

Abstract
Abstract: The protein quality control network (pQC) plays critical roles in maintaining protein and cellular homeostasis, especially during stress. Lon is a major pQC AAA+ protease, conserved from bacteria to human mitochondria. It is the principal enzyme that degrades most unfolded or damaged proteins. Degradation by Lon also controls cellular levels of several key regulatory proteins. Recently, our group determined that Escherichia coli Lon, previously thought to be an obligate homo‐hexamer, also forms a dodecamer. This larger assembly has decreased ATPase activity and displays substrate‐specific alterations in degradation compared with the hexamer. Here we experimentally probe the physical hexamer–hexamer interactions and the biological roles of the Lon dodecamer. Using structure prediction methods coupled with mutagenesis, we identified a key interface and specific residues within the Lon N domain that participates in an intermolecular coiled coil unique to the dodecamer. With this knowledge, we made a Lon variant (LonVQ) that forms a dodecamer with increased stability, as determined by analytical ultracentrifugation and electron microscopy. Using this altered Lon, we characterize the Lon dodecamer's activities using a panel of substrates. Lon dodecamers are clearly functional, and complement critical lon‐ phenotypes but also exhibit altered substrate specificity. For example, the small heat shock proteins IbpA and IbpB are only efficiently degraded well by the hexamer. Thus, by elucidating the intermolecular contacts connecting the hexamers, we are starting to illuminate how dodecamer formation versus disassembly can alter Lon function under conditions where controlling specific activities and substrate preferences of this key protease may be advantageous.
Keywords: AAA+ family, electron microscopy, crosslinking, analytical ultracentrifugation, coiled coils, regulated proteolysis
Abbreviations
- AUC
analytical ultracentrifugation
- EM
electron microscopy
- Ibp
inclusion body binding protein
- sHSP
small heat shock protein
Introduction
The protein quality control (pQC) network is integral for maintaining cellular homeostasis and viability during and after stress by preventing the accumulation of potentially toxic unfolded, damaged, and aggregated proteins. This system is composed of several enzymatic machines, including protein chaperones, disaggregases, and proteases. One of the central proteases responsible for pQC is Lon, as it degrades the majority of unfolded proteins in bacteria and is thought to perform a similar function in human mitochondria.1, 2 Lon is a highly conserved member of the AAA+ (ATPases associated with diverse cellular activities) protease family, which also includes ClpAP, ClpXP, FtsH, HslUV, and the 26S proteasome.3 In bacteria, Lon also modulates levels of several short‐lived regulatory proteins induced during cellular stresses, including heat shock and DNA damage.4, 5, 6 In humans, mitochondrial Lon antagonizes aging by degrading oxidatively damaged proteins,7 and Lon is identified as a potential therapeutic target to treat both lymphoma and bladder cancer.8, 9
Lon assembles into a barrel‐shaped homo‐hexamer with the proteolytic active sites sequestered in an internal chamber, largely inaccessible to folded proteins; this architecture serves to prevent degradation of non‐substrate proteins. After substrate binding, Lon uses the power of ATP hydrolysis coupled to conformational changes to unfold substrates (if necessary), and then to translocate the polypeptide chain through a central pore into the degradation chamber.10, 11 Lon often recognizes unfolded proteins via short peptide sequences (degrons) exposed in the unfolded protein that are buried and inaccessible in the folded version. The beta‐20 peptide (or β‐20), isolated from unfolded β‐galactosidase, is an example of this type of degron.12 Lon also degrades multiple other classes of substrates. In many of these instances, Lon recognizes folded proteins through unstructured peptide degrons displayed on the protein surface, such as in an exposed loop or near the termini. For example, Lon specifically recognizes the last 20 amino acids of the cell division inhibitor SulA and also the first 21 residues of the superoxide response regulator SoxS.5, 13 In contrast, Lon can also bind certain folded protein domains lacking any known degron peptide; this class of substrate is represented by the α‐crystallin domains of the small heat shock proteins (sHSPs, IbpA, and IbpB in bacteria).4 Thus, the pool of Lon substrates and the mechanisms of recognition for degradation are both important and diverse.
Each Lon monomer contains three functional sub‐regions: the N domain, AAA+ ATPase module, and a protease domain. The ATPase and protease domains are the most well‐conserved regions of Lon. Interestingly, the ~300 residue N domain is the most variable region in primary sequence among Lon homologs, and it is also the least well understood in function.14 Multiple lines of evidence indicate a role of the N domain in mediating recognition and processing of certain Lon substrates.15, 16, 17, 18 However, the precise binding interface and molecular interactions that underlie recognition of most Lon substrates remain unknown. Furthermore, deletion of portions of the N domain can alter or ablate both ATPase and protease activity, indicating the N domain also plays a critical role in assembly and/or control of the entire machine.14, 19 More recently, we discovered an additional role for the Lon N domain in the Escherichia coli enzyme: to mediate the higher‐order assembly of a Lon dodecamer.20
In this previous study, we showed that Lon homo‐hexamers associate in a head‐to‐head fashion to form a large, ellipsoid‐shaped dodecamer based on an electron microscopy reconstruction.20 Modeling of the individual structural regions of Lon strongly suggested that this association is mediated by intermolecular interactions between N domains. Protein association constants and in vivo concentration measurements indicate that the ratio of Lon dodecamer to hexamer is likely close to equal during balanced growth and increases when lon gene expression is induced under stress (e.g. heat shock).20 In vitro analysis reveals that increasing dodecamer population correlates with approximately 10‐fold lower ATPase activity as well as slower degradation of certain substrates.20 Other reports identified multiple binding partners that modulate Lon activity in various ways, including DNA, inorganic polyphosphate, and protein adaptors.21, 22, 23, 24 Therefore, Lon binding to itself via dodecamer formation may represent an additional mechanism by which intermolecular interactions control Lon activity or tune substrate preference in addition to these other well‐studied binding effectors. In light of these findings, we designed experiments to better understand the structural basis of dodecamer formation and possible biological roles of the Lon dodecamer during different, potentially stressful, growth conditions that require pQC.
Here, we report mutagenesis and chemical crosslinking studies that identify a precise segment of the Lon N domain in mediating dodecamer assembly. Importantly, we find a Lon variant carrying mutations in this region that preferentially forms dodecamers. The Lon V217A/Q220A (LonVQ) variant, in which residues Val217 and Gln220 that are present in a region predicted to form intermolecular coiled coils between hexamers were mutated to alanine, showed an increased propensity to form dodecamers both in solution and by EM analysis. This LonVQ variant was analyzed biochemically as well as expressed in vivo. Our results indicate that the dodecamer is active, although it exhibits alterations in substrate selection and/or degradation. Similar changes in substrate choice are observed both in vivo and in vitro, strongly suggesting that LonVQ exists largely in dodecamer form under standard growth conditions. Cells expressing only LonVQ are healthier than Lon‐deficient strains during normal growth and perform similarly to wild‐type Lon in a panel of in vivo bioassays except for degradation of small heat shock proteins. Thus, we conclude that the dodecamer successfully completes many of the Lon protease's important regulatory functions while modifying substrate choice, perhaps to better manage protein quality control under conditions such as UV, heat, and oxidative stress.
Results
Identification of N domain interactions underlying Lon dodecamer formation
Our previous work identified that Lon forms a dodecamer and suggested that intermolecular N domain interactions were likely responsible. This model of Lon dodecamer assembly was constructed using low‐resolution transmission electron microscopy (EM) image analysis.20 The resulting ellipsoidal electron density map was sufficient to model two barrel‐shaped hexamers at the distal ends of the dodecamer corresponding to the Lon ATPase and protease modules [Fig. 1(a)]. The two barrels were then bridged by six extended helical structures, which were modeled as six N domain dimers forming end‐to‐end interactions that mimic two‐stranded, antiparallel coiled coils [Fig. 1(a)]. Although the model informs that the Lon N domains are primarily responsible for dodecamer formation, the specific intermolecular interactions necessary to stabilize this large assembly could not be obtained due to the low resolution of the EM map (~35–40 Å).20 Therefore, to identify the specific residue contacts that underlie dodecamer assembly, we carried out bioinformatics analysis followed by scanning mutagenesis and crosslinking studies targeted at residues predicted to participate in N domain interactions.
Figure 1.
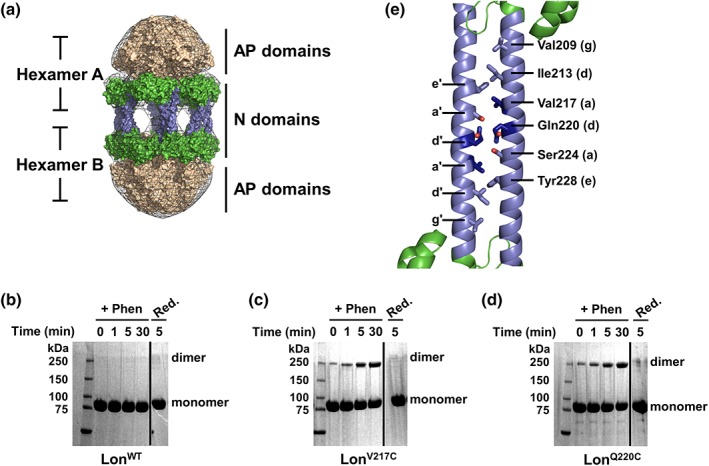
The Lon dodecamer forms via putative N domain coiled‐coil interactions. (a) Low‐resolution electron microscopy 3D reconstruction presents a model for dodecamer formation via the association of Lon N domains through antiparallel coiled coils. Lon N domain dimers are in green with the predicted coiled‐coil region colored blue, the hexameric ATPase and protease domains are colored tan, and the electron density map is shown as a gray mesh. Details for model reconstruction were described previously20. (b–d) Copper phenanthroline catalyzed crosslinking of LonWT and Lon variants analyzed by SDS‐PAGE. Lon proteins (20 μM) were incubated with catalyst (+Phen) for the indicated times, then reduced with 10 mM β‐mercaptoethanol and 5 mM DTT (Red.). LonWT (b) shows no crosslinking in the presence of phenanthroline whereas LonV217C (c) and LonQ220C (d) both show a time‐dependent increase in crosslinking with oxidizing agent. All crosslinking was abolished by addition of reducing agent. Solid line indicates a portion of the gel that was removed for clarity. (e) Bioinformatics analysis was used to create a theoretical model of the Lon N domain antiparallel coiled‐coil interactions (cartoon representation, colored as in (a). The residues chosen for mutagenesis (shown in stick representation) were mapped on the E. coli Lon N domain structure (PDB 3LJC). Two Lon N domain monomers were manually modeled in an antiparallel conformation to mimic the potential interaction surface and residues Val217 and Gln220 are highlighted in dark blue. The lower case letters denote the coiled‐coil heptad position as displayed in Fig. S1.
Inspection of the isolated E. coli Lon N domain crystal structure reveals an extended α‐helix (Residues 189–242), which contains more than one region with a high probability of forming coiled‐coil interactions based on primary sequence analysis.25, 26 Using the Lon EM dodecamer model, we constrained our bioinformatics predictions to be dimeric, antiparallel, coiled coils. Candidate residues for subsequent cysteine mutagenesis and crosslinking were identified from several models predicted using Drawcoil 1.0, including Val209, Ile213, Val217, Gln220, Ser224, and Tyr228 (Fig. S1).27 We purified Lon variants in the presence of reducing agent, then rapidly diluted into Lon reaction buffer containing ATPγS (a non‐hydrolyzable ATP analog) and the oxidizing catalyst copper phenanthroline, and monitored crosslinking over time by SDS‐PAGE. In the presence of the oxidizing catalyst, wild‐type Lon (LonWT), which has six cysteine residues but none located in the predicted coiled‐coil region, showed no disulfide‐based crosslinking [Fig. 1(b)]. In contrast, we identified two cysteine variants from the six candidates, Val217C (LonV217C) and Gln220C (LonQ220C), that reproducibly yielded fast and robust intermolecular disulfide crosslinking [Fig. 1(c,d)]. Dimer formation increased over time, suggesting these two residues are likely participating in the antiparallel coiled‐coil interface between two Lon hexamers. Additionally, low levels of oligomerization were detected for both variants even without an oxidizing catalyst, indicating that the absence of a reducing agent alone is sufficient for spontaneous disulfide‐bond formation. Finally, treatment of the oxidized samples with reducing agent resulted in a loss of the higher molecular weight species in SDS‐PAGE, confirming that the cysteine‐based disulfide crosslinking was responsible for the formation of the SDS‐resistant dimers. A model of an antiparallel coiled coil that is consistent with our results and the corresponding bioinformatics helical wheel diagram are shown in Figures 1(e) and S1.
Mutation of Val217 and Gln220 shifts Lon population toward dodecamer
To further probe the role of residues Val217 and Gln220 in dodecamer formation and stabilization, both residues were mutated to alanines, and the resulting variant (Lon V217A/Q220A, henceforth LonVQ) was characterized biophysically using analytical ultracentrifugation (AUC). Our previous study showed LonWT sediments as a mixture of oligomer assemblies, and the percentage of dodecamer in the population increases with concentration.20 Due to the interconversion between hexamer and dodecamer, all future references to Lon concentration will be in monomer equivalents. Both LonWT and LonVQ were assayed using sedimentation velocity AUC at multiple concentrations in the presence of ATPγS (Fig. 2). The previously calculated dissociation constant between dodecamer and hexamer for LonWT was approximately 1–3 μM (monomer equivalents). Interestingly, LonVQ also sediments as a mixture of hexamer and dodecamer, however, this variant exhibited a higher percentage of dodecamer populated compared with LonWT at all concentrations tested. Importantly, at lower concentrations (e.g. 3 and 1.5 μM) where LonWT is presumed to be predominately hexamer, LonVQ is still predominately dodecamer and the relative amount of dodecamer increases with increasing concentration. At the concentration where LonWT approximately equally populated the hexamer and dodecamer, LonVQ was calculated to be over 75% dodecamer. Furthermore, at the highest concentration assayed, LonVQ was more than 90% dodecamer. Using these data, the LonVQ variant was calculated to have a dissociation constant of ~0.5 ± 0.1 μM, about 5‐fold tighter than that of LonWT (see Materials and Methods). Therefore, the LonVQ variant exists predominately as a dodecamer in solution, even at concentrations where LonWT exists mostly as a hexamer.
Figure 2.
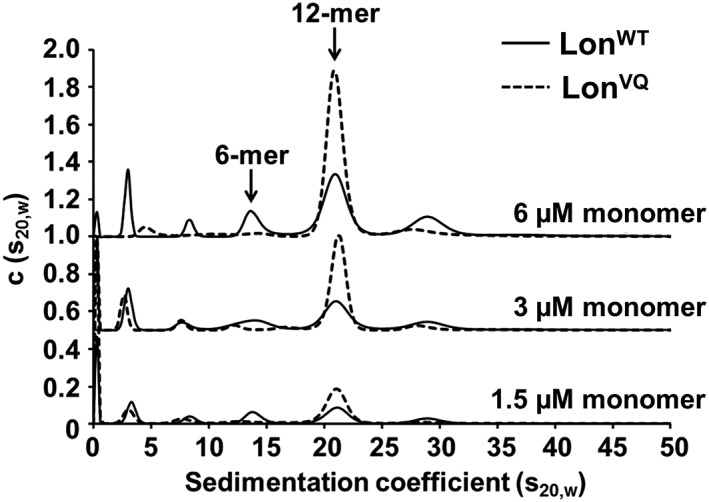
LonVQ preferentially forms dodecamers in solution. Sedimentation velocity analytical ultracentrifugation of LonWT (solid line) and LonVQ (dashed line). Sedimentation analysis shows the LonVQ variant forms a higher percentage of dodecamers in solution. The population of dodecamer increases with concentration for both Lon variants; however, at each concentration shown, there is a higher dodecamer population for LonVQ compared with LonWT. The c(s20,w) values for each concentration series were offset by a standard value for clarity.
Next, we analyzed the oligomeric propensity of the LonVQ variant using EM (Fig. 3). Both LonWT and LonVQ samples were diluted to the same starting concentration, which populates primarily dodecamers for both variants (11 μM monomer). The samples were incubated at 37°C with ATP and MgCl2, then the reaction quenched with ATPγS. Samples were diluted to 340 nM, immediately immobilized on grids, and stained with uranyl acetate before imaging. Individual hexamer and dodecamer particles were chosen over multiple images and quantified. LonWT EM images displayed approximately 43% hexamer and 57% dodecamer particles. Under the same experimental conditions and protein concentrations, the LonVQ particles were approximately 21% hexamer and 79% dodecamer. Although this procedure is not a true equilibrium experiment as time of dilution, the grid surface, and a capacity of being able to more easily identify some types of particles than others could all influence the quantification. Nonetheless, this EM characterization confirms the crosslinking experiments presented above in identifying Lon residues Val217 and Gln220 as being involved in dodecamer stabilization.
Figure 3.

Electron microscopy analysis and quantification of LonVQ dodecamer formation. Negative stain electron micrographs of LonWT (left) and LonVQ (right). Under the same experimental conditions and upon immobilization on the grids, LonVQ forms a higher percentage of dodecamers. Representative dodecamer and hexamer particles are denoted with white and black arrows, respectively. Inset left: representative class images of dodecamers (top) and hexamers (bottom) based on the LonWT negative stain particles.20 Inset right: zoomed in view of raw LonVQ particles. The three particles on the left in white boxes are dodecamers and the particle on the right in the black box is a representative hexamer. Scale bar shown represents 100 nm.
LonVQ displays biochemical activities comparable to the LonWT dodecamer
Previous studies revealed that Lon's intrinsic ATPase activity (not stimulated by protein substrate) decreases with increasing protein concentration and the coincident dodecamer formation.20 Based on these basal ATPase activities, the Lon dodecamer hydrolyzes ATP approximately 10‐fold slower than the hexamer. We assayed ATPase activity for LonVQ at concentrations in which LonWT was either predominately hexamer or predominately dodecamer (each with the same number of monomer equivalents) [Fig. 4(a)]. At lower concentrations where LonWT is mostly hexamer (0.75 μM), LonVQ displayed an approximately 1.5‐fold slower basal ATPase activity. As the concentration of Lon increases, so does the amount of dodecamer present in the sample. At 12 μM Lon monomer, where the majority of LonWT is predicted to be dodecamer, LonVQ had nearly identical ATPase activity compared with LonWT. Notably, at monomer concentrations at or below the dissociation constant for LonWT, LonVQ had depressed ATPase activity compared with LonWT. These activity assays thus confirm our previous biophysical studies that reveal that LonVQ has a tighter hexamer–dodecamer equilibrium. We also assayed substrate‐stimulated ATPase rates using an unfolded substrate with a Lon‐specific degron (Fig. S2). While both LonWT and LonVQ displayed higher ATPase activity in the presence of substrate, there was still a concentration‐dependence in activity, indicating that substrate‐binding does not lead to dodecamer dissociation.
Figure 4.
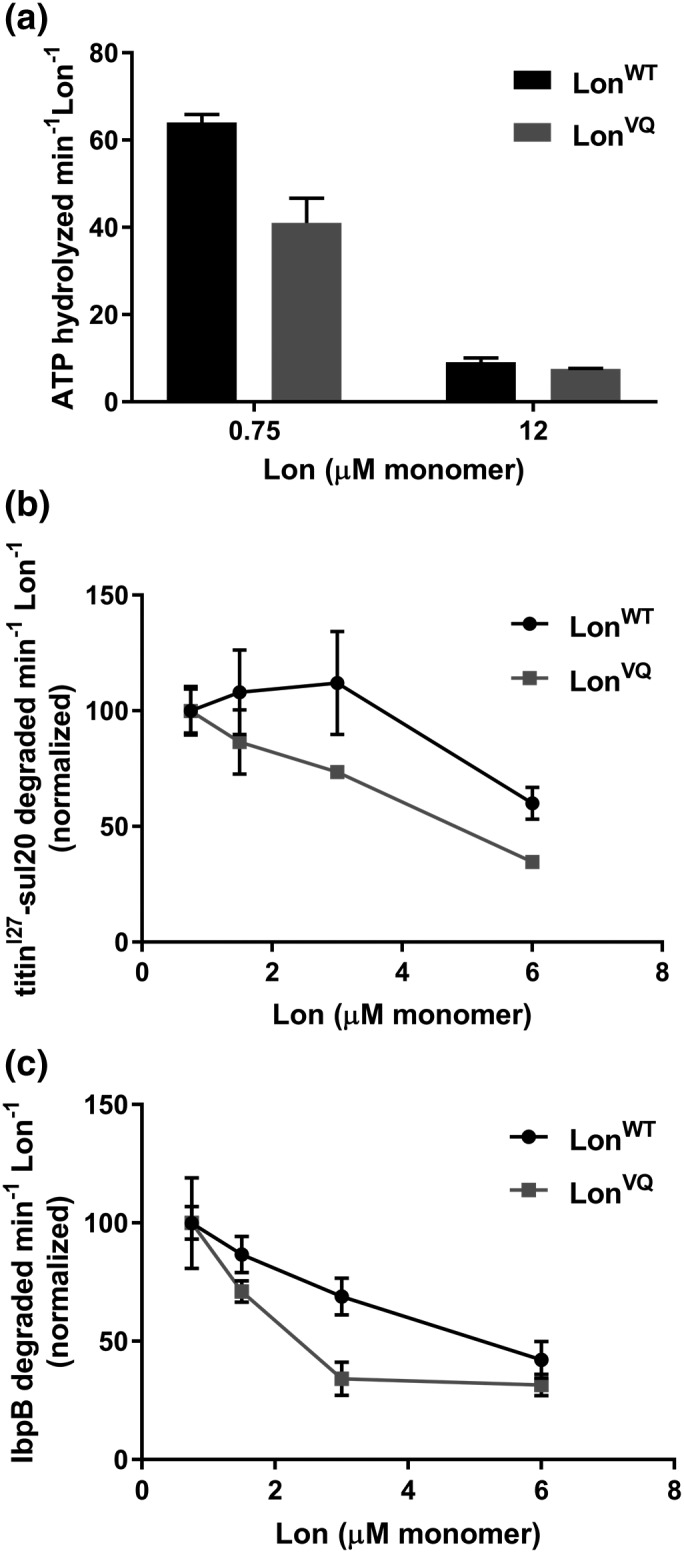
LonVQ displays diminished ATPase and degradation activity compared with LonWT. (a) ATP hydrolysis activity of LonWT (dark gray bars) and LonVQ (light gray bars). At lower concentrations where LonWT is predominately hexameric, there is approximately 1.5‐fold higher activity for LonWT compared with LonVQ. However, at higher concentrations where both proteins are expected to be predominantly dodecamer, the ATP hydrolysis activity is comparable. The values reported are from three independent experiments performed in triplicate, and the error bars represent SEM. (b and c) Degradation of 40 μM 35S‐titinI27‐sul20 (b) and 35S‐IbpB (c) by LonWT and LonVQ was monitored by generation of acid‐soluble radioactive peptides. The indicated Lon concentrations are in monomer equivalents. Degradation rates were normalized in comparison to the rate at 0.75 μM. Each experiment was performed in triplicate and error bars represent SEM (curved lines connecting data points do not represent statistically‐based fitting).
We also assayed in vitro degradation of two classes of radiolabeled model Lon substrates by monitoring generation of acid‐soluble peptides with time over a range of LonWT or LonVQ concentrations that would contain different percentages of dodecamer. First, we monitored the degradation of SulA as a representative of small folded substrates. SulA is induced during the SOS response to DNA‐damage to allow for DNA repair prior to cell division. After stress, Lon specifically degrades SulA via recognition of a short unstructured C‐terminal tail (sul20, residues 150–169) with a K m of approximately 40 μM.5, 12, 28 We appended the SulA degron onto the C‐terminus of the I27 domain of human titin (titinI27‐sul20, approximately 14 kDa). Increasing concentrations of LonVQ caused a larger decrease in proteolytic activity compared with LonWT, and at the highest concentration assayed, the LonVQ degradation rate was significantly (1.8‐fold) slower than LonWT [Fig. 4(b)]. We also assayed processing of the sHSP IbpB, which readily forms oligomers (tetramers and greater) under our experimental conditions, making it a larger substrate than the titinI27 domain. LonWT recognizes IbpB via its folded α‐crystallin domain with a K m of 16 μM.4 LonVQ was able to degrade the oligomeric IbpB in vitro, however at a much slower rate than LonWT [Fig. 4(c)]. Again, increasing LonVQ concentration had a greater effect on diminishing degradation activity for IbpB compared with LonWT, similar to the processing of the titinI27‐sul20 substrate. The concentration range over which LonVQ showed slowed degradation was significantly lower than was observed with LonWT. Thus, as with the intrinsic ATPase rates, protein degradation rates, especially of IbpB, appear to serve as a surrogate for measuring the amount of the Lon dodecamer population at each concentration. These observations support the conclusion that LonVQ hexamers and dodecamers behave similarly to LonWT except for exhibiting a shifted hexamer–dodecamer equilibrium constant to a lower (tighter) value.
LonVQ is altered in recognition of dodecamer‐sensitive substrates in vivo
We established in vitro that a hallmark of the Lon dodecamer is a diminished rate of IbpB degradation (this work and Refs. [20]). Therefore, using this phenotype as a measure of dodecamer population, we sought to assay the assembly and activity of LonVQ in vivo by monitoring the interaction of Lon with IbpA, a paralog IbpB and also a substrate of Lon,4 after heat shock. IbpA is induced to help prevent toxic aggregation of unfolded proteins during heat stress.29, 30 Lon recognizes the folded α‐crystallin domain of IbpA leading to degradation of IbpA after heat stress, allowing for release of unfolded client proteins so they are available for refolding by folding chaperones.4 While both IbpA and IbpB are Lon substrates, we were constrained to follow only IbpA degradation in vivo due to the poor specificity of our IbpB antibodies. Although IbpA monomers are small (~16 kDa), they form large oligomers under heat stress, similar to IbpB.31, 32 Escherichia coli cultures expressing LonWT, LonVQ (integrated into the normal chromosomal locus) or with the endogenous lon gene deleted and replaced with a kanamycin‐resistant cassette (Δlon) were grown at 37°C and then stressed by shifting the cultures to 45°C. Cellular levels of IbpA were monitored at the indicated time points via Western blot (Fig. 5). LonWT degrades IbpA slowly over time under heat stress (t 1/2~ 30 min) with approximately 70% of the initial protein lost 45 min post heat shock. Conversely, IbpA degradation is severely reduced when LonVQ was expressed, with less than 15% of total IbpA degraded after the same time period. As a control that IbpA is normally a Lon substrate under these conditions, we observed that IbpA was stable throughout the time course when endogenous lon was deleted. In our previous report, we identified that cellular levels of LonWT increase ~1.2‐fold under heat stress.20 However, under these conditions, there is still approximately 45% LonWT hexamer available to degrade IbpA. With the LonVQ variant, because it is largely dodecamer both at 37°C and 45°C, we would expect an intermediate phenotype between LonWT (a mixture of hexamers and dodecamers) and Δlon strains, which is indeed what we found. Thus, we suggest that the increased dodecamer population in LonVQ disrupts the ability of Lon to degrade IbpA in vivo. This degradation pattern by LonVQ, taken together with our in vitro analysis demonstrating that a major signature of the Lon dodecamer is its slow degradation of IbpB [Fig. 4(c)], suggests that LonVQ is largely in the dodecamer form under these growth conditions in vivo, whereas LonWT has a comparatively higher hexamer population.
Figure 5.
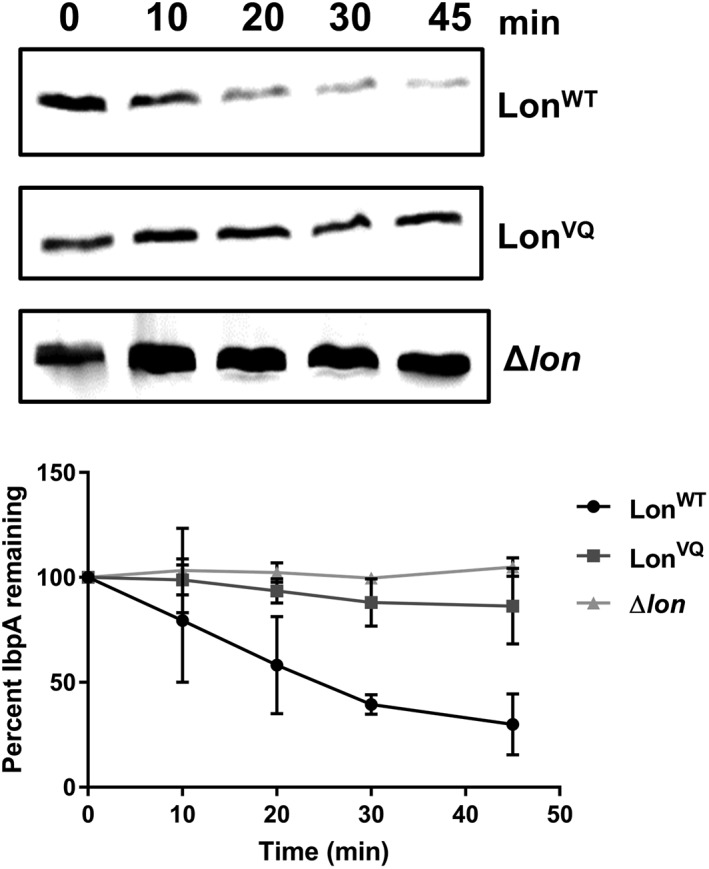
In vivo degradation of IbpA by Lon upon heat shock. Representative Western blots of E. coli cell extracts during heat shock (45°C) monitoring loss of the IbpA protein over time. Bottom. Protein bands were quantified and the percentage of IbpA protein remaining when LonWT (circles), LonVQ (squares), or Δlon (triangles) was expressed is plotted as a function of time. The experiment was performed in triplicate and error bars represent SEM (lines connecting data points are not indicative of statistically based fitting).
LonVQ is active in vivo, indicating dodecamers are a functional form of Lon
Having developed combined in vitro and in vivo evidence that LonVQ is largely behaving as a dodecamer, we went on to investigate possible in vivo function(s) of the dodecamer. We started with bioassays for classically‐studied substrates. First, we monitored Lon degradation of RcsA, a 24 kDa transcription factor, which normally suppresses colonic acid exopolysaccharide production.33 Using the same set of isogenic lon strains, we assayed growth on minimal media agar plates [Fig. 6(a)]. After 4 days, there was robust overproduction of colonic acid in the Δlon strain, evidenced by mucoid colonies, indicating this strain is deficient in RcsA degradation. However, both the LonWT‐ and LonVQ‐expressing strains were non‐mucoid, indicating that the Lon dodecamer is competent to degrade RcsA in vivo and thus properly regulate exopolysaccharide production.
Figure 6.
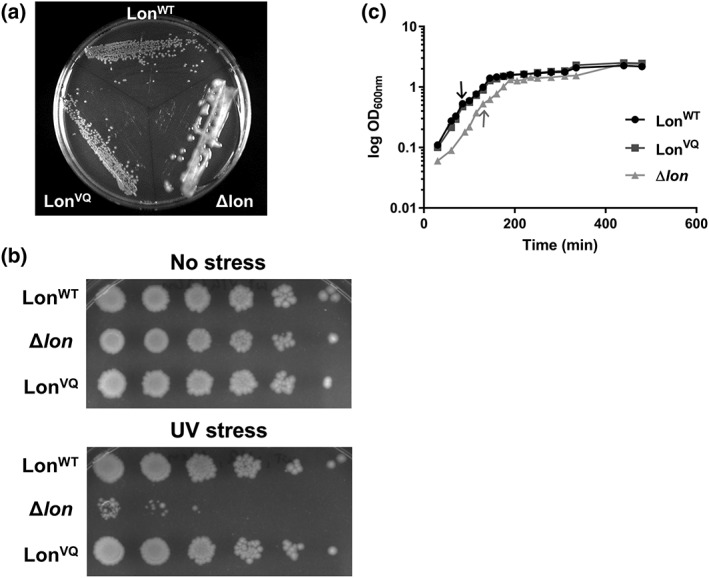
LonVQ is functional in vivo. (a) W3110 E. coli cells expressing either LonWT or LonVQ from the endogenous lon locus or with the genomic lon copy deleted and replaced with a kanamycin‐resistant cassette (Δlon) were grown in culture then plated on M9 minimal media agar plates. Lon degradation of RcsA reduces colonic acid synthesis. Stabilization of RcsA in Δlon strain results in a mucoid phenotype. (b) In vivo bioassay monitoring inactivation of the cell division inhibitor protein SulA by Lon after UV stress. LonWT is able to inactivate SulA which allows for cell division and growth after UV irradiation (top row). However, when Lon is deleted (middle row), cells are unable to recover from UV damage. Expression of LonVQ from the endogenous locus is also able to allow for cell growth recovery after UV stress similar to LonWT. Each column, from left to right, represents a 10‐fold serial dilution. (c) Growth curves of isogenic lon W3110 strains. The Δlon strain grew significantly slower than LonWT and LonVQ (student's unpaired t test, P > 0.0005 and 0.005 for LonWT and LonVQ, respectively) at 37°C; however, all strains had similar growth rates at 45°C (temperature shift indicated by black arrow for LonWT and LonVQ or gray arrow for Δlon). Each semi‐log plot is the average of two independent cultures and error bars represent SEM.
We next assayed in vivo inactivation of the cell division inhibitor SulA (19 kDa). DNA damage was introduced by a pulse of UV irradiation and then surviving cells were allowed to grow overnight on LB agar plates [Fig. 6(b)]. In the case of the Δlon strain, cells had little or no ability to form colonies after irradiation. However, strains expressing either LonWT or LonVQ were able to rescue the growth‐arrest phenotype induced by DNA damage. This assay suggests that the LonVQ dodecamer is capable of interacting with SulA in vivo in a manner leading to timely SulA inactivation and resumption of cell division. It should be noted that this assay may report on the binding of Lon to SulA, and not necessarily SulA degradation, as a catalytically inactive Lon variant in which the active‐site serine is mutated to alanine (LonS679A) is also able to suppress the growth inhibition caused by SulA.18
Finally, we monitored the growth of these strains during heat stress [Fig. 6(c)]. Cultures were grown in exponential phase to an OD600 of 0.3, then transferred to 45°C and OD600 measurements taken at the indicated intervals. At 37°C, the Δlon strain grew significantly slower than the other two strains and never established a true exponential phase [Fig. 6(c)]. In contrast, there was no appreciable difference between the growth rates of the lon WT and lon VQ strains, indicating that Lon dodecamers can perform the functions needed for wild‐type growth efficiency at normal temperatures. Interestingly, however, after increasing temperature, the growth rates for all three strains were slower but their doubling times were similar within experimental error (~43 min). These data suggest that immediately after shifting to high temperature, loss of Lon activity is not deleterious to viability. Therefore, the lower activity of Lon dodecamers may be advantageous, as its formation prevents degradation of certain substrates, such as IbpA, IbpB, and perhaps others. A switch to low temperature and recovery from heat stress would then require the activity of the Lon hexamer to degrade these regulatory proteins that are no longer needed.
Discussion
Lon protease plays a major role in maintaining pQC in bacteria as the primary protease responsible for degrading unfolded or damaged proteins that result from various cellular stresses. Previous studies characterized Lon to function as a homo‐hexamer with the proteolytic active sites sequestered in a cavity that is accessed through a small central pore. More recently, our group found that Lon hexamers associate in a head‐to‐head orientation to form a larger ellipsoidal‐shaped dodecamer. Using the available crystal structures of individual Lon domains25, 34 together with bioinformatics, we proposed and then tested if this association was mediated by antiparallel coiled coils between opposing Lon N domains, canonical interaction motifs that underlie oligomerization of many proteins.35 In vitro we demonstrated that the dodecamer is increasingly populated with increasing Lon concentration. Based on the determined K D for assembly and the cellular Lon concentration, we predicted that Lon exists in equilibrium between the hexamer and dodecamer forms in wild‐type cells in vivo.20 Importantly, the Lon dodecamer displays decreased ATPase activity as well as differences in substrate processing in vitro compared with the Lon hexamer. As such, we sought to further characterize Lon to advance understanding of the dodecamer form and uncover possible biological functions of this assembly state during pQC and stress responses.
The major impediment to elucidating the biological function of the Lon dodecamer is the inability to isolate a homogeneous population of this assembly using wild‐type protein. Further limiting experimental approaches, the precise molecular contacts required for dodecamer formation were unknown. Therefore, we employed computation‐directed mutagenesis, protein–protein crosslinking, and biochemical analysis to identify a variant of Lon that prefers the dodecameric state. We identified that Lon N domain residues Val217 and Gln220 are featured at the oligomerization interface based on the results of cysteine mutagenesis and disulfide‐based crosslinking. Interestingly, when both residues are mutated to alanine, presumably removing wild‐type hydrophobic or polar interactions between the antiparallel coils, the dodecamer is stabilized compared with that of LonWT. These mutations result in an increased percentage of dodecamer at physiological protein concentrations, as observed by analytical ultracentrifugation. This increased propensity to form dodecamers was confirmed using electron microscopy. We hypothesize the alanine mutations may have disrupted the natural balance of intermolecular interactions that allow for rapid interconversion between the hexameric and dodecameric forms in favor of a tighter dodecamer interface. As predicted from its increased dodecamer stability, LonVQ is less active at low concentrations compared with LonWT, an additional indicator of a larger dodecamer population. Importantly, based on the hexamer–dodecamer K d of 0.5 μM for LonVQ, this newly identified variant is predicted to exist as majority dodecamer in vivo, as the intracellular Lon concentration was determined to be approximately 2.5 μM monomer.20
It should be noted that although the LonVQ variant is largely dodecamer at all concentrations assayed in vitro, these samples are not homogeneous, as a small fraction of hexamers remain. This mixture is evidenced by the concentration dependence of the ATPase activity and in vitro substrate degradation rates, a behavior that was also seen for LonWT, albeit over a higher concentration range. Additionally, quantification of EM particles reports approximately 20% of the total LonVQ population is hexameric after immobilization on grids. Nevertheless, the combination of these mutations yielded a Lon variant with an approximate 5‐fold tighter dissociation constant for dodecamer formation compared with LonWT. Therefore, we have improved our understanding of how the dodecamer is formed by identifying (i) the N domain antiparallel coiled‐coil register, (ii) a critical part of the intermolecular interaction interface that mediates dodecamer assembly as well as (iii) generating a molecular tool to more clearly test the activities and in vivo functions of the dodecamer.
A previous study identified a separate mutation in the Lon N domain that also yields a Lon population that is primarily dodecamer.17 It was reported that the Glu240 to Lys mutation (LonE240K) results in a Lon dodecamer dissociation constant of 2.6 nM, an approximately 1000‐times tighter interaction than that of LonWT.17 Residue 240 is located in the second of the two predicted coiled‐coil regions in the Lon N domain (coiled‐coil Region 1 is comprised of Residues 185–228 and Region 2 is composed of Residues 230–278). We chose to focus on mutations solely in the first predicted coiled‐coil region for multiple reasons. First, the LonE240K variant has altered substrate specificity as evidenced by the finding that the recognition and the degradation of the RcsA substrate by LonE240K are diminished while SulA substrate recognition is unaffected.15 Also, biochemical evidence suggested the LonE240K dodecamer might be allosterically regulated in a different manner compared with the LonWT dodecamer. For example, LonE240K dodecamers degraded the model Lon substrate casein nearly as efficiently as the LonWT hexamer, whereas dodecamers formed by LonWT or LonVQ subunits behave similarly and degrade this substrate very slowly.17 Finally, other reports identified that the N domain region containing Residues 240–252 is important for proper ATPase activity, substrate translocation, and degradation.19 Therefore, we chose to focus this study on N domain residues upstream of this allosteric regulatory region in order to minimize potential confounding effects that could interfere with the unambiguous elucidation of dodecamer function.
Among Lon homologs, the N domain is the most variable region, compared with the ATPase and protease domains. Despite lower sequence homology, dimerization of N domains via coiled‐coil interactions seems to be a conserved feature among Lon homologs.15, 34, 36, 37 However, there may be additional regions other than the segment probed in this study that could mediate antiparallel coiled‐coil formation.15, 36 Interestingly, E. coli Val217 is conserved among proteobacteria whereas Gln220 is slightly more variable. Therefore, it will be worthwhile to determine if other Lon homologs also form larger assemblies. If so, dodecamer formation may be a conserved mechanism to regulate Lon protease function. Based on experiments with Bacillus subtilis Lon, it was suggested this homolog also forms larger assemblies in solution as the N domains dimerize via coiled‐coil interaction.34 Preliminary experiments in our lab also detected a dodecamer‐like species with this full‐length protein.
There is increasing evidence to support the theory that coiled‐coil interactions may be a conserved feature of several AAA+ pQC enzymes. Although there is likely some diversity in the orientation of these interactions, it seems higher‐order assembly is a mechanism for modulating activity. For instance, the coiled‐coil middle domains (MD) of the ClpB disaggregase interact in a head‐to‐tail orientation, leading to suppression of ATPase activity.38, 39 Disruption of these interactions by binding of the DnaK co‐chaperone, in turn, alleviates the suppression, allowing for normal ClpB activity.40 Another AAA+ chaperone ClpC was recently shown to also oligomerize via coiled‐coil MD interactions, but in this instance in a head‐to‐head orientation, as we find for E. coli Lon.41 Binding of the MecA adaptor to the MD results in conformational changes that break the coiled‐coil interactions leading to stabilization of active ClpC hexamers. The differentiating feature of the Lon dodecamer is that it does not appear to be an inactive “storage form” of Lon preventing degradation of substrates, and we did not identify substrate interactions that altered assembly. For example, previous negative stain EM micrographs, taken in the presence of a SulA degron peptide, still contained a significant dodecamer population.20 Also, the LonVQ dodecamer variant is sufficiently active in a series of in vivo bioassays, including suppression of extracellular capsular polysaccharide production and re‐establishing cell division during recovery from DNA damage. Rather, our results suggest that the Lon dodecamer may be unable to degrade larger substrates like the oligomers formed by the Ibps,42 but still able to degrade smaller (<25 kDa) substrates relatively well, such as RcsA and SulA (this work) as well as small degron‐tagged model proteins.20 The Lon dodecamer's inability to degrade IbpA during growth at high temperature in vivo may be important to allow efficient IbpA binding to aggregating client proteins during heat shock. Then, once cells return to a lower stress environment, the Ibp–client complexes are likely preferential substrates for disaggregation and refolding. Thus, unlike ClpB or ClpC, Lon dodecamer formation may be a way to tune, but not preclude, substrate recognition and subsequent degradation in the presence of cellular stress. Because Lon has such a large number of substrates that must be degraded under specialized conditions, such tuning rather than inactivation may be advantageous to cells.
Several questions about the role of Lon dodecamer formation in vivo remain to be addressed. For instance, could certain substrate and small molecule binding sites located on the N domain become occluded upon dodecamer assembly? Also, could the altered “resting” ATPase activity of the dodecamer play a role in slowing or preventing substrate processing during cellular stress when certain Lon substrates are needed for their anti‐aggregation function? These questions can now be dissected using this LonVQ dodecamer‐favoring variant using a combined in vivo and in vitro approach. Commensurate with our understanding of the biological significance of the Lon dodecamer, the next step in understanding Lon function in vivo is to develop variants of Lon that form obligate hexamers to compare to our current data. However, this endeavor may prove to be precarious, especially if dodecamer formation is a key mechanism for controlling deleterious proteolysis of critical Lon substrates in response to specific environmental conditions.
Materials and Methods
Protein expression and purification
Wild‐type Lon and Lon N domain variants were expressed and purified as previously described.20 Briefly, Lon was cloned into the pBAD33 vector and all Lon variants were produced with the QuikChange site‐directed mutagenesis protocol (Agilent) using specific primers containing the desired mutations. Proteins were grown in TB media at 37°C and expression induced with 0.2% l‐arabinose for 3–4 h followed by centrifugation to collect cell pellets. For purification, cells were resuspended in phosphate binding buffer (100 mM potassium phosphate, pH 6.5, 10% glycerol, 1 mM DTT, 1 mM EDTA) and lysed via French Press. After incubation with benzonase (25U), the lysate was clarified by centrifugation and the supernatant incubated with phosphocellulose resin (Whatman). The bound protein was eluted with 400 mM potassium phosphate, concentrated and subjected to gel filtration in high salt buffer (GE Superose 6 10/300, 25 mM Hepes pH 7.5, 2 M NaCl, 10% glycerol, 1 mM DTT, 1 mM EDTA). The appropriate protein fractions were pooled and concentrated while exchanging into Lon Storage Buffer (50 mM Hepes pH 7.5, 150 mM NaCl, 10% glycerol, 0.1 mM TCEP, 1 mM EDTA) then frozen in liquid nitrogen and stored at −80°C. All Lon concentrations are expressed in monomer equivalents using the extinction coefficient of 46,300 M−1 cm−1 at 280 nm. 35S‐labeled substrates were expressed and purified as previously described.4
Disulfide crosslinking
The Lon cysteine variants were exchanged into pre‐crosslinking buffer (50 mM Hepes, pH 7.6, 150 mM NaCl, 10% glycerol, 0.1 mM TCEP) using a PD‐10 desalting column (GE Healthcare) and then concentrated (Amicon). Before starting the reaction, all proteins were diluted (approximately 1.3–3.2‐fold) to 200 μM in the pre‐crosslinking buffer. The reaction began by diluting Lon to 20 μM in crosslinking buffer (50 mM Hepes, pH 7.6, 150 mM NaCl, 10% glycerol) along with 2.5 mM ATPγS, 5 mM MgCl2, and crosslinking catalyst containing 5 μM cupric sulfate and either 10 μM phenanthroline or 1% DMSO. All reactions were carried out at 22°C and were quenched with 10 mM EDTA at the indicated time points. After 30 min, the samples were treated with 5 mM DTT and 10 mM β‐mercaptoethanol and incubated at 95°C for 5 min to reduce disulfide bonds formed during the oxidation reaction. Samples were mixed with SDS‐PAGE loading dye without reducing agent and efficiency of crosslinking analyzed by electrophoresis on a 4–15% acrylamide gel (Bio‐Rad).
Negative stain electron microscopy
Lon protein samples were first diluted to 1 mg/mL in EM buffer (50 mM Hepes pH 7.5, 150 mM NaCl, 0.1 mM TCEP, 0.01 mM EDTA) and filtered with a 0.1 μM inorganic filter (Whatman). Prior to setting up grids, Lon was incubated with ATP and MgCl2 at 37°C. After 5 min, the reaction was quenched with ATPγS. Lon was diluted 0.03 mg/mL in dilution buffer (50 mM Hepes pH 7.5, 150 mM NaCl, 0.1 mM TCEP, 0.01 mM EDTA, 10 mM MgCl2, 5 mM ATPγS) immediately prior to spotting on grid. The diluted Lon samples were immobilized on copper mesh grids (Pacific Grid‐Tech) washed with dilution buffer and the grids were subsequently stained with 2% uranyl acetate. Images were obtained with the FEI Technai Spirit Transmission Electron Microscope. For quantification of hexamer versus dodecamer population, a total of 233 and 193 particles were chosen for LonWT and LonVQ, respectively.
Analytical ultracentrifugation
LonWT and LonVQ protein samples were exchanged into AUC buffer (50 mM Hepes pH 7.5, 150 mM NaCl, 0.1 mM TCEP, 0.01 mM EDTA) via overnight dialysis. Both proteins were filtered and diluted to the indicated concentrations using dialysis buffer and each sample, along with the corresponding buffer reference, was supplemented with 1 mM MgCl2 and 0.1 mM ATPγS. Sedimentation velocity centrifugation using interference optics was performed with the Beckman Optima XL‐1 analytical ultracentrifuge at 20°C and 20,000 rpm. Data were analyzed with Sedfit to calculate the continuous distribution of sedimentation coefficients from 1S to 60S at a resolution of 200 scans per concentration with a confidence level (F‐ratio) of 0.95.43 Calculations were performed using a density of 1.00831, a viscosity of 0.010475, and a Lon partial specific volume of 0.7431 as determined by SEDNTERP (Biomolecular Interaction Technologies Center at the University of New Hampshire; http://bitcwiki.sr.unh.edu/index.php/Main_Page). The dissociation constants for the dodecamer to hexamer transition for LonWT and LonVQ were calculated in KaleidaGraph (Synergy Software) with the equation y = x/(x + K d), where y is the fraction of dodecamer and x is the concentration of Lon monomer.
ATP hydrolysis activity assays
ATP hydrolysis activity was monitored at 37°C using an NADH‐coupled assay.44 Briefly, Lon protein samples were diluted serially using Lon storage buffer. The ATP regeneration mix was prepared with Lon reaction buffer (25 mM Tris pH 8, 100 mM KCl, 10 mM MgCl2, 1 mM DTT), 0.7 mM NADH, 4 mM ATP, 2% DMSO, 4 mM phosphoenolpyruvate, 13 U/mL pyruvate kinase, and 17 U/mL lactate dehydrogenase and was warmed to 37°C. The protein was added to a 384‐well clear‐bottom plate (Corning) in triplicate and the reaction was started by adding ATP regeneration mix to each well with a repeat pipettor. The loss of absorbance at 340 nm over time was monitored using a UV/Vis spectrometer (Molecular Devices).
SulA inactivation assay
Escherichia coli strain W3110 expressing wild‐type Lon, Δlon::kanR or lon::lonV217A/Q220A from the endogenous Lon promoter were grown in LB broth to an OD600 of 0.9–1.3, diluted into fresh LB broth to an OD600 of 0.25, and 10‐fold serial dilutions were prepared. Ten microliters of each dilution were spotted onto an LB‐agar plate. The plate was exposed to 254 nm UV light for 5 s and incubated overnight in the dark at 37°C.
IbpA in vivo degradation assay
In vivo degradation assays were performed as previously described.4 Briefly, LB cultures were grown at 37°C in a shaking water bath to an OD600 of 0.2. The culture was then transferred to 45°C for 30 min. A 900 μL aliquot of the culture was added to 100 μL 100% trichloroacetic acid (TCA) for the 0 min time point sample. Spectinomycin (400 μg/mL) was added, and samples were taken at 15 min intervals for 60 min while maintaining the cultures at 45°C. For each sample, OD600 was determined and then TCA was added to a final concentration of 10%. Samples were centrifuged and the pellets were rinsed with acetone and suspended in 2× Tris‐tricine sample buffer to normalize the cell density. An equal volume of each sample was run on a 12% Tris‐tricine polyacrylamide gel. IbpA levels were detected using the anti‐IbpA antibody (1:2000 dilution) and were quantified using ImageQuant software (GE Health Sciences).
IbpB and titinI27‐sul20 in vitro degradation assay
In vitro degradation reactions using 40 μM radiolabeled substrates were performed as previously described.20 IbpB degradation reactions contained 60% (vol/vol) IbpB storage buffer (50 mM Hepes‐KOH [pH 8], 600 mM potassium glutamate, 20% sucrose, 0.1 mM TCEP), 5% (vol/vol) Lon storage buffer, 5 mM MgCl2, 5 mM KCl, and 2% (vol/vol) DMSO. The degradation reactions were initiated by the addition of an ATP‐regeneration system, containing a final concentration of 4 mM ATP, 100 mg/mL creatine kinase, and 10 mM creatine phosphate. Degradation was monitored at 37°C by the formation of TCA‐soluble radioactive peptides, as previously described.4, 45 Degradation of titinI27‐sul20 was carried out at 37°C in buffer containing 25 mM Tris–HCl, pH 8.0, 100 mM KCl, 10 mM MgCl2, 1 mM DTT, 2 mM ATP, 100 mg/mL creatine kinase, and 10 mM creatine phosphate. Kinetics were determined using a mixture of 5% (mol/mol) 35S‐labeled substrate and 95% (mol/mol) unlabeled substrate, as previously described.45
Mucoid phenotype assay
LB cultures (30 mL) were inoculated with 500 μL of overnight W3110 starter culture, either wild type, or Δlon::kanR or lon::lonV217A/Q220A and grown at 37°C to an OD600 of 0.8–1.0. Cultures were diluted with LB to OD600 0.25, which was further diluted 1:1000 with LB and 10 μL plated on M9 minimal media agar plates supplemented with 1 mM magnesium sulfate, 0.05% thiamine hydrochloride, 0.004% casamino acids, and 0.2% glucose. Plates were incubated at 37°C for approximately 18 h, then at 22°C for an additional 72 h before imaging.
W3110 E. coli growth curves
Duplicate LB cultures (50 mL) were inoculated with 500 μL of overnight starter culture of W3110 wild type, Δlon::kanR or lon::lonV217A/Q220A and grown at 37°C to OD600 of 0.3. At this point, cultures were transferred to a 45°C water bath with shaking and samples removed at the indicated time points for OD measurements.
Supporting information
Appendix S1: Supplementary Material
Acknowledgments
This investigation was supported in part by a grant from The Jane Coffin Childs Memorial Fund for Medical Research (61‐1529) and a Burroughs Wellcome Postdoctoral Enrichment Program Fellowship Award 1015092 (B.L.B), a National Institutes of Health National Research Service Award postdoctoral Fellowship F32GM094994 (E.F.V.), and the Howard Hughes Medical Institute. T.A.B. and S.K. are employees of the Howard Hughes Medical Institute. Analytical Ultracentrifugation data was collected at the MIT Biophysical Instrumentation Facility for the Study of Complex Macromolecular Systems (NSF‐0070319). Electron microscopy data were obtained at the W.M. Keck Biological Imaging Facility, Whitehead Institute.
Summary statement Lon protease degrades several critical regulatory proteins and maintains protein quality control. We characterized a variant of Escherichia coli Lon that preferentially forms a dodecamer; an assembly that has distinct activities and substrate recognition compared with the Lon hexamer. This variant served as a molecular probe to investigate the biological roles of the dodecamer–hexamer equilibrium in vivo and in vitro. This work thus highlights new aspects of the complex molecular processes that control Lon activity.
Tejas Kalastavadi's current address is Department of Studies in Genetics and Genomics, University of Mysore, Manasagangotri, Mysore 570006, India.
James Z. Chen's current address is Department of Biochemistry and Molecular Biology, Oregon Health and Science University, Portland, OR, USA.
The authors declare no competing interests.
References
- 1. Gottesman S (1996) Proteases and their targets in Escherichia coli . Annu Rev Genet 30:465–506. [DOI] [PubMed] [Google Scholar]
- 2. Wang N, Gottesman S, Willingham MC, Gottesman MM, Maurizi MR (1993) A human mitochondrial ATP‐dependent protease that is highly homologous to bacterial Lon protease. Proc Natl Acad Sci USA 90:11247–11251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gottesman S (2003) Proteolysis in bacterial regulatory circuits. Annu Rev Cell Dev Biol 19:565–587. [DOI] [PubMed] [Google Scholar]
- 4. Bissonnette SA, Rivera‐Rivera I, Sauer RT, Baker TA (2010) The IbpA and IbpB small heat‐shock proteins are substrates of the AAA+ Lon protease. Mol Microbiol 75:1539–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ishii Y, Sonezaki S, Iwasaki Y, Miyata Y, Akita K, Kato Y, Amano F (2000) Regulatory role of C‐terminal residues of SulA in its degradation by Lon protease in Escherichia coli . J Biochem 127:837–844. [DOI] [PubMed] [Google Scholar]
- 6. Neher SB, Villen J, Oakes EC, Bakalarski CE, Sauer RT, Gygi SP, Baker TA (2006) Proteomic profiling of ClpXP substrates after DNA damage reveals extensive instability within SOS regulon. Mol Cell 22:193–204. [DOI] [PubMed] [Google Scholar]
- 7. Bota DA, Van Remmen H, Davies KJ (2002) Modulation of Lon protease activity and aconitase turnover during aging and oxidative stress. FEBS Lett 532:103–106. [DOI] [PubMed] [Google Scholar]
- 8. Bernstein SH, Venkatesh S, Li M, Lee J, Lu B, Hilchey SP, Morse KM, Metcalfe HM, Skalska J, Andreeff M, Brookes PS, Suzuki CK (2012) The mitochondrial ATP‐dependent Lon protease: a novel target in lymphoma death mediated by the synthetic triterpenoid CDDO and its derivatives. Blood 119:3321–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu Y, Lan L, Huang K, Wang R, Xu C, Shi Y, Wu X, Wu Z, Zhang J, Chen L, Wang L, Yu X, Zhu H, Lu B (2014) Inhibition of Lon blocks cell proliferation, enhances chemosensitivity by promoting apoptosis and decreases cellular bioenergetics of bladder cancer: potential roles of Lon as a prognostic marker and therapeutic target in baldder cancer. Oncotarget 5:11209–11224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sauer RT, Baker TA (2011) AAA+ proteases: ATP‐fueled machines of protein destruction. Annu Rev Biochem 80:587–612. [DOI] [PubMed] [Google Scholar]
- 11. Gur E, Vishkautzan M, Sauer RT (2012) Protein unfolding and degradation by the AAA+ Lon protease. Protein Sci 21:268–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gur E, Sauer RT (2008) Recognition of misfolded proteins by Lon, a AAA(+) protease. Genes Dev 22:2267–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shah IM, Wolf RE Jr (2006) Sequence requirements for Lon‐dependent degradation of the Escherichia coli transcription activator SoxS: identification of the SoxS residues critical to proteolysis and specific inhibition of in vitro degradation by a peptide comprised of the N‐terminal 21 amino acid residues. J Mol Biol 357:718–731. [DOI] [PubMed] [Google Scholar]
- 14. Roudiak SG, Shrader TE (1998) Functional role of the N‐terminal region of the Lon protease from Mycobacterium smegmatis . Biochemistry 37:11255–11263. [DOI] [PubMed] [Google Scholar]
- 15. Ebel W, Skinner MM, Dierksen KP, Scott JM, Trempy JE (1999) A conserved domain in Escherichia coli Lon protease is involved in substrate discriminator activity. J Bacteriol 181:2236–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Iyer LM, Leipe DD, Koonin EV, Aravind L (2004) Evolutionary history and higher order classification of AAA+ ATPases. J Struct Biol 146:11–31. [DOI] [PubMed] [Google Scholar]
- 17. Wohlever ML, Baker TA, Sauer RT (2013) A mutation in the N domain of Escherichia coli lon stabilizes dodecamers and selectively alters degradation of model substrates. J Bacteriol 195:5622–5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wohlever ML, Baker TA, Sauer RT (2014) Roles of the N domain of the AAA+ Lon protease in substrate recognition, allosteric regulation and chaperone activity. Mol Microbiol 91:66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cheng I, Mikita N, Fishovitz J, Frase H, Wintrode P, Lee I (2012) Identification of a region in the N‐terminus of Escherichia coli Lon that affects ATPase, substrate translocation and proteolytic activity. J Mol Biol 418:208–225. [DOI] [PubMed] [Google Scholar]
- 20. Vieux EF, Wohlever ML, Chen JZ, Sauer RT, Baker TA (2013) Distinct quaternary structures of the AAA+ Lon protease control substrate degradation. Proc Natl Acad Sci USA 110:E2002–E2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chung CH, Goldberg AL (1982) DNA stimulates ATP‐dependent proteolysis and protein‐dependent ATPase activity of protease La from Escherichia coli . Proc Natl Acad Sci USA 79:795–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuroda A, Nomura K, Ohtomo R, Kato J, Ikeda T, Takiguchi N, Ohtake H, Kornberg A (2001) Role of inorganic polyphosphate in promoting ribosomal protein degradation by the Lon protease in E. coli . Science 293:705–708. [DOI] [PubMed] [Google Scholar]
- 23. Mukherjee S, Bree AC, Liu J, Patrick JE, Chien P, Kearns DB (2015) Adaptor‐mediated Lon proteolysis restricts Bacillus subtilis hyperflagellation. Proc Natl Acad Sci USA 112:250–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lu B, Liu T, Crosby JA, Thomas‐Wohlever J, Lee I, Suzuki CK (2003) The ATP‐dependent Lon protease of Mus musculus is a DNA‐binding protein that is functionally conserved between yeast and mammals. Gene 306:45–55. [DOI] [PubMed] [Google Scholar]
- 25. Li M, Gustchina A, Rasulova FS, Melnikov EE, Maurizi MR, Rotanova TV, Dauter Z, Wlodawer A (2010) Structure of the N‐terminal fragment of Escherichia coli Lon protease. Acta Cryst D66:865–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Delorenzi M, Speed T (2002) An HMM model for coiled‐coil domains and a comparison with PSSM‐based predictions. Bioinformatics 18:617–625. [DOI] [PubMed] [Google Scholar]
- 27. Grigoryan G, Keating AE (2008) Structural specificity in coiled‐coil interactions. Curr Opin Struct Biol 18:477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gur E, Sauer RT (2009) Degrons in protein substrates program the speed and operating efficiency of the AAA+ Lon proteolytic machine. Proc Natl Acad Sci USA 106:18503–18508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Allen SP, Polazzi JO, Gierse JK, Easton AM (1992) Two novel heat shock genes encoding proteins produced in response to heterologous protein expression in Escherichia coli . J Bacteriol 174:6938–6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Laskowska E, Wawrzynow A, Taylor A (1996) IbpA and IbpB, the new heat‐shock proteins, bind to endogenous Escherichia coli proteins aggregated intracellularly by heat shock. Biochimie 78:117–122. [DOI] [PubMed] [Google Scholar]
- 31. Arrigo AP, Suhan JP, Welch WJ (1988) Dynamic changes in the structure and intracellular locale of the mammalian low‐molecular‐weight heat shock protein. Mol Cell Biol 8:5059–5071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bentley NJ, Fitch IT, Tuite MF (1992) The small heat‐shock protein Hsp26 of Saccharomyces cerevisiae assembles into a high molecular weight aggregate. Yeast 8:95–106. [DOI] [PubMed] [Google Scholar]
- 33. Torres‐Cabassa AS, Gottesman S (1987) Capsule synthesis in Escherichia coli K‐12 is regulated by proteolysis. J Bacteriol 169:981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Duman RE, Lowe J (2010) Crystal structures of Bacillus subtilis Lon protease. J Mol Biol 401:653–670. [DOI] [PubMed] [Google Scholar]
- 35. Lupas A (1996) Coiled coils: new structures and new functions. Trends Biochem Sci 21:375–382. [PubMed] [Google Scholar]
- 36. Lee AY, Hsu CH, Wu SH (2004) Functional domains of Brevibacillus thermoruber lon protease for oligomerization and DNA binding: role of N‐terminal and sensor and substrate discrimination domains. J Biol Chem 279:34903–34912. [DOI] [PubMed] [Google Scholar]
- 37. Djuranovic S, Hartmann MD, Habeck M, Ursinus A, Zwickl P, Martin J, Lupas AN, Zeth K (2009) Structure and activity of the N‐terminal substrate recognition domains in proteasomal ATPases. Mol Cell 34:580–590. [DOI] [PubMed] [Google Scholar]
- 38. Carroni M, Kummer E, Oguchi Y, Wendler P, Clare DK, Sinning I, Kopp J, Mogk A, Bukau B, Saibil HR (2014) Head‐to‐tail interactions of the coiled‐coil domains regulate ClpB activity and cooperation with Hsp70 in protein disaggregation. Elife 3:e02481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oguchi Y, Kummer E, Seyffer F, Berynskyy M, Anstett B, Zahn R, Wade RC, Mogk A, Bukau B (2012) A tightly regulated molecular toggle controls AAA+ disaggregase. Nat Struct Mol Biol 19:1338–1346. [DOI] [PubMed] [Google Scholar]
- 40. Seyffer F, Kummer E, Oguchi Y, Winkler J, Kumar M, Zahn R, Sourjik V, Bukau B, Mogk A (2012) Hsp70 proteins bind Hsp100 regulatory M domains to activate AAA+ disaggregase at aggregate surfaces. Nat Struct Mol Biol 19:1347–1355. [DOI] [PubMed] [Google Scholar]
- 41. Carroni M, Franke KB, Maurer M, Jager J, Hantke I, Gloge F, Linder D, Gremer S, Turgay K, Bukau B, Mogk A (2017) Regulatory coiled‐coil domains promote head‐to‐head assemblies of AAA+ chaperones essential for tunable activity control. Elife 6:e30120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shearstone JR, Baneyx F (1999) Biochemical characterization of the small heat shock protein IbpB from Escherichia coli . J Biol Chem 274:9937–9945. [DOI] [PubMed] [Google Scholar]
- 43. Schuck P (2000) Size‐distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J 78:1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lindsley JE (2001) Use of a real‐time, coupled assay to measure the ATPase activity of DNA topoisomerase II. Methods Mol Biol 95:57–64. [DOI] [PubMed] [Google Scholar]
- 45. Gottesman S, Roche E, Zhou Y, Sauer RT (1998) The ClpXP and ClpAP proteases degrade proteins with carboxy‐terminal peptide tails added by the SsrA‐tagging system. Genes Dev 12:1338–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supplementary Material