

Abstract
RNA signals located downstream of stop codons in eukaryotic mRNAs can stimulate high levels of translational readthrough by the ribosome, thereby giving rise to functionally distinct C-terminally extended protein products. Although many readthrough events have been previously discovered in Nature, a broader description of the stimulatory RNA signals would help to identify new reprogramming events in eukaryotic genes and provide insights into the molecular mechanisms of readthrough. Here, we explore the RNA reprogramming landscape by performing in vitro translation selections to enrich RNA readthrough signals de novo from a starting randomized library comprising >1013 unique sequence variants. Selection products were characterized using high-throughput sequencing, from which we identified primary sequence and secondary structure readthrough features. The activities of readthrough signals, including three novel sequence motifs, were confirmed in cellular reporter assays. Then, we used machine learning and our HTS data to predict readthrough activity from human 3’-untranslated region sequences. This led to the discovery of >1.5% readthrough in four human genes (CDKN2B, LEPROTL1, PVRL3, and SFTA2). Together, our results provide valuable insights into RNA-mediated translation reprogramming, offer tools for readthrough discovery in eukaryotic genes, and present new opportunities to explore the biological consequences of stop codon readthrough in humans.
Graphical Abstract
The ribosome coordinates protein synthesis from mRNA templates according to a nearly universal set of decoding rules.1 While the majority of mRNA substrates conform to standard decoding with high fidelity,2 in some cases, the translation program can be altered by cis-acting elements located within the mRNA transcript or nascent polypeptide chain.3 Each phase of protein synthesis is subject to revision, with examples including non-AUG4 and IRES-mediated5 initiation, site-specific ribosomal frameshifting,6 co-translational 2A peptide cleavage,7 and stop codon readthrough.8 These “translation reprogramming” mechanisms serve to expand the coding capacity of individual genes by allowing for the synthesis of multiple protein products from a single eukaryotic mRNA and also enable co-translational regulation of gene expression.9
One common form of translation reprogramming is programmed stop codon readthrough, wherein termination codons are interpreted as sense codons in response to local RNA signals.10,11 The fidelity of translation termination typically restricts basal readthrough levels to approximately 0.1%,12 with the UGA stop codon demonstrating the greatest extent of readthrough.12–14 However, in the presence of efficient RNA signals, readthrough levels exceeding 15% can occur.15,16 In many cases, the C-terminally extended readthrough product possesses biological activities distinct from that of the standard translation product. Moreover, the two proteins are synthesized at a fixed ratio that matches the efficiency of the readthrough signal, and this ratio is often critical to maintaining biological function.17
Like many translation reprogramming phenomena, programmed readthrough is particularly common in viruses, which employ this mechanism to regulate the synthesis of essential downstream genes like polymerases.18–20 A prominent example is found in the tobacco mosaic virus (TMV), which achieves 5–10% readthrough using the hexanucleotide sequence CAAUUA situated directly downstream of a UAG stop codon.21 Other viruses achieve readthrough using RNA structural elements. For example, a downstream hairpin structure stimulates readthrough in the Colorado tick fever virus (CTFV),22 whereas an RNA pseudoknot is responsible for readthrough in the murine leukemia virus (MLV)23,24
Beyond viruses, programmed readthrough is now recognized as a mechanism of translation regulation and protein augmentation in endogenous genes of fungi and animals.25–29 In these organisms, readthrough has been identified using phylogenetic analyses,30,31 ribosome profiling,32 genome-based computational approaches,27,33 and nucleotide sequence screens.13,34 This has led to the notable discovery of widespread readthrough in drosophila30,32 and mosquitos35 as well as readthrough in a small set of human genes that contain a UGA stop codon paired with a CUAG sequence motif.15,16 As a result of these studies, many intriguing proposals have been put forth regarding the biological roles of readthrough in cellular organisms.36 While these studies have been both foundational and illuminating, their applicability to the discovery of additional readthrough events is restricted to those that are evolutionarily conserved or specific to particular tissues or organisms or utilize signals that are underrepresented across genomes. As a result, many readthrough events that do not satisfy the conditions mentioned above likely remain unrecognized.
Given that much of our understanding about readthrough signals follows from fortuitous discoveries in nature, we hypothesized that numerous RNA signals might exist that have yet to be identified. Therefore, a broad and unbiased exploration of RNA sequence space could uncover a larger spectrum of readthrough motifs and features that, when used in conjunction with widely available genome sequencing data, could aid the discovery of readthrough events in eukaryotic organisms. Furthermore, the discovery of new readthrough motifs could provide additional insights for unraveling the molecular mechanism of this translation—reprogramming phenomenon, which remains largely unresolved.
Here, we combine in vitro RNA selections, high-throughput sequencing (HTS) analysis, and machine learning to characterize readthrough-promoting RNA features and identify readthrough signals in human genes. First, an mRNA display selection for readthrough was established and applied to a library containing ~1013 randomized starting RNA sequences. Transcripts enriched by this selection were then characterized by HTS. Recovered motifs were subsequently validated in yeast and human cell culture assays. Then, the HTS data were further used to train an additive classification model that nominates readthrough activity from input 3’-untranslated region (UTR) transcript sequences. This approach identified six human 3’-UTRs that promote significant stop codon readthrough (>1.5%) in human cell assays.
MATERIALS AND METHODS
mRNAs Display Template Construction.
The Inactive-RT and MLV-PK in vitro selection constructs were assembled from synthesized oligonucleotides (IDT) and cloned into the pCR-TOPO vector using the Topo-TA system (Invitrogen) (Tables S1–S3). Following sequence verification, constructs were amplified via polymerase chain reaction (PCR) from plasmids using Vent polymerase (NEB) to generate double-stranded DNA (dsDNA) templates for in vitro transcription. For library constructs, ultramer oligonucleotides were synthesized (IDT, 4 nmol) and purified by urea denaturing polyacrylamide gel electrophoresis (PAGE). PCR amplification of the control library ultramer oligonucleotide was performed using CT-for and RT-rev primers with Vent polymerase. PCR amplification of the RT library was performed using RT-for and RT-rev primers with Vent polymerase in a 10 mL scale reaction (50 pmol of total input ssDNA library, ~3 × 1013 molecules) for 12 cycles. The RT library PCR mixture was extracted with phenol and chloroform. DNA was precipitated with NaOAc and EtOH, pelleted by centrifugation, and then dissolved in RNase-free doubly distilled H2O. The DNA concentration was quantified by in-gel ethidium fluorescence. For all constructs, in vitro RNA transcription was performed using T7 RNA polymerase (NEB), and the RNA products were purified by urea denaturing PAGE and electroelution. For the RT library, the first round of transcription was performed using 200 pmol of the input dsDNA library at a 1 mL scale; subsequent transcriptions contained 20 pmol of dsDNA at a 100 μL scale. Purified RNA products were ligated to phospho-dA27 dCdC-puromycin (TriLink Biotechnologies) by splint ligation with T4 DNA ligase (NEB) and oligonucleotide AVA95 as previously described.37 Ligation products were purified from nonligated RNA by urea denaturing PAGE and electroeluted to yield mRNA display templates for in vitro translation selections.
In Vitro Translation Selection.
mRNA display templates were translated at a concentration of 200 nM in 40% rabbit reticulocyte lysate (nuclease-treated, Promega) supplemented with 0.5 mM Mg(OAc)2, 100 mM KCl, and 1× amino acid mix. Round 1 of selection was performed at the 1 mL scale, and subsequent translations were carried out at the 100 μL scale. Reaction mixtures were incubated at 30 °C for 1 h, followed by treatment with 0.38 volume of fusion salt mix (made by combining 312 μL of 2.5 M KCl and 68 μL of 1 M MgCl2), and incubated at room temperature for 15 min. mRNA–peptide fusions were FLAG purified using anti-FLAG M2 Affinity Gel (Sigma) as described previously38 and then purified with Ni-NTA resin (Qiagen) as described previously.37 Following purification, mRNA–peptide fusions were buffer exchanged into reverse transcription buffer [50 mM Tris (pH 8.3), 75 mM KCl, 3 mM MgCl2, and 20 mM DTT] using 30000 molecular weight cutoff spin concentrators (Amicon). cDNA was reverse transcribed from purified mRNA–peptide fusions with Superscript II (Life Technologies) in the presence of RNasin (Promega) using primer AVA95 for 50 min at 42 °C. The reverse transcription products were subjected to a second FLAG purification, amplified with GoTaq polymerase (Promega) in a 0.5 mL reaction mixture, and purified using a PCR cleanup kit (Qiagen) to yield dsDNA for subsequent rounds of selection or cloning.
High-Throughput Sequencing and Processing.
Products from round 3 of the selection were prepared for HTS by a two-step PCR amplification process. Three first-round PCRs were performed using reverse primer AVA320 and one of three forward primers (AVA314–AVA316) containing one, two and three randomized nucleotide insertions, respectively, to produce staggered sequencing reads. The pooled first-round PCR products were then amplified with AVA319 and AVA321 to add Truseq adaptors and indices. DNA quality was assessed by agarose gel electrophoresis and chip-based capillary electrophoresis (Bioanalyzer, Agilent). DNA sequencing was performed on the Illumina HiSeq platform at the Columbia Genome Center, which generated approximately 65 million raw sequencing reads. Raw reads from the fastq file were processed into length 75 nucleotide strings representing the full library region, and FLAG-containing sequences were separated from the readthrough sequence pool (see bash scripts at https://github.com/szairis/readthru). The readthrough data set was reduced to a set of unique sequences with corresponding read counts. Then, after the set of all unique sequences with abundances of ≥45 (corresponding to >0.0001% of total reads) had been taken, sequences derived from more abundant library sequences through point mutations were removed. The resulting final readthrough data set contains ~38000 unique sequences that account for >70% of the read mass in the readthrough data set.
Statistical Correlation Analysis.
For pairs of position-specific sequence substrings, which can be analyzed at a chosen length and position, a 2 × 2 contingency table (rows, string 1, NOT string 1; columns, string 2, NOT string 2) is constructed for every possible observed substring pairing using the aggregate position-specific substring frequencies from the readthrough data set. The resulting contingency tables are evaluated for statistically significant associations using Fisher’s exact test and Pearson’s χ2 test, providing χ2, φ, p value, and odds ratio (OR) test statistics in a tabular output format. p values were adjusted for multiple comparisons.
Plasmid Construction.
Yeast dual-FP plasmids (Table S3) were derived from the previously reported p425-dual-FP plasmid (AmpR, LEU2, 2 μm).39 CMV-dual-FP plasmids (Table S3) for HEK293T assays were constructed from pH2B-mCherry (Addgene 20972) and pH2B-GFP (Addgene 11680) using standard molecular cloning techniques. All readthrough inserts (Table S3) were assembled from synthesized oligonucleotides (IDT) or were ordered as synthesized gblock dsDNA fragments (IDT) and then cloned into NheI/AatII-digested p425-dual-FP or BamHI/NheI-digested pCMV-dual-FP by the Gibson Assembly method (NEB). Plasmids encoding the human vitamin D receptor (VDR) with expression driven by the CMV promoter were constructed using synthesized gblock dsDNA fragments (IDT) and the parent plasmid pH2B-GFP (Addgene 11680).
Yeast Dual-FP Readthrough Assays.
Yeast dual-FP assays were performed using strain Fy251 (ATCC 96098). p425-dual-FP plasmids were transformed using the lithium acetate/PEG transformation method40 and selected on SC (2% glucose, -Leu) agar plates. Six or more individual colonies were selected, grown overnight in liquid SC (2% glucose, -Leu) medium at 30 °C and 250 rpm to high density, diluted into fresh medium at a starting OD600 of 0.2, and then grown for 8 h at 30 °C in a 96-well plate format at 800 rpm. Readthrough activity was quantified by measurement of yEGFP and mCherry fluorescence signals (Infinite M200 plate reader, Tecan) and side-by-side comparison to a control strain harboring the full-length EGFP-mCherry fusion construct (average of three colonies).
Machine Learning.
A model was trained to discriminate readthrough promoting 3’-UTR contexts from decoy contexts. This implies a mapping from the feature encoding of a UTR context X to a binary label y representing either a true readthrough element versus a decoy. Decision trees are transparent in the interactions between features in X, enabling biological interpretation of model performance. Decision trees were used as base learners in a gradient-boosted ensemble to achieve excellent classification performance and robustness to overfitting.41 No upfront dimensionality reduction was performed prior to classifier training. We use the binomial deviance loss function with a maximum decision tree depth of 5 and a learning rate of 0.1 as implemented in the scikit-learn library.42 The training data consists of 11000 UTR contexts represented as vectors of 30 features (24 sequence and 6 structural), 1000 of which are positive examples (readthrough promoting) and 10000 of which are negative examples (flag decoys). The classifier is trained for 100 rounds of boosting under 10-fold stratified cross validation (CV) and achieves a mean test split AUROC of 0.991.
HEK293T Dual-FP Readthrough Assays.
HEK293T cells (ATCC CRL-3216) were cultured in Dulbecco’s modified Eagle’s medium DMEM (Dibco) supplemented with 10% HI-FBS and 1% Pen-strep at 37 °C and 5% CO2. For transfections, cells were grown in six-well plates to a confluence of 70% and then transfected using X-tremeGENE (Sigma-Aldrich) according to the manufacturer’s protocol (1 μg of plasmid, 3 μL of X-tremeGENE, and Opti-MEM to a final volume of 200 μL). Cells were allowed to grow at 37 °C for an additional 16–24 h, trypsinized and washed with PBS, and then analyzed. Readthrough activity was quantified by measurement of EGFP and mCherry fluorescence signals and comparison to cells transfected with the full-length EGFP-mCherry fusion plasmid. Reporter validation was performed by transfecting different ratios of the full-length EGFP-mCherry fusion plasmid and the inactive control plasmid.
Western Blots.
Three plasmid variants were constructed and analyzed to assess readthrough in human VDR: (1) human VDR isoform A with an N-terminal 3xFLAG tag, its natural TGA termination codon, and no further natural 3’-UTR sequence; (2) human VDR isoform A with an N-terminal 3xFLAG tag, its natural TGA termination codon, and the first 201 nucleotides of the VDR 3’-UTR; and (3) human VDR isoform A containing an N-terminal 3xFLAG tag, mutation of the natural TGA termination codon to a TGG codon, and the first 201 nucleotides of the VDR 3’-UTR. Vectors were transfected into HEK239T cells as described above and then lysed with RIPA buffer (ThermoFisher) according to the manufacturer’s protocol. Proteins were resolved on a 10% polyacrylamide gel (Bio-Rad) and then dry-transferred to a nitrocellulose membrane (Invitrogen). Blots were blocked with 3% bovine serum albumin in TBST and then incubated with the 1:1000 HRP-conjugated anti-FLAG antibody (Sigma) in TBST. After six rounds of washes with TBS, antibody-bound bands were visualized with CN/DAB reagent (ThermoFisher) according to the manufacturer’s protocol.
RESULTS
An in Vitro Selection for Stop Codon Readthrough.
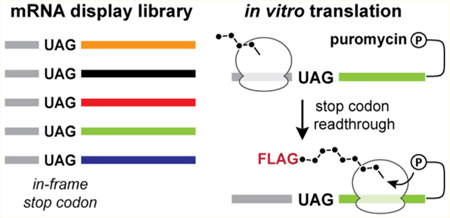
To enrich eukaryotic readthrough motifs de novo by in vitro selection, we designed a strategy based on mRNA display.43 The translation stage of this process takes place in a cell lysate and thereby integrates the expansive libraries that are accessible to classical in vitro RNA selections44–46 with the biochemical complexity of the cellular environment. In mRNA display, libraries of RNA sequence variants are translated and become covalently linked to their peptide products via a 3’-tethered puromycin motif (Figure 1A). This RNA–peptide linkage relies on a distance-dependent puromycin fusion reaction that occurs only when the ribosome reaches the 3’-end of the mRNA template.37 Therefore, mRNA display can in principle select for translation reprogramming signals within the RNA that enable the ribosome to bypass deliberately placed in-frame termination codons (Figure 1B). Our prior work demonstrated that this concept can be exploited to identify eukaryotic −1 programmed ribosomal frameshifting motifs from large libraries of RNA sequence variants.39
Figure 1.
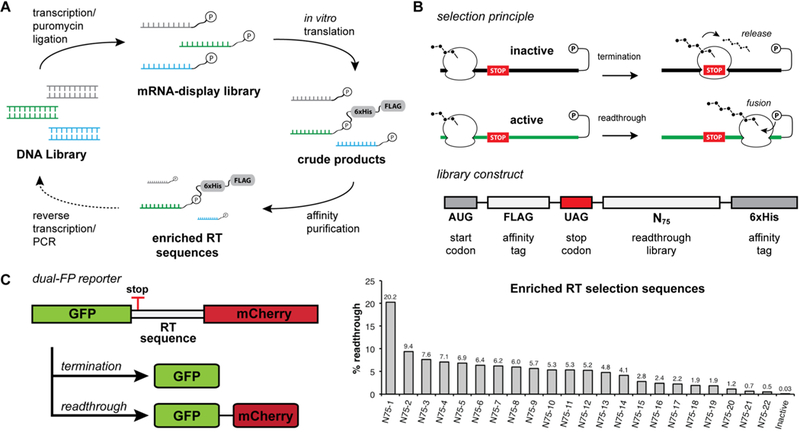
In vitro selection for stop codon readthrough. (A) mRNA display in vitro selection cycle. mRNA is transcribed from the DNA library and then ligated to a puromycin-containing DNA oligonucleotide. The mRNA display library is translated in rabbit reticulocyte lysate, and translation products are affinity purified. Enriched sequences are reverse transcribed and PCR amplified for subsequent rounds of selection. (B) Selection principle and library selection construct. During the mRNA display selection, translation termination at an internal stop codon prevents the formation of the mRNA–peptide fusion and leads to the release of peptides containing affinity tags. Stop codon readthrough allows for translation of the full mRNA template and subsequent fusion of peptide affinity tags to the mRNA template that promotes readthrough. The library selection construct encodes an open reading frame containing an N-terminal FLAG tag, an in-frame UAG stop codon, a library of 75 randomized nucleotides, and a C-terminal hexahistidine tag. (C) Postselection readthrough library sequences were analyzed using a dual-fluorescent protein (dual-FP) reporter assay in Saccharomyces cerevisiae. Readthrough efficiency was determined by comparing the GFP:mCherry fluorescence ratio to a no-stop GFP-mCherry control set to 100%.
Before performing a library selection for readthrough, we first carried out validation studies to establish enrichment of readthrough signals by mRNA display. Consistent with prior studies,47 a single round of mRNA display selection on a pool of randomized open reading frames resulted in depletion of stop codon-containing mRNAs (Figure S1A). When constructs were designed to contain a single deliberately placed in-frame stop codon, mRNA display selection resulted in the substantial enrichment of sequences containing mutations to the stop codon (Figure S1B). Lastly, upon translation in the same reaction as inactive sequences, an ~30-fold single-round enrichment was observed for mRNAs containing the MLV readthrough stimulating pseudoknot23,48 placed downstream of the stop codon (Figure S1C). Together, these results support the idea that mRNA display selection is capable of enriching active readthrough elements from a pool composed largely of inactive sequences by subverting translation termination.
Following validation of the selection platform, we next set out to enrich readthrough motifs de novo from randomized RNA sequences. We designed a library to contain 75 randomized nucleotides directly downstream of a UAG stop codon (Figure 1B, UAG-N75). This design allows for enrichment of short readthrough-stimulating RNA sequences as well as RNA structural elements composed of many downstream nucleotides. Although the theoretical library size of 475 (~1045) cannot be covered by the ~1013 unique RNA molecules submitted to the first round of in vitro translation, 1013 sequence variants theoretically allow for saturation coverage of any combination of 22 nucleotides (422 ≈ 1013) within the 75-nucleotide random sequence window (see Materials and Methods). The amber (UAG) stop codon was chosen on the basis of its intermediate termination efficiency by comparison to UAA and UGA.49 The construct also encodes N-terminal and C-terminal affinity tags for selection by purification.
After two rounds of selection on the UAG-N75 mRNA display library, we did not observe enrichment for any particular motifs when a sampling of library members was analyzed by sequencing. However, after the third round of selection, we recovered several library members containing the previously reported CARYYA motif.34 This suggested that readthrough-promoting sequences had been enriched. To examine the readthrough activity of the selection products in living eukaryotic cells, postselection sequences were evaluated in a dual-fluorescent protein (dual-FP) reporter assay in Saccharomyces cerevisiae (Figure 1C). A substantial percentage of library members showed readthrough activity, with some displaying efficiencies as high as 20%. Notably, while many sequences were found to contain a CARYYA motif, the range of overall readthrough efficiencies suggested that factors beyond the hexanucleotide motif were influencing activity.
HTS Reveals Primary Sequence Readthrough Motifs.
After confirming the readthrough activity of postselection library members in cellular assays, we set out to characterize the landscape of enriched readthrough motifs by deep sequencing analysis. From the pool of selection products, approximately 66 million 100 bp sequencing reads were generated and processed into a set of unique sequences with corresponding read counts. Within this data set, the 10000 most abundant sequences accounted for roughly half of all sequencing reads (Figure S2).
Interestingly, initial inspection of the data revealed that a significant fraction of sequences encoded methionine residues followed by FLAG-like peptide sequences (Figure S3). Presumably, enrichment occurred due to initiation by ribosomes that scanned past the first AUG and the programmed UAG stop codon, followed by synthesis of a FLAG-like peptide and affinity enrichment by anti-FLAG antibodies. Sequences that met defined criteria for FLAG-like epitopes were excluded from the readthrough pool and compiled into a set of decoy selection products for subsequent comparative analysis (see Materials and Methods). After these and additional filtering steps were implemented to remove redundant sequences, approximately 38000 unique readthrough sequences remained that collectively accounted for roughly half of the total sequencing reads.
When analyzing individual library nucleotide frequencies in the aggregate readthrough data, we observed a striking enrichment for particular bases in the first nine positions (Figure 2A). Specifically, C was found in position 1 in 79% of all readthrough sequences and 98% of sequences ranked in the top 5% by abundance. This is consistent with the known decrease in eukaryotic termination efficiency when stop codons are followed by a cytosine. Notable enrichments were also observed for positions 2–6 and 9 with nucleotides A, A, U/C, U/C, A, and G, respectively. Beyond these first nine nucleotides, the library displayed a clear triplet periodicity in nucleotide frequency (Figure 2A), consistent with a translated open reading frame.50
Figure 2.
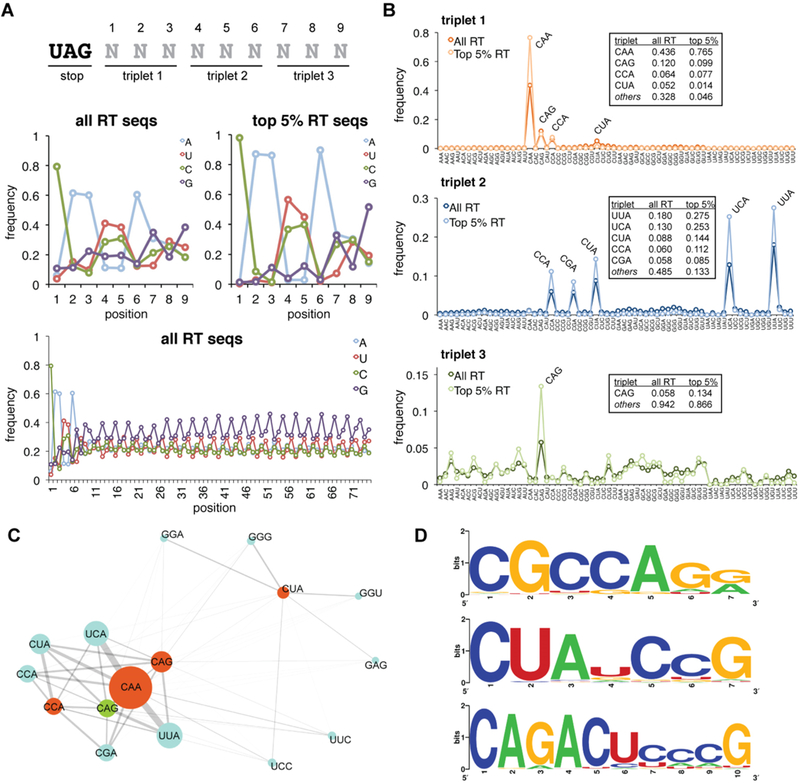
Analysis of primary sequence readthrough features from HTS data. (A) Position-specific nucleotide frequencies within the randomized library region are shown for the full readthrough (RT) data set and the group of sequences ranked in the top 5% based on read abundance. The first nine nucleotide positions are displayed as well as the frequencies across the full library region. (B) Analysis of triplet nucleotide frequencies. Position-specific triplet nucleotide frequencies are displayed for the full readthrough data set and the top 5%, for comparison. Numerical frequencies are tabulated for the most frequently observed triplets. (C) Connectivity map displaying associations between nucleotide triplets at positions 1 (orange), 2 (blue), and 3 (green). The triplet abundance is illustrated by the diameter of the circle; the strength of association is represented by the connector weight. (D) Sequence logos for additional readthrough motifs identified. Logos were generated from the set of sequences with a Hamming distance of ≤1 for CGCCAGR and CUAUCCG and a Hamming distance of ≤2 for CAGACUCCCG.
Equally striking was the enrichment of extended triplet nucleotide sequences (Figure 2B). In triplet position 1, CAA was observed in nearly 44% of all sequences and 76% of sequences in the top 5%. CAG, CCA, and CUA accounted for many of the remaining position 1 triplets. Similarly, triplet position 2 was enriched with YYA and CGA sequences. Lastly, triplet position 3 showed enrichment, albeit less pronounced, for CAG.
Downstream of the first nine nucleotides, in-frame triplet nucleotide frequencies were consistent throughout the library and likely reflect codon preferences in the rabbit reticulocyte lysate translation system (Figure S4). To mitigate potential bias arising from codon usage, we normalized position-specific sequence frequencies to their mean library prevalence in the appropriate reference reading frame (Figure S5). Assessing triplet position 3 after this normalization, we observed more pronounced enrichment for AUN, ANG, and GCN triplets and depletion of CGN and GGN triplets.
Next, to identify extended sequence motifs, we examined the association between individual nucleotides and triplet substrings in a position-specific manner using Fisher’s exact test and Pearson’s χ2 test (see Materials and Methods). Significant associations were observed between position 1 triplets CAA, CAG, and CCA and position 2 triplets UUA, UCA, CUA, CCA, and CGA. While all combinatorial pairings of these triplets were observed with high frequency, stronger associations were observed between particular triplets (Figure S6). Moreover, CAA was also associated with UUC and UCC triplets in position 2. Hereafter, we will refer to this family of hexanucleotide sequences as the Expanded-TMV (E-TMV) motif based on the resemblance to the TMV viral readthrough signal.21
Overall, the E-TMV motif was observed in 50% of all recovered sequences. E-TMV sequences were also specifically associated with CAG, AUN, ANG, and GCN triplets in position 3, while non-E-TMV motif sequences were not (Figure S5B). Lastly, and quite remarkably, the overall most frequently observed nine-nucleotide sequence substring, CAAUUACAG, exactly matches the first nine nucleotides of the naturally occurring TMV readthrough signal.
The E-TMV motif does not contain sequences starting with CUA, the remaining high-frequency position 1 triplet. Instead, we found that one or two guanine residues typically followed CUA (Figure 2C), with the most common downstream triplet being GGC. Notably, this conforms to the previously reported CUAG motif, which has been found to promote high-efficiency readthrough of UGA stop codons.15 Despite the few nucleotide constraints in this motif, its relative absence among the top-ranked sequences suggests that it promotes lower-efficiency readthrough of the UAG stop codon or that it requires additional elements to promote high-efficiency readthrough.
Other statistically meaningful pairings led to the identification of novel extended motifs (Figure 2D). A strong association between CAG and ACU ultimately led to the identification of a broader motif exemplified by its CAGACUCCCG mode sequence. Similarly, we identified enrichment for a motif with the consensus CGCCAGR (Figure S7). To the best of our knowledge, these motifs have not been previously associated with stop codon readthrough events. We also observed modest enrichment for sequences conforming to CUANCCG. It is noteworthy that while this motif has not been explicitly described previously, the CUAUCCG sequence is found adjacent to a UAG stop codon that undergoes readthrough in the Yarrowia lipolytica GAPDH gene.26
To characterize the readthrough activity of sequence motifs in cells, we evaluated a series of dual-FP constructs in S. cerevisiae (Figure 3A). As anticipated, the CAAUUA signal led to high levels of readthrough, but its activity was substantially influenced by the downstream nucleotides at positions 7–9 (RT-2–RT-4). These results are consistent with our HTS data, which showed enrichment for GCN and CAG codons but underrepresentation of GGA following CAAUUA (Figure S5B). Other members of the E-TMV motif promoted readthrough at levels that agree with their library enrichment.
Figure 3.
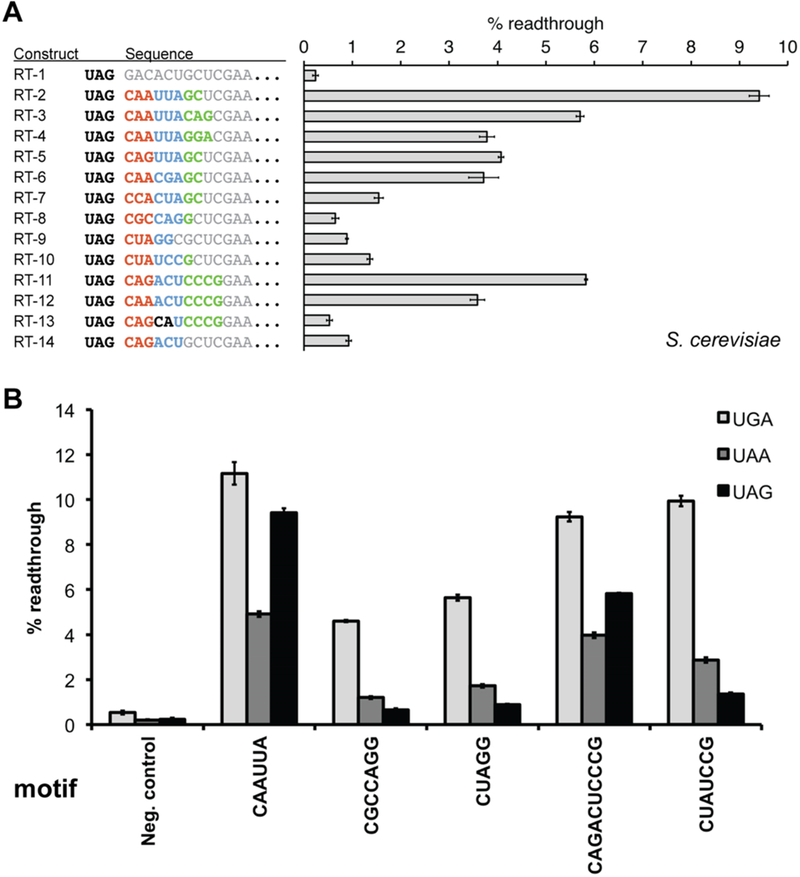
Assays of primary sequence readthrough motifs in yeast. (A) Readthrough sequence motifs recovered from the in vitro selection were evaluated in yeast using the dual-FP reporter assay within a fixed sequence context. RT-1 represents the negative control containing a randomly generated sequence. Sequence elements constituting the RNA motif are shown in color, whereas surrounding fixed sequence context is colored gray. (B) Comparison of primary sequence readthrough motif activities across each of the three stop codons. For all constructs, the readthrough efficiency was determined by comparing the GFP:mCherry fluorescence ratio to a no-stop GFP-mCherry control set to 100%. Neg. control is identical in sequence to RT-1, with only the stop codon substitutions. Error bars represent the standard error of the mean for six or more individual colony replicates.
It is noteworthy that the new CAGACUCCCG sequence motif promoted substantial readthrough in our assay (Figure 3A, RT-11). When its fourth and fifth nucleotides were transposed (RT-13) or nucleotides 7–10 were omitted (RT-14), we observed a significant reduction in readthrough activity (11- or 6-fold, respectively). Alternatively, substitution of A for G at position 3 led to a mild decrease in the level of readthrough (Figure 3A, RT-12). Finally, CGCCAGG (RT-8), CUAGG (RT-9), and CUAUCCG (RT-10) motifs promoted low but meaningful levels of UAG readthrough in yeast.
To evaluate primary sequence motifs with each of the three termination codons, we next measured readthrough activity for one member from each motif class with UAG, UGA, and UAA stop codons (Figure 3B). Across the board, the UGA codon displayed the highest readthrough levels, consistent with prior studies.12–14,49 Motifs that were found to be less active with the UAG codon (CUAG, CGCCAGG, and CUAUCCG) displayed robust readthrough activity with UGA (>4% in all cases). Although the UAA stop codon displayed the lowest readthrough levels for CAAUUA and CAGACUCCCG, UAA was read through at higher levels than UAG for the other motifs tested. Thus, readthrough activity can depend on the pairing between the particular sequence motif and the identity of the stop codon.
Structural Elements in Readthrough Sequences.
In our analysis of primary sequence motifs, we identified enrichment of sequence substrings located in a window located approximately 8–15 nucleotides downstream of the stop codon (Figure 4A and Figure S8). We discovered that these nucleotide strings were complementary to downstream regions of fixed sequence corresponding to the linker in the selection construct. This finding was highly suggestive of RNA secondary structure formation and, furthermore, that enrichment for structural elements had occurred in the in vitro readthrough selection. Indeed, RNA structure predictions for several top-ranked library members revealed extensive secondary structure formation that engaged these linker sequences (Figure S9).
Figure 4.
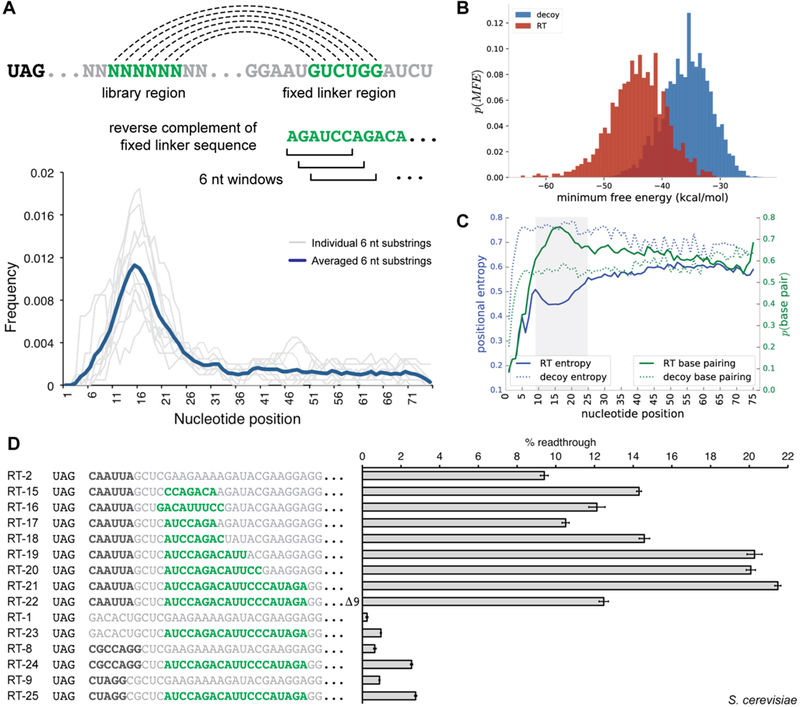
Analysis of readthrough-stimulating secondary structures from HTS data. (A) Sequence substrings complementary to downstream fixed linker regions have the potential for secondary structure formation; six-nucleotide windows were scanned across the library region of readthrough sequences in search of defined complementary sequence substrings. Density plots for individual six-nucleotide substrings (gray lines) and the averaged density for all evaluated substrings (dark blue) are displayed. (B) Secondary structures for sequences in the readthrough (RT) and FLAG decoy data set were predicted with RNAfold; minimum free energies (MFEs) are shown plotted as a histogram for the two populations. (C) Plots of position-specific base pairing probabilities and positional entropies averaged over a subset of sequences from the readthrough and decoy FLAG data sets. The shaded region represents an area of increased base pairing probability and decreased positional entropy when compared to the remaining library region. (D) Dual-FP reporter assays in yeast evaluating the influence of secondary structure on readthrough activity. Primary sequence readthrough motifs are indicated by bold black type. Sequence with secondary structure potential is indicated by bold green type. Error bars represent the standard error of the mean for six or more biological replicates.
To globally evaluate the presence of structural elements within enriched library members, we predicted the secondary structures for the readthrough and FLAG decoy sequence sets using RNAfold.51 By comparison to the FLAG decoy set, the distribution of readthrough sequences demonstrated significantly lower free energies of folding, implying an increased overall secondary structure formation level (Figure 4B). When analyzing structural predictions for an ensemble of the 1000 top-ranked readthrough sequences in a position-specific manner, we observed a peak in base pair probability, with a corresponding trough in positional entropy, at nucleotide positions 9–25 downstream of the stop codon (Figure 4C, shaded region). These results strongly suggest that structures organizing in this region promote readthrough and are consistent with prior studies that found increased levels of RNA structural elements downstream of stop codons in viral genes that undergo readthrough.52
To interrogate the influence of RNA secondary structures on readthrough activity in cells, we evaluated a series of constructs encoding hairpins of varying length using the yeast dual-FP reporter (Figure 4D). Inclusion of enriched hairpin-promoting sequence elements increased readthrough efficiency in a stem-length-dependent manner. Readthrough levels reached 21.5% for the longest stem paired with the CAAUUA motif (Figure 4D, RT-21), compared to ~9.5% readthrough in the construct lacking the hairpin (Figure 4D, RT-2). The enhancement in readthrough activity was significantly reduced when the downstream complementary linker sequence was deleted (Figure 4D, RT-22). Hairpins also enhanced readthrough when paired with weaker primary sequence motifs (Figure 4D, RT-24 and RT-25) and even stimulated 1% readthrough in the absence of any upstream sequence motif (Figure 4D, RT-23). From these data, one can conclude that primary sequence motifs and secondary structures act synergistically to stimulate stop codon readthrough.
Machine Learning-Nominated Readthrough in Human Genes.
Given the complex interplay of sequence and structural features that govern readthrough activity, we pursued a machine learning approach to aid in the identification of readthrough events from 3′-UTR sequences. We therefore constructed a feature space that included the identities of nucleotides in the first six positions following the stop codon as well as various features relating to predicted secondary structures (Figure 5A).
Figure 5.
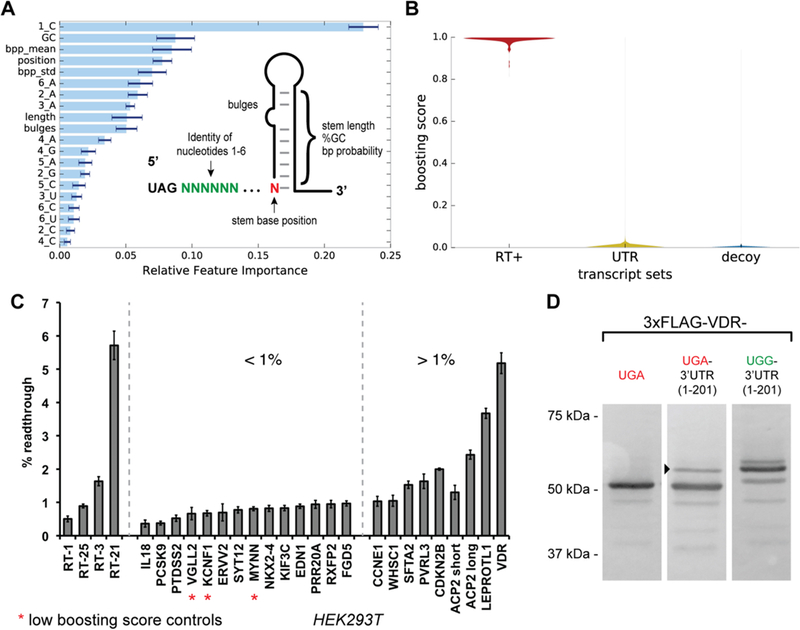
Machine learning prediction of readthrough activity from human transcripts and validation in HEK293T cell assays. (A) The machine learning feature space was constructed from position-specific nucleotide identities and secondary structure attributes. The relative importance of a feature for the additive classifier after training on the readthrough and decoy sequence sets is shown. (B) Violin plot showing the distribution of trained classifier boosting scores for the readthrough (RT+) sequence set, the human 3′-UTR transcript set, and the FLAG decoy sequence set. (C) Selected synthetic readthrough constructs and human 3′-UTRs evaluated for stop codon readthrough in HEK293T cells using the dual-FP reporter assay. Error bars represent the standard error of the mean for three or more independent transfection replicates. (D) Analysis of VDR stop codon readthrough in HEK293T cells. Three constructs were evaluated: full-length VDR containing its native UGA stop codon and no native 3′-UTR sequence, full-length VDR containing its native UGA stop codon and the first 201 nucleotides of its 3′-UTR, and full-length VDR containing a UGA to UGG mutation to its native stop codon and the first 201 nucleotides of its 3′-UTR. The arrowhead indicates the putative readthrough product.
A gradient-boosted ensemble of decision trees was trained on our binary classification task of true readthrough elements versus FLAG decoys. The most informative sequence features were 1_C, 2_A, 3_A, and 6_A, while the most informative structural features were the GC content, stem starting position, and mean base pairing probability of a predicted stem (Figure 5A). The importance of an aggregate feature over the entire ensemble of trees was calculated, and no evidence of overfitting was observed on the basis of the monotonic decrease in test set loss as a function of boosting rounds (Figure S10).
A benefit of our feature space construction is that it generalizes to RNA sequences outside of our randomized library. Thus, we collected all annotated human 3′-UTR coordinates from the UCSC table browser53 and extracted their corresponding nucleotide sequence from the hg19 genome assembly using bedtools.54 UTR sequences were trimmed to the 135 nucleotides 3′ to the stop codon, and hairpin structure prediction was performed using RNAfold. Position-specific nucleotide information and predicted structures were combined into the same feature vector representation described for the model training described above. The trained classifier was then applied to the featurized human 3′-UTRs, which classified sequences as likely or unlikely to display readthrough using decision trees (Figure S11). As expected, the vast majority of human transcripts are predicted to have no readthrough, while a narrow tail of higher-scoring UTRs are interesting from the perspective of uncovering novel regulation of translation in humans (Figure 5B).
To assay the readthrough activity of chosen 3′-UTRs in a human cell culture, we constructed a mammalian dual-fluorescent protein reporter plasmid (pCMV-dualFP) analogous to the yeast dual-FP reporter. We first validated the reporter by co-transfecting negative and positive control constructs in various proportions and by recapitulating the readthrough activity of several previously reported human 3′-UTR sequences15 (Figure S12). In addition, we confirmed readthrough activity for a series of synthetic constructs, which were also found to demonstrate appreciable readthrough (Figure 5C).
Next, we selected human 3′-UTRs to evaluate for readthrough on the basis of classifier scores and biological interest or as controls. From our sampled human 3′-UTR set, six sequences were found to promote significant levels of readthrough ranging from 1.5 to 5.1%. Among the highest-scoring human 3′-UTRs was ACP2 [score of 0.97 (Supporting Information)], which contains a primary sequence readthrough signal (CAACCA) and a predicted downstream hairpin structure that is aptly situated to promote readthrough. Previously, readthrough was predicted in ACP2 on the basis of a phylogenetic analysis, although initial human cell culture assays revealed only low levels of readthrough from a construct where the stem region was not included.15 We found that mutation of a stop codon within the stem allowed for inclusion of the structured sequence in the reporter, leading to 2.4% readthrough (Figure 5C, ACP2) and an ~2-fold increase in readthrough activity over that of the truncated construct lacking the stem (Figure 5C, trACP2). A similar result has been reported for the full-length ACP2 construct that included the hairpin element.16
Other 3′-UTRs were evaluated on the basis of the presence of novel readthrough motifs that were found from our in vitro selection. Significantly, we identified 2% readthrough for CDKN2B, which contains the novel CGCCAGR motif. In addition, we observed 3.5% readthrough for LEPROTL1 and ~1.5% readthrough for SFTA2 and PVRL3, each of which contains an E-TMV primary sequence motif. Among the sequences that contain the enriched CUAG motif, we chose to explore the 3′-UTR from the vitamin D receptor gene (VDR), as it had the highest boosting score within this group (boosting score in the top 1% of human genes). When evaluated in the 293T dual-FP assay, we found that the VDR 3′-UTR promoted 5.1% readthrough of its cognate UGA stop codon, consistent with a recent report in HeLa cell assays.55
Each of the 3′-UTR sequences evaluated would produce an extended protein product in the context of its natural gene. To determine if stop codon readthrough does indeed lead to stable extended proteins in cells, we evaluated VDR and its readthrough-extended product that contains an additional 67 amino acid residues. We expressed N-terminal 3xFLAG-tagged VDR followed by a UGA stop codon and 201 downstream nucleotides from the VDR 3′-UTR. A Western blot of transfected 293T cells demonstrated a 51 kDa band corresponding to normal full-length VDR as well as an ~58 kDa band corresponding to the predicted readthrough product (Figure 5D and Figure S13). The fainter, higher-migrating band observed in the UGG control (Figure 5D) could represent a posttranslational modification of the C-terminal extension or a frameshift event within the canonical 3′-UTR that leads to the production of a slightly longer protein product (predicted 1 kDa larger). Overall, these results demonstrate that stop codon readthrough can produce an extended VDR protein that is stable in cells.
DISCUSSION
We have applied a combination of in vitro RNA selections and high-throughput sequencing to characterize RNA features that contribute to stop codon readthrough in eukaryotes. Our data illustrate that many primary sequence motifs and secondary structure elements can act independently or in concert to promote efficient stop codon readthrough during eukaryotic translation. The E-TMV primary sequence motif, which contains the naturally occurring TMV viral motif, emerged as the dominant solution to stop codon readthrough in our selection system. This motif likely prevailed because of its small size and high readthrough efficiency. Other known motifs, such as CUAG, were also identified, but the relatively low enrichment is consistent with our assays demonstrating weaker readthrough of the UAG stop codon compared to UGA and UAA codons. Among the novel sequence motifs that we discovered, a notable 10-nucleotide sequence, CAGACUCCCG, was found to promote high readthrough activity across all stop codons when assayed in yeast cells. Other motifs, such as CGCCAGR and CUAUCCG, were found to be most active with UGA and UAA stop codons.
In contrast to prior readthrough context studies, our in vitro selection enabled the search of an expansive RNA sequence space within a mammalian cell lysate. Using a selection strategy based on mRNA display to enrich readthrough signals de novo, we explored a library of 1013 sequence variants containing a 75-nucleotide randomized region downstream of the stop codon. While it is possible that the selection could have enriched frameshift signals in addition to readthrough signals, the absence of canonical slippery site sequences upstream of the stop codon and the requirement for in-frame translation to produce the C-terminal hexahistidine tag likely limited the enrichment of frameshift sequences. Ultimately, our selection led to the discovery of novel readthrough motifs that were eventually identified in human 3′-UTRs. Our HTS data set then made it possible to define a high-resolution map of sequence and structural constraints for a very large number of readthrough signals. As a result, our data reflect a broad range of possible readthrough promoting elements. While our in vitro selection data reflect readthrough of the UAG stop codon, a similar approach could be used in the future to evaluate readthrough signals for UAA and UGA stop codons.
Our results may also provide insights into the molecular mechanisms of readthrough in eukaryotes. High-resolution cryo-electron microscopy structures have been reported for eukaryotic release factor 1 (eRF1) bound to the ribosome in active recognition of termination codons.56 These structures reveal that eRF1 recognition is accompanied by contraction of the mRNA, stacking of the +2 and +3 stop codon purines, and movement of the +4 nucleotide into the ribosomal A site. This process should require a change in downstream mRNA structure or positioning and thus suggests that interactions outside of the A site that restrict mRNA conformational flexibility might inhibit translation termination.
In evaluating eukaryotic 18S rRNA in the proximity of the A site and mRNA tunnel, we found conserved sequences in helix 1 and helix 18 that are complementary to two of the primary sequence readthrough motifs, CARYYA and CAGACU, that emerged from our selection (Figure S14A). If these primary sequence readthrough motifs form stable interactions with the 40S subunit, they may promote readthrough by impeding the requisite mRNA conformational changes that accompany eRF1 stop codon recognition. Secondary structures organizing in the proximity of the entryway of the mRNA tunnel might also interfere with eRF1-induced mRNA movement and synergize with primary sequence readthrough motifs (Figure S14B). Further biochemical studies will be necessary to evaluate if such interactions exist and whether they are responsible for readthrough phenomena.
With our large collection of readthrough signal examples containing diverse RNA sequence and structural features, we developed a machine learning approach to rank 3′-UTRs based on their likelihood of promoting readthrough. Our RNA-centric approach is complementary to other methods that have been explored such as phylogenetic analysis and ribosome profiling. First, our method does not depend on phylogenetic conservation of peptide extensions and thus can be applied for discovery of nonconserved readthrough events. Second, our data and discovery approach are potentially applicable to many eukaryotic organisms, whereas ribosome profiling experiments must be repeated for each new organism or cell type that is examined. Moreover, many genes are not expressed within particular cell lines or are present at levels below the threshold of readthrough detection by profiling. By contrast, our trained ensemble classifier should be directly applicable to readthrough discovery in eukaryotic organisms where genome or transcriptome sequencing data are available. In combination with other methods, our approach should accelerate the identification of readthrough events in eukaryotes.
Stop codon readthrough produces C-terminally extended proteins that may differ substantially from their standard translation counterparts. In the human 3′-UTRs examined in this study that were found to undergo readthrough, peptide extensions ranging from 7 to 71 amino acids are predicted. Our analysis of transfected human VDR constructs demonstrates that readthrough-extended protein is expressed in human cells, though it remains to be seen whether endogenous transcripts also generate readthrough products. If so, readthrough extensions could function as protein–protein interaction mediators, subcellular targeting signals, post-translational modification substrates, or even independent protein domains. Further biological studies will be necessary to determine the biological significance of stop codon readthrough in these and other cellular genes.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Ruben L. Gonzalez for helpful discussions. Sequencing was performed using the Illumina Hi-Seq platform at the Columbia Genome Center.
Funding
This research was supported by funding from the National Institutes of Health (Grants R01 R01AI110794 to V.W.C., F30 CA174357 to A.V.A., U54 CA193313 to R.R., and TL1 TR000082 to S.Z.).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.8b01280.
Figures S1–S14 and DNA sequences and plasmids used in this study (Tables S1–S6) (PDF)
Accession Codes
Vitamin D receptor (human), UniProt entry P11473.
The authors declare no competing financial interest. Sequencing data, unique sequences and read counts, and data processing and analysis scripts can be found at https://github.com/szairis/readthru.
REFERENCES
- (1).Crick FH, Barnett L, Brenner S, and Watts-Tobin RJ (1961) General nature of the genetic code for proteins. Nature 192, 1227–1232. [DOI] [PubMed] [Google Scholar]
- (2).Zaher HS, and Green R (2009) Fidelity at the Molecular Level: Lessons from Protein Synthesis. Cell 136, 746–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Gesteland RF, and Atkins JF (1996) Recoding: Dynamic Reprogramming of Translation. Annu. Rev. Biochem. 65, 741–768. [DOI] [PubMed] [Google Scholar]
- (4).Kearse MG, and Wilusz JE (2017) Non-AUG translation: a new start for protein synthesis in eukaryotes. Genes Dev. 31, 1717–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Komar AA, and Hatzoglou M (2011) Cellular IRES-mediated translation. Cell Cycle 10, 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Atkins JF, Loughran G, Bhatt PR, Firth AE, and Baranov PV (2016) Ribosomal frameshifting and transcriptional slippage: From genetic steganography and cryptography to adventitious use. Nucleic Acids Res. 44, 7007–7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ryan MD, and Drew J (1994) Foot-and-mouth disease virus 2A oligopeptide mediated cleavage of an artificial polyprotein. EMBO J. 13, 928–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Beier H, and Grimm M (2001) Misreading of termination codons in eukaryotes by natural nonsense suppressor tRNAs. Nucleic Acids Res. 29, 4767–4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Baranov PV, Atkins JF, and Yordanova MM (2015) Augmented genetic decoding: global, local and temporal alterations of decoding processes and codon meaning. Nat. Rev. Genet. 16, 517–529. [DOI] [PubMed] [Google Scholar]
- (10).Bertram G, Innes S, Minella O, Richardson J, and Stansfield I (2001) Endless possibilities: translation termination and stop codon recognition. Microbiology 147, 255–269. [DOI] [PubMed] [Google Scholar]
- (11).Harrell L, Melcher U, and Atkins JF (2002) Predominance of six different hexanucleotide recoding signals 3′ of read-through stop codons. Nucleic Acids Res. 30, 2011–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).McCaughan KK, Brown CM, Dalphin ME, Berry MJ, and Tate WP (1995) Translational termination efficiency in mammals is influenced by the base following the stop codon. Proc. Natl. Acad. Sci U. S. A. 92, 5431–5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bonetti B, Fu L, Moon J, and Bedwell DM (1995) The Efficiency of Translation Termination is Determined by a Synergistic Interplay Between Upstream and Downstream Sequences in Saccharomyces cerevisiae. J. Mol. Biol. 251, 334–345. [DOI] [PubMed] [Google Scholar]
- (14).Dabrowski M, Bukowy-Bieryllo Z, and Zietkiewicz E (2015) Translational readthrough potential of natural termination codons in eucaryotes – The impact of RNA sequence. RNA Biol. 12, 950–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Loughran G, Chou M-Y, Ivanov IP, Jungreis I, Kellis M, Kiran AM, Baranov PV, and Atkins JF (2014) Evidence of efficient stop codon readthrough in four mammalian genes. Nucleic Acids Res. 42, 8928–8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Loughran G, Howard MT, Firth AE, and Atkins JF (2017) Avoidance of reporter assay distortions from fused dual reporters. RNA 23, 1285–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Shehu-Xhilaga M, Crowe SM, and Mak J (2001) Maintenance of the Gag/Gag-Pol ratio is important for human immunodeficiency virus type 1 RNA dimerization and viral infectivity. J. Virol. 75, 1834–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Weiner AM, and Weber K (1973) A single UGA codon functions as a natural termination signal in the coliphage Qβ coat protein cistron. J. Mol. Biol. 80, 837–855. [DOI] [PubMed] [Google Scholar]
- (19).Pelham HR (1978) Leaky UAG termination codon in tobacco mosaic virus RNA. Nature 272, 469–471. [DOI] [PubMed] [Google Scholar]
- (20).Firth AE, and Brierley I (2012) Non-canonical translation in RNA viruses. J. Gen. Virol. 93, 1385–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Skuzeski JM, Nichols LM, Gesteland RF, and Atkins JF (1991) The signal for a leaky UAG stop codon in several plant viruses includes the two downstream codons. J. Mol. Biol. 218, 365–373. [DOI] [PubMed] [Google Scholar]
- (22).Napthine S, Yek C, Powell ML, Brown TDK, and Brierley I (2012) Characterization of the stop codon readthrough signal of Colorado tick fever virus segment 9 RNA. RNA 18, 241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wills NM, Gesteland RF, and Atkins JF (1991) Evidence that a downstream pseudoknot is required for translational readthrough of the Moloney murine leukemia virus gag stop codon. Proc. Natl. Acad. Sci. U. S. A. 88, 6991–6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Green L, and Goff SP (2015) Translational readthrough promoting drugs enhance pseudoknot-mediated suppression of the stop codon at the Moloney murine leukemia virus gag-pol junction. J. Gen. Virol. 96, 3411–3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).True HL, and Lindquist SL (2000) A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature 407, 477–483. [DOI] [PubMed] [Google Scholar]
- (26).Freitag J, Ast J, and Bölker M (2012) Cryptic peroxisomal targeting via alternative splicing and stop codon read-through in fungi. Nature 485, 522–525. [DOI] [PubMed] [Google Scholar]
- (27).Schueren F, Lingner T, George R, Hofhuis J, Dickel C, Gärtner J, and Thoms S (2014) Peroxisomal lactate dehydrogenase is generated by translational readthrough in mammals. eLife 3, No. e03640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Stiebler AC, Freitag J, Schink KO, Stehlik T, Tillmann BAM, Ast J, and Bölker M (2014) Ribosomal Readthrough at a Short UGA Stop Codon Context Triggers Dual Localization of Metabolic Enzymes in Fungi and Animals. PLoS Genet. 10, No. e1004685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Eswarappa SM, Potdar AA, Koch WJ, Fan Y, Vasu K, Lindner D, Willard B, Graham LM, DiCorleto PE, and Fox PL (2014) Programmed Translational Readthrough Generates Antiangiogenic VEGF-Ax. Cell 157, 1605–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Jungreis I, Lin MF, Spokony R, Chan CS, Negre N, Victorsen A, White KP, and Kellis M (2011) Evidence of abundant stop codon readthrough in Drosophila and other metazoa. Genome Res. 21, 2096–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Namy O, Duchateau Nguyen G, Hatin I, Denmat SH-L, Termier M, and Rousset J-P (2003) Identification of stop codon readthrough genes in Saccharomyces cerevisiae. Nucleic Acids Res. 31, 2289–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Dunn JG, Foo CK, Belletier NG, Gavis ER, and Weissman JS (2013) Ribosome profiling reveals pervasive and regulated stop codon readthrough in Drosophila melanogaster. eLife 2, No. e01179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Namy O, Duchateau-Nguyen G, and Rousset J-P (2002) Translational readthrough of the PDE2 stop codon modulates cAMP levels in Saccharomyces cerevisiae. Mol. Microbiol. 43, 641–652. [DOI] [PubMed] [Google Scholar]
- (34).Namy O, Hatin I, and Rousset J-P (2001) Impact of the six nucleotides downstream of the stop codon on translation termination. EMBO Rep. 2, 787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Jungreis I, Chan CS, Waterhouse RM, Fields G, Lin MF, and Kellis M (2016) Evolutionary Dynamics of Abundant Stop Codon Readthrough. Mol. Biol. Evol. 33, 3108–3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Yordanova MM, Loughran G, Zhdanov AV, Mariotti M, Kiniry SJ, O’Connor PBF, Andreev DE, Tzani I, Saffert P, Michel AM, Gladyshev VN, Papkovsky DB, Atkins JF, and Baranov PV (2018) AMD1 mRNA employs ribosome stalling as a mechanism for molecular memory formation. Nature 553, 356–360. [DOI] [PubMed] [Google Scholar]
- (37).Liu R, Barrick JE, Szostak JW, and Roberts RW (2000) Optimized synthesis of RNA-protein fusions for in vitro protein selection. Methods Enzymol 318, 268–293. [DOI] [PubMed] [Google Scholar]
- (38).Seelig B (2011) mRNA display for the selection and evolution of enzymes from in vitro-translated protein libraries. Nat. Protoc. 6, 540–552. [DOI] [PubMed] [Google Scholar]
- (39).Anzalone AV, Lin AJ, Zairis S, Rabadan R, and Cornish VW (2016) Reprogramming eukaryotic translation with ligand-responsive synthetic RNA switches. Nat. Methods 13, 453–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Gietz RD, and Schiestl RH (2007) High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 31–34. [DOI] [PubMed] [Google Scholar]
- (41).Friedman J, Hastie T, and Tibshirani R (2000) Additive logistic regression: a statistical view of boosting (With discussion and a rejoinder by the authors). Ann. Stat. 28, 337–407. [Google Scholar]
- (42).Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, Vanderplas J, Passos A, Cournapeau D, Brucher M, Perrot M, and Duchesnay É (2011) Scikit-learn: Machine Learning in Python. Journal of Machine Learning Research 12, 2825–2830. [Google Scholar]
- (43).Roberts RW, and Szostak JW (1997) RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc. Natl. Acad. Sci. U. S. A. 94, 12297–12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Ellington AD, and Szostak JW (1990) In vitro selection of RNA molecules that bind specific ligands. Nature 346, 818–822. [DOI] [PubMed] [Google Scholar]
- (45).Tuerk C, and Gold L (1990) Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249, 505–510. [DOI] [PubMed] [Google Scholar]
- (46).Robertson DL, and Joyce GF (1990) Selection in vitro of an RNA enzyme that specifically cleaves single-stranded DNA. Nature 344, 467–468. [DOI] [PubMed] [Google Scholar]
- (47).Cho G, Keefe AD, Liu R, Wilson DS, and Szostak JW (2000) Constructing high complexity synthetic libraries of long ORFs using In Vitro selection. J. Mol. Biol. 297, 309–319. [DOI] [PubMed] [Google Scholar]
- (48).Houck-Loomis B, Durney MA, Salguero C, Shankar N, Nagle JM, Goff SP, and D’Souza VM (2011) An equilibrium-dependent retroviral mRNA switch regulates translational recoding. Nature 480, 561–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Cridge AG, Crowe-McAuliffe C, Mathew SF, and Tate WP (2018) Eukaryotic translational termination efficiency is influenced by the 3′ nucleotides within the ribosomal mRNA channel. Nucleic Acids Res. 46, 1927–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Tsonis AA, Elsner JB, and Tsonis PA (1991) Periodicity in DNA coding sequences: Implications in gene evolution. J. Theor. Biol. 151, 323–331. [DOI] [PubMed] [Google Scholar]
- (51).Lorenz R, Bernhart SH, Höner zu Siederdissen C, Tafer H, Flamm C, Stadler PF, and Hofacker IL (2011) ViennaRNA Package 2.0. Algorithms Mol. Biol. 6, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Firth AE, Wills NM, Gesteland RF, and Atkins JF (2011) Stimulation of stop codon readthrough: frequent presence of an extended 3′ RNA structural element. Nucleic Acids Res. 39, 6679–6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, and Kent WJ (2004) The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 32, 493D–496D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Quinlan AR, and Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Loughran G, Jungreis I, Tzani I, Power M, Dmitriev RI, Ivanov IP, Kellis M, and Atkins JF (2018) Stop codon readthrough generates a C-terminally extended variant of the human vitamin D receptor with reduced calcitriol response. J. Biol. Chem. 293, 4434–4444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Brown A, Shao S, Murray J, Hegde RS, and Ramakrishnan V (2015) Structural basis for stop codon recognition in eukaryotes. Nature 524, 493–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.