

Abstract
Adoption of electronic informed consent (eConsent) for research remains low despite evidence of improved patient comprehension, usability, and workflow processes compared to paper. At our institution, we implemented an eConsent workflow using REDCap, a widely used electronic data capture system. The goal of this study was to evaluate the extent to which the REDCap eConsent solution adhered to federal guidance for eConsent. Of 29 requirements derived from sixteen recommendations from the United States Office for Human Research Protections (OHRP) and Food and Drug Administration (FDA), the REDCap eConsent solution supported 24 (86%). To the best of our knowledge, this is among the first studies to evaluate an eConsent approach’s support for federal guidance. Findings suggest use of REDCap may help other institutions overcome barriers to eConsent adoption, and that OHRP and FDA expand guidance to recommend eConsent solutions integrate with enterprise clinical and research information systems.
Introduction
Experts from academic medical centers and the biopharmaceutical industry have identified electronic informed consent (eConsent) as beneficial to research stakeholders including patients, healthcare organizations, and sponsors. (1,2) Studies have demonstrated numerous benefits of eConsent compared to the standard paper-based approach, including improved patient comprehension, usability, and workflow processes.(3–7) Despite these benefits, adoption of eConsent for research in academic medical centers is low due to barriers including funding and system selection.(8) In contrast, REDCap, an electronic data capture system maintained by Vanderbilt University, is free for use by academic medical centers and has seen widespread adoption at more than 2,500 institutions worldwide.
To overcome common barriers to adoption of eConsent, our institution implemented a REDCap-based workflow intended to mimic an existing paper-based approach. Although the literature describes use of REDCap for eConsent (7), to our knowledge no studies have evaluated REDCap for eConsent’s ability to replace paper and support federal guidance for electronic informed consent.(9) Because of the widespread adoption of REDCap, REDCap-based eConsent approaches have the potential to generalize broadly. The goal of this study was to evaluate a REDCap-based eConsent approach’s usage, support for local requirements, and adherence to federal guidance.
Methods
Setting
Weill Cornell Medicine (WCM), located in New York City, consists of Weill Cornell Medical College, Weill Cornell Graduate School of Medical Sciences, and Weill Cornell Physician Organization. The WCM Physician Organization consists of over 900 physicians to serving more than 2 million patients at over 20 clinics across the metro area. Physicians have academic appointments in Weill Cornell Medical College and admitting privileges to NewYork-Presbyterian Hospital (NYP), a teaching hospital with over 2,600 beds. In addition to its Clinical and Translation Science Center (CTSC) funded by the National Center for Advancing Translation Sciences (NCATS), WCM and NYP formed the Joint Clinical Trials Office (JCTO) to further clinical research by sharing infrastructure. The JCTO supports a large number of clinical trials, such as collaborating with the Center for Advanced Digestive Care (CADC) and the Division of Hematology and Medical Oncology (HemOnc).
WCM Information Technologies & Services (ITS) supports clinical trials by providing a variety of services, such as maintaining a clinical trial management system (CTMS) (10) and a biospecimen information management system (BIMS).(11) WCM physicians use EpicCare Ambulatory in outpatient clinics and Allscripts Sunrise Clinical Manager in inpatient settings, which are maintained by separate information technology teams from WCM and NYP, respectively.
In 2016, CADC and HemOnc were conducting longitudinal registry studies, in their respective disease areas, that included the collection of biospecimens and request to contact for future studies. The paper-based consent workflow involved manual data entry of demographics and elections. To streamline processes, JCTO engaged ITS to determine if an electronic informed consent form (ICF) solution could be implemented in-house based upon their requirements. Key requirements included a static electronic ICF that mirrored the traditional paper ICF with an electronic signature and compliance with institutional (e.g., ITS security) and legal (e.g., HIPAA) guidelines. The ITS pilot using REDCap for electronic ICF began on June 1, 2016 and ended on November 30, 2016.
System description
To support CADC and HemOnc research consent requirements, we deployed a REDCap project for each study to enable patients to provide, update, and withdrawal consent, as well as study teams to track participant status and maintain versions of informed consent forms and approved research protocols. The sections that follow describe the REDCap-based workflows.
Providing initial consent
Figure 1 illustrates the workflow for participants to provide initial consent to a study. Of note, the workflow assumes that a physician and patient discussed study participation, the patient expressed interest in participating, and the physician notified the research coordinator to discuss initial consent with the patient based on the patient’s expression of interest.
Figure 1.

Workflow required by research coordinator and participant for informed consent using REDCap on a tablet.
After logging into a tablet computer, a research coordinator opened a web browser and navigated to a REDCap survey for the study. As shown in Figure 2, a survey form contained fields for a research coordinator to transcribe the patient’s medical record number (MRN) and name from the EHR into form fields and select the clinic site where the consent activity occurred. After entering the demographics, the research coordinator then handed the tablet to the patient for review. Of note, the REDCap survey form displayed a stamp approved by the WCM Institutional Review Board (IRB) including study approval and expiration dates.
Figure 2.
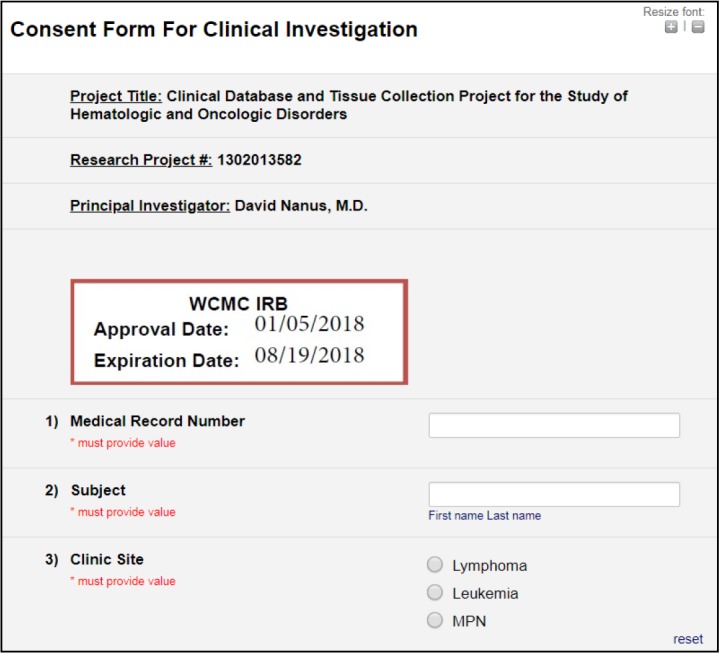
Initial screen displaying fields for participant demographics.
The patient then read the consent form text, scrolling down as necessary, until reaching a section asking the participant to check a box indicating he or she has read the informed consent. Upon clicking the “Submit” button, the browser then displayed a password-protected REDCap Survey Queue page, which prevented a participant from advancing without assistance from a research coordinator. Clicking the “Submit” button also generated an automated email notification to a clinic site-specific email distribution list (determined by the clinic site selected in the survey form) to inform a research coordinator, who may or may not be in the same room as the patient depending on clinic workflow and patient preferences, that the patient completed reading the informed consent. The participant then handed the tablet to the research coordinator.
If the patient indicated he or she did not want to participate in the study, the research coordinator then requested removal of the patient’s record from the REDCap project by emailing research ITS support. The process mimicked the paper shredding of an informed consent form for a patient who started the informed consent process and ultimately elected not to participate.
If the patient indicated he or she wanted to participate in the study, the research coordinator then entered the password into the password-protected REDCap Survey Queue page. With the correct password, the browser then displayed a new survey form consisting of the participant’s demographics (entered originally in the previous survey form) to facilitate identity verification followed by the informed consent form text, checkboxes for consent elections, and a signature field (Figure 3). The research coordinator then handed the tablet to the participant, and the research coordinator and participant reviewed the informed consent form text together and the research coordinator answered questions from the participant. Upon making elections and signing, the participant clicked “ Submit,” which concluded input from the participant. The research coordinator then generated a PDF of the participant’s signed informed consent form and printed or emailed a copy to the participant depending on participant’s preference.
Figure 3.

Consent election options and signature.
Updating consent elections
After initially consenting to participate, a participant had the ability to change his or her consent elections. For example, a subject initially consented to all elections, but in subsequent clinic visits decided he or she no longer wanted investigators to share tissue or clinical data with researchers at outside institutions. For a participant to update elections, a research coordinator navigated to a separate survey form via the password-protected REDCap Survey Queue page that displayed checkboxes elections and handed the tablet to the participant to make updates similar to the process described in Figure 1.
Withdrawing consent
To support withdrawal of informed consent, we configured an additional survey form within the REDCap project. In the event that a subject notified the study team that s/he wished to withdraw from the study, a research coordinator navigated to the survey form via the password-protected REDCap Survey Queue page, as shown in Figure 4. On the survey, the research coordinator then documented the participant’s decision to withdraw and reason for withdrawal, as well as the research coordinator’s identity and the date. Upon completion of the form documenting withdrawal of consent, the record became locked, preventing study coordinators from further editing. Completion of the withdrawal survey triggered an email to the clinic listserv as described above.
Figure 4.

Withdrawal decision and reason.
Maintaining versions of informed consent forms
When IRB protocol amendments, IRB continuing reviews, and IRB-approved informed consent form changes occurred, research team administrators and research IT staff coordinated to copy the contents of the REDCap project to maintain history of versions. First, we copied the current REDCap project, which contained all existing consent records, into a new REDCap project, which we identified using the protocol name and date range of its validity. Second, we archived the new project in REDCap to maintain data integrity for the data range for which the consent was valid. Third, we updated the current REDCap project’s IRB stamp to reflect the new approval and expiration dates. New study participants consented to the current project, not an archived project. The archived project copy served as a version of the prior ICF, as per the pilot’s workflow, while the current REDCap project was the location of the current ICF. If a participant who consented to a now-archived version of the ICF wished to update his or her consent, the participant followed the previously-described update process using the current REDCap project; the archived project copy maintained a record of the initial consent. To determine the version of a protocol to which a patient consented, study team members compared the date when a patient consented as documented in REDCap against the date of the protocol revision, as documented in the name of the archived REDCap project. Subsequent updates to the ICF would yield further copies of current live REDCap project to become archived project copies.
Hardware and security
To access REDCap for consent, research coordinators and participants used Apple iPad tablets managed by WCM ITS. All iPads had restricted settings through the MobileIron security management application and the web@work secure browser. Of note, web@work ensured that bookmarks for the CADC and HemOnc REDCap survey links always appeared and that users could only visit REDCap URLs.
Evaluation
We measured REDCap for eConsent with regard to usage, support for local requirements, and adherence to federal guidance for electronic informed consent. To measure usage, we determined the number of initial consents, re-elections, withdrawals, and projects created to support new versions of informed consent and IRB protocols during the six-month pilot period. To measure support for local requirements, the pilot’s success was based on replicating the research team’s current paper ICF workflow, which includes following local best practices and adhering to federal guidelines. To measure compliance with federal guidance, two of the authors (CC, ES) identified 29 specific requirements derived from the sixteen questions in the the Office for Human Research Protection (OHRP) and the Food and Drug Administration (FDA) guidance on the use of electronic informed consent (9) and reviewed the approach to determine how our methodology adhered to or diverged from these specific requirements. We resolved disagreements through consensus. In a prior investigation, the study team used a similar approach to study biospecimen information management system support for best practices (11).
Results
Usage
During the first twelve months of system availability, a total of 434 patients enrolled in the two studies. Only one update of consent options occurred via the re-election process, and no withdrawals occurred. New versions of the ICF and/or IRB protocol required the creation of five total new REDCap projects for archival purposes. Data dictionaries for both CADC and HemOnc projects are available on GitHub: https://github.com/wcmc-research-informatics/econsentredcap
Support for local requirements
Adherence to federal guidance
Of the 29 requirements we specifically identified as derived from federal guidance, the use of REDCap for electronic consent specifically adhered to 24 (86%), as detailed in Table 2.
Table 2.
Adherence to requirements derived from federal guidance on electronic informed consent
| Requirement Number | Question and Requirement Description | Adherence (yes/no) |
|---|---|---|
| Q1: How should information in the eConsent be presented to the subject? | ||
| 1 | eConsent must contain everything required by 45 CFR 46.116 (General requirements for informed consent) and 21 CFR 50.25 (Elements of informed consent) | yes |
| 2 | eConsent must be easy to navigate, allowing user to go forward and back and to stop and continue at a later time | yes |
| 3 | Subjects should have the option to use paper-based or electronic IC methods completely or partially throughout the process | yes |
| Q2: How and where may the eConsent process be conducted? | ||
| 4 | Process must be conducted at study site or remotely. If remote, must include mechanism to ensure person electronically signing is the person participating or their LAR. | yes |
| 5 | eConsent must be legally binding, regardless of onsite or off-site | yes |
| 6 | eConsent must include mechanism to ensure person electronically signing is the participant or their LAR | yes |
| Q3: How and when should questions from subjects be answered? | ||
| 7 | Questions should be answered via in person discussions or combination of electronic messaging, telephone calls, video conferencing, or live chat with remotely located investigator or study personnel. | yes |
| 8 | eConsent should have methods in place to ensure the process allows subjects the opportunity to consider whether or not to participate and to ask questions | yes |
| 9 | When live chat or video is used, there should be reminders to conduct the discussion in a private location | no |
| 10 | Subjects should be given a description of how and when they will receive answers to their questions, and how they can communicate back in the event that they sustain a research-related injury | yes |
| Q4: What steps may be taken to facilitate the subject’s understanding of the information being presented? | ||
| 11 | eConsent content should be appropriate for intended audience based on age, language, and comprehension level | yes |
| Q5: What steps may be taken to convey additional information, including significant new findings, to the subject during the course of the research? | ||
| 12 | eConsent must contain a statement that significant new findings or updates/amendements that may affect the subject’s willingness to continue will be communicated to the subject or their LAR and that they’ll have the opportunity to ask questions and sign an updated eConsent | yes |
| Q6: How can electronic signatures be used to document eConsent? | ||
| 13 | eConsent must comply with all 21 CFR 11 (Electronic Records; Electronic Signatures) requirements | yes |
| Q7: What methods may be used to verify the identity of the subject who will be electronically signing an eConsent for FDA-regulated clinical investigations? | ||
| 14 | eConsent must comply with 21 CFR 11 (Electronic Records; Electronic Signatures) | yes |
| Q8: What special considerations should be given to the use of eConsent for pediatric studies? | ||
| 15 | eConsent must comply with 45 CFR 46 (General requirements for informed consent) and 21 CFR 50 (Elements of informed consent) | no |
| 16 | IRB must determine that there are sufficient provisions for soliciting the assent of children | no |
| Q9: Should subjects receive a copy of their eConsent and have easy access to the materials and information presented to them in their eConsent? | ||
| 17 | Subjects must be given a copy of of the signed informed consent form unless requirement for documentation has been waived. | yes |
| 18 | If the eConsent uses multimedia to convey information related to research, hyperlinks should be provided on printed paper copies and accessible until study completion | no |
| Q10: What steps can be taken to ensure privacy, security, and confidentiality of the eConsent information? | ||
| 19 | eConsent must be secure with restricted access as per 21 CFR 11 and should include methods to ensure confidentiality regarding identity, subject participation, and personal information after IC has been obtained | yes |
| 20 | If the entity holding PII is a covered entity or BAA of one, eConsent must adhere to HIPAA rules regarding privacy, security, and breach notifications | yes |
| Q11: Can HIPAA authorizations for research, which are frequently combined with informed consent documents, be obtained electronically? | ||
| 21 | When covered entities seek authorization forms, they must provide the participant with a copy of the signed authorization form | yes |
| Q12: What eConsent materials should the investigator submit to the IRB? | ||
| 22 | All forms, electronic and paper, must be submitted to the IRB, along with any modifications | yes |
| 23 | Investigators should discuss plans for using eConsent with IRB before finalizing development | yes |
| Q13: What are the IRB’s responsibilities in the eConsent process? | ||
| 24 | IRB must review and have authority to approve, require modifications to, or disapprove all research activities. IRB must approve and review eConsent and any amendments | yes |
| 25 | IRB must approve and review eConsent and any amendments, as well as maintain/retain copies of any materials | yes |
| 26 | IRB should review any optional questions or methods used to gauge subject comprehension and ensure that eConsent materials are usable, as well as maintain copies of any study-related information | yes |
| Q14: What eConsent documentation does FDA require for submission with applications? | ||
| 27 | eConsent documentation must be available for federal review for INDs. IDEs must include eConsent documentation. Documentation must be the same materials that will be presented to subjects. | yes |
| Q15: What steps can be taken to ensure the system archives the eConsent materials appropriately for FDA-regulated clinical investigations? | ||
| 28 | eConsent should incorporate procedures to ensure documents can be archived appropriate and retrieved easily in compliance with applicable FDA regulations. | yes |
| Q16: What materials or documents will FDA require during an inspection? | ||
| 29 | When sites are inspected by FDA, FDA must be granted access to site-specific versions of the EIC along with all amendments and subject-signed forms. | yes |
Discussion
We implemented a REDCap-based electronic informed consent methodology for research involving biospecimens and evaluated its usage, support for local requirements, and adherence to federal guidance. In an initial pilot period, the methodology saw extensive use with studies centered on biospecimen storage. The methodology supported all local requirements and adhered to 86% of federal guidance for electronic informed consent.
To our knowledge, this case report is among the first to demonstrate use of REDCap as a replacement for traditional paper-based informed consent forms for multiple longitudinal studies in an academic medical center. While the initial requirements obtained from users may not constitute an exhaustive list of measures for an enterprise-level electronic consent tracking system, it offers a basis for measuring the validity and value of the approach. Additionally, to the best of our knowledge, this is the first description of how to use REDCap from the vantage point of a study coordinator and a participant. Combined with the REDCap resources posted on GitHub, this offers a comprehensive description that may be valuable to practitioners in other settings. The methodology described herein may generalize to other settings and spur adoption of electronic informed consent, addressing the low rates of adoption identified in a recent study (3). The method described in this report may serve as a “how-to” for others.
As the goal of our pilot was to address implementation of the technology infrastructure, these requirements do not represent a failure of this technique to adhere to federal guidance but rather domains of guidance where compliance depends on the extent to which institutional policy and individual study workflows follow federal standards.
The CADC and HemOnc studies have continued to use the REDCap eConsent forms after the end of the pilot, and additional clinic locations have adopted the approach beyond the initial sites. While previous studies investigating the use of REDCap for electronic informed consent focused on short-timeframe intervention-based studies (7, 12), this study evaluated the use of a similar approach for longitudinal, biospecimen-oriented studies, necessitating a focus on implementing robust techniques for tracking and maintaining consent and withdrawal over extended time periods. Moreover, the approach’s ability to support changes in elections and protocol amendments over time is a key strength for registries in contrast to time-sensitive clinical trials with few likely changes after initial consent (12). In addition to being highly customizable, REDCap logs all record creation and removal as it is designed to be 21 CFR Part 11 capable. Based on results of this pilot and others (7,12), REDCap appears capable of supporting research consent needs for prospective clinical trials and longitudinal studies as well as retrospective studies.
This study had limitations. First, we did not seek to evaluate the multimedia interactivity of the REDCap-based eConsent format with regard to participant comprehension, usability, and engagement (4,5,13,14). However, our objective was to mimic the existing paper-based ICF workflow using REDCap – not to create or evaluate an interactive multimedia experience for consent – as well as to assess REDCap’s support for local requirements and adherence to federal guidance. Second, the development of our evaluation methodology had intrinsic limitations; the interpretation of the sixteen questions provided in federal guidance documentation to a yes/no format may have involved a loss of nuance. Future work will evaluate how to address areas where the REDCap-based solution failed to satisfy federal guidance as well as usability of the eConsent approach for patients and staff.
A vital element of the development of the REDCap eConsent approach was the close collaboration between the JCTO, HemOnc, CADC, and ITS personnel. The development of the initial set of requirements involved weekly meetings and there were monthly status update meetings during the duration of the pilot. This provided a forum for continuous improvements of the approach.
The results described here suggest that REDCap is well-suited to the development and implementation of an electronic informed consent platform. It is possible that the REDCap consortium may, based on the proof of concept described here, wish to create dedicated functionality to support electronic informed consent. However, some institutions may find this impracticable; for example, institutions with 21 CFR Part 11 validation of their current REDCap instance may be unable to upgrade to a new version without losing validation. These institutions may be able to adopt some or all of our approach, and the cost of building new or extending existing REDCap infrastructure may be a lower cost alternative compared to investment in a dedicated research consent management system. Future cost-benefit analyses can determine the value of REDCap versus other options for expanding eConsent to more studies at an institution.
To further the extent to which this platform addresses these challenges, a potential benefit of developing REDCap as an enterprise-level electronic consent tracking system is leveraging the existing capabilities for importing and exporting data. We plan to extend our existing implementation of the REDCap Dynamic Data Pull (DDP) plugin to automatically retrieve data from the EHR, such as patient demographics based on entry of an MRN into the live REDCap eConsent project, to reduce research coordinator data entry and improve participant identity management (15). Future work also includes automated transmission of the participant enrollment status from REDCap to our institution’s clinical trials management system (CTMS) and the EHR. These system integrations have the potential to streamline workflows and increase efficacy of the research consent process.
Table 1.
Measures for supporting local requirements
| Requirement Description | Adherence(yes/no) |
|---|---|
| Researchers are able to successfully enroll participants | yes |
| Participants receive a copy of the completed consent form | yes |
| Participants are able to update elections | yes |
| Participants are able to withdraw | yes |
| IRB amendments are successfully implemented | yes |
| eConsentcomplies with local guidance (e.g., security) | yes |
| eConsent complies with federal guidance (e.g., HIPAA) | yes |
Acknowledgements
This study received support from NewYork-Presbyterian Hospital (NYPH) and Weill Cornell Medical College (WCMC), including the Clinical and Translational Science Center (CTSC) (UL1 TR000457) and Joint Clinical Trials Office (JCTO). This study also received support from the Michael Wolk Heart Foundation.
References
- 1.TransCelerate BioPharma Inc. eConsent: Implementation Guidance [Internet] 2017. [cited 2018 Aug 8]. Available from: http://www.transceleratebiopharmainc.com/wp-content/uploads/2017/11/eConsent-Implementation-Guidance.pdf.
- 2.Clinical Trials Transformation Inititative. CTTI Recommendations: Informed Consent [Internet] 2015. [cited 2018 Aug 9]. Available from: https://www.ctti-clinicaltrials.org/files/ctti-informedconsent-recs.pdf.
- 3.Simon CM, Klein DW, Schartz HA. Traditional and electronic informed consent for biobanking: a survey of U.S. biobanks. Biopreserv Biobank. 2014 Dec;12(6):423–429. doi: 10.1089/bio.2014.0045. [DOI] [PubMed] [Google Scholar]
- 4.Friedlander JA, Loeben GS, Finnegan PK, Puma AE, Zhang X, de Zoeten EF. A novel method to enhance informed consent: a prospective and randomised trial of form-based versus electronic assisted informed consent in paediatric endoscopy. J Med Ethics. 2011 Apr;37(4):194–200. doi: 10.1136/jme.2010.037622. [DOI] [PubMed] [Google Scholar]
- 5.Rowbotham MC, Astin J, Greene K, Cummings SR. Interactive informed consent: randomized comparison with paper consents. PLoS One. 2013 Mar 6;8(3):e58603. doi: 10.1371/journal.pone.0058603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chalil Madathil K, Koikkara R, Obeid J, Greenstein JS, Sanderson IC, Fryar K. An investigation of the efficacy of electronic consenting interfaces of research permissions management system in a hospital setting. Int J Med Inform. 2013 Sep;82(9):854–863. doi: 10.1016/j.ijmedinf.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frelich MJ, Bosler ME, Gould JC. Research Electronic Data Capture (REDCap) electronic Informed Consent Form (eICF) is compliant and feasible in a clinical research setting. Int J Clin Trials. 2015 Aug 13;2(3):51. [Google Scholar]
- 8.Schwartze J, Haarbrandt B, Rochon M, Wagner M, Haux R, Kleinschmidt T. Design and implementation of an informed consent process for a standardized health information exchange solution on the example of the lower saxony bank of health. Stud Health Technol Inform. 2013;192:318–322. [PubMed] [Google Scholar]
- 9.U.S. Department of Health and Human Services, Office for Human Research Protections (OHRP), U.S. Department of Health and Human Services, Food and Drug Administration (FDA) Use of Electronic Informed Consent: Questions and Answers Guidance for Institutional Review Boards, Investigators, and Sponsors [Internet] 2016. [cited 2018 Jul 10]. Available from: https://www.fda.gov/downloads/drugs/guidances/ucm436811.pdf.
- 10.Campion TR, Blau VL, Brown SW, Izcovich D, Cole CL. Implementing a clinical research management system: one institution’s successful approach following previous failures. AMIA Jt Summits Transl Sci Proc. 2014 Apr 7;2014:12–17. [PMC free article] [PubMed] [Google Scholar]
- 11.Chen C, Wulff RT, Sholle ET, Roboz GJ, Kraemer DA, Campion TR. Evaluating generalizability of a biospecimen informatics approach: support for local requirements and best practices. AMIA Jt Summits Transl Sci Proc. 2018 May 18;2017:55–62. [PMC free article] [PubMed] [Google Scholar]
- 12.Haussen DC, Doppelheuer S, Schindler K, Grossberg JA, Bouslama M, Schultz M. Utilization of a smartphone platform for electronic informed consent in acute stroke trials. Stroke. 2017 Oct 6;48(11):3156–3160. doi: 10.1161/STROKEAHA.117.018380. [DOI] [PubMed] [Google Scholar]
- 13.Abujarad F, Alfano S, Bright TJ, Kannoth S, Grant N, Gueble M. Building an Informed Consent Tool Starting with the Patient: The Patient-Centered Virtual Multimedia Interactive Informed Consent (VIC) AMIA Annu Symp Proc. 2017;2017:374–383. [PMC free article] [PubMed] [Google Scholar]
- 14.Simon CM, Klein DW, Schartz HA. Interactive multimedia consent for biobanking: a randomized trial. Genet Med. 2016 Jan;18(1):57–64. doi: 10.1038/gim.2015.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campion TR, Sholle ET, Davila MA. Generalizable middleware to support use of redcap dynamic data pull for integrating clinical and research data. AMIA Jt Summits Transl Sci Proc. 2017 Jul 26;2017:76–81. [PMC free article] [PubMed] [Google Scholar]