

Abstract
Proteins are dynamic, fluctuating between multiple conformational states. Protein dynamics, spanning orders of magnitude in time and space, allow proteins to perform specific functions. Moreover, under certain conditions, proteins can morph into a different set of conformations. Thus, a complete understanding of protein structural dynamics can provide mechanistic insights into protein function. Here, we review the latest developments in methods used to determine protein ensemble structures and to characterize protein dynamics. Techniques including X-ray crystallography, cryogenic electron microscopy, and small angle scattering can provide structural information on specific conformational states or on the averaged shape of the protein, whereas techniques including nuclear magnetic resonance, fluorescence resonance energy transfer (FRET), and chemical cross-linking coupled with mass spectrometry provide information on the fluctuation of the distances between protein domains, residues, and atoms for the multiple conformational states of the protein. In particular, FRET measurements at the single-molecule level allow rapid resolution of protein conformational states, where information is otherwise obscured in bulk measurements. Taken together, the different techniques complement each other and their integrated use can offer a clear picture of protein structure and dynamics.
Keywords: Conformational dynamics, Integrative structural biology, Distance restraint, Ensemble averaging, Nuclear magnetic resonance (NMR)
1. Introduction
X-ray crystallography, nuclear magnetic resonance (NMR), and cryogenic electron microscopy (cryoEM) are the three main tools used in structural biology. In recent years, we have witnessed the increasing popularity of cryoEM, which can solve the structures of large biological macromolecules frozen in vitreous conditions. These structural biology techniques have provided us with numerous atomic resolution structures of proteins and other biological macromolecules. As of March 31st, 2019, 150 000 structures have been deposited in the protein data bank (PDB, http://www.rcsb.org). However, a static picture of a protein may not provide sufficient information to understand how the protein works. Structural dynamics, more relevantly, shows enzymes, transporters, signaling proteins, and others in action (Henzler-Wildman and Kern, 2007).
The structural dynamics of a protein arise from the vibrations and rotations of the chemical bonds occurring in femto-second timescale, and the fluctuation of the orientations of bond vectors occurring in pico-second to nano-second (ps–ns) timescale. These high-frequency dynamics can be coupled, giving rise to the collective movement occurring at micro-second to milli-second (µs–ms) or even slower timescales. Protein collective motion involves many residues, for example, the opening and closing of protein domains (Tang et al., 2007), and is intimately related to protein function (Bahar et al., 2010). A protein adopting a particular conformation is associated with a certain free energy (Bryngelson et al., 1995). At ambient temperature, the thermal kinetic energy allows the protein to overcome the energy barrier and interconvert between different conformational states (Fig. 1a).
Fig. 1.

An illustration of a protein energy landscape
(a) The conformational state (A or B) with the lowest free energy is the most populated. Yet all the states are stochastically accessible at ambient temperature. (b) Different cellular conditions, including but not limited to post-translational modifications, local pH, macromolecular crowding, and liquid droplet formation, can all lead to altered energy landscapes of protein conformational states
The population of each conformational state is determined by its free energy, and the distribution is governed by the Boltzmann probability. As a result, the predominant lowest-energy conformational state is the most likely to be captured and visualized by X-ray crystallography. Traditionally, protein crystals are flash frozen in liquid nitrogen to avoid radiation damage during crystallographic data collection. As the protein is structurally fixed in the crystal lattice, only local conformational fluctuations of the protein are allowed. Such dynamics can now be studied using room-temperature X-ray crystallography (Fraser et al., 2011).
The kinetics of the interconversion between protein conformational states A and B are dictated by the energy barrier (Fig. 1a), as described by the Arrhenius equation. Thus, due to stochastic thermal fluctuations, it may only take a protein molecule µs–ms to overcome the large energy barrier. CryoEM deduces three-dimensional shapes from two-dimensional projection images and classifies the structure of the protein into a set of conformational states (Bai et al., 2015). However, upon freezing in liquid ethane, the energy landscape of the protein is likely perturbed, and the relative populations of the conformational states likely differ from those under ambient conditions. More critically, the interconversion dynamics are completely lost at −188 °C. Therefore, though powerful as a structural biology tool, cryoEM may only provide snapshots of the many alternative conformational states of proteins and protein complexes.
The interconversion among different conformational states allows the protein to perform specific functions. Indeed, a protein must undergo structural changes for catalysis, ligand binding, and signal transduction (Henzler-Wildman and Kern, 2007). More importantly, the energy landscape of the protein can be altered under different cellular conditions (Fig. 1b) and upon post-translational modifications. For example, ubiquitin, a 76-residue signaling protein, mainly adopts a relaxed state with a common ubiquitin fold, and rarely samples a C-terminal β-strand-retracted state even at 45 °C (Gladkova et al., 2017). However, phosphorylation by ubiquitin kinase PINK1 at residue Ser65 increases the population of the retracted state by more than 100-fold. Moreover, the relative populations of relaxed and retracted states of the phosphorylated ubiquitin can change with pH, as the phosphorylation-enriched retracted state becomes more populated at a slightly basic pH (Dong et al., 2017). In recent years, molecular crowding and the formation of liquid droplets have been increasingly recognized as factors that perturb protein structural dynamics and enable certain protein functions (Kuznetsova et al., 2014; Elbaum-Garfinkle et al., 2015). Importantly, the concentration of the phase-separated protein in the droplet can reach 400 mg/mL or higher (Brady et al., 2017). Such high protein concentrations would promote fleeting protein–protein interactions with binding affinities in the millimolar range (Xing et al., 2014). Hence, protein liquid droplets may only form with multi-valent and weak interactions between proteins and protein domains (Li et al., 2012). Taken together, structural dynamics involves an ensemble of structures adopted by a protein or protein complex over time.
2. NMR is inherently integrative
NMR has been used to characterize proteins and other biological macromolecules in solution. Unlike X-ray crystallography and cryoEM, solution NMR is uniquely suited to characterize protein structural dynamics at near physiological conditions. In recent years, NMR has also been used to determine protein structures in the cell (Sakakibara et al., 2009; Luchinat and Banci, 2016; Pan et al., 2016).
It has been more than 40 years since the first protein structure was determined by NMR (Wagner and Wüthrich, 1978). Yet, the key concept of NMR structure determination remains largely the same. This is because NMR is inherently integrative, and can always incorporate additional experimental data as a new energy term. In NMR structure determination, the structure is optimized to make the calculated values based on the structural model agree best with the experimental inputs. The goodness of the fit is evaluated with a pseudo-energy term, which is minimized with simulated annealing.
Typical inputs for NMR structure calculation include nuclear Overhauser enhancement (NOE) cross-peaks, chemical shift values, scalar-coupling constants, and residual dipolar couplings. They can be readily measured and used in structure calculation software, such as Xplor-NIH (Schwieters et al., 2018). In addition, with a paramagnetic probe site-specifically attached to an otherwise diamagnetic protein, paramagnetic NMR parameters, exemplified by paramagnetic relaxation enhancement (PRE), can be measured (Iwahara et al., 2007; Clore and Iwahara, 2009). With the latest NMR developments, more and more energy terms from different types of experimental data have been incorporated. For example, small angle X-ray scattering (SAXS) and small angle neutron scattering (SANS) can provide an overall silhouette of the protein in solution (Schwieters and Clore, 2014). Probability of protein dihedral angles from the statistics of high-precision structures can also be used as a knowledge-based potential (Bermejo et al., 2012).
3. Distance matters
When a protein exists in a particular conformation, each atom, residue, and domain of the protein is separated from one another by certain distances. When the protein changes conformation, a new set of distances emerge. Just like protein dynamics are hierarchical, the distance between the domains can also be considered as the sum of inter-residue and inter-atomic distances (Fig. 2a). On the other hand, if all the distances can be measured, the particular protein structure or the ensemble of protein structures can be deduced. Yet, it should be noted that protein structure determination based on NMR restraints is an under-determined problem, as the structure solution accounting for all the experimental data is unlikely to be unique. Thus, Bayesian inference has been applied for NMR structure determination, which assigns a statistical probability to the structural model (Rieping et al., 2005; MacCallum et al., 2015).
Fig. 2.
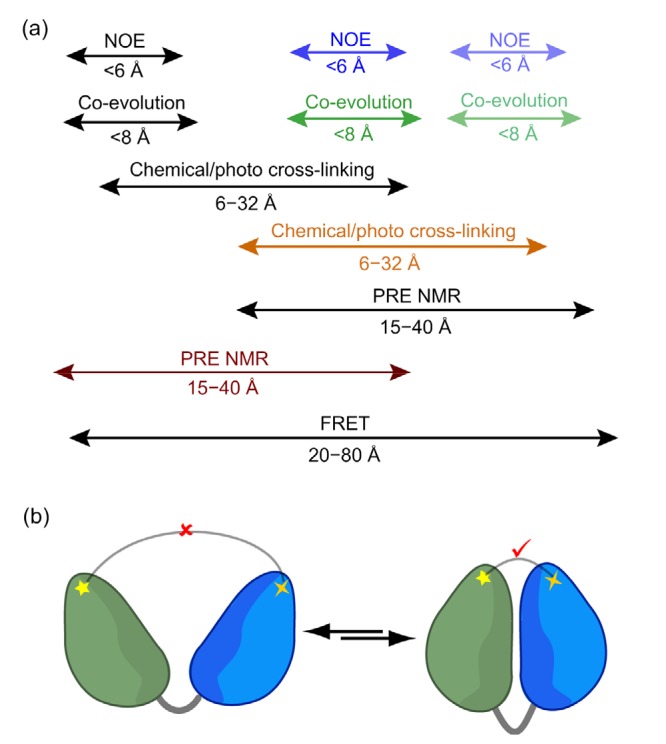
Distance information between protein domains, residues, and atoms, which can be obtained from a multitude of experimental inputs
(a) The distance restraints with different length-scales are hierarchical and can help to reconstruct a complete structural picture of the protein. (b) With protein dynamics present, the distance restraints are ensemble-averaged, and sometimes manifest as the most compact conformation(s). This scheme illustrates that chemical cross-linking may only occur when this two-domain protein adopts a closed conformation, in which the distance between cross-linked residues is shorter than the length of the cross-linker. NOE: nuclear Overhauser enhancement; PRE: paramagnetic relaxation enhancement; FRET: fluorescence resonance energy transfer
Many distance measurements in NMR and in other biophysical methods are based on dipolar interactions that follow the <r −6>distance relationship. The NOE effect arises from the dipolar interactions between two protons separated by less than 6 Å. The PRE effect arises from dipolar interaction between a proton and an unpaired electron in the site-specifically attached paramagnetic probe. Since an electron has a much larger magnetic dipole moment than a proton, the PRE can provide a distance relationship of up to 40 Å (Iwahara et al., 2003; Liu et al., 2016). Fluorescence resonance energy transfer (FRET) also arises from dipolar interactions between two fluorophores which have been site-specifically attached to a protein or protein complex, and can provide distance information in the range of 20 to 80 Å. The fluorescence signal has a much lower detection limit than the NMR signal, and as a result, FRET can be measured at the single-molecule level (Lerner et al., 2018). How single-molecule FRET (smFRET) can aid the analysis of protein structural dynamics will be discussed in the next section.
Methods have also been devised to obtain the distance restraints for protein structure calculation without introducing an NMR or fluorescent probe. For example, chemical cross-linking coupled with mass spectrometry (CXMS) allows the identification of pairs of residues closer than the lengths of the cross-linkers used (Yang et al., 2012). It should be noted that the cross-links may only “crawl” along the protein surface; therefore, solvent accessible distances instead of Euclidean distances should be calculated and used as distance restraints (Matthew Allen Bullock et al., 2016; Ding et al., 2017). Using only the distance restraints from cross-linking, it is now possible to obtain de novo protein structures (Brodie et al., 2017). On the other hand, with genomics information becoming available for different species, internal residues that are in contact can also be deduced from evolutionary couplings (Tang et al., 2015). Co-evolution of proximal residues assumes that mutations are correlated to minimize perturbations to the protein structure. Taken together, the distance relationships from various types of experimental data are multi-tiered and hierarchical, and the accurate measurement of these distances lays the foundation for structural characterization of proteins and protein complexes (Fig. 2a).
4. Resolving multiple conformational states with multiple methods
Proteins are dynamic. As a result, the distance restraint obtained may not arise from a single conformation, but is likely an average from multiple conformational states adopted simultaneously by the protein (Fig. 2b). For a typical bulk measurement, such as NMR and SAXS/SANS, the data are collected for many billions of protein molecules, and are therefore ensemble-averaged over all the conformational states adopted by the protein. Thus, it is an ill-posed inverse problem to decompose experimental data and to assign the relative contributions to each conformational state. In particular, this is the case for PRE NMR measurements. Owing to the <r −6>distance dependence, a proton in a conformation with a short distance to the paramagnetic probe experiences a disproportionally large effect when compared to the overall observed PRE data, since the PRE effect rapidly tapers at longer distances. As a result, the characterization of protein dynamics based on PRE NMR data alone can be biased towards the most compact conformational state (Clore and Iwahara, 2009). Therefore, it is important to refine the ensemble structures against different types of experimental data. Such an integrative approach will not only yield a more accurate and complete picture of protein structure and dynamics, and the various types of data with different information content and different ways of ensemble averaging can also provide cross-validation (Fig. 3).
Fig. 3.

Use of an integrative hybrid approach to fully characterize protein structure and dynamics
NMR: nuclear magnetic resonance; FRET: fluorescence resonance energy transfer; CXMS: chemical cross-linking coupled with mass spectrometr; SAXS: small angle X-ray scattering; SANS: small angle neutron scattering
Unlike bulk measurements, single-molecule techniques inspect only one molecule at a time. Therefore, single-molecule detection is not ensemble-averaged, and from the smFRET profile the number of interconverting conformational states and their relative populations can be readily obtained. Equipped with such information, bulk measurements, like PRE NMR, can be defined without resorting to complicated calculations. For example, the cross-links identified in CXMS represent the compact conformational state(s) of the protein, in which the separation between the cross-linked residues is shorter than the length of the cross-linker (Gong et al., 2015; Ferber et al., 2016; Ding et al., 2017). SAXS data, on the other hand, contain averaged shape information of all the conformational states. Liu et al. (2018) used smFRET in conjunction with SAXS and CXMS data to rapidly characterize the three interconverting structures of K63-linked di-ubiquitin.
5. Along came the time dimension
Characterization of a set of interconverting conformational states or ensemble structure of a protein provides information on their conformational dynamics. Here, only the snapshots and their relative populations are visualized. To fully understand protein structural dynamics, we also need to know the timescale of the dynamics and the pathway of the dynamic interconversion.
NMR is uniquely suited to characterize protein dynamics since it is inherently integrative, and a plethora of techniques exist that can define timescale information of protein dynamics (Sekhar and Kay, 2013). By examining the relaxation properties of NMR spins, and using relaxation dispersion and off-resonance spin-lock methods, ps‒ns and µs‒ms timescale dynamics can be identified. To analyze protein dynamics with slower timescales, chemical exchange saturation transfer, Z-axis exchange experiments, and real-time two-dimensional spectroscopy data can be collected.
The smFRET can also provide information on the timescales of protein dynamics. It allows the identification of multiple interconverting conformational states, and provides kinetic information for the exchange between any two of the conformational states (Kalinin et al., 2010). Thus, smFRET can chart the pathway of conformational dynamics, that is, how the protein preferentially switches from one state to another (Peulen et al., 2017). Such information can be difficult to obtain using NMR alone.
With more experimental data on protein structure and dynamics becoming available, physics and/or knowledge-based computational simulation methods have also greatly improved over the past 10 years, and are increasingly used along with experimental analysis (MacCallum et al., 2015). Based on Newtonian laws, simulations can recapitulate the essential dynamics of protein systems (Dror et al., 2012). Moreover, simulations can predict structural outcomes which can then be experimentally validated. Simulations can also provide information on the detailed interconversion process between protein conformational states, which may not be obtainable from experiments.
6. Concluding remarks
Today’s biological research systems are becoming increasingly complex. To tackle these problems and to determine the structures of these systems, researchers have resorted to the “non-conventional” methods in addition to X-ray crystallography, NMR, and cryoEM. Structures obtained using an integrative approach may not provide an atomic resolution picture, yet it still gives us mechanistic insights. Recently, a database called PDB-Dev (https://pdb-dev.wwpdb.org) was established for curating structures determined using integrative hybrid methods (Sali et al., 2015). However, the strength of using NMR and other biophysical methods jointly should go beyond determining a single structure of the protein, and can excel in providing an ensemble of protein structures with a time stamp. The various types of experimental and computational data complement each other, and as a result, a clear and complete picture of protein structural dynamics emerges.
Footnotes
Project supported by the National Key R&D Program of China (No. 2018YFA0507700)
Contributors: Both authors conceived the idea. Qing-fen YANG wrote the initial draft and Chun TANG revised the manuscript.
Compliance with ethics guidelines: Qing-fen YANG and Chun TANG declare that they have no conflict of interest.
This article does not contain any studies with human or animal subjects performed by either of the authors.
References
- 1.Bahar I, Lezon TR, Bakan A, et al. Normal mode analysis of biomolecular structures: functional mechanisms of membrane proteins. Chem Rev. 2010;110(3):1463–1497. doi: 10.1021/cr900095e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bai XC, McMullan G, Scheres SHW. How cryo-EM is revolutionizing structural biology. Trends Biochem Sci. 2015;40(1):49–57. doi: 10.1016/j.tibs.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 3.Bermejo GA, Clore GM, Schwieters CD. Smooth statistical torsion angle potential derived from a large conformational database via adaptive kernel density estimation improves the quality of NMR protein structures. Protein Sci. 2012;21(12):1824–1836. doi: 10.1002/pro.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brady JP, Farber PJ, Sekhar A, et al. Structural and hydrodynamic properties of an intrinsically disordered region of a germ cell-specific protein on phase separation. Proc Natl Acad Sci USA. 2017;114(39):E8194–E8203. doi: 10.1073/pnas.1706197114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brodie NI, Popov KI, Petrotchenko EV, et al. Solving protein structures using short-distance cross-linking constraints as a guide for discrete molecular dynamics simulations. Sci Adv. 2017;3(7):e1700479. doi: 10.1126/sciadv.1700479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bryngelson JD, Onuchic JN, Socci ND, et al. Funnels, pathways, and the energy landscape of protein folding: a synthesis. Proteins. 1995;21(3):167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 7.Clore GM, Iwahara J. Theory, practice, and applications of paramagnetic relaxation enhancement for the characterization of transient low-population states of biological macromolecules and their complexes. Chem Rev. 2009;109(9):4108–4139. doi: 10.1021/cr900033p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding YH, Gong Z, Dong X, et al. Modeling protein excited-state structures from “over-length” chemical cross-links. J Biol Chem. 2017;292(4):1187–1196. doi: 10.1074/jbc.M116.761841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dong X, Gong Z, Lu YB, et al. Ubiquitin S65 phosphorylation engenders a pH-sensitive conformational switch. Proc Natl Acad Sci USA. 2017;114(26):6770–6775. doi: 10.1073/pnas.1705718114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dror RO, Dirks RM, Grossman JP, et al. Biomolecular simulation: a computational microscope for molecular biology. Annu Rev Biophys. 2012;41:429–452. doi: 10.1146/annurev-biophys-042910-155245. [DOI] [PubMed] [Google Scholar]
- 11.Elbaum-Garfinkle S, Kim Y, Szczepaniak K, et al. The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc Natl Acad Sci USA. 2015;112(23):7189–7194. doi: 10.1073/pnas.1504822112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferber M, Kosinski J, Ori A, et al. Automated structure modeling of large protein assemblies using crosslinks as distance restraints. Nat Methods. 2016;13(6):515–520. doi: 10.1038/nmeth.3838. [DOI] [PubMed] [Google Scholar]
- 13.Fraser JS, van den Bedem H, Samelson AJ, et al. Accessing protein conformational ensembles using room-temperature X-ray crystallography. Proc Natl Acad Sci USA. 2011;108(39):16247–16252. doi: 10.1073/pnas.1111325108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gladkova C, Schubert AF, Wagstaff JL, et al. An invisible ubiquitin conformation is required for efficient phosphorylation by PINK1. EMBO J. 2017;36(24):3555–3572. doi: 10.15252/embj.201797876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gong Z, Ding YH, Dong X, et al. Visualizing the ensemble structures of protein complexes using chemical cross-linking coupled with mass spectrometry. Biophys Rep. 2015;1(3):127–138. doi: 10.1007/s41048-015-0015-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henzler-Wildman K, Kern D. Dynamic personalities of proteins. Nature. 2007;450(7172):964–972. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- 17.Iwahara J, Anderson DE, Murphy EC, et al. EDTA-derivatized deoxythymidine as a tool for rapid determination of protein binding polarity to DNA by intermolecular paramagnetic relaxation enhancement. J Am Chem Soc. 2003;125(22):6634–6635. doi: 10.1021/ja034488q. [DOI] [PubMed] [Google Scholar]
- 18.Iwahara J, Tang C, Clore GM. Practical aspects of 1H transverse paramagnetic relaxation enhancement measurements on macromolecules. J Magn Reson. 2007;184(2):185–195. doi: 10.1016/j.jmr.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kalinin S, Valeri A, Antonik M, et al. Detection of structural dynamics by FRET: a photon distribution and fluorescence lifetime analysis of systems with multiple states. J Phys Chem B. 2010;114(23):7983–7995. doi: 10.1021/jp102156t. [DOI] [PubMed] [Google Scholar]
- 20.Kuznetsova IM, Turoverov KK, Uversky VN. What macromolecular crowding can do to a protein? Int J Mol Sci. 2014;15(12):23090–23140. doi: 10.3390/ijms151223090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lerner E, Cordes T, Ingargiola A, et al. Toward dynamic structural biology: two decades of single-molecule Förster resonance energy transfer. Science. 2018;359(6373):eaan1133. doi: 10.1126/science.aan1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li PL, Banjade S, Cheng HC, et al. Phase transitions in the assembly of multivalent signalling proteins. Nature. 2012;483(7389):336–340. doi: 10.1038/nature10879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Z, Gong Z, Dong X, et al. Transient protein–protein interactions visualized by solution NMR. Biochim Biophys Acta. 2016;1864(1):115–122. doi: 10.1016/j.bbapap.2015.04.009. [DOI] [PubMed] [Google Scholar]
- 24.Liu Z, Gong Z, Cao Y, et al. Characterizing protein dynamics with integrative use of bulk and single-molecule techniques. Biochemistry. 2018;57(3):305–313. doi: 10.1021/acs.biochem.7b00817. [DOI] [PubMed] [Google Scholar]
- 25.Luchinat E, Banci L. A unique tool for cellular structural biology: in-cell NMR. J Biol Chem. 2016;291(8):3776–3784. doi: 10.1074/jbc.R115.643247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.MacCallum JL, Perez A, Dill KA. Determining protein structures by combining semireliable data with atomistic physical models by Bayesian inference. Proc Natl Acad Sci USA. 2015;112(22):6985–6990. doi: 10.1073/pnas.1506788112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matthew Allen Bullock J, Schwab J, Thalassinos K, et al. The importance of non-accessible crosslinks and solvent accessible surface distance in modeling proteins with restraints from crosslinking mass spectrometry. Mol Cell Proteomics. 2016;15(7):2491–2500. doi: 10.1074/mcp.M116.058560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pan BB, Yang F, Ye YS, et al. 3D structure determination of a protein in living cells using paramagnetic NMR spectroscopy. Chem Commun. 2016;52(67):10237–10240. doi: 10.1039/c6cc05490k. [DOI] [PubMed] [Google Scholar]
- 29.Peulen TO, Opanasyuk O, Seidel CAM. Combining graphical and analytical methods with molecular simulations to analyze time-resolved FRET measurements of labeled macromolecules accurately. J Phys Chem B. 2017;121(35):8211–8241. doi: 10.1021/acs.jpcb.7b03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rieping W, Habeck M, Nilges M. Inferential structure determination. Science. 2005;309(5732):303–306. doi: 10.1126/science.1110428. [DOI] [PubMed] [Google Scholar]
- 31.Sakakibara D, Sasaki A, Ikeya T, et al. Protein structure determination in living cells by in-cell NMR spectroscopy. Nature. 2009;458(7234):102–105. doi: 10.1038/nature07814. [DOI] [PubMed] [Google Scholar]
- 32.Sali A, Berman HM, Schwede T, et al. Outcome of the first wwPDB hybrid/integrative methods task force workshop. Structure. 2015;23(7):1156–1167. doi: 10.1016/j.str.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwieters CD, Clore GM. Using small angle solution scattering data in Xplor-NIH structure calculations. Prog Nucl Magn Reson Spectrosc. 2014;80:1–11. doi: 10.1016/j.pnmrs.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwieters CD, Bermejo GA, Clore GM. Xplor-NIH for molecular structure determination from NMR and other data sources. Protein Sci. 2018;27(1):26–40. doi: 10.1002/pro.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sekhar A, Kay LE. NMR paves the way for atomic level descriptions of sparsely populated, transiently formed biomolecular conformers. Proc Natl Acad Sci USA. 2013;110(32):12867–12874. doi: 10.1073/pnas.1305688110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang C, Schwieters CD, Clore GM. Open-to-closed transition in apo maltose-binding protein observed by paramagnetic NMR. Nature. 2007;449(7165):1078–1082. doi: 10.1038/nature06232. [DOI] [PubMed] [Google Scholar]
- 37.Tang YF, Huang YJ, Hopf TA, et al. Protein structure determination by combining sparse NMR data with evolutionary couplings. Nat Methods. 2015;12(8):751–754. doi: 10.1038/nmeth.3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wagner G, Wüthrich K. Dynamic model of globular protein conformations based on NMR studies in solution. Nature. 1978;275(5677):247–248. doi: 10.1038/275247a0. [DOI] [PubMed] [Google Scholar]
- 39.Xing Q, Huang P, Yang J, et al. Visualizing an ultra-weak protein–protein interaction in phosphorylation signaling. Angew Chem Int Ed Engl. 2014;53(43):11501–11505. doi: 10.1002/anie.201405976. [DOI] [PubMed] [Google Scholar]
- 40.Yang B, Wu YJ, Zhu M, et al. Identification of cross-linked peptides from complex samples. Nat Methods. 2012;9(9):904–906. doi: 10.1038/nmeth.2099. [DOI] [PubMed] [Google Scholar]