

Watch a video presentation of this article
Autoimmune hepatitis (AIH), a chronic hepatic necroinflammatory disorder, occurs mostly in women. AIH is characterized by prominent interface hepatitis and varying degrees of lobular hepatitis. Laboratory studies show elevated aminotransferase values, hypergammaglobulinemia, and serologically demonstrable tissue‐directed autoantibodies. The clinical presentation of autoimmune hepatitis has been reviewed in this edition of Clinical Liver Disease.1 Liver biopsy is almost always mandated to establish the diagnosis and estimate the prognosis.2, 3
Immune mechanisms contribute to other liver diseases, including acute and chronic hepatitis caused by hepatotropic viruses A, B and C, primary biliary cirrhosis (PBC), primary sclerosing cholangitis (PSC), and drug‐induced chronic hepatitis as well as alcoholic liver disease, Wilson's disease, and perhaps alpha‐1‐antitrypsin deficiency. However, the conditions principally considered to be autoimmune liver diseases are autoimmune hepatitis (including drug‐induced autoimmune hepatitis), PBC, PSC, and autoimmune cholangitis/cholangiopathy.
AIH should be considered in any patient with unexplained elevated serum aminotransferase values, particularly because a timely diagnosis and appropriate therapy can be of great value in suppressing disease activity. Untreated, AIH universally leads to cirrhosis and its complications, including death, and there is a low but significant incidence of hepatocellular carcinoma. Remission, both clinical and biochemical, is achieved by as many as 85% of patients, and the need for transplantation can be significantly reduced.4 Establishing the correct diagnosis can be challenging because of the heterogeneity of the clinical presentation and the absence of a specific diagnostic test. Simple scoring systems5 rely principally on the clinical history and an evaluation of autoantibodies, but they also include an evaluation of liver biopsy samples.
Histopathology
Biopsy allows an assessment of the inflammatory activity and the severity of fibrosis, which is almost always seen, often with the initial biopsy. Importantly, neither aminotransferase values nor immunoglobulin G levels reflect the degree of tissue damage. An adequate tissue sample is vital, and cores with a total length of at least 2.5 cm and with at least 10 portal tracts are needed.6, 7 Although the histological features listed in the modified staging system are generally integral to establishing the diagnosis, considerable variation and other features can be seen.
Differential diagnoses include viral hepatitides (in practice the most common consideration), other immune liver disorders, drug reactions (in which eosinophils are generally more prominent), alcoholic liver disease (with fat and Mallory hyaline), alpha‐1‐antitrypsin deficiency and Wilson disease (both of which can be histochemically or biochemically demonstrated), and other diagnoses. In recent years, it has been recognized that the biopsy appearance of chronic hepatitis E can resemble autoimmune hepatitis with a lymphoplasmacytic portal infiltrate but generally with less prominent interface and lobular inflammation. It has long been recognized that biopsy findings for acute hepatitis A can also resemble those for autoimmune hepatitis, but clinical and serological features generally establish the correct diagnosis. In most cases, histological features as well as histochemistry and immunohistochemistry make specific identification feasible (Table 1).
Table 1.
| Hepatitis B | Modest interface hepatitis |
| Ground‐glass cells | |
| Immunostain‐demonstrable hepatitis B core and surface antigens | |
| Hepatitis C | Modest interface hepatitis |
| Lymphohistiocytic infiltrate with lymphoid aggregates, including germinal centers | |
| Few, scattered acidophilic bodies | |
| Hepatitis E | Lymphoplasmacytic portal infiltrate |
| Modest interface hepatitis | |
| Mild or no lobular hepatitis | |
| Alcoholic liver disease | Steatosis |
| Balloon hepatocytes | |
| Mallory hyaline | |
| Pericellular, zone 3 (chicken‐wire) fibrosis | |
| Wilson disease | Increased numbers of glycogenated nuclei |
| Rarely, visible zone 1 copper granules | |
| Copper‐associated protein demonstrable with Victoria blue or orcein stains | |
| Copper assay for quantitation | |
| Genetic hemochromatosis | Iron granules visible in zone 1 hepatocytes |
| Iron stain (e.g., Prussian blue) | |
| Iron assay for quantitation | |
| Alpha‐1‐antitrypsin deficiency | Globules potentially seen in zone 1 hepatocytes with hematoxylin‐eosin |
| Globules easily seen with diastase–periodic acid Schiff and/or immunostaining | |
| Primary biliary cirrhosis | Interface hepatitis minimal |
| Characteristic bile duct lesion | |
| Progressive bile duct loss | |
| Granulomas | |
| Cholate stasis | |
| Bile ductular reaction (biliary interface hepatitis) | |
| Increased copper and/or copper‐associated protein | |
| Sclerosing cholangitis | Interface hepatitis unusual in stage 1 disease |
| Interface hepatitis can be prominent in stage 2 disease. | |
| Mild bile duct lesion | |
| Periductal concentric laminar fibrosis (onion skin) | |
| Bile duct loss with smudgy fibrosis | |
| Variable, often mild ductular reaction | |
| Drug reaction | Portal neutrophils and eosinophils |
| Plasma cells usually not prominent | |
| Fibrosis usually not seen |
In autoimmune hepatitis, a low‐magnification image strongly suggests the diagnosis because of prominent interface and zone 1 lobular hepatitis (Fig. 1); this picture is unusual for the various conditions listed previously. When interface hepatitis is absent or mild, AIH is unlikely, and care should be taken to prevent unnecessary therapy. An irregularly distributed and relatively intense portal infiltrate with either periportal or (in cases with bridging fibrosis or cirrhosis) paraseptal interface hepatitis is typical (Fig. 1, 2. Hepatocyte necrosis (acidophilic bodies and apoptotic bodies) is seen in periportal areas as well as the rest of the lobule. Plasma cells with accompanying eosinophils and lymphocytes are characteristic, but they are not always the dominant cells, and in the appropriate clinical setting, the diagnosis can be made with only modest numbers.8 In some cases, lymphocytes are dominant instead. When this occurs, plasma cells in small clusters at the interface support the diagnosis (Fig. 3). Pseudoacini (rosettes) are seen with significant lobular involvement and regenerative activity (Fig. 4). Rosettes and plasma cells are typical but are not pathognomonic or consistently seen. Furthermore, plasma cells and rosettes occur with other liver diseases. Zone 1 (periportal) emperipolesis, the engulfing of lymphocytes by hepatocytes, occurs in the interface hepatitis area. There may be giant‐cell transformation (giant‐cell hepatitis).
Figure 1.
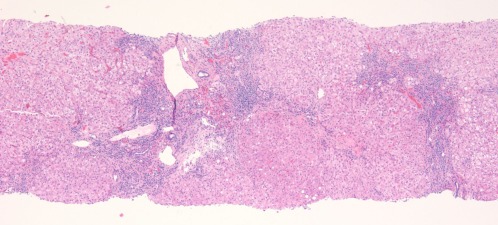
AIH. This low‐magnification image (hematoxylin‐eosin, ×40) shows expanded portal tracts with effacement of the interface by a lymphoplasmacytic infiltrate including many plasma cells. The connective tissue stain shows early fibrosis and regenerative activity with 2‐cell‐thick liver plates and early nodule formation.
Figure 2.
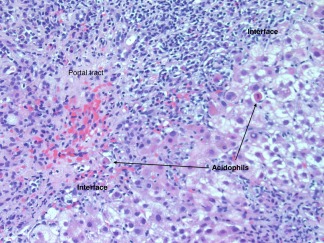
AIH. This medium‐magnification image (hematoxylin‐eosin, ×200) shows a portal tract with an intense lymphoplasmacytic infiltrate effacing the interface with rosette formation and hepatocyte necrosis (acidophilic bodies).
Figure 3.
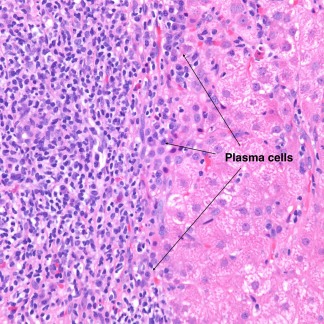
AIH. This high‐magnification image (hematoxylin‐eosin, ×400) shows a predominantly lymphocytic portal infiltrate with clusters of plasma cells at the interface.
Figure 4.
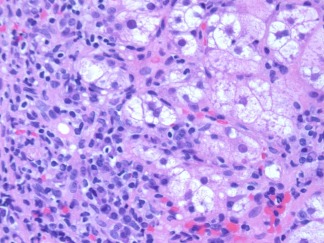
AIH. This image (hematoxylin‐eosin, ×400) shows rosettes.
Figure 5.
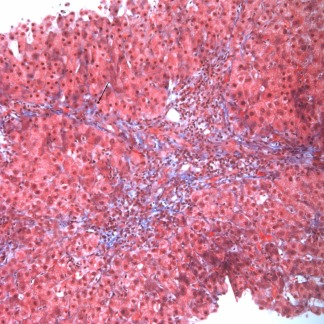
AIH. This first‐biopsy image (Masson trichrome, ×200) shows fibrosis with early bridge formation (arrow).
Figure 6.
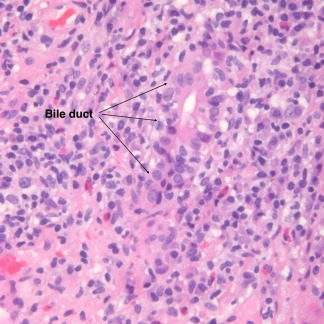
AIH with overlap syndrome. This image (hematoxylin‐eosin, ×200) shows nonsuppurative cholangitis consistent with PBC.
Severe inflammation extends beyond the periportal zone with parenchymal collapse and, not uncommonly, bridging necrosis, especially with acute clinical relapse and when AIH is acute and fulminant.9 Portal‐to‐portal or portal‐to‐central fibrosis and cirrhosis are seen (Fig. 5).
Key histological features contributing positively to a score establishing the diagnosis of AIH according to the revised International Autoimmune Hepatitis Group modified staging system4 are (1) interface hepatitis (+3), which is the most important; (2) a lymphoplasmacytic infiltrate (+1); and (3) rosette formation (+1). Other features also help to establish the diagnosis. For example, AIH differs from chronic hepatitis C in having more severe lobular inflammation and necrosis as well as greater numbers of plasma cells, more marked interface hepatitis, and broad areas of parenchymal collapse (Table 2). Zone 3 (centrilobular) necrosis is well described in AIH but is often inadequately recognized.10 Zone 3 necrosis without fulminant hepatitis can lead to erroneous diagnoses such as ischemia/hypoxia and toxic/drug injury. Biliary changes are uncommon in AIH and are almost always indicative of some other disorder. Isolated bile duct injury, however, can be seen and does not exclude AIH.11 When anti‐nuclear antibody values are significantly increased in association with biopsy‐demonstrable bile duct injury (Fig. 6), the diagnosis of AIC is likely. Negative score findings are the absence of these three findings (−5), biliary changes (−3), and features suggesting an alternative etiology (−3).
Table 2.
Histopathological Features Most Useful in Differentiating AIH From Chronic Hepatitis C Virus
| Feature | AIH | Hepatitis C Virus |
|---|---|---|
| Lobular inflammation/necrosis | +−+++ | +/− |
| Plasma cells | +−+++ | 0−+ |
| Interface hepatitis | +−+++ | 0−++ |
| Parenchymal collapse | +−+++ | 0 |
| Steatosis | 0−+ | +−+++a |
| Portal lymphoid aggregates | 0−+ | +−+++ |
| Germinal center formation in lymphoid aggregates | 0 | 0−++ |
| Bile duct injury† | 0−+ | +−++ |
This table has been adapted with permission from Biopsy Interpretation of the Liver, 2nd ed.12
Steatosis is particularly seen with genotype 3 hepatitis C virus.
Bile duct injury is rarely seen in AIH but is characteristic of AIC.11
0 = no change, += minimal or mild change, ++= moderate change, +++= marked change.
There are no known direct microscopic correlates for the various identifiable autoantibodies. For example, the biopsy findings for liver/kidney microsome (LKM)–associated AIH are similar to those for other forms (Fig. 7). When overlap syndromes (e.g., AIH + PBC, AIH + PSC, and AIH + AIC) occur, atypical histological changes can be seen (Fig. 6).12, 13 Rarely, granulomas are seen. Other coincidental disorders, such as hepatitis C, alcoholic liver disease, human immunodeficiency virus positivity, and iron storage disease, affect morphology. Increasingly, drug effects must be considered. AIH‐like microscopic changes caused by drugs are generally resolved with the cessation of medication, but chronic drug‐induced AIH also occurs. Fibrosis and cirrhosis are distinctly unusual in drug‐induced AIH, but cholestasis, portal neutrophils, and eosinophils are likely. The experience of the reviewing pathologist can also affect the ability to establish the diagnosis.
Figure 7.
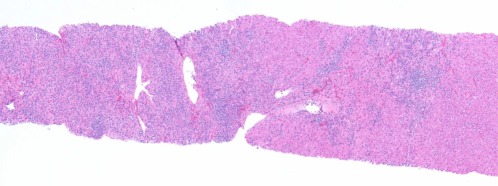
LKM AIH. Similar to Fig. 1, this low‐magnification image (hematoxylin‐eosin, ×20) shows a marked lymphoplasmacytic infiltrate with effacement of the interface.
Fulminant AIH is uncommon and is morphologically indistinguishable from other forms of massive/submassive necrosis.12, 13
Cirrhosis in AIH generally shows a greater degree of inflammation than cirrhosis due to other causes. Septa are easily recognized, as are areas of prior parenchymal collapse, and the developing nodules vary greatly in size. Dysplastic nodules can be seen, as can small hepatocellular carcinomas.
Key features of AIH are summarized in Table 3.
Table 3.
Key Histopathology Features of AIH
| 1. Liver biopsy shows a moderate to severe necroinflammatory process with prominent portal inflammation, interface hepatitis, a lymphoplasmacytic infiltrate including many plasma cells, and acinar transformation of hepatocytes (rosettes). |
| 2. Plasma cells are not always the dominant inflammatory cells and may be prominent only at the interface. |
| 3. Fibrosis/cirrhosis is often seen on first biopsy. |
| 4. It may present as acute, fulminant liver failure with massive or submassive necrosis, including centrilobular (zone 3) necrosis. |
| 5. Autoimmune liver disease variants may show features of more than one immune disorder (overlap syndromes). |
| 6. AIC is a distinct disorder histologically resembling PBC (without anti‐mitochondrial antibodies in serum and with anti‐nuclear antibodies). |
Abbreviations
- AIC
autoimmune cholangitis/cholangiopathy
- AIH
autoimmune hepatitis
- LKM
liver/kidney microsome
- PBC
primary biliary cirrhosis
- PSC
primary sclerosing cholangitis.
Potential conflict of interest: Nothing to report.
References
- 1. Lucey MR, Vierling J. Clinical presentation and natural history of autoimmune hepatitis. Clin Liver Dis 2014;9‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liberal R, Grant CR, Mieli‐Vergani G, Vergani D. Autoimmune hepatitis: a comprehensive review. J Autoimmun 2013;41:126‐139. [DOI] [PubMed] [Google Scholar]
- 3. Weiler‐Norman C, Sebode M, Lohse AW. Autoimmune hepatitis 2103 and beyond. Minerva Gastroenterol Dietol 2013;59:133‐141. [PubMed] [Google Scholar]
- 4. Strassburg CP. Autoimmune hepatitis. Dig Dis 2013;31:155‐163. [DOI] [PubMed] [Google Scholar]
- 5. Alvarez F, Berg PA, Bianchi FB, Bianchi L, Burroughs AK, Cancado EL, et al. International Autoimmune Hepatitis Group report: a review of criteria for diagnosis of autoimmune hepatitis. J Hepatol 1999;31:929‐938. [DOI] [PubMed] [Google Scholar]
- 6. Bedossa P, Dargere D, Paradis V. Sampling variability of liver fibrosis in chronic hepatitis C. Hepatology 2003;38:1449‐1457. [DOI] [PubMed] [Google Scholar]
- 7. Rousselet MC, Michalak S, Dupré F, Croué A, Bedossa P, Saint‐André JP, et al. Sources of variability in histological scoring of chronic viral hepatitis. Hepatology 2005;41:257‐264. [DOI] [PubMed] [Google Scholar]
- 8. Vergani D, Longhi MS, Bogdanos DP, Ma Y, Mieli‐Vergani G. Autoimmune hepatitis. Semin Immunopathol 2009;31:421‐435. [DOI] [PubMed] [Google Scholar]
- 9. Czaja AJ. Acute and acute severed (fulminant) autoimmune hepatitis. Dig Dis Sci 2013;58:897‐914. [DOI] [PubMed] [Google Scholar]
- 10. Zen Y, Notsumata K, Tanaka N, Nakanuma Y. Hepatic centrilobular zonal necrosis with positive antinuclear antibody: a unique subtype or early disease in autoimmune hepatitis. Hum Pathol 2007;38:1669‐1675. [DOI] [PubMed] [Google Scholar]
- 11. Ludwig J, Czaja AJ, Dickson ER, LaRusso NF, Weisner RH. Manifestations of nonsuppurative cholangitis in chronic hepatobiliary disease: morphologic spectrum, clinical correlations and terminology. Liver 1984;4:105‐116. [DOI] [PubMed] [Google Scholar]
- 12. Geller SA. Autoimmune hepatitis and related disorders In: Geller SA, Petrovic LM, eds. Biopsy Interpretation of the Liver. 2nd ed Philadelphia, PA: Lippincott Williams & Wilkins; 2009:120‐135. [Google Scholar]
- 13. Washington MK, Manns MP. Autoimmune hepatitis In: Burt A, Portmann B, Ferrell L, eds. MacSween's Pathology of the Liver. 6th ed Edinburgh, United Kingdom: Churchill Livingstone; 2012:467‐490. [Google Scholar]