area of a molecule that is solvent exposed and typically measured using a probe radius of 1.4 Å, which is the radius of water. To facilitate the analysis, we have also calculated the difference plot (WT truncation) [Supporting Information Fig. S3(B)]. In this plot, negative values correspond to residues that are more solvent exposed in the truncation mutant than in the WT. Two observations could be made from this analysis: First, the truncation mutant exposes more overall surface area to the solvent than the corresponding residues in the WT (i.e., most values are negative), and second a more pronounced effect (i.e., differences larger than 0.2 nm2) is observed for the following, mostly hydrophobic, residues: G1207, I1230, Q1238, R1239, V1240, G1241, L1242, L1243, K1250, L1258, I1269, D1270, L1279, and Q1281. In particular, six consecutive residues (Q1238, R1239, V1240, G1241, L1242, and L1243) demonstrate an average increase of 0.34 nm2 in their SASA. Together, these observations provide a model to explain the higher aggregation tendency of the W1282X truncation mutant and perhaps the selection of other ERAD substrates for Hsp104‐dependent degradation.
Figure 4.
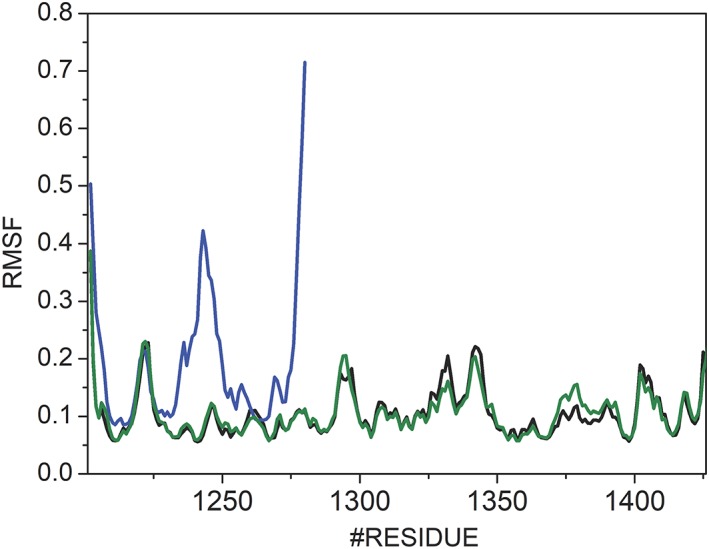
The W1282X termination codon destabilizes the NBD2 of CFTR. Average RMSF values over three 500 ns simulations are shown. Black, blue, and green lines correspond to the WT, W1282X, and N1303K construct, respectively. Low RMSF values are indicative of a more stable construct.
Development of a new apolipoprotein B yeast expression system
In the absence of sufficient lipids in the ER, ApoB synthesis and translocation into the ER are slowed, which allows for chaperone recognition, ubiquitin conjugation, and degradation by the 26S proteasome.25, 43 Because ApoB is a large (~540 kDa) amphipathic protein, we hypothesized that cytosolic factors must prevent aggregation as it is being retrotranslocated from the protective environment of the ER to the cytosol. It has also been suggested that ER‐associated lipid droplets (LDs) could interact with ApoB and prevent aggregation during ERAD.44, 45, 46 Although data suggest that LDs are dispensable for the ERAD of select substrates,47 some aggregation‐prone proteins in the cytosol interact with LDs en route to their destruction.48, 49
In prior work, we used the yeast Saccharomyces cerevisiae to model the ApoB degradation pathway with the ApoB29 isoform, which is still regulated by ERAD (see below), and identified chaperones that affect turnover, including Hsp70, Hsp90, Hsp110, and protein disulfide isomerase.27, 50, 51 However, ApoB was expressed under the control of the GAL promoter, which necessitated a shift from growth in glucose, an optimal carbon source, to galactose, a nonoptimal carbon source. Because yeast replication is compromised in galactose, we were concerned that this stress‐inducing condition might confound data on the potential roles that disaggregases play during ERAD. Therefore, we coopted the chimeric GEV transcription factor to regulate ApoB expression. GEV contains a Gal4 DNA‐binding domain, an Estrogen (e.g., β‐estradiol)‐binding domain, and a portion of the VP16 transcription factor [Fig. 5(A)]. As previously described,52 GEV is constitutively expressed and inactive, but in the presence of β‐estradiol, GEV promotes transcription of genes under GAL control.
Figure 5.
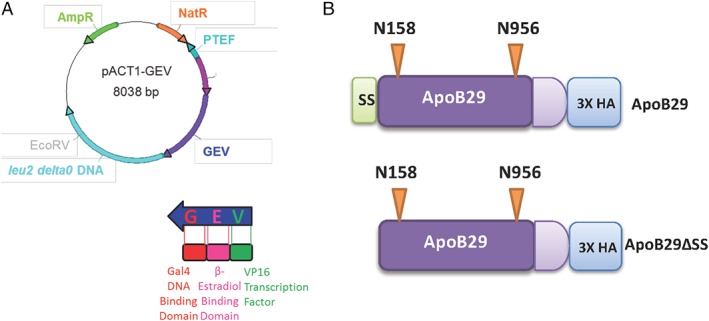
Design of a new ApoB yeast expression system. (A) Diagram of the pACT1‐GEV yeast integration vector. (B) Linear map of the signal sequence (ss) containing (top) and signal sequence deficient (ApoBΔSS; bottom) ApoB29 inserts, which were ligated into a GAL‐inducible plasmid. The positions of the two N‐linked glycosylation (orange inverted triangles) and the triple HA tag are also shown (image is not to scale).
After introducing a GEV expression system into recipient yeast, we determined the time course of ApoB expression after β‐estradiol addition. As noted above, for these studies, we used a shorter form of the apolipoprotein, ApoB29, that still retains significant lipid‐binding properties, undergoes regulated degradation, and is expressed at high levels in yeast.27, 53 We then constructed versions of ApoB29 that either contained or lacked the signal sequence [Fig. 5(B)]. Several disease‐causing mutations in ApoB, which cause hypolipidemia, reside in the signal sequence, which reduces protein translocation into the ER.54, 55, 56 This phenomenon could be recapitulated in yeast.57 We found that an HA epitope‐tagged form of the signal sequence‐containing (ApoB) and a signal sequence‐deficient derivative (ApoBΔSS) of the protein were observed as early as 30 min after β‐estradiol addition [Fig. 6(A); Supporting Information Fig. S4(A)]. In contrast, a 4 h incubation is required to induce ApoB in the presence of galactose.27 As hoped, yeast also continued to grow at normal rates in the presence of β‐estradiol [Supporting Information Fig. S4(B)] as they remained in glucose‐containing media.
Figure 6.
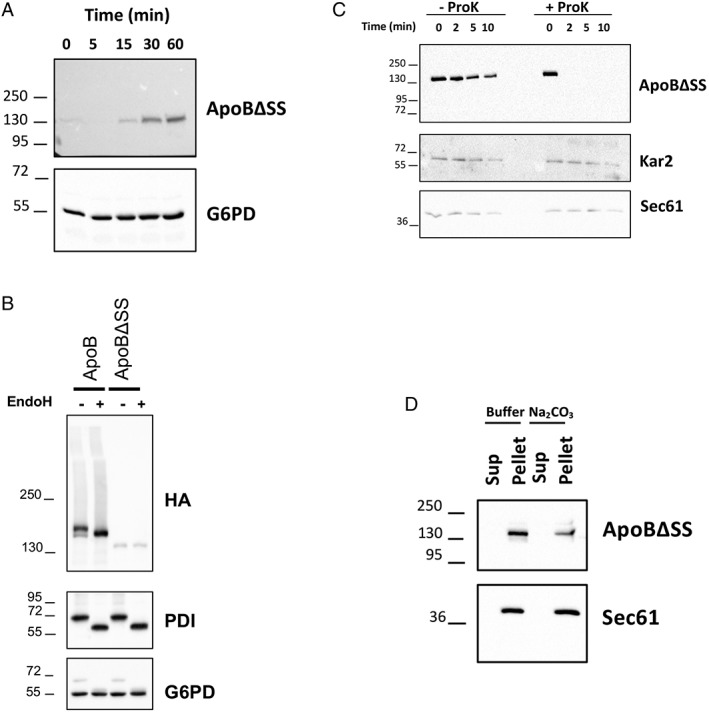
The signal sequence‐containing ApoB species translocates into the ER, whereas the signal sequence‐deficient protein remains tightly associated with the external surface of the ER. (A) Rapid expression of the signal sequence‐deficient protein (ApoBΔSS) after addition of β‐estradiol. An identical pattern of expression was observed when the signal sequence‐containing species was examined (Supporting Information Fig. S4A). G6PD served as a loading control. (B) Lysates from cells expressing ApoB and ApoBΔSS were treated with buffer or EndoH, as indicated, proteins were resolved by SDS‐PAGE, and ApoB (HA), the glycosylated PDI protein (as a positive control), and G6PD (as a negative control) were detected by Western blotting. (C) ER‐derived microsomes from cells expressing ApoBΔSS were treated with buffer or proteinase K (ProK), as indicated, and the stability of the proteins as well as the ER lumenal protein Kar2 and the integral ER membrane protein Sec61 (as negative controls) were examined by western blotting. (D) A crude membrane fraction from cells expressing ApoBΔSS was treated with buffer or sodium carbonate, and after centrifugation, the residence of the protein as well as Sec61 (as a positive control) in the supernatant (Sup) or pellet fraction was analyzed by Western blotting.
We next asked if ApoB29 and ApoBΔSS associate with the ER and whether the signal sequence supported ER translocation. Lysates from yeast containing each form of ApoB29 were treated with Endoglycosidase H [Fig. 6(B); EndoH], which removes N‐linked glycans. Only the signal sequence‐containing ApoB species shifted to a lower molecular weight, and the apparent shift of ~6 kDa was consistent with the expected acquisition of two N‐linked glycans [Fig. 5(B)].58 As a control, the ER resident glycosylated enzyme, Pdi1, also shifted to a lower molecular weight after lysates were treated with EndoH. In contrast, the migration of rabbit anti‐glucose 6 phosphate dehydrogenase (G6PD), an unglycosylated protein, was unaffected. Next, we focused on ApoB29 lacking the signal sequence because it was robustly targeted for ERAD and was aggregation‐prone (see below). Limited proteolysis was conducted on ER‐derived microsomes prepared from yeast expressing ApoBΔSS. We found that ApoBΔSS was rapidly proteolyzed [Fig. 6(C)], confirming that the substrate failed to enter the ER. Sec61, an ER‐resident integral membrane protein, and Kar2, an ER luminal chaperone, were protease resistant. Finally, we asked whether ApoBΔSS, even though it lacked a signal sequence, was associated with the ER membrane. Robust ER association was anticipated based on its fractionation with ER‐derived microsomes [Fig. 6(B)] and its overall hydrophobic character. As anticipated, ApoBΔSS was resistant to carbonate extraction, which is used to extract proteins that peripherally associate with biological membranes,59 as was Sec61 [Fig. 6(D)]. Taken together, we have established a new, superior ApoB expression system and show that a signal sequence directs ApoB into the yeast ER, where the protein is glycosylated. In contrast, ApoB lacking the signal sequence associates with the exterior of the ER.
ApoB is absent from LDs
LDs were proposed to facilitate the retrotranslocation of integral membrane ERAD substrates,60 and previous work suggested that ApoB resides at or near LDs in transfected cells that normally lack this protein.44, 45, 46 To determine if ApoB occupies LDs in yeast, which may maintain protein solubility and obviate the requirement for Hsp104, we isolated LDs [Fig. 7(A)].61 For this analysis, we used cells containing an integrated GFP‐tagged copy of Tgl3, which resides in LDs,62 and that expressed both the signal sequence‐containing and signal sequence‐deficient versions of ApoB. We first noted that Tgl3 was present in the LD fraction, as anticipated [Fig. 7(B)]. In contrast, Vph1, a vacuolar protein, and Sec61 resided instead in the membrane fraction (pellet). ApoB and ApoBΔSS (upper and lower bands, respectively) also resided in the membrane fraction but were absent from LDs.
Figure 7.
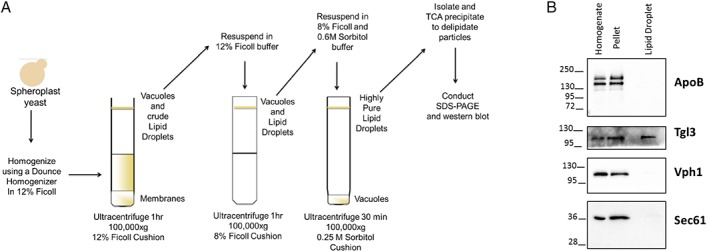
Yeast LDs lack exogenously expressed ApoB. (A) Protocol for the isolation of highly enriched LDs in yeast. (B) Aliquots from the LD isolation protocol were examined by Western blotting using the indicated antibodies. Yeast expressed both translocated (upper band) and untranslocated (lower band) ApoB and aliquots were removed before (Homogenate) and after (Pellet) a centrifugation step that was used to isolate an initial membrane fraction as well as from the final LD fraction. In this experiment, ApoB expression was induced in the presence of galactose, as previously described.27 The Tgl3 LD‐resident lipase served as a positive control. Note that this LD fraction lacked contaminating vacuolar (Vph1) as well as ER (Sec61) membranes.
Hsp104 facilitates the proteasomal degradation of ApoB lacking a signal sequence
To determine whether ApoB and ApoBΔSS were targeted for ERAD, we next conducted cycloheximide chase assays. Based on preliminary experiments, we found that only the ApoBΔSS species was unstable (data not shown but see Fig. 8) and posit that robust insertion of the signal sequence‐containing species into the ER prevents retrotranslocation. This is in contrast to full length ApoB100, which is an ERAD substrate in the absence of sufficient lipids due to translational pausing.63 Therefore, we focused on the fate of ApoBΔSS, which associates with the exterior surface of the ER (Fig. 6). To determine whether ApoBΔSS was degraded by the proteasome, we again conducted cycloheximide chase assays but in both WT and pdr5Δ yeast, which prevents efflux of the MG132 proteasome inhibitor. As shown in Figure 8(A), ApoBΔSS degradation was slowed in the presence of MG132. In contrast, degradation was unaffected in yeast lacking Pep4, which is the major vacuolar protease (data not shown). Next, we asked whether Hsp104 plays a role in ApoBΔSS degradation and discovered that turnover was inhibited in hsp104Δ yeast [Fig. 8(B)]. Consistent with these data, expression of ApoBΔSS also led to the formation of Hsp104 puncta in cells, particularly when the proteasome was inhibited [Fig. 2(C)]. Our results suggest that the degradation of disease‐associated forms of ApoB, which inefficiently translocate into the ER in some patients with hypolipidemia, requires the activity of an Hsp104‐like enzyme.
Figure 8.
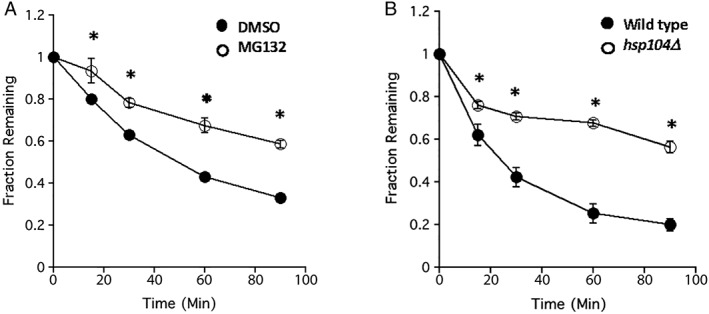
Untranslocated, ER‐associated ApoB is degraded in a proteasome‐ and Hsp104‐dependent manner in yeast. (A) Yeast lacking the multidrug efflux pump, Pdr5, were incubated in the presence of vehicle (DMSO) or the proteasome inhibitor, MG132, and after addition of cycloheximide, aliquots were taken at the indicated times and the relative amount of ApoBΔSS remaining was determined by Western blotting. Data represent the means of eight independent experiments, ±SEM, and were standardized to the amount of protein at the 0 min time point. (B) WT and hsp104Δ yeast expressing ApoBΔSS were subject to a cycloheximide chase analysis, as above. Data represent the means of 10–11 independent experiments, ±SEM, and were standardized to the amount of protein at the 0 min time point. * denotes P < 0.05.
Discussion
The ER is responsible for folding, post‐translationally modifying, and assembling one third of the proteome in a eukaryotic cell.64, 65, 66, 67, 68 However, many proteins that enter the secretory pathway fold poorly and/or require long periods of time before they mature and exit the ER for delivery to their final destinations. Stochastic errors in gene expression or stress responses further compromise protein folding in the ER. Protein misfolding is further exacerbated when proteins harbor disease‐associated mutations, and many of these substrates are handled by the ERAD machinery.6 Similarly, misfolded proteins in the cytosol are directed to quality control systems, and aggregation‐prone species are recognized by specialized AAA‐ATPases, which have been best described in yeast.14, 69 In contrast, only a few disease‐associated substrates, for example, the Z variant of alpha‐1 antitrypsin (ATZ), assemble into highly ordered oligomers within the lumen of the ER, which are then destroyed in the lysosome (in humans) or vacuole (in yeast).70, 71, 72
In this study, we developed and characterized an ER‐associated substrate that contains a hydrophobic lipid assembly domain (ApoBΔSS) and an ER‐tethered aggregation‐prone NBD (W1282X) that resides in the cytosol. We then showed that the Hsp104 disaggregase is required for the efficient degradation of these substrates. Importantly, deletion of Hsp104 does not generally impair the disposal of substrates via the ubiquitin‐proteasome system, indicating that the phenomena we report in this study do not arise from a general, nonspecific stress response.73, 74 Therefore, we suggest that disease‐associated forms of ApoB that compromised protein translocation as well as the W1282X CFTR mutant might aggregate or form inclusions in human cells, affect protein homeostasis (i.e., “proteostasis”), and contribute to disease onset. It is likely that their destruction will require the action of either a protein disaggregase or an alternate disposal mechanism. Notably, our previous data suggested that Hsp104 acted before Cdc48‐mediated retrotranslocation during the ERAD of aggregation‐prone substrates in yeast.22 It will therefore be important to define how p97 operates with putative alternate disaggregases in human cells.
Which factors might handle these ER‐associated substrates in humans? To date, several candidates exist in human cells. The prime candidate is the Hsp70‐Hsp40‐Hsp110 system. Work from Bukau and colleagues indicates that aggregates formed from model substrates as well as disease‐associated aggregates are effectively solubilized by this system.69 After solubilization, the substrate either folds to its native state or is targeted to the proteasome and destroyed. Data in bacteria and yeast suggest that most aggregates in the cytosol fold after interacting with Hsp104 and associated chaperones.14 Another candidate, RuvBL1/2, is also a AAA+‐ATPase, and is best described as a DNA‐dependent helicase75; however, modulating the levels of this enzyme in yeast or mammalian cells impacts the generation and dissolution of protein aggregates.76 Other, more specialized factors with disaggregase‐like activities have also been identified.69
It is possible that protein inclusions associated with the ER are handled by the autophagy machinery. Early data on the Z variant of ATZ—a soluble protein that resides exclusively in the ER—suggested that the substrate was targeted for autophagy after it oligomerized in the ER.72 More recently, it was shown that ATZ was delivered for lysosomal degradation after transport through the secretory pathway but was then recognized in an autophagy‐like manner, that is, one that required an ER‐phagy receptor complex.72 It is noteworthy that aggregated forms of ApoB, which arise when cells are treated with oxidizing agents such as polyunsaturated fatty acids,77 are delivered from the Golgi via autophagolysosomes for degradation in the lysosome.78 Similarly, degradation and refolding of the W1282X mutant appear to be modulated by small molecules that regulate the autophagy pathway as well as proteasome activity.35 Based on these and other emerging data,79 autophagy‐like pathways may represent a back‐up system for substrates ineffectively routed to the ERAD pathway in human cells.
Finally, it is likely that other ER‐associated proteins—especially those which display misfolded or large hydrophobic domains in the cytosol—will require ERAD back‐up systems to prevent cellular toxicity. It is also important to note that aggregation‐prone, disaggregase‐requiring ERAD substrates may represent the exception rather than the rule. To date, several proteins, including KWS (a membrane protein with misfolded domains in both the ER and cytosol)80 and CTG* (a GFP‐tagged, membrane tethered form of a soluble ERAD substrate),22 the WT and N1303K chimeras, and the signal sequence‐containing ApoB29 species (this work), were degraded in an Hsp104‐independent manner in yeast. However, we anticipate that the number of ERAD substrates linked to human diseases will rise over time due to the ever‐expanding availability of human genome sequence databases and better links between clinical and genomic databases. Many of these substrates might also display misfolded, hydrophobic domains in the cytosol. It will be vital to consider whether they too will require protein disaggregases for elimination, or whether ER‐phagy or noncanonical forms of autophagy will be required. This information will be critical as drugs are sought to correct defects associated with misfolded ER proteins.
Materials and Methods
Yeast strains, strain construction, and growth conditions
Unless noted otherwise, yeast strains were grown at 30°C using standard conditions.81 To express ApoB, yeast strains were first made β‐estradiol inducible by linearizing pACT1‐GEV (a kind gift from Dr. A. O'Donnell, University of Pittsburgh) using EcoRV (New England Biolabs) according to the manufacturer's specifications. The linearized plasmid was transformed into log‐phase yeast cells (OD600 = 0.4–0.6) and integrated at leu2Δ0. Transformants were selected on rich media supplemented with glucose and containing 0.1 mg/mL Nourseothricin (Nat) (Werner Bioagents, Jena, Germany). Positive transformants were struck three successive times onto media supplemented with Nat to ensure successful integration.
Yeast strains used in this study include: WT strains MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 and MATalpha his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0, as well as the Pdr5‐deficient strain MATalpha, ade2‐1 can1‐100, his3‐11,15, leu2‐3,112, trp1‐1, ura3‐1, pdr5::KANMX4 and the vacuolar protease‐deficient strain MATalpha, ade2‐1 can1‐100, his3‐11,15, leu2‐3,112, trp1‐1, ura3‐1, pep4::KANMX (Dharmacon, Lafayette, CO); MATalpha, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, TGL3::GFP‐HIS3MX6;62 MATalpha ura3‐52 lys2‐801 ade2‐101 trp1‐Δ63 his3‐Δ200 leu2‐Δ1 and MATalpha ura3‐52 lys2‐801 ade2‐101 trp1‐Δ63 his3‐Δ200 leu2‐Δ1 hsp104::LEU2;82 CDC48 (yPC9815), MATa, ura3‐52, his3Δ200, leu2‐3,2–112, trp1‐1, lys2‐801 and MATa, ura3‐52, his3Δ200, leu2‐3,2–112, trp1‐1, lys2‐801, cdc48‐6;83 and MATa, pdr5Δ::KANMX, can1Δ::STE2pr‐Sp_his5, lyp1Δ, ura3Δ0, met15Δ0, HSP104‐GFP::LEU2 (a gift from Thomas Nystrom).
Plasmids and plasmid construction
ApoB29 containing a heterologous signal sequence (“ApoB”27) or lacking the signal sequence (ApoBΔSS) were constructed as follows. Plasmid pYES2‐ApoB29ΔSS was constructed by polymerase chain reaction (PCR) amplification of ApoB29 (amino acid 126–1371) in pSLW1‐B2927 with forward (5′‐GGGAATATTAAGCTTGGTACATGTCCAGGTATGAGCTCAAG) and reverse (5′‐AGTGGATCCGAGCTCGGTACTCATTAAGCGTAATCTGGAAC) primers. The purified PCR product and Kpn1 digested pYES2 plasmid (Thermo Fisher Scientific) were fused using Gibson assembly (New England Biolabs, Ipswich, MA) and transformed into Escherichia coli. To generate the ApoB expression vector, the full prepro‐ApoB29 insert was generated using PCR overlap extension84 followed by yeast gap repair cloning as described.85 The following primers were used to amplify the prepro sequence (amino acids 1–82): forward (5′‐TCGGACTACTAGCAGCTGTAATACGACTCACTATAGGGAATATTAAGCTTATGAGATTTCCTTCAATTTTTACT) and reverse (5′‐ACCAGGCTGACATTTTCCTTCACTAGTGATACCC), and ApoB29 (amino acids 33–1371), forward (5′‐GGGTATCACTAGTGAAGGAAAATGTCAGCCTGGT) and reverse (5′‐ATCTGCAGAATTCCAGCACACTGGCGGCCGTTACTAGTGGATCCGAGCTCGTCGACTCATTAAGCGTAATCTG) from pSLW1‐B29.27 The assembled ApoB PCR product and Kpn1 digested pYES2 plasmid were then transformed into yeast using standard methods and plated on selective media. Transformants were screened for ApoB expression in yeast by inducing protein expression with 2% galactose for 5 h at 30°C.
The CFTR chimera series was derived from Chimera A21 and expressed under the control of the TEF promoter in the pRS416 yeast expression vector.86 To create the chimera, overlap PCR (see above) was performed to fuse tm1‐2 of Chimera A to NBD2 in CFTR while removing an EcoRI site in the CFTR NBD2 with a silent mutation to allow for cloning into pRS416. Because the tm1‐2 of Chimera A contains an internal triple HA tag, the plasmids also contain sequences to express triply HA‐tagged proteins. To introduce the analogous W1282X truncation, primer oMP01 (5′‐GCGCCCGGGATGAACTTTTTAAGTTTTAAGAC‐3′) was used in combination with oMP21 (5′‐CGCCTGCAGTTACTGTTGCAAAGTTATTG‐3′). To introduce the analogous N1303K mutation in Chimera CFTR, QuikChange II site‐directed mutagenesis (Agilent) was performed with oMP22 (5′‐GGAACATTTAGAAAAAAATTGGATCCCTATG‐3′) and oMP23 (5′‐CATAGGGATCCAATTTTTTTCTAAATGTTCC‐3′). The WT CFTR chimera expression vector was produced using the Q5 site‐directed mutagenesis kit (New England Biolabs) to revert N1303K to the WT NBD2 nucleotide sequence with oCG284 (5′‐TTAGAAAAAACTTGGATCCCTATG‐3′) and oCG285 (5′‐ATGTTCCAGAAAAAATAAATACTTTC‐3′). The resulting plasmids, which are engineered to express the WT, W1282X, and N1303K chimeras are named pMP13 through pMP15, respectively. The full coding sequence of each construct was confirmed by DNA sequencing (Genewiz, South Plainfield, NJ).
Antibodies and quantitative Western blotting
Antibodies used in this study include: horseradish peroxidase (HRP) conjugated anti‐HA (Roche Applied Science, 3F10) used at a 1:5000 dilution; G6PD (Sigma) diluted to 1:5000; donkey anti‐rabbit HRP (GE Healthcare) used at 1:5000; rabbit anti‐protein disulfide isomerase (PDI; a kind gift from Dr. Vlad Denic, Harvard University) diluted to 1:5000; rabbit anti‐Kar287 diluted to 1:5000; rabbit anti‐Sec61 (raised again peptide LVPGFSDLM and purified by Cocalico Biologicals, Stevens, PA) diluted to 1:1000; mouse monoclonal anti‐GFP (Roche) used at 1:1000; monoclonal anti‐mouse HRP (Cell Signaling Technology, Danvers, MA) diluted to 1:5000; mouse monoclonal anti‐Vph1 (Abcam, 10D7A7B2) diluted to 1:5000; rabbit anti‐Hsp104 (a kind gift from Dr. John Glover, University of Toronto) diluted to 1:1000; rabbit anti‐Ssa127 used at 1:5000; and rabbit anti‐Sse127 diluted to 1:5000.
After sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) and transfer to nitrocellulose, proteins were visualized using the Supersignal West Pico Chemiluminescent substrate (ThermoFisher Scientific) or Supersignal West Femto Maximum Sensitivity chemiluminescent substrate (ThermoFisher Scientific, Pittsburgh, PA). Images were obtained and quantified using either a Kodak 440CF Image station and the associated Kodak 1D software (Eastman Kodak, Rochester, NY) or a BioRAD Universal Hood II Imager and ImageJ software version 1.48v (National Institutes of Health).
Fluorescence imaging to detect protein aggregates
A yeast strain expressing Hsp104‐GFP and lacking a multidrug resistance pump (pdr5Δ)40 was transformed with an empty vector (pRS416‐TEF), or vectors engineered for the expression of Chimera A*, W1282X, or ApoBΔSS (also see above).22 For ApoBΔSS, the GEV expression system was introduced into the strain, prior to transformation. Cultures were grown in synthetic complete media lacking uracil and diluted to early log phase the next day, prior to plating onto a polylysine‐coated glass bottom microwell dish (MatTek, Ashland, MA). Expression of ApoBΔSS was induced by the addition of β‐estradiol 1.5 h prior to imaging. Cells were imaged prior to treatment and 1 h after the addition of a final concentration of 100 μM MG132 and incubation at 37°C, essentially as described.22 Images were taken using a Nikon A1 point‐scanning confocal and an Apo TIRF 100X oil immersion objective, NA = 1.49. Z‐stacks were deconvolved using NIS‐Elements v5.10.01 (Nikon, Melville, NY), and a single plane is displayed.
Limited proteolysis and carbonate extraction
Medium‐scale microsomes88 were prepared from yeast expressing ApoB as published, and 200 mg of total microsomes were combined with Buffer 88 (20 mM 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid (HEPES), pH 6.8, 150 mM KOAc, 250 mM sorbitol, and 5 mM MgOAc) on ice. The 0 min time point was removed, mixed with a final concentration of 30% trichloroacetic acid (TCA), and stored on ice. The remainder of the sample was incubated with 0.04 mg/mL Proteinase K (Sigma) on ice. Aliquots were taken at the indicated time points, combined with TCA as above and stored on ice. All samples were then centrifuged at 13,000 rpm in a microfuge (10 min at 4°C), washed with acetone, air dried, and resuspended in TCA sample buffer (80 mM Tris, pH 8, 8 mM ethylenediaminetetraacetic acid (EDTA), 120 mM DTT, 3.5% SDS, 0.29% glycerol, 0.08% Tris base, 0.01% bromophenol blue). Samples were heated 37°C for 20 min and resolved by SDS‐PAGE followed by Western blotting.
Carbonate extraction was conducted on yeast expressing ApoB essentially as described.89 This procedure has long been used as a means to remove peripherally associated proteins bound to membranes.59 In brief, transformed yeast were grown at 30°C until log phase in synthetic minimal media lacking uracil and supplemented with 2% glucose. ApoB was induced using 300 nM β‐estradiol for 2 h at 30°C. Yeast were harvested and resuspended in 20 mM HEPES, pH 7.4, 50 mM KOAc, 2 mM EDTA, and 0.1M sorbitol plus protease inhibitors and lysed using glass beads. Unbroken cells were removed by low‐speed centrifugation and the supernatant was centrifuged to isolate a crude membrane fraction. After washing, the isolated membranes were incubated with either 0.1M Na2CO3 or Buffer 88 supplemented with protease inhibitors and incubated on ice for 30 min. The treated membranes were then centrifuged at 50,000g in SW55 Ti rotor for 1 h at 4°C, and the supernatant and pellet samples were isolated and retained. The pellets were resuspended in 0.1M Na2CO3 or buffer and centrifuged at 60,000g in SW55 rotor for 10 min at 4°C, and the final pellets were resuspended in TCA sample buffer using a mechanical pestle. The supernatants were mixed with TCA, incubated on ice for 15 min, and centrifuged at 14,000 rpm for 10 min at 4°C in a microfuge, and the resulting pellets were resuspended in TCA sample buffer using a mechanical pestle. All samples were incubated at 37°C for 20 min followed by SDS‐PAGE and Western blotting.
Protein degradation assays
To measure ApoB29 degradation using the β‐estradiol inducible system, yeast transformed with the desired ApoB expression plasmid (see above) were grown overnight in synthetic minimal media lacking uracil but supplemented with 2% glucose at 30°C. The cultures were diluted and grown to log phase, and the ApoB protein was induced using 300 nM β‐estradiol for 2 h at 30°C. Equal optical densities (ODs) of yeast were harvested and then resuspended to 5 OD600/mL in synthetic minimal media lacking uracil but supplemented with 2% glucose and 300 nM β‐estradiol. Cycloheximide chase assays were conducted at 30°C as described previously.27, 51 After protein synthesis was stopped using 50 μg/mL cycloheximide, 2 OD600 units of cells were harvested at the indicated times. Total protein was precipitated with TCA90 and resolved by SDS‐PAGE followed by Western blotting.
To determine the turnover rates of the CFTR chimeras, cycloheximide chases were performed as described previously.21, 91 In brief, cells were grown to logarithmic phase in synthetic complete media lacking uracil and incubated at 25°C. When the Hsp104 mutant strain was examined, the cells were then shifted to and grown at 37°C, while the Cdc48 mutant strain was shifted to 39°C. Cycloheximide was added to a final concentration of 150 μg/mL to each culture, the cells were lysed with 3.3M NaOH, 15% 2‐mercaptoethanol, 1.1 mM phenylmethylsulfonyl fluoride (PMSF), 2.2 mM leupeptin, and 0.77 mM pepstatin A, and then treated with TCA to a final concentration of 12%. The protein pellets were resuspended in TCA sample buffer and samples were incubated at 37°C. Resuspended pellets were analyzed by SDS‐PAGE and immunoblotting.
Isolation of yeast LDs
LDs were isolated essentially as described,61 starting from a 2 L culture of yeast that expressed a GFP‐tagged form of Tgl3 (a kind gift from G. Daum, Graz University of Technology). After ApoB was induced in a log‐phase yeast culture using galactose for 5 h at 30°C, the cells were harvested, resuspended in 100 mM Tris, pH 9.4 supplemented with 10 mM DTT, and incubated at room temperature for 15 min before being harvested. The treated cells were resuspended in 0.7M sorbitol, 75% yeast extract and peptone, 0.5% glucose, 10 mM Tris, and pH 7.4 supplemented with 5 mM DTT. After lyticase treatment for 15 min at 30°C, spheroplasts were overlaid onto a cushion of 0.8M sucrose, 1.5% Ficoll 400, 20 mM HEPES, pH 7.4, and centrifuged in an HB‐6 rotor at 6000 rpm for 10 min at 4°C. The pelleted spheroplasts were resuspended in 20 mM KPO4, pH 7.4, and 1.2M sorbitol and centrifuged in an HB‐6 rotor at 6000 rpm for 5 min at 4°C. The washed spheroplasts were next resuspended in 10 mM MES‐Tris, pH 6.9, 12% Ficoll 400, and 0.2 mM EDTA plus protease inhibitors and lysed using a Dounce homogenizer with 15 strokes on ice, and the homogenate was diluted with 1 volume of buffer plus protease inhibitors and recentrifuged. This supernatant, which contained the broken cell lysate, was overlaid with an equal volume of buffer plus protease inhibitors and centrifuged in a SW‐28 swinging bucket rotor at 28,000 rpm for 1 h at 4°C. The top floating layer was again mixed with 1 volume of buffer plus protease inhibitors using a Dounce homogenizer with 10 strokes on ice. This sample was overlaid with an equal volume of 10 mM MES‐Tris, pH 6.9, 8% Ficoll 400, and 0.2 mM EDTA plus protease inhibitors and centrifuged in a SW‐28 swinging bucket rotor at 28,000 rpm for 1 h at 4°C. The final top floating layer, which contained LDs as well as vacuole remnants, was gently combined with an equal volume of 10 mM MES‐Tris, pH 6.9, 0.6M sorbitol, 8% Ficoll 400, and 0.2 mM EDTA plus protease inhibitors. Samples were overlaid with an equal volume of 10 mM MES‐Tris, pH 6.9, 0.25M sorbitol, and 0.2 mM EDTA plus protease inhibitors and centrifuged in a SW‐28 rotor at 28,000 rpm for 30 min at 4°C. Highly enriched LDs were isolated and mixed with TCA to a final concentration 10%. Samples were incubated on ice for 20 min and spun at 14,000 rpm in a microfuge for 15 min at 4°C. The final pellets were washed in acetone, recentrifuged, air dried, and resuspended in TCA sample buffer using a mechanical pestle before being subject to SDS‐PAGE and Western blotting.
Assays for endoglycosidase H sensitivity, ubiquitination, and detergent solubility
Expression of the ApoB protein was induced in transformed yeast with 300 nM β‐estradiol for 2 h at 30°C. Equal amounts of protein were harvested and TCA precipitated, as described above. Samples were treated in the presence or absence of Endoglycosidase H (Roche) for 2 h at 37°C according to the manufacturer's specifications. Samples were subsequently resolved by SDS‐PAGE and analyzed by Western blotting.
To compare the level of ubiquitination for the CFTR chimeras, a modified in vivo ubiquitination assay was performed.22 In brief, 30 ODs of transformed yeast were grown in selective media supplemented with 2% glucose at 20°C–22°C and incubated for 1 h in the presence of 100 μM copper sulfate to induced expression of myc‐tagged ubiquitin from an introduced plasmid. The cells were harvested and lysed by glass bead lysis in RIPA buffer (10 mM Tris‐Cl pH 8.0, 140 mM NaCl, 1 mM EDTA, 1% Triton‐X100, 0.1% sodium deoxycholate, 0.1% SDS, 1 mM PMSF, 2 mM leupeptin, and 0.7 mM pepstatin A), and unbroken cells were removed by centrifugation. The supernatant was collected, the beads were washed and combined with the first supernatant, and the lysed material was next centrifuged at 18,000g for 10 min at 4°C. The supernatants were collected and the amount of solubilized protein was assessed by measuring the A280. Next, equal amounts of total protein were immunoprecipitated with HA‐conjugated beads, and after incubation overnight, the beads were washed and the immunoprecipitated protein was resolved by SDS‐PAGE and analyzed by Western blotting. To monitor the biochemical ubiquitination and retrotranslocation of the W1282X chimera, microsomes containing the substrate (see above) were subjected to an in vitro assay, as published.88
To monitor the detergent solubility of the CFTR‐derived chimeras, ER‐enriched microsomes were isolated via a large‐scale microsome isolation protocol92 and a modified detergent solubility assay was used.22 Reactions containing ER‐enriched microsomes (at a final protein concentration of 0.5 μg/mL), DDM (at a final concentration of 0.5 mM), and phosphate buffered saline (PBS) supplemented with 250 mM sorbitol were incubated for 30 min at 20°C–22°C. As controls, the ER‐enriched microsomes were incubated in the absence of detergent or in the presence of a strong ionic detergent. Next, the reactions were centrifuged for 10 min at 18,000g and at 4°C. The supernatants were moved to fresh tubes and the pellets were resuspended in an equal volume of PBS with sorbitol. Both fractions were then treated with TCA at a final concentration of 12% for 30 min on ice. Samples were again centrifuged and the pellets were resuspended in TCA sample buffer. These final samples were incubated at 37°C for 30 min, centrifuged briefly, and subjected to SDS‐PAGE and analyzed by Western blotting.
Sucrose gradient analysis of an aggregation‐prone integral membrane protein
To investigate the aggregation state of the membrane‐embedded CFTR chimeras, WT yeast strains were transformed with expression vectors for the indicated protein and grown in selective media, as above. A modified version of a previously described flotation assay41 was then performed with ~50 ODs of cells that were collected and lysed in 20 mM HEPES, pH 7.4, 50 mM KOAc, 2 mM EDTA, 0.1M sorbitol, 1 mM of freshly added DTT, 20 μM MG132, 10 mM NEM, and Roche Complete EDTA‐Free PI cocktail in the presence of glass beads. The lysate was centrifuged for 5 min at 300g and at 4°C to remove unbroken cells, and the beads were washed with Buffer 88, which was collected and pooled with the lysates. These combined samples were recentrifuged, as above, the resulting supernatants were collected, and a portion was mixed with a 1.9M sucrose flotation buffer in 50 mM HEPES pH 7.4, 150 mM NaCl, 5 mM EDTA, freshly added 1 mM DTT, 1 mM PMSF, and Roche Complete EDTA‐Free PI cocktail. Gradients were set up as follows: 600 μL of the 1.9M sucrose flotation buffer on the bottom, 800 μL sample that was mixed with this buffer, 1.2 mL of a 0.8M in sucrose flotation buffer, and 1 mL of a 0.6M sucrose in flotation buffer. The gradients were then centrifuged at 77,000g for 16 h at 4°C, and 12 fractions were removed from the top of the gradient with a pipette and placed in fresh tubes. Each sample was incubated on ice for 30 min with a final volume of 12% TCA, the mixture was centrifuged in a microfuge at the top speed for 10 min at 4°C, and the pellets were resuspended in TCA sample buffer by mechanical disruption as described above. All samples were then analyzed by SDS‐PAGE and Western blotting.
MD simulations
The initial structure for the simulation was taken from the 3GD7.pdb file in which the NBD2 is in a dimer incompatible conformation, including ATP and Mg2+. The sequence of NBD2 was mutated back to the WT and the mutated residues were further optimized using Discovery Studio.93 Two constructs were created depending on the boundaries of the sequence: (1) WT NBD2 and N1303K with the boundaries 1202–1427 (226 residues), and (2) W1282X, with 1202–1281 boundaries (80 residues). Prior to simulation, the proper protonation state of titratable residues was assigned using the prepare protein protocol in discovery studio. All systems were first subjected to minimization with the steepest descent algorithm.94 Following minimization, the number of particles, volume, temperature (NVT) and the isothermal‐isobaric ensemble (NPT) equilibration for 100 ps each were performed. The MD simulations were carried out using GROMACS (version 4.5.5)95, 96 with the AMBER99SB‐ILDN force field97 and TIP4P water98 at 300 K with a time step of 2 fs using the leapfrog algorithm.99, 100 The cutoff of 10 Å was set for van der Waals and Coulomb interactions. Long‐range electrostatic interactions were calculated using particle mesh Ewald electrostatics.100, 101 The LINCS algorithm for bond lengths constrain was used.102 Periodic boundary conditions were applied in all directions. For each construct, three separate, 500 ns simulations were performed, each initiated from a different random seed for a total of 1.5 μs.
Per‐residue RMSF, per‐residue SASA, and total Rg were calculated for the common part of all constructs (residues 1202–1281) using the appropriate routines in GROMACS. The secondary structure of the proteins was calculated by the DSSP analysis,42 using pattern recognition hydrogen bond and geometric features for secondary structure determination.
Supporting information
Figure S1 Representative DSSP plots for residues 1202–1282 of (A) Wild type (WT) and (B) W1282X NBD2 as a function of simulation time (500 ns simulation). In these plots residue numbers are given on the y‐axis and simulation time on the x‐axis. Red, blue, green, yellow, black, and white color correspond to b‐sheet, a‐helix, bend, turn, b‐bridge, and coil secondary structure respectively.
Figure S2. Average Rg values over three 500 ns simulations for residues 1202–1282 are shown. Black, blue and green lines correspond to the wild type, 1282X, and N1303K constructs, respectively. Low Rg values are indicative of a more compact construct.
Figure S3. (A) SASA values averaged over three 500 ns simulations for residues 1202–1282 are shown. Black and blue lines correspond to the wild type and 1282X constructs, respectively. Low SASA values are indicative of a less exposed surface area. (B) Average differences plots (WT‐1282X) over three 500 ns simulations are shown. Negative values indicate a more exposed SASA in the truncation mutant.
Figure S4. (A) Induction time‐course for full‐length ApoB29 expression in yeast transformed with the pYES2‐ApoB29 expression vector. ApoB29 expression was monitored following the addition of β‐estradiol at the indicated time‐points. G6PD serves as a loading control. (B) Overnight yeast cultures transformed with an empty vector (Vector) or ApoBΔSS were diluted to OD600 = 0.05 and growth was monitored over the indicated times in the presence (+) or absence (−) of β‐estradiol.
Acknowledgments
We wish to thank A. O'Donnell, A. Mittal, V. Denic, J. Glover, G. Daum, and P. Thibodeau for reagents, advice, and technical assistance.
Lynley M. Doonan, Christopher J. Guerriero, and G. Michael Preston contributed equally to this work.
Significance statement: Misfolded proteins can aggregate, which impairs cellular homeostasis and can give rise to disease. Although yeast express an enzyme that dissolves aggregates, humans lack this protein. We show that human aggregation‐prone proteins associated with the endoplasmic reticulum are substrates for the yeast disaggregase. These data suggest that a human analog of the yeast disaggregase exists or that another mechanism removes aggregates associated with the endoplasmic reticulum.
References
- 1. Berner N, Reutter KR, Wolf DH (2018) Protein quality control of the endoplasmic reticulum and ubiquitin‐proteasome‐triggered degradation of aberrant proteins: yeast pioneers the path. Annu Rev Biochem 87:751–782. [DOI] [PubMed] [Google Scholar]
- 2. Wu X, Rapoport TA (2018) Mechanistic insights into ER‐associated protein degradation. Curr Opin Cell Biol 53:22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zattas D, Hochstrasser M (2015) Ubiquitin‐dependent protein degradation at the yeast endoplasmic reticulum and nuclear envelope. Crit Rev Biochem Mol Biol 50:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Christianson JC, Ye Y (2014) Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat Struct Mol Biol 21:325–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ruggiano A, Foresti O, Carvalho P (2014) Quality control: ER‐associated degradation: protein quality control and beyond. J Cell Biol 204:869–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guerriero CJ, Brodsky JL (2012) The delicate balance between secreted protein folding and endoplasmic reticulum‐associated degradation in human physiology. Physiol Rev 92:537–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chiti F, Dobson CM (2017) Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu Rev Biochem 86:27–68. [DOI] [PubMed] [Google Scholar]
- 8. Vincenz‐Donnelly L, Holthusen H, Korner R, Hansen EC, Presto J, Johansson J, Sawarkar R, Hartl FU, Hipp MS (2018) High capacity of the endoplasmic reticulum to prevent secretion and aggregation of amyloidogenic proteins. EMBO J 37:337–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rousseau E, Dehay B, Ben‐Haiem L, Trottier Y, Morange M, Bertolotti A (2004) Targeting expression of expanded polyglutamine proteins to the endoplasmic reticulum or mitochondria prevents their aggregation. Proc Natl Acad Sci U S A 101:9648–9653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grumati P, Dikic I, Stolz A (2018) ER‐phagy at a glance. J Cell Sci 131:jcs217364. [DOI] [PubMed] [Google Scholar]
- 11. Smith M, Wilkinson S (2017) ER homeostasis and autophagy. Essays Biochem 61:625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nillegoda NB, Bukau B (2015) Metazoan Hsp70‐based protein disaggregases: emergence and mechanisms. Front Mol Biosci 2:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zolkiewski M, Zhang T, Nagy M (2012) Aggregate reactivation mediated by the Hsp100 chaperones. Arch Biochem Biophys 520:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mogk A, Bukau B, Kampinga HH (2018) Cellular handling of protein aggregates by disaggregation machines. Mol Cell 69:214–226. [DOI] [PubMed] [Google Scholar]
- 15. Neuwald AF, Aravind L, Spouge JL, Koonin EV (1999) AAA+: a class of chaperone‐like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res 9:27–43. [PubMed] [Google Scholar]
- 16. Meyer H, Bug M, Bremer S (2012) Emerging functions of the VCP/p97 AAA‐ATPase in the ubiquitin system. Nat Cell Biol 14:117–123. [DOI] [PubMed] [Google Scholar]
- 17. Wickner RB, Edskes HK, Son M, Bezsonov EE, DeWilde M, Ducatez M (2018) Yeast prions compared to functional prions and amyloids. J Mol Biol 430:3707–3719. [DOI] [PubMed] [Google Scholar]
- 18. Zhao M, Brunger AT (2016) Recent advances in deciphering the structure and molecular mechanism of the AAA+ ATPase N‐ethylmaleimide‐sensitive factor (NSF). J Mol Biol 428:1912–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Parsell DA, Kowal AS, Singer MA, Lindquist S (1994) Protein disaggregation mediated by heat‐shock protein Hsp104. Nature 372:475–478. [DOI] [PubMed] [Google Scholar]
- 20. Schirmer EC, Glover JR, Singer MA, Lindquist S (1996) HSP100/Clp proteins: a common mechanism explains diverse functions. Trends Biochem Sci 21:289–296. [PubMed] [Google Scholar]
- 21. Guerriero CJ, Reutter KR, Augustine AA, Preston GM, Weiberth KF, Mackie TD, Cleveland‐Rubeor HC, Bethel NP, Callenberg KM, Nakatsukasa K, Grabe M, Brodsky JL (2017) Transmembrane helix hydrophobicity is an energetic barrier during the retrotranslocation of integral membrane ERAD substrates. Mol Biol Cell 28:2076–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Preston GM, Guerriero CJ, Metzger MB, Michaelis S, Brodsky JL (2018) Substrate insolubility dictates Hsp104‐dependent endoplasmic‐reticulum‐associated degradation. Mol Cell 70:242–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Preston GM, Brodsky JL (2017) The evolving role of ubiquitin modification in endoplasmic reticulum‐associated degradation. Biochem J 474:445–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Doonan LM, Fisher EA, Brodsky JL (2018) Can modulators of apolipoproteinB biogenesis serve as an alternate target for cholesterol‐lowering drugs? Biochim Biophys Acta Mol Cell Biol Lipids 1863:762–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ginsberg HN, Fisher EA (2009) The ever‐expanding role of degradation in the regulation of apolipoprotein B metabolism. J Lipid Res 50:S162–S166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Segrest JP, Jones MK, De Loof H, Dashti N (2001) Structure of apolipoprotein B‐100 in low density lipoproteins. J Lipid Res 42:1346–1367. [PubMed] [Google Scholar]
- 27. Hrizo SL, Gusarova V, Habiel DM, Goeckeler JL, Fisher EA, Brodsky JL (2007) The Hsp110 molecular chaperone stabilizes apolipoprotein B from endoplasmic reticulum‐associated degradation (ERAD). J Biol Chem 282:32665–32675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sosnay PR, Siklosi KR, Van Goor F, Kaniecki K, Yu H, Sharma N, Ramalho AS, Amaral MD, Dorfman R, Zielenski J, Masica DL, Karchin R, Millen L, Thomas PJ, Patrinos GP, Corey M, Lewis MH, Rommens JM, Castellani C, Penland CM, Cutting GR (2013) Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat Genet 45:1160–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang W, Hong JS, Rab A, Sorscher EJ, Kirk KL (2016) Robust stimulation of W1282X‐CFTR channel activity by a combination of allosteric modulators. PLoS One 11:e0152232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zainal Abidin N, Haq IJ, Gardner AI, Brodlie M (2017) Ataluren in cystic fibrosis: development, clinical studies and where are we now? Expert Opin Pharmacother 18:1363–1371. [DOI] [PubMed] [Google Scholar]
- 31. Rapino D, Sabirzhanova I, Lopes‐Pacheco M, Grover R, Guggino WB, Cebotaru L (2015) Rescue of NBD2 mutants N1303K and S1235R of CFTR by small‐molecule correctors and transcomplementation. PLoS One 10:e0119796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lewis HA, Buchanan SG, Burley SK, Conners K, Dickey M, Dorwart M, Fowler R, Gao X, Guggino WB, Hendrickson WA, Hunt JF, Kearins MC, Lorimer D, Maloney PC, Post KW, Rajashankar KR, Rutter ME, Sauder JM, Shriver S, Thibodeau PH, Thomas PJ, Zhang M, Zhao X, Emtage S (2004) Structure of nucleotide‐binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J 23:282–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vernon RM, Chong PA, Lin H, Yang Z, Zhou Q, Aleksandrov AA, Dawson JE, Riordan JR, Brouillette CG, Thibodeau PH, Forman‐Kay JD (2017) Stabilization of a nucleotide‐binding domain of the cystic fibrosis transmembrane conductance regulator yields insight into disease‐causing mutations. J Biol Chem 292:14147–14164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Atwell S, Brouillette CG, Conners K, Emtage S, Gheyi T, Guggino WB, Hendle J, Hunt JF, Lewis HA, Lu F, Protasevich II, Rodgers LA, Romero R, Wasserman SR, Weber PC, Wetmore D, Zhang FF, Zhao X (2010) Structures of a minimal human CFTR first nucleotide‐binding domain as a monomer, head‐to‐tail homodimer, and pathogenic mutant. Protein Eng Des Sel 23:375–384. [DOI] [PubMed] [Google Scholar]
- 35. Liu Q, Sabirzhanova I, Yanda MK, Bergbower EAS, Boinot C, Guggino WB, Cebotaru L (2018) Rescue of CFTR NBD2 mutants N1303K and S1235R is influenced by the functioning of the autophagosome. J Cyst Fibros 17:582–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mutyam V, Libby EF, Peng N, Hadjiliadis D, Bonk M, Solomon GM, Rowe SM (2017) Therapeutic benefit observed with the CFTR potentiator, ivacaftor, in a CF patient homozygous for the W1282X CFTR nonsense mutation. J Cyst Fibros 16:24–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, Decker CJ, Miller M, McCartney J, Olson ER, Wine JJ, Frizzell RA, Ashlock M, Negulescu PA (2011) Correction of the F508del‐CFTR protein processing defect in vitro by the investigational drug VX‐809. Proc Natl Acad Sci U S A 108:18843–18848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Oliver KE, Han ST, Sorscher EJ, Cutting GR (2017) Transformative therapies for rare CFTR missense alleles. Curr Opin Pharmacol 34:76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nakatsukasa K, Huyer G, Michaelis S, Brodsky JL (2008) Dissecting the ER‐associated degradation of a misfolded polytopic membrane protein. Cell 132:101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hill SM, Hao X, Liu B, Nystrom T (2014) Life‐span extension by a metacaspase in the yeast Saccharomyces cerevisiae . Science 344:1389–1392. [DOI] [PubMed] [Google Scholar]
- 41. Nakatsukasa K, Kamura T (2016) Subcellular fractionation analysis of the extraction of ubiquitinated polytopic membrane substrate during ER‐associated degradation. PLoS One 11:e0148327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen‐bonded and geometrical features. Biopolymers 22:2577–2637. [DOI] [PubMed] [Google Scholar]
- 43. Brodsky JL, Fisher EA (2008) The many intersecting pathways underlying apolipoprotein B secretion and degradation. Trends Endocrinol Metab 19:254–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ohsaki Y, Cheng J, Fujita A, Tokumoto T, Fujimoto T (2006) Cytoplasmic lipid droplets are sites of convergence of proteasomal and autophagic degradation of apolipoprotein B. Mol Biol Cell 17:2674–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fujimoto T, Ohsaki Y (2006) Proteasomal and autophagic pathways converge on lipid droplets. Autophagy 2:299–301. [DOI] [PubMed] [Google Scholar]
- 46. Ohsaki Y, Cheng J, Suzuki M, Fujita A, Fujimoto T (2008) Lipid droplets are arrested in the ER membrane by tight binding of lipidated apolipoprotein B‐100. J Cell Sci 121:2415–2422. [DOI] [PubMed] [Google Scholar]
- 47. Olzmann JA, Kopito RR (2011) Lipid droplet formation is dispensable for endoplasmic reticulum‐associated degradation. J Biol Chem 286:27872–27874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cole NB, Murphy DD, Grider T, Rueter S, Brasaemle D, Nussbaum RL (2002) Lipid droplet binding and oligomerization properties of the Parkinson's disease protein alpha‐synuclein. J Biol Chem 277:6344–6352. [DOI] [PubMed] [Google Scholar]
- 49. Scherzer CR, Feany MB (2004) Yeast genetics targets lipids in Parkinson's disease. Trends Genet 20:273–277. [DOI] [PubMed] [Google Scholar]
- 50. Gusarova V, Caplan AJ, Brodsky JL, Fisher EA (2001) Apoprotein B degradation is promoted by the molecular chaperones hsp90 and hsp70. J Biol Chem 276:24891–24900. [DOI] [PubMed] [Google Scholar]
- 51. Grubb S, Guo L, Fisher EA, Brodsky JL (2012) Protein disulfide isomerases contribute differentially to the endoplasmic reticulum‐associated degradation of apolipoprotein B and other substrates. Mol Biol Cell 23:520–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McIsaac RS, Silverman SJ, McClean MN, Gibney PA, Macinskas J, Hickman MJ, Petti AA, Botstein D (2011) Fast‐acting and nearly gratuitous induction of gene expression and protein depletion in Saccharomyces cerevisiae . Mol Biol Cell 22:4447–4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Patel SB, Grundy SM (1995) Heterologous expression of apolipoprotein B carboxyl‐terminal truncates: a model for the study of lipoprotein biogenesis. J Lipid Res 36:2090–2103. [PubMed] [Google Scholar]
- 54. Boerwinkle E, Chan L (1989) A three codon insertion/deletion polymorphism in the signal peptide region of the human apolipoprotein B (APOB) gene directly typed by the polymerase chain reaction. Nucleic Acids Res 17:4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cladaras C, Hadzopoulou‐Cladaras M, Nolte RT, Atkinson D, Zannis VI (1986) The complete sequence and structural analysis of human apolipoprotein B‐100: relationship between apoB‐100 and apoB‐48 forms. EMBO J 5:3495–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Xu CF, Tikkanen MJ, Huttunen JK, Pietinen P, Butler R, Humphries S, Talmud P (1990) Apolipoprotein B signal peptide insertion/deletion polymorphism is associated with Ag epitopes and involved in the determination of serum triglyceride levels. J Lipid Res 31:1255–1261. [PubMed] [Google Scholar]
- 57. Sturley SL, Talmud PJ, Brasseur R, Culbertson MR, Humphries SE, Attie AD (1994) Human apolipoprotein B signal sequence variants confer a secretion‐defective phenotype when expressed in yeast. J Biol Chem 269:21670–21675. [PubMed] [Google Scholar]
- 58. Harazono AKN, Kawanishi T, Hayakawa T (2005) Site‐specific glycosylation analysis of human apolipoprotein B100 using LC/ESI MS/MS. Glycobiology 15:447–462. [DOI] [PubMed] [Google Scholar]
- 59. Fujiki Y, Hubbard AL, Fowler S, Lazarow PB (1982) Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J Cell Biol 93:97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ploegh HL (2007) A lipid‐based model for the creation of an escape hatch from the endoplasmic reticulum. Nature 448:435–438. [DOI] [PubMed] [Google Scholar]
- 61. Leber R, Zinser E, Zellnig G, Paltauf F, Daum G (1994) Characterization of lipid particles of the yeast, Saccharomyces cerevisiae . Yeast 10:1421–1428. [DOI] [PubMed] [Google Scholar]
- 62. Athenstaedt K, Daum G (2003) YMR313c/TGL3 encodes a novel triacylglycerol lipase located in lipid particles of Saccharomyces cerevisiae . J Biol Chem 278:23317–23323. [DOI] [PubMed] [Google Scholar]
- 63. Zhou M, Fisher EA, Ginsberg HN (1998) Regulated co‐translational ubiquitination of apolipoprotein B100. A new paradigm for proteasomal degradation of a secretory protein. J Biol Chem 273:24649–24653. [DOI] [PubMed] [Google Scholar]
- 64. Braakman I, Bulleid NJ (2011) Protein folding and modification in the mammalian endoplasmic reticulum. Annu Rev Biochem 80:71–99. [DOI] [PubMed] [Google Scholar]
- 65. Brodsky JL, Skach WR (2011) Protein folding and quality control in the endoplasmic reticulum: recent lessons from yeast and mammalian cell systems. Curr Opin Cell Biol 23:464–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Araki K, Nagata K (2012) Protein folding and quality control in the ER. Cold Spring Harb Perspect Biol 4:a015438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Molinari M, Hebert DN (2015) Glycoprotein maturation and quality control. Semin Cell Dev Biol 41:70. [DOI] [PubMed] [Google Scholar]
- 68. Feige MJ, Hendershot LM (2011) Disulfide bonds in ER protein folding and homeostasis. Curr Opin Cell Biol 23:167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Nillegoda NB, Wentink AS, Bukau B (2018) Protein disaggregation in multicellular organisms. Trends Biochem Sci 43:285–300. [DOI] [PubMed] [Google Scholar]
- 70. Kruse KB, Brodsky JL, McCracken AA (2006) Characterization of an ERAD gene as VPS30/ATG6 reveals two alternative and functionally distinct protein quality control pathways: one for soluble Z variant of human alpha‐1 proteinase inhibitor (A1PiZ) and another for aggregates of A1PiZ. Mol Biol Cell 17:203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hidvegi T, Mukherjee A, Ewing M, Kemp C, Perlmutter DH (2011) The role of autophagy in alpha‐1‐antitrypsin deficiency. Methods Enzymol 499:33–54. [DOI] [PubMed] [Google Scholar]
- 72. Fregno I, Fasana E, Bergmann TJ, Raimondi A, Loi M, Solda T, Galli C, D'Antuono R, Morone D, Danieli A, Paganetti P, van Anken E, Molinari M (2018) ER‐to‐lysosome‐associated degradation of proteasome‐resistant ATZ polymers occurs via receptor‐mediated vesicular transport. EMBO J 37:e99259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Metzger MB, Maurer MJ, Dancy BM, Michaelis S (2008) Degradation of a cytosolic protein requires endoplasmic reticulum‐associated degradation machinery. J Biol Chem 283:32302–32316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lee DH, Goldberg AL (2010) Hsp104 is essential for the selective degradation in yeast of polyglutamine expanded ataxin‐1 but not most misfolded proteins generally. Biochem Biophys Res Commun 391:1056–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gribun A, Cheung KL, Huen J, Ortega J, Houry WA (2008) Yeast Rvb1 and Rvb2 are ATP‐dependent DNA helicases that form a heterohexameric complex. J Mol Biol 376:1320–1333. [DOI] [PubMed] [Google Scholar]
- 76. Zaarur N, Xu X, Lestienne P, Meriin AB, McComb M, Costello CE, Newnam GP, Ganti R, Romanova NV, Shanmugasundaram M, Silva ST, Bandeiras TM, Matias PM, Lobachev KS, Lednev IK, Chernoff YO, Sherman MY (2015) RuvbL1 and RuvbL2 enhance aggresome formation and disaggregate amyloid fibrils. EMBO J 34:2363–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Pan M, Cederbaum AI, Zhang YL, Ginsberg HN, Williams KJ, Fisher EA (2004) Lipid peroxidation and oxidant stress regulate hepatic apolipoprotein B degradation and VLDL production. J Clin Invest 113:1277–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Amengual J, Guo L, Strong A, Madrigal‐Matute J, Wang H, Kaushik S, Brodsky JL, Rader DJ, Cuervo AM, Fisher EA (2018) Autophagy is required for sortilin‐mediated degradation of apolipoprotein B100. Circ Res 122:568–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Forrester A, De Leonibus C, Grumati P, Fasana E, Piemontese M, Staiano L, Fregno I, Raimondi A, Marazza A, Bruno G, Iavazzo M, Intartaglia D, Seczynska M, van Anken E, Conte I, De Matteis MA, Dikic I, Molinari M, Settembre C (2018) A selective ER‐phagy exerts procollagen quality control via a Calnexin‐FAM134B complex. EMBO J 38:e99847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Vashist S, Ng DT (2004) Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J Cell Biol 165:41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Adams A, Gottschling DE, Kaiser CA, Stearns T. Methods in yeast genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 1997. [Google Scholar]
- 82. Glover JR, Lindquist S (1998) Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell 94:73–82. [DOI] [PubMed] [Google Scholar]
- 83. Rape M, Hoppe T, Gorr I, Kalocay M, Richly H, Jentsch S (2001) Mobilization of processed, membrane‐tethered SPT23 transcription factor by CDC48(UFD1/NPL4), a ubiquitin‐selective chaperone. Cell 107:667–677. [DOI] [PubMed] [Google Scholar]
- 84. Longtine MS, McKenzie A 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR (1998) Additional modules for versatile and economical PCR‐based gene deletion and modification in Saccharomyces cerevisiae . Yeast 14:953–961. [DOI] [PubMed] [Google Scholar]
- 85. Bessa D, Pereira F, Moreira R, Johansson B, Queiros O (2012) Improved gap repair cloning in yeast: treatment of the gapped vector with Taq DNA polymerase avoids vector self‐ligation. Yeast 29:419–423. [DOI] [PubMed] [Google Scholar]
- 86. Sikorski RS, Hieter P (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae . Genetics 122:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Brodsky JL, Schekman R (1993) A Sec63p‐BiP complex from yeast is required for protein translocation in a reconstituted proteoliposome. J Cell Biol 123:1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Nakatsukasa K, Brodsky JL (2008) The recognition and retrotranslocation of misfolded proteins from the endoplasmic reticulum. Traffic 9:861–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Buck TM, Jordahl AS, Yates ME, Preston GM, Cook E, Kleyman TR, Brodsky JL (2017) Interactions between intersubunit transmembrane domains regulate the chaperone‐dependent degradation of an oligomeric membrane protein. Biochem J 474:357–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Zhang Y, Nijbroek G, Sullivan ML, McCracken AA, Watkins SC, Michaelis S, Brodsky JL (2001) Hsp70 molecular chaperone facilitates endoplasmic reticulum‐associated protein degradation of cystic fibrosis transmembrane conductance regulator in yeast. Mol Biol Cell 12:1303–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zhang Y, Michaelis S, Brodsky JL (2002) CFTR expression and ER‐associated degradation in yeast. Methods Mol Med 70:257–265. [DOI] [PubMed] [Google Scholar]
- 92. Nakatsukasa K, Brodsky JL (2010) In vitro reconstitution of the selection, ubiquitination, and membrane extraction of a polytopic ERAD substrate. Methods Mol Biol 619:365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Accelrys Discovery Studio Modeling Environment , A.S.I.S.D., Dassault Systèmes BIOVIA, Discovery Studio, San Diego: Dassault Systèmes, 2005–2009.
- 94. Marquardt D (1963) An algorithm for least‐squares estimation of nonlinear parameters. J Soc Indust Appl Math 11:431–441. [Google Scholar]
- 95. Hess B, Kutzner C, van der Spoel D, Lindahl E (2008) GROMACS 4: algorithms for highly efficient, load‐balanced, and scalable molecular simulation. J Chem Theory Comput 4:435–447. [DOI] [PubMed] [Google Scholar]
- 96. Berendsen H, Vanderspoel D, Vandrunen R (1995) Gromacs—a message‐passing parallel molecular‐dynamics implementation. Comput Phys Commun 91:43–56. [Google Scholar]
- 97. Lindorff‐Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE (2010) Improved side‐chain torsion potentials for the Amber ff99SB protein force field. Proteins 78:1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Jorgensen WLCJ, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935. [Google Scholar]
- 99. Hockney RW, Eastwood JW (1974) Quiet high resolution computer models of a plasma. J Comput Phys 14:148–158. [Google Scholar]
- 100. Essmann UPL, Berkowitz ML, Darden T, Lee H, Pedersen LG (1995) A smooth particle mesh Ewald method. J Chem Phys 103:8577–8593. [Google Scholar]
- 101. Darden TYD, Pedersen L (1993) Particle mesh Ewald—an n.log(N) method for Ewald sums in large systems. J Chem Phys 98:10089–10092. [Google Scholar]
- 102. Hess BH, Berendsen H, Fraaijen J (1997) Lincs: a linear constraint solver for molecular simulations. J Comput Chem 18:1463–1472. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Representative DSSP plots for residues 1202–1282 of (A) Wild type (WT) and (B) W1282X NBD2 as a function of simulation time (500 ns simulation). In these plots residue numbers are given on the y‐axis and simulation time on the x‐axis. Red, blue, green, yellow, black, and white color correspond to b‐sheet, a‐helix, bend, turn, b‐bridge, and coil secondary structure respectively.
Figure S2. Average Rg values over three 500 ns simulations for residues 1202–1282 are shown. Black, blue and green lines correspond to the wild type, 1282X, and N1303K constructs, respectively. Low Rg values are indicative of a more compact construct.
Figure S3. (A) SASA values averaged over three 500 ns simulations for residues 1202–1282 are shown. Black and blue lines correspond to the wild type and 1282X constructs, respectively. Low SASA values are indicative of a less exposed surface area. (B) Average differences plots (WT‐1282X) over three 500 ns simulations are shown. Negative values indicate a more exposed SASA in the truncation mutant.
Figure S4. (A) Induction time‐course for full‐length ApoB29 expression in yeast transformed with the pYES2‐ApoB29 expression vector. ApoB29 expression was monitored following the addition of β‐estradiol at the indicated time‐points. G6PD serves as a loading control. (B) Overnight yeast cultures transformed with an empty vector (Vector) or ApoBΔSS were diluted to OD600 = 0.05 and growth was monitored over the indicated times in the presence (+) or absence (−) of β‐estradiol.