

Abstract
The binding specificities of an individual’s antibody repertoire contain a wealth of biological information. They harbor evidence of environmental exposures, allergies, ongoing or emerging autoimmune disease processes, and responses to immunomodulatory therapies, for example. Highly multiplexed methods to comprehensively interrogate antibody-binding specificities have therefore emerged in recent years as important molecular tools. Here we provide a detailed protocol for performing “Phage ImmunoPrecipitation Sequencing” (PhIP-Seq), which is a powerful method for analyzing antibody repertoire binding specificities in high throughput and at low cost. The methodology uses oligonucleotide library synthesis (OLS) to encode proteomic-scale peptide libraries for display on bacteriophage. These libraries are then immunoprecipitated using an individual’s antibodies, for subsequent analysis using high-throughput DNA sequencing. We have used PhIP-Seq to identify novel self-antigens associated with autoimmune disease, to characterize the self-reactivity of broadly neutralizing HIV antibodies, and in a large international cross-sectional study of exposure to hundreds of human viruses. Compared with alternative array-based techniques, PhIP-Seq is far more scalable in terms of sample throughput and cost per analysis. Cloning and expression of recombinant proteins is not required (versus protein microarrays), and peptide lengths are limited only by DNA synthesis chemistry (up to 90 amino acid peptides, versus the typical 8–12 amino acid length limit of synthetic peptide arrays). Compared with protein microarrays, however, PhIP-Seq libraries lack discontinuous epitopes and post translational modifications. To increase the accessibility of PhIP-Seq, we provide detailed instructions for the design of phage displayed peptidome libraries, their immunoprecipitation using serum antibodies, deep sequencing-based measurement of peptide abundances, and statistical determination of peptide enrichments that reflect antibody-peptide interactions. Once a library has been constructed, PhIP-Seq data can be obtained for analysis in under a week.
Keywords: Bacteriophage display, serology, humoral immunity, autoantibodies, proteomics, synthetic biology, Phage ImmunoPrecipitation Sequencing, PhIP-Seq, peptidome, synthetic peptidome, peptide library
EDITORIAL SUMMARY:
Phage ImmunoPrecipitation Sequencing (PhIP-Seq) is a high-throughput method for analyzing antibody repertoire binding specificities. Phage-displayed oligonucleotide libraries encoding peptidomes are immunoprecipitated using an individual’s antibodies and analysed by high-throughput DNA sequencing.
Introduction
Development of the protocol.
Phage ImmunoPrecipitation sequencing (PhIP-Seq) is a powerful technology platform that overcomes many previous limitations of comprehensive antibody binding analysis.1–5 PhIP-Seq combines oligonucleotide library synthesis (OLS)6 with high-throughput DNA sequencing analysis of phage-displayed libraries. The synthetic oligonucleotide libraries are designed to encode peptide tiles that together span a library of protein sequences (entire proteomes, for example). The result is a comprehensive and normalized (uniform in abundance) representation of the encoded peptides, which we refer to as the “peptidome(s)”. Deep DNA sequencing of phage-displayed peptidomes permits the quantification of each peptide’s antibody-dependent enrichment, relative to other library peptides, spike-in standards, other antibody containing samples, and/or negative control samples lacking antibodies (Figure 1). Sample multiplexing is achieved using barcoded PCR primers, which are employed during preparation of the sequencing library. This dramatically reduces the per-sample DNA sequencing cost, thereby enabling the analysis of large sample sets. Importantly, the streamlined protocol presented here can be easily performed by hand or automated for high throughput sample processing using liquid handling robotics. Compared with protein microarrays,7, 8 PhIP-Seq is not restricted to proteins that have been cloned and can be expressed recombinantly. However, phage displayed peptidomes lack many of the conformational epitopes present on full length proteins. Compared with peptide microarrays,9 PhIP-Seq features longer, higher quality peptides. However, due to the cost and effort of constructing new phage libraries, programmable peptide arrays may be more appropriate for screening sample-individualized peptide libraries. For projects involving large numbers of samples, the per-sample analysis cost of PhIP-Seq is roughly two orders of magnitude less expensive compared to microarray-based alternatives.
Figure 1.
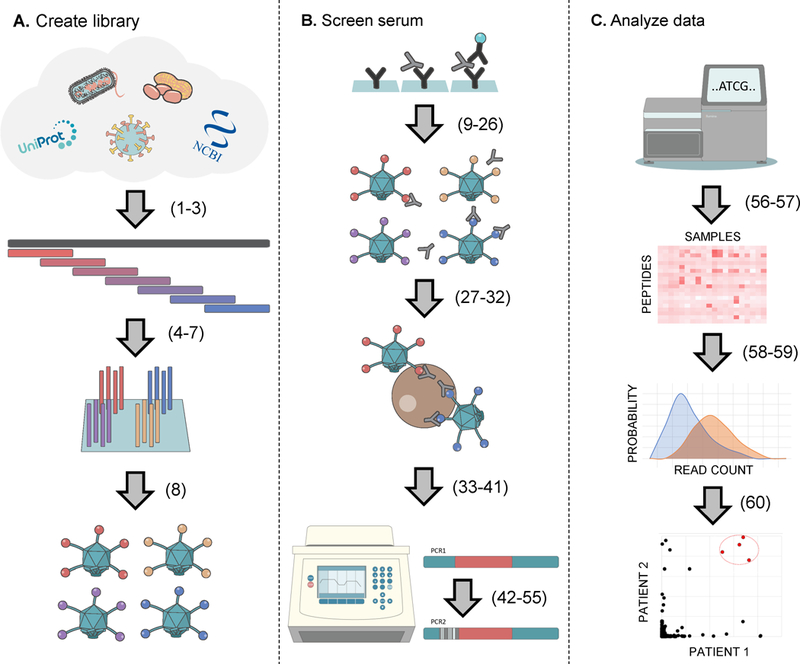
Overview of the PhIP-seq methodology. Procedure step numbers are indicated in parentheses. A. A protein database is downloaded or designed. The pepsyn software is used to tile the protein sequences with overlapping peptide sequences. The oligonucleotide library encoding the peptide sequences is synthesized. The oligonucleotide library is PCR amplified with adapters for cloning into the phage display vector of choice. B. ELISA is used to quantify each sample’s IgG content for normalizing amount of antibody input into each phage binding reaction. Antibodies and their bound phage are captured using protein A/G coated magnetic beads. The library of peptide encoding DNA sequences are amplified by PCR directly from the immunoprecipitate. A second round of hemi-nested PCR is used to add sample-specific barcodes and sequencing adapters to the PCR1 product. Barcoded amplicons are pooled for sequencing on an Illumina instrument. C. Fastq sequencing files are demultiplexed and aligned to the reference sequences to obtain a count matrix. Statistical analysis of the count matrix is performed to determine peptide enrichments. Project specific analysis of peptide enrichments (e.g. identification of a common autoantigen) can then be carried out.
Overview of the protocol.
From start to finish, the key stages involved in a PhIP-Seq project include: computational design of the peptidome(s) (Steps 1–6), construction of the phage library (Steps 7–8), quantification of samples’ immunoglobulin content (Steps 9–26), phage-antibody complex formation and immunoprecipitation (Steps 27–41), preparation of DNA sequencing libraries (Steps 42–55), deep sequencing and data analysis (Steps 56–60). Detailed protocols for the construction and expansion of phage libraries can be found elsewhere (Novagen T7Select® System Manual for example). Once a phage library is successfully constructed and expanded, it can be used for analysis of large samples sets and re-expanded at almost no cost.
Additional applications of the protocol.
Beyond performing PhIP-Seq analysis of serum antibodies, we have also used it to identified the epitopes of monoclonal antibodies,4 as well as binding partners of recombinant proteins.1 In addition to identifying cognate antibody targets, we have utilized PhIP-Seq to dissect the fine specificity of antibody binding with variant epitope libraries designed to contain informative non-synonymous mutations and/or truncations.5, 10 For antibody isotype-specific analyses, this protocol can be easily adapted to incorporate streptavidin coated magnetic beads prepared with biotinylated isotype-specific capture antibodies. These and other related applications may require significant deviation from the protocol presented here, along with assay-specific optimization.
Limitations.
It is important that users of PhIP-Seq understand its limitations. Phage displayed peptide libraries may lack the conformational structure required to detect important binding specificities, due to the limited length of synthetic oligonucleotide library encoded peptides (currently up to 90-aa, for example).1, 11 Disulfide linkages and post translational modifications will also be absent from typical T7 phage particles, which are produced in the cytoplasm of E. coli. Since antibodies frequently target conformational and modified epitopes, PhIP-Seq may frequently fail to identify the targets of monoclonal antibodies, compared with those of polyclonal antibodies, which often harbor a subset of specificities that recognize linear epitopes. Depending on the experimental context, PhIP-Seq may therefore perform optimally in combination with alternative methodologies intended to interrogate more ‘native’ protein antigens, such as protein microarray analysis,12, 13 ParalleL Analysis of Translated ORFs (PLATO)14, 15 and/or immunoprecipitation followed by mass spectrometry16.
Experimental Design
Design and Construction of a Bacteriophage Library.
After downloading or constructing the protein sequence database or translated open reading frame (ORF) database to be encoded, we use the pepsyn Python package (https://github.com/lasersonlab/pepsyn) to design our oligonucleotide library, including the following processes: (i) splitting the protein sequences into peptide tiles of chosen length and chosen length of overlap, (ii) selection of representative peptide sequences from peptide clusters of similarity greater than a chosen threshold, (iii) reverse translation of the selected peptide tiles with an optimized E. coli codon usage algorithm, (iv) addition of forward and reverse PCR primer binding sequences to the resulting DNA sequences, and (v) removal of restriction cloning sites (aside from those intended) by silent codon substitution (Figure 2A). The resulting DNA sequences are then provided to a DNA manufacturer for oligonucleotide library synthesis (“OLS”). We have previously purchased libraries from Agilent Technologies, Inc, who employs ink jet printing technology for synthesis. Alternative vendors include Twist Bioscience, for example.
Figure 2.

Bioinformatics workflows. Procedure step numbers are indicated. A. Pepsyn workflow. Workflow for designing a peptide library. We only provide an outline of the protocol as this stage will likely be customized depending on your library/preferences. B. PhIP-stat workflow for PhIP-Seq data analysis.
The optimal lengths and overlaps of the peptide tiles are governed by considerations related in part to the manufacturing of the oligonucleotide library. There are two main tradeoffs in terms of tile length. Longer peptides will contain greater secondary structure, which is an important aspect of many antibody-epitope interactions. However, longer oligonucleotides will contain more mutations per peptide, and thus may reduce the overall quality of the library. A second consideration relates to assessment of polyclonal responses. Observing multiple, non-overlapping enriched peptides from the same protein may provide increased confidence in an antigen-driven response, versus a single, potentially cross-reactive antibody specificity. The length of the overlaps determines both the density of the tiles (i.e. how many tiles per length of protein), as well as the size of the smallest epitopes contained in the library. There is thus another tradeoff in terms of library size (and thus the cost to construct and sequence it), versus the coverage of antigenic space.
Upon receipt of the synthetic oligonucleotide pool, standard PCR (we prefer the Herculase II DNA Polymerase from Agilent) is used for amplification, prior to restriction cloning into a phage vector of choice (we have used a derivative of the T7Select 10–3b, mid-copy system called T7-FNS2), according to the manufacturer’s instructions (Novagen T7Select® System Manual). We have preferentially employed the T7Select 10–3b mid-copy system for PhIP-Seq, primarily because lytic bacteriophage libraries are expected to exhibit less bias compared to trans-membrane secretion systems (such as M13, for example). The mid-copy system permits display of up to ~1,000 amino acid long peptides at a copy number of 5–15 per particle. The drawback of the T7Select system is that libraries must be packaged using an expensive extract, which is also less efficient compared to electroporation into host bacterial cells.
The success of any PhIP-Seq project will depend upon the quality of the starting phage library. Aside from the library design and fidelity of the oligonucleotide library synthesis, the quality of the library is also determined by clonal dropout, skewing, titer, and presence of contaminants. ‘Dropout’ refers to the loss of peptide library members due to insufficient coverage of the library during construction of the initial, unexpanded phage library. We recommend scaling the library construction steps such that each library member is always represented on average by at least 100 infectious particles. For example, packaging of the T7 gDNA ligation reaction should result in at least 107 plaques for a library containing 105 unique peptides. This applies to library expansion too. Library skewing refers to changes in relative abundance of individual library members due to differing growth kinetics and stochastic fluctuation. There are a variety of factors that determine how a particular displayed peptide will influence the kinetics of the phage clone’s growth. Even small differences in phage replication efficiency can result in significant differences in a clone’s final representation within the expanded library, especially after serial expansions. Phage clones that express truncated peptides due to nonsense mutations may have a growth advantage over their unmutated counterparts, especially for longer or toxic peptides, thus reducing the representation of the corresponding unmutated library members. Phage library degradation due to skewing is minimized primarily by expanding the library on solid media (rather than in liquid culture), and by avoiding unnecessary serial passage of the library. The expanded screening library should have a plaque forming unit (pfu) concentration (titer) that provides each library member with a representation of at least 105 pfu per ml. This means that for a library of complexity 105, an absolute minimal titer of 1010 pfu/ml must be achieved in the expanded library. For the T7Select 10–3b mid copy system, we typically obtain titers ~1011 by centrifugally concentrating log phase host bacterial cells to an optical density (at 600 nm) of ~4, just prior to performing plate amplification of the library (otherwise according to the manufacturer’s instructions). Finally, it is important to remove particulate (including bacterial cells) from the expanded phage library lysate by centrifugation and to prevent further growth of bacteria by addition of a second antibiotic, to which the host cells are sensitive. We store our final expanded phage peptidome library aliquots indefinitely at −80 °C after addition of 10% DMSO.
Library Quality Control.
The quality of each new, or newly expanded, phage library should be assessed in two ways prior to screening. First, several plaques (we recommend at least 20) should be individually picked and the inserts analyzed by Sanger sequencing to assess the fidelity of the oligonucleotide synthesis (as described in the Novagen T7Select® System Manual). Clones expressing mutated or truncated inserts may have a growth advantage over intact inserts, so it is important to pick from a representative range of physical plaque sizes in order to avoid unintentionally underestimating the quality of the library due to biased plaque selection. Second, the library should be analyzed using deep sequencing1, 5 in order to assess the baseline distribution of the clonal frequency and completeness of the library. Ideally, 90% of the library should fall within one log of clonal frequency, and at least 90% of the library should be observed at a sequencing depth of at least 10 reads per clone.
Protein A/G-based immunoprecipitation.
For convenience and optimal performance, we suggest using protein A/G coated magnetic magnetic beads as the capture matrix for PhIP-Seq experiments. In order to obtain reproducible inter-sample data, it is important that the amount of IgG antibody input be uniform and below the binding capacity of the capture matrix. Steps 9–26 of this protocol are therefore devoted to the measurement of each sample’s IgG concentration, to ensure appropriate IgG input.
Antibody isotype-specific immunoprecipitations.
For antibody isotype-specific immunoprecipitations, we recommend pre-coating M-280 streptavidin Dynabeads (Invitrogen, cat# 11205D) with an amount of capture antibody that is two to four times the binding capacity of the beads. This will minimize bead aggregation, which can dramatically reduce target antibody capture efficiency. For isotype-specific antibody capture experiments, we typically immobilize ~1 ug of capture antibody, for capture of at most 1 ug of target antibody. Aside from these bead preparation protocol variations, the remainder of the Procedure will apply equally well.
Controls.
Within-experiment negative controls (and for 96-well plate runs, preferably within plate negative controls) are extremely important for obtaining interpretable results. Such controls depend on the experimental design. For example, profiling antibodies to identify antibody-phenotype associations should include analysis of individuals without the phenotype, but who are otherwise well matched. Technical positive and negative controls are also important to include when possible. Positive controls may include monoclonal antibodies or previously analyzed samples, for example. We strongly suggest including a set of negative controls that lack antibody input, to obtain important background binding information for each phage clone. Sequencing of the unenriched input library is required to determine the clonal distribution of the starting library and to use the computational pipeline provided here. To this end, we typically add ~1E7 pfu of starting library to a PCR1 reaction.
Sequencing the Library.
A variety of high throughput (‘next generation’) DNA sequencing platforms now exist for the analysis of DNA libraries. PhIP-seq can in principle be adapted to any such platform, provided that the number of sequencing reads per library member is sufficient for quantification of peptide enrichment (ideally ~10 reads per clone on average). We have primarily utilized the Illumina HiSeq and NextSeq instruments, as they provide the lowest per-read cost. The PCR primer sequences presented here therefore include the Illumina sequencing adapters (suitable for both single and paired-end flow cells). Substitution with different platform-specific adapters should be straightforward. In addition, we present PCR primers that are specific for one of our T7-FNS2 derived libraries (VirScan), but these are easily replaced with alternative, library-specific primers. The sequencing primer provided here is also specific for the VirScan library, but any appropriately designed sequencing primer can be used instead. The use of a sequencing primer that results in balanced base incorporation for at least the first 5 sequencing cycles is necessary for Illumina instruments to resolve clusters. Use of a PhIX spike-in (at >30%) can help with cluster resolution for biased libraries when this is not possible.
A second DNA barcode may be incorporated into the PCR2 forward primer for “dual indexing” (e.g. AATGATACGGCGACCACCGAGATCTACACxrefXXGGAGCTGTCGTATTCCAGTC, where the eight X’s represent the eight nucleotides of the i5 index). This allows combinatorial (i5 + i7) barcoding of PCR products, thus increasing the level of potential sample multiplexing.17 Of note, on certain Illumina instruments (e.g. NextSeq 500), i5 is sequenced in the reverse direction such that it requires a custom i5 sequencing primer (AGCATCACACCTGACTGGAATACGACAGCTCC).
Data Analysis.
Analysis of high throughput DNA sequencing data requires an informatics pipeline, which can be implemented on a high performance computing cluster. The analytical stages include: (i) demultiplexing and alignment to the reference sequence database, (ii) tabulation of aligned sequences, (iii) statistical evaluation of each peptide’s enrichment within each sample, and (iv) interpretation of peptides’ enrichments, the first three steps of which are illustrated in Figure 2B. For stage (iii), we have previously reported the use of a generalized Poisson distribution as a null model.1 Conceptually, it is important to understand that sequencing-based quantitation of library member abundance is governed by sampling statistics of count data. More abundant clones will be sampled more deeply compared to less abundant clones, meaning that differences in relative abundance can be measured more accurately for more abundant clones. For example, a 10-fold enrichment can be much more reproducibly measured for a clone that is sequenced hundreds of times in the control condition, versus a clone that is sequenced only once or twice in the control condition. Our statistical model therefore takes this into account when comparing enrichments among differentially abundant clones.
How can measures of phage clone enrichment be correlated to more familiar concepts such as assay dynamic range, signal-to-noise, antibody titer, and so on? Unfortunately, there is no simple way to convert the statistical assessment of PhIP-seq enrichments into parameters that are traditionally applied to single-plex assays, which typically produce chemical signals of a continuous (non-discrete) nature. For each individual target peptide, one could envision constructing a sample dilution standard curve to plot the PhIP-seq enrichment p-value against the signal from a corresponding ELISA assay, for example. However, such an exercise may be of little value, as the relative impact of differences in antibody abundance, affinity, or avidity are expected to differ in a nonlinear way between these two types of measurements.
After quantifying phage peptide enrichments, project-specific considerations will determine the best approach to the interpretation of their significance. For example, we have utilized permutation analyses of cross-sectional case-control studies to set false discovery rate thresholds on lists of candidate disease-associated autoantibodies.2 Longitudinal studies, on the other hand, may entail intra-patient pairwise sample comparisons. It should also be emphasized that PhIP-seq is primarily a hypothesis generating tool, and that absence of peptide enrichment cannot be interpreted as absence of the corresponding antibody specificity. We therefore suggest confirming PhIP-seq discoveries via at least one or two orthogonal assays. In the case of autoantigen confirmation, we have utilized mammalian cells that express full length, epitope-tagged proteins.1, 3 Western blotting for the epitope tag can be used to assess the abundance of the tagged protein in the immunoprecipitate. ELISA assay using commercially available proteins is another possibility. For validation of anti-viral antibodies, clinically validated antibody tests are available for a variety of human pathogens.
Materials
Equipment
E-max Precision Microplate Reader (Molecular Devices)
96-well ELISA plates (Thermo Fisher Scientific)
Thermolyne Labquake Rotator (Barnstead)
2.0 ml, PP, Pyramid-Bottom, Non- sterile 96-well plates (Cell Treat)
Full skirted PCR plate (Bio-Rad)
Silicone 96-well plate sealing gaskets (Thermo Fisher Scientific)
MicroAmp Optical Adhesive Film (Thermo Fisher Scientific)
96-well magnet (Agilent) or Magnetic Particle Concentrator (for 1.5 ml tube, Thermo Fisher Scientific)
Software
Prism (Version 6, GraphPad Software)
Python (pepsyn requires Python 3.6+; phip-stat works with Python 2.7 and Python 3)
pepsyn for oligo design tools (https://github.com/lasersonlab/pepsyn) CRITICAL The pepsyn package is under active development; check the README on the GitHub site for the latest protocols.
(optional) cd-hit for clustering oligos to reduce redundancy (http://weizhongli-lab.org/cd-hit/)
phip-stat for processing of PhIP-seq raw data (https://github.com/lasersonlab/phip-stat) CRITICAL The phip-stat package is under active development; check the README on GitHub for the most up-to-date protocol. Example data is available in the GitHub repository in the examples/ directory.
bowtie for alignment (http://bowtie-bio.sourceforge.net/index.shtml)
(optional) HPC cluster with batch job scheduler like LSF, Grid Engine, SLURM, etc.
Equipment Setup
Input data
This protocol assumes the existence of a text file called input_orfs.fasta that contains the full protein library sequences in fasta format. We recommend the sequence identifiers for each protein sequence should be a simple, unique name, ideally without spaces or other punctuation other than underscores, periods, or dashes. Most tools in the pepsyn package accept “-” as the input and output files, which will read/write fasta data from stdin and stdout. This facilitates the modular integration of various tools into processing pipelines using the Unix pipe functionality. The protocol below is just one possible example that illustrates this principle, and the separate processing parts can be easily be swapped or varied. The computations are generally fast, allowing rapid iteration on designs. The commands below are executed in a bash shell.
Software Installation and Setup
We recommend using the Anaconda/Miniconda Python distribution for all Python work. It is easy to install and comes with a modern package manager (conda) to manage local Python environments. It can also install other non-Python software (such as cd-hit and bowtie).
Install miniconda into your home directory on your local machine or cluster using the following commands:
curl https://repo.continuum.io/miniconda/Miniconda3-latest-Linux-x86_64.sh \ > miniconda3.sh bash miniconda3.sh -b -p $HOME/miniconda3
(See the ContinuumIO documentation at https://conda.io/docs/installation.html for a Windows-compatible command.) If desired, add the new Python distribution to your PATH to make sure it is set as the default distribution by setting the following commands:
export PATH=“$HOME/miniconda3/bin:$PATH”
in your .bash_profile configuration file (using the correct location of the conda installation).
Install the Python packages required for pepsyn and phip-stat using the following commands:
conda install -y numpy scipy biopython click tqdm
Finally, you can use conda to install bowtie and cd-hit as well. First add the “bioconda” channel to your conda installation using the following commands:
conda config --add channels conda-forge conda config --add channels defaults conda config --add channels r conda config --add channels bioconda
Then install bowtie and cd-hit using the following commands:
conda install -y bowtie cd-hit
The tools should now be available for use from your PATH. Using conda, it is very easy to switch between Python 2 and 3, or different custom environments.
Using phip-stat to process the raw data, many users will be working on an HPC cluster with a job scheduler. Typically on such a cluster, one would submit a job for batch execution like so:
bsub –q expressalloc –W 0:20 my_command
which will execute my_command somewhere on the cluster assuming you use the LSF scheduler.
CRITICAL: Consult with your local HPC cluster for guidance on submitting many jobs concurrently. Steps that are relatively computationally intensive and parallelizable are pointed out in the Procedure.
Reagents
CRITICAL: Prepare all solutions using deionized water. Prepare and store all reagents at room temperature, 25 °C (unless otherwise indicated).
Capture Antibody: Goat anti-human IgG-UNLB (store at 4°C, Southern Biotech catalogue # 2040–01)
Detection Antibody: Goat F (ab’) 2 Anti-Human IgG-HRP (store at 4°C, Southern Biotech catalogue # 2042–05)
Human IgG ELISA Standards (store at −20°C, Life Technologies Life Technologies catalogue # 02–7102)
One Step Turbo TMB ELISA (Store at 4°C. Thermo Fisher Scientific catalogue # 34022)
Stop Solution: 2 N or 1 Molar H2SO4 (Store at room temperature. Sigma Aldrich catalogue # 258105–100ml)
Dynal Protein A and Protein G Beads (store at 4°C, Invitrogen catalogue # 10002D Dynabeads Protein A, and catalogue # 10004D Dynabeads Protein G)
Serum samples for study. CAUTION: Serum samples must be analyzed in compliance with IRB approved Human Subject Research guidelines. The example data shown here were obtained from de-identified donors under the JHU Human Subject Research exemption IRB00049327.
Caution: Wear a one-time use splash shield, a mask, a biohazard suit and surgical gloves while handling human serum samples. Dispose after single use. Serum samples are stored in cryovials at −80 C for long-term usage. The diluted serum sample (1:1 million dilution) can be stored at 4°C for two days.
PCR primers, Integrated DNA Technologies: see Table 1 and Supplementary Table 1.
Table 1.
Primer sequences for PhIP-seq. Primers for PCRs 1–3 can be used for any PhIP-seq project which is based on the T7Select 10–3b FNS2 vector1, 5. Underlined sequences are adapter sequences, and thus do not participate in the initial round of PCR. The bold sequence is the barcode (“index”) that uniquely defines the sample. We have designed and tested 96 of these sequences; an example (X=1) is shown here. The remaining 95 ad_min_BCX_P7 primer sequences can be found in Supplementary Table 1. The T7-VirScan_SP sequencing primer is specific for analysis of the VirScan library. The Read 2 Illumina Index Read Primer (“Index_SP”) is a standard Illumina primer and available for use from most high throughput DNA sequencing core facilities free of charge.
| Step | Primer Name | Sequence |
|---|---|---|
| PCR1 (Step 42) | T7-Pep2_PCR1_F | 5’-ATA AAG GTG AGG GTA ATG TC-3’ |
| PCR1 (Step 42) | T7-Pep2_PCR1_R+ad_min | 5’-CTG GAG TTC AGA CGT GTG CTC TTC CGA TCA GTT ACT CGA GCT TAT CGT C-3’ |
| PCR2 (Step 45 | T7-Pep2_PCR2_F_P5 | 5’-AAT GAT ACG GCG ACC ACC GAG ATC TAC ACG GAG CTG TCG TAT TCC AGT C-3’ |
| PCR2 (Step 48) | ad_min_BCX_P7 (X is 1 to 96) | 5’-CAA GCA GAA GAC GGC ATA CGA GAT GAC TGA CTG TGA CTG GAG TTC AGA CGT GTG CTC-3’ |
| PCR3 (Step 52) | P5 | 5’-AAT GAT ACG GCG ACC ACC GA-3’ |
| PCR3 (Step 52) | P7.2 | 5’-CAA GCA GAA GAC GGC ATA CGA-3’ |
| Sequencing, Read 1 (Step 55) | T7-VirScan_SP | 5’-GGT GTG ATG CTC GGG GAT CCA GGA ATT CCG CTG CGT-3’ |
| Sequencing, Read 2 (Step 55) | Index_SP | 5’-GAT CGG AAG AGC ACA CGT CTG AAC TCC AGT CAC-3’ |
Herculase II Polymerase (store at −20 °C, Agilent catalogue # 600679)
DNA Clean & Concentrator kit (Store at room temperature. Zymo Research catalogue # D4005)
Agarose, Type I EEO (Store at room temperature. Sigma-Aldrich catalogue # 9012–36-6)
KAPA Library Quantification Kit (Store at −20 °C, KAPA Biosystems, catalog #KK4828)
NucleoSpin Gel and PCR Clean-up (Store at room temperature. Macherey-Nagel catalogue # 740609.50)
T7Select Packaging Kit (store at −80°C, EMD Millipore catalog # 70014–3) for library construction
T7Select 10–3b DNA (store at 4°C, EMD Millipore catalog # 70548) for library construction
NaHCO3 (Sigma Aldrich catalogue # S5761)
Na2CO3 (Sigma Aldrich catalogue # 223530)
PBS (ThermoFisher Scientific catalogue #
10010023)
Fetal bovine serum (Corning catalogue # 35–011-CV)
150 mM NaCl (Sigma Aldrich catalogue # S7653)
50 mM Tris-HCl (Sigma Aldrich catalogue # RES3098T-B7)
0.1% NP-40 (Sigma Aldrich catalogue # 492016)
Reagent set up
ELISA Coating Buffer:
Dissolve 2.93 g NaHCO3 and 1.5 g Na2CO3, in 900 ml of deionized water, pH to 9.5 (pH indicated is critical), adjust final volume of the buffer to 1 L. Store at room temperature for up to one month
ELISA Wash Buffer:
Mix 0.5 ml of Tween-20 in 1L PBS. Store at room temperature for up to one month.
ELISA Blocking Buffer
Mix 5% fetal bovine serum in 1X PBS. Make fresh and store at 4°C for no more than one week.
Magnetic Bead Wash Buffer:
Supplement 1x PBS with 0.02% Tween-20. Store at room temperature for up to one month.
IP Wash Buffer:
Make 150 mM NaCl (8.76 g/L), 50 mM Tris-HCL (7.88 g/L) and 0.1% NP-40 (1 ml/L), pH to 7.5 (pH indicated is critical). Store at 4°C for up to one month.
Procedure
Synthetic Peptidome Library Design. Timing: 1 day
-
1Generate two sets of peptide sequences: one set that tiles across the whole protein and a separate set that is comprised of the C-terminal peptides, using the following commands in the pepsyn software package.
TILESIZE=56 OVERLAP=28 cat input_orfs.fasta \ | pepsyn x2ggsg - - \ | pepsyn tile -l $TILESIZE -p $OVERLAP - - \ | pepsyn disambiguateaa - - \ > orf_tiles.fasta cat input_orfs.fasta \ | pepsyn x2ggsg - - \ | pepsyn ctermpep -l $TILESIZE --add-stop - - \ | pepsyn disambiguateaa - - \ > cterm_tiles.fasta
Note how the commands are stitched together into a pipeline, each one reading fasta data and writing fasta data, allowing for flexible and modular pipelines during the design phase. The first command (pepsyn x2ggsg) eliminates stretches of Xs by replacing them with glycine-serine linker sequence. The next command (either pepsyn tile or pepsyn ctermpep) chops up each ORF into short tiles with specified length. The tile version generates overlapping sequences, while ctermpep only takes the last amino acids of the sequences (i.e., “C-terminal peptide”). Finally, disambiguateaa removes ambiguous IUPAC amino acid codes (e.g., B for aspartic acid or asparagine) by generating all possible peptides. The peptides are written into orf_tiles.fasta and cterm_tiles.fasta. Note that we have elected to add amber stop codons to the C-terminal peptides to allow flexibility in whether native stop codons are incorporated into the peptide or not.
CRITICAL STEP: You can find usage notes by adding -h to any command (e.g., pepsyn -h or pepsyn tile -h). There are numerous additional tools that perform helpful operations in peptide design (e.g., pepsyn clip for trimming sequences, pepsyn builddbg for building a De Bruijn graph on k-mers).
-
2The resulting files may contain peptides that are identical or highly similar to each other. Eliminate some of this redundancy using the cd-hit tool, similar to what is done in the UniProt database, using the following commands:
cd-hit -i orf_tiles.fasta -o orf_tiles_clustered.fasta \ -c 0.95 -G 0 -A 50 -M 0 -T 1 -d 0 cd-hit -i cterm_tiles.fasta -o cterm_tiles_clustered.fasta \ -c 0.95 -G 0 -aL 1.0 -aS 1.0 -M 0 -T 1 -d 0
In this particular case, we are clustering the peptide tiles to 95% (-c 0.95) local identity (-G 0) while controlling the alignment coverage (-A 50 requires the alignment to cover at least 50 amino acids). The C-terminal peptides are aligned more stringently to ensure that the final residues of the ORF are not lost (-aL 1.0 –aS 1.0 requires 100% of each sequence to be aligned with possible mismatches). Specifying –M 0 allows unlimited memory, -T 1 specifies one CPU thread, and –d 0 ensures sequence names are not truncated. See cd-hit documentation for more options (cd-hit.org). The clustered peptides are written to orf_tiles_clustered.fasta and cterm_tiles_clustered.fasta.
-
3The rest of the peptide processing is the same for the C-terminal and tiled peptides. Use the following commands to first concatenate the files (cat). Because the results of the last step could generate some peptide sequences shorter than 56 amino acids, also pad the peptides to make them uniform length (pad).
cat orf_tiles_clustered.fasta cterm_tiles_clustered.fasta \ | pepsyn pad -l $TILESIZE --c-term - - \ > protein_tiles.fasta
The final peptide tiles are combined in protein_tiles.fasta.
-
4To this point, we have been manipulating amino acid sequences. Now, reverse-translate the peptide sequences into DNA sequences using the revtrans command. This command randomly chooses codons according to the E. coli frequency table, dropping any codons that are more rare than a given frequency threshold. We use exclusively the amber stop codon. Add prefix/suffix sequences that will be used for PCR/cloning. Finally, search for any restriction sites that will be used for cloning within the coding sequence and recode them as necessary. The final oligonucleotides are presented in oligos.fasta.
PREFIX=AGGAATTCCGCTGCGT SUFFIX=GCCTGGAGACGCCATC PREFIXLEN=${#PREFIX} SUFFIXLEN=${#SUFFIX} FREQTHRESH=0.01 cat protein_tiles.fasta \ | pepsyn revtrans --codon-freq-threshold $FREQTHRESH --amber-only - - \ | pepsyn prefix -p $PREFIX - - \ | pepsyn suffix -s $SUFFIX - - \ | pepsyn recodesite --site EcoRI –site HindIII --clip-left $PREFIXLEN \ --clip-right $SUFFIXLEN --codon-freq-threshold $FREQTHRESH \ --amber-only - - \ > oligos.fasta ?TROUBLESHOOTING -
5Finally, verify that the library is free of any EcoRI or HindIII sites, using the following command:
pepsyn findsite --site EcoRI --clip-left 3 oligos.fasta pepsyn findsite --site HindIII oligos.fasta
-
6Generate a bowtie index now, in preparation for aligning sequencing data later. Generate a reference fasta file that contains just the DNA tiles without the adaptors using the following command:
pepsyn clip --left $PREFIXLEN --right $SUFFIXLEN oligos.fasta oligos-ref.fasta
Then create the bowtie index (or index for whichever aligner you prefer, such as bwa, bowtie2, or kallisto, among others) called “mylibrary” as follows:bowtie-build -q oligos-ref.fasta bowtie_index/mylibrary
Construction and expansion of the phage screening library TIMING: 3 weeks
-
7
Send the oligos.fasta file to a DNA synthesis company for manufacture of the oligonucleotide library. PAUSE POINT The oligonucleotide library should be aliquoted and stored frozen at −80° C indefinitely.
-
8
Perform library PCR using primers with cloning sites and binding sites for the PREFIX/SUFFIX sequences appended to the oligonucleotide library. Follow procedures for standard cloning of PCR product into bacteriophage for display using published protocols (e.g. Novagen T7Select® System Manual).[Nat biotech paper and VirScan paper] PAUSE POINT The expanded library should be aliquoted and stored in 10% DMSO at −80° C until used. CRITICAL STEP: We recommend centrifuging the expanded library for two hours at 4°C at 3,000 x g and carefully moving the supernatant to a new container prior to freezing, in addition to the centrifugation specified in step 28. CRITICAL STEP: The quality of each new phage library should be assessed by Sanger sequencing of 20 or more randomly selected plaques (to confirm the fidelity of the oligonucleotide synthesis), and by Illumina sequencing to assess the distribution of the clonal frequency (See Experimental Design).
Serum IgG Quantification by ELISA Timing: 1 day
-
9
If working with more than a few samples, randomly assign each sample to a position on a 96-well plate. This will reduce the potential for positional artifacts. Dilute each sample 1:100 in PBS, to a final volume of 200 μl, in a non-tissue culture treated round bottom 96-well plate. Caution: Whenever working with human serum, wear a one-time use splash shield, a mask a biohazard suit and surgical gloves while handling human serum samples. Dispose after single use.
-
10
Dilute unlabeled IgG capture antibody to a final concentration of 2 μg/ml in ELISA Coating Buffer and add 50 μl to each well of an enhanced binding ELISA plate.
-
11
Wrap the ELISA plate with Saran™ wrap and incubate on a flat surface at 4°C overnight.
-
12
Splash out the capture antibody coating solution and wash the ELISA plate 3 times with 150 μl ELISA Wash Buffer.
-
13
Block the ELISA plate by adding 150 μl of ELISA Blocking Buffer to each well.
-
14
Wrap the ELISA plate with Saran™ wrap and incubate on flat surface at 37 °C for at least 1 hr.
-
15
Wash the plate 3 times with 150 μl ELISA Wash Buffer. Just prior to addition of samples and standards, blot plate against a clean paper towel.
-
16
Dilute human IgG (hIgG) standard to 100 ng/ml in ELISA Blocking Buffer and perform six 1:3 serial dilutions in ELISA Blocking Buffer. At the same time that samples are added to the ELISA plate (Step 17), add 50 μl of diluted standard per well, to the empty wells reserved for use as negative controls. We typically include 8 such wells per 96 well plate, one of which serves as a blank and contains blocking buffer only.
-
17
For total human IgG quantitation, serially dilute sera 1:100 two additional times (for a final dilution of 1:1,000,000) in ELISA Blocking Buffer. Add 50 μl of the diluted sample to each well of the ELISA plate at the same time as the IgG standards (Step 16).
-
18
Wrap the ELISA plate with Saran™ wrap and incubate on flat surface at 37 °C for 1 hr.
-
19
Wash the plate 5 times with 150 μl ELISA Wash Buffer. Just prior to addition of detection antibody, blot plate against a clean paper towel.
-
20
Dilute hIgG-HRP detection antibody 1:5,000 in ELISA Blocking Buffer. Add 50 μl to each well of the ELISA plate.
-
21
Wrap the ELISA plate with Saran™ wrap and incubate on flat surface at room temperature for at least 30 min.
-
22
Wash the plate 5 times with 150 μl ELISA Wash Buffer, and blot against a clean paper towel.
-
23
Add 50 μl of TMB substrate to each well. Incubate from 3 – 30 minutes at room temperature until color becomes somewhat dark in the highest IgG standard well, but not yet colored in the blank standard well.
?TROUBLESHOOTING
-
24
Add 50 μl of Stop Solution to each well in the same order as the TMB substrate and at the same speed.
-
25
Read the optical absorbance for each well at 450 nm using a plate reader.
-
26
Prepare an XY table in a graphing program and generate a standard curve graph. Interpolate the sample X-values by nonlinear regression analysis using a “one-site binding” model. To do this, the net OD values for all serum samples are obtained by subtracting the OD of blank well from their original OD values. Then, these OD values are analyzed using Prism software with the Single Site Binding Saturation Regression Analysis to estimate the serum IgG concentration (X-values in ng/ml) of each sample for 1:1,000,000 dilution. Multiply the X-values (ng/ml) by 10,000 to obtain the sample concentration in the 1:100 dilution plate (created in Step 9). Calculate the volume of the 1:100 sample dilution that will contain 2 μg of IgG.
?TROUBLESHOOTING
Pause Point: After IgG quantification, diluted human serum samples from Step 9 can be stored refrigerated at 4°C until proceeding to immunoprecipitation, but not for longer than a couple of days.
Peptide-Antibody Complex Formation and Immunoprecipitation Timing: 2 days
CRITICAL: Steps 27–41 can be performed in single 1.5 ml Eppendorf® tubes for a small number of samples, or in 96-well plate format for larger numbers of samples. Here, we refer only to the 96-well plate format. If performing screens in 1.5 ml Eppendorf® tubes, the Magnetic Particle Concentrator (Thermo Fisher Scientific) can be used rather than the 96-well plate magnet.
-
27
Thaw phage library or libraries from Step 8 and combine into a large enough vessel for all screens that will be performed in the current run. We recommend peptide-antibody complex formation volumes of 1 ml, and an input of 105 pfu per individual phage library member. After addition of phage library (or libraries), make up remaining volume by adding PBS (without divalent cation). Optionally add IP spike-ins (e.g. control antibodies and/or control phage clones) at this time.
-
28
Centrifuge phage library mixture for two hours at 4°C at 3,000 xg and carefully move supernatant to new container, being careful not to disturb any pelleted material (even if no visible pellet). Pelleted material may include cell debris or precipitate that may interfere with the assay and so should be discarded.
-
29
Mix phage very well by pipetting up and down with a serological pipette and distribute 1 ml to each well of a 2 ml deep well plate.
-
30
Add 2 μg of serum IgG from the 1:100 dilution, in PBS (volume calculated in Step 26) to the corresponding wells of the deep well plate containing the phage mix. We suggest running multiple negative controls without antibody so that antibody-dependent enrichments may be quantified by comparison. Optionally, if screening a large number of samples, this step is best automated to avoid error.
CRITICAL STEP: It is important that the amount of input antibody is below the binding capacity of the magnetic beads. If in excess, soluble antibody will compete with bound antibody for specific interactions with target phage, reducing enrichment efficiency and thus sensitivity.
-
31
Rotate mixtures end-over-end in the cold room at 4°C for about 18 hours. If screening in 96-well plate format, wells must be tightly sealed (e.g. with a 96-well silicone matt gasket seal) to avoid cross-contamination. CRITICAL STEP: We have alternatively performed this step at 37 °C for 1 hour with roughly similar results.
-
32
Centrifuge IP mixtures at 1000 x g for 1 minute to remove liquid from gasket seal.
-
33
Wash 20 μl of Protein A and 20 μl of Protein G coated magnetic beads per IP 3 times in Bead Wash Buffer and resuspend in the same volume of 1X PBS.
-
34
For capture of human IgG, add 20 μl of prewashed Protein A coated Dynabeads and 20 μl of prewashed Protein G-coated Dynabeads to each tube or well.
-
35
Again rotate mixtures end-over-end in the cold room for about 4 hours. If screening in 96-well plate format, wells must be tightly sealed (e.g. with a silicone matt gasket seal) to avoid cross-contamination. If peptide-antibody complex formation was performed at 37 °C, this step can also be performed at 37 °C for 30 minutes during end-over-end rotation.
-
36
Centrifuge mixtures at 900xg for 2 minutes at room temperature to remove volume from gasket seal (and to pellet beads if necessary given that the geometry of many magnets would not efficiently pellet beads in deep wells). Optional: Steps 37–41 can be automated. We have implemented the bead washing steps on the BioMek FX and the Agilent Bravo liquid handling robots with similar results.
-
37
Remove and discard supernatant from the pelleted beads, but leave ~100 μl in each well. Use this volume to resuspend the beads and transfer to a full-skirted PCR plate.
-
38
Place plate on 96-well magnet and allow beads to collect. Remove as much supernatant as possible without aspirating beads.
-
39
Immediately resuspend beads in 170 μl of IP Wash Buffer. If using a liquid handling robot, bead resuspension is best accomplished by combined pipetting and light vortexing CRITICAL STEP: strong vortexing may shear the immunoprecipitated phage particles off the beads), until the bead suspension is uniform. This typically requires about 20 cycles of automated pipetting, or about 10 cycles of pipetting by hand.
-
40
Repeat Steps 38 and 39 once more for a total of two bead washes. Performing additional washes or raising the salt concentration can increase the wash stringency as desired for specific projects.
CRITICAL STEP: It is important that the DNA polymerase used for PCR1 be insensitive to residual detergent in the wash buffer. If a sensitive polymerase must be used, a final bead wash lacking detergent should be performed.
-
41
Repeat Step 38 once more so that only the collected beads remain in the wells.
Pause Point: Beads can now be stored frozen (−20 °C to −80 °C) indefinitely until proceeding to PCR1.
Preparation of Peptidome Library DNA for Sequencing Timing: 1 day
-
42.Make enough 1x PCR1 master mix for one 20 μl reaction (19 μl reaction plus ~1 μl of immunoprecipitate and bead volume) per IP as follows:
Component Volume (ul) Final concentration H2O 14.5 5x Herculase Buffer 4 1x dNTPs 0.2 1 mM PhIP-seq_PCR1_F 0.05 0.25 μM PhIP-seq_PCR1_R 0.05 0.25 μM Herculase II Polymerase 0.2 Total 19 CRITICAL STEP: In addition to the sample and mock IPs, devote at least one PCR1 reaction to sequencing of the input library. To this end, use 1 μl of the input phage library as the template for PCR1. This will generate the set of input counts used later in the analysis.
-
43
If frozen, remove IP plate or tubes from Step 41 from freezer and allow beads to come to room temperature. Resuspend beads (~1 μl, the residual volume of beads) into 19 μl of PCR1 master mix from Step 42 and transfer to a full-skirted PCR plate.
-
44Perform thermocycling as follows:
Cycle number Denature Anneal Extend 1 95°C, 2 min 2–21 95°C, 20s 58°C, 30s 72°C, 30s 22 72°C, 3min Pause Point: Either proceed immediately to PCR2 or store PCR1 reactions at −80 °C indefinitely.
-
45Make enough PCR2 master mix for one 20 μl reaction (once primers and template have been added in Step 48) per IP, as follows:
Component Volume (ul) Final concentration H2O 8.55 5x Herculase Buffer 4 1x dNTPs 0.2 1 mM T7-Pep2_PCR2_F_P5 0.05 0.25 μM Herculase II 0.2 Total 13 -
46
Distribute 13 μl of PCR2 master mix to each well of a full-skirted 96-well plate.
-
47
If frozen, thaw PCR1 product from Step 44 and keep on ice.
-
48
To each 13 μl of PCR2 master mix from Step 46, add 2 μl of PCR1 from Step 44 or 47, and 5 μl of the appropriate ad_min_BCX_P7 barcoding reverse primer (from 1 μM stock concentration). Mix well.
-
49Perform thermocycling as follows:
Cycle number Denature Anneal Extend 1 95 °C, 2 min 2–21 95 °C, 20s 58 °C, 30s 72 °C, 30s 22 72 °C, 3min Each DNA library now contains a unique, sample-specifying DNA barcode (or “index”).
Pause Point: Stop and store PCR2 product at −80 °C indefinitely.
-
50
Pool barcoded PCR2 products. If PCR2 proceeds to primer depletion (as evidenced by laddering effect observed on 2% agarose gel), then it can safely be assumed that the amount of PCR2 product will be relatively uniform across all samples. In this case, mix the same volume of PCR2 from each sample (e.g. 5 μl). If, however, the amount of PCR2 is expected to be substantially different between samples, one might want to normalize each sample’s representation in the final pool so as to ensure uniform sampling of each library. For accurate quantification of PCR2 products (before or after pooling), we recommend using the KAPA Library Quantification Kit according to the manufacturer’s instructions.
?TROUBLESHOOTING
-
51
Perform PCR2 product cleanup using DNA Clean & Concentrator columns from Zymo Research according to the manufacturer’s instructions. Repeat this step to clean up the PCR2 product a second time.
-
52If gel purification is desired prior to deep sequencing, perform a PCR3 (a single PCR cycle with replenished primer) to produce a single clear band on the gel. To do so, prepare enough PCR3 master mix for ten 20 μl reactions, as follows:
Component Volume (μl) Final concentration H2O 12.6 5x Herculase Buffer 4 1 x dNTPs 0.2 1 mM P5 0.5 1 μM P7.2 0.5 1 μM Herculase II 0.2 Template (pooled, column purified PCR2 product from Step 50) 200 ng Total 20 -
53Perform thermocycling as follows:
Cycle number Denature Anneal Extend 1 95 °C, 2 min 2 95 °C, 20s 58 °C, 30s 72 °C, 30s 3 °C, 3min -
54
All library DNA should now be non-laddered dsDNA, which will appear as a single sharp band on a 2% agarose gel (Figure 3). Extract the PCR3 product from the gel using the NucleoSpin Gel and PCR Clean-up kit from Macherey-Nagel according to the manufacturer’s instructions. If multiple peptidome libraries are being analyzed simultaneously (e.g. the human peptidome and the human virome), it may be desirable to sequence them separately or to differing depths. In this case, gel electrophoresis of PCR3 may separate differently sized libraries. PCR3 products can then be separately isolated and quantified, prior to mixing together in a ratio that will determine their relative sampling depth. SPRI beads could be used as an alternative to gel purification, however, this is not a method we have tested.
?TROUBLESHOOTING
-
55
Submit purified PCR3 libraries to a core facility for quantification and deep sequencing. Be sure to provide the custom sequencing primer (e.g. T7-VirScan_SP). For libraries that lack diversity in the first several bases downstream of the sequencing primer (due to adapter sequence, for example), it may be necessary to spike in a base-balanced library, such as the PhiX standard control that is used by Illumina for quality control (at up to 30% molar ratio). The library can be sequenced using a 50-cycle, single-end protocol. It is essential that the sequencing run includes the i7 index read (“Read 2”), and that it be of sufficient length (we use 8 nucleotide barcode sequences) in order to link the sample identity with each peptidome library sequence (Figure 4).
Figure 3.

Primer-depleted, pooled VirScan PCR2 products run at a higher molecular weight than expected (shown on a 2% agarose gel in lithium borate).
Lane 1: 1Kb Plus DNA Ladder (Invitrogen)
Lane 2: empty
Lane 3: Primer-depleted VirScan PCR2 product.
Lane 4: Product of VirScan PCR3, without replenishment of primers.
Lane 5: Product of VirScan PCR3, with replenishment of primers of primers (P5 and P7.2).
Figure 4.

Organization of bacteriophage genome, primer binding sites and PCR products. The peptide coding sequence, originally derived from the oligonucleotide library, is cloned into the T7 genome as a C-terminal fusion with the 10B capsid protein. PCR1 (steps 42–44): T7-Pep2_PCR1_F is used as the outside PCR1 primer. T7-Pep2_PCR1_R+ad_min is used as the reverse PCR1 primer, and to add the minimal adapter required for subsequent addition of the sample barcode during PCR2. PCR2 (steps 45–49): The product of PCR1 is used as the template for PCR2. T7-Pep2_PCR2_F_P5 is used as the forward PCR2 primer, and to add the required Illumina P5 adapter (and optionally the i5 dual index, not shown). The set of primers called ad_min_BCX_P7 (where X defines the sample-specific DNA barcode) are used individually as the reverse PCR2 primers, to add the sample-specific DNA barcode, and to add the required Illumina P7 adapter. After pooling PCR2 products from all the samples, a single round of PCR3 is performed (steps 52–53), using the P5 and P7.2 primers, which ensures the DNA libraries are fully double-stranded. Illumina sequencing (step 55): T7-VirScan_SP is the Read 1 sequencing primer used to obtain the peptide coding sequence. Index_SP is the standard Illumina Multiplex Single End Read 2 sequencing primer used to obtain the sample-specific barcode. The sequences generated from the Illumina sequencing run are shown as dashed lines: Read 1 obtains the first 50 bases of the peptide coding sequence and Read 2 is the 8 cycle index read
Processing the Raw PhIP-seq Data Timing: several hours
-
56Separately align each sample’s .fastq using the bowtie short read aligner, with the following command:
mkdir -p workdir/alns bowtie -n 3 -l 100 --best --nomaqround --norc -k 1 -p 4 --quiet \ bowtie_index/mylibrary workdir/reads/sample1.fastq.gz \ workdir/alns/sample1.aln
CRITICAL STEP: See bowtie’s documentation (bowtie-bio.sourceforge.net) for additional alignment options. With many samples, the commands can be submitted to a batch job scheduler such as LSF, Grid Engine, or SLURM that are commonly available in scientific computing environments.
CRITICAL STEP: This step is computationally expensive and we recommend submitting a job for each file to a batch system on a cluster.
CAUTION: Take care with correctly specifying the path to the bowtie index. If the bowtie index is called index/mylibrary.1.ebwt (along with the additional files), then you should specify index/mylibrary. Note that the backslashes above mean line-continuation.
?TROUBLESHOOTING
-
57Aggregate each alignment file into a count vector for that sample, using the following command:
phip compute-counts -i workdir/alns -o workdir/counts \ -r path/to/input/counts.tsv
The command line flags are: -i, input directory; -o, output directory; -r, reference file containing the input counts for the library. Each count file generated by the command above will contain one column for the input counts (specified with -r) and another column for the counts in that sample (specified with -i). Therefore, this step requires the input counts generated in step 42. Alternatively (and if input counts are not available), aggregated counts from negative control samples can also be used with the -r flag. The input counts are necessary for the statistical model used to compute the enrichment scores. Since this current step is relatively light-weight, it is performed locally.
?TROUBLESHOOTING
-
58Generate (–log10) p-values from the counts by fitting a Generalized Poisson model and computing a significance score for each pair of count values. Specifically, we model the count value Yi for peptide i as
Yi ~ GeneralizedPoisson(λ(Xi), θ(Xi))
where the functions λ(x) = a x + b and θ(x) = c are fit empirically to the observed data. For each possible input value x, we compute the maximum likelihood estimates for λ, θ using the counts of all peptides with x reads, and regress the λ’s and θ’s against the input counts to get estimated λ and θ as a function of x. The scores can be generated by running the following command:phip compute-pvals -i workdir/counts/sample1.tsv \ -o workdir/mlxp/sample1.mlxp.tsv
Here, -i is a file containing sample counts and -o is the destination file containing the MLXP values (Note: “mlxp” is short for “minus log10 p-val”).
CRITICAL STEP: This step is computationally expensive and we recommend submitting a job for each file to a batch system on a cluster.
?TROUBLESHOOTING
-
59Alternatively, merge the count values into a single tab-delimited file to make it easier to analyze as a single matrix with the following command: .
phip merge-columns -i workdir/mlxp -o mlxp.tsv -p 1
Here, -i is directory containing MLXP files and -o points to the merged MLXP file containing the full matrix.
This will merge the 2nd column (zero-indexed) of each file together; it assumes the first column is the join key. This step can also be parallelized on a batch scheduler like the alignment step.
-
60Load the resulting tab-delimited file into Python or R as a dataframe for further analysis (e.g. the Python pandas library (https://pandas.pydata.org/) or the R tidyverse (https://www.tidyverse.org/)). In python, the command would be:
import pandas as pd df = pd.read_csv(‘mlxp.tsv’, sep=‘\t’, header=0)
TROUBLESHOOTING
Troubleshooting advice can be found in Table 2.
Table 2:
Troubleshooting
| Step | Problem | Possible Reason | Solution |
|---|---|---|---|
| 4 | Oligo library sequences not generated | Illegal characters in header or protein sequence. | Remove illegal characters |
| 23 | Low ELISA signal | Poor binding of capture antibody. TMB expired. | Make sure proper ELISA plates are being used, TMB not expired. |
| 26 | ELISA data not within dynamic range | TMB was developed for too long or not long enough. | Increase or reduce the TMB development time. |
| 50, 54 | No PCR product | Incorrect primers were used. Incorrect thermocycling conditions were used. dNTPs expired. | Carefully repeat with fresh reagents. |
| 56 | Crash or program freeze. | This step essentially rewrites the entire data set and thus may use more disk space than is available. | Ensure your filesystem has enough disk space. |
| 57 | Too much of library is missing | Library was bottlenecked during construction or became too skewed during expansion. | Reconstruct library |
| 58 | Extreme, unreproducible enrichments | Contamination by host bacterial cells | More stringent centrifugation to remove cells, addition of antibiotic to prevent growth |
| 58 | Unreproducible enrichments | Sample cross-contamination. This can usually be determined by examining overlapping hits between samples. | Avoid antibody cross-contamination, PCR1 cross-contamination, PCR2 barcode cross-contamination |
Timing
Steps 1–6, peptide library design: 1 day
Steps 7–8, construction and expansion of the phage screening library: 3 weeks
Steps 9–26, IgG quantification by ELISA: 1 day
Steps 27–41, antibody binding and immunoprecipitation: 2 days
Steps 42–55, DNA sequencing library preparation: 1 day
Steps 56–60, PhIP-Seq data processing: several hours
Anticipated Results
The results of any PhIP-Seq experiment completely depend on the samples and libraries used for the analysis. We have observed the number of both autoantibody and viral antibody specificities to increase with the age of the donor. Nearly all human serum samples we have analyzed contain antibodies to Rhinovirus A peptides, and most adults harbor antibodies that bind several Epstein Barr Virus, as well as Herpes Simplexvirus 1 peptides. Known autoantibodies can be detected with variable success. For example, TRIM21 (“Ro52”) antibodies can be detected in ~90% of Sjögren’s Syndrome patients who are seropositive by the clinical ELISA assay, whereas we tend not to detect clinically confirmed anti-insulin antibodies present in type 1 diabetics. PhIP-seq is in many cases less sensitive than optimized single-plex assays, but has the advantage of being much more comprehensive in assessing antibody binding specificities.
The sections below provide additional details about key results generated in the course of a PhIP-seq project.
Output of the peptide library design software
The ultimate output of the library design phase is a fasta file containing oligo sequences (Step 4) that will be sent out for synthesis. The design phase can use multiple pepsyn tools in a pipeline, and we recommend inspecting the results of intermediate steps to ensure they correspond to expected results. For example, are all of the expected ORF sequences represented? Are the peptides/oligos the expected length? Are the number of oligos allowable given your budget? It is crucial to check the final library design, as synthesis is costly and will waste time if it has to be repeated. Especially important is to ensure the length of the resulting oligos is as expected, and to test whether designed oligos contain restriction sites that may pose a problem during cloning (Step 8).
Quality control of the input phage library
Following deep sequencing of the input library and read count tabulation (Step 57), ideally, 90% of the library should fall within one log of clonal frequency. If sequencing depth is of 10 or more reads per library member is achieved, >90% of the library should be sequenced at least once.
IgG ELISA data
The standard curve data (Step 26) should be visually inspected, along with the data from the samples, to ensure that sample measurements are within the dynamic range of the assay (e.g. significantly above background and below saturation of the standard curve). For typical serum samples, IgG concentration should be roughly 10 μg/μl. However, samples stored frozen for a long time will tend to concentrate due to sublimation, compared to fresh serum. The volume of the 1:100 diluted serum samples required for 2 μg, should therefore be about 20 μl, but may be substantially more or less. If the calculation is significantly different than expected, however, there may be a problem with the standards or the calculation.
Amplification of the sequencing libraries
The 20 cycles recommended for PCR1 (Step 44) do not produce high concentration amplicon. Nevertheless, a weak PCR1 band (507 nt for VirScan) can usually be visualized on an agarose gel after extended exposure. Fewer than the recommended 20 cycles of PCR2 are sufficient to produce enough library for sequencing. However, we typically perform 20 cycles of PCR2 (Step 49) to ensure complete primer depletion, and thus equimolar amplification yield, among all samples. There is thus no need to separately quantify the PCR2 amplicons from each reaction prior to pooling. Such “overamplified” libraries, however, run as non fully dsDNA structures (including pseudoconcatemers) on agarose gels. Gel extraction thus requires a single round of primer replenished PCR3 (Step 53), which produces fully dsDNA product at the expected molecular weight (Figure 3).
Sequencing data processing
You should expect to successfully align at least 70% of your raw reads (Step 56). Lower alignment rate can indicate the use of the wrong index, poor sequencing quality, or a high rate of synthesis error in your oligonucleotide library.
Enrichment analysis
It is important to run several types of control samples, especially during the initial establishment of the PhIP-seq platform. Negative controls (no antibody input, “mock IPs”) should be run alongside samples in every experiment. We typically reserve 4 to 8 wells on a 96 well plate for such controls. These data should reveal relatively few enriched peptides; reproducible enrichments may reflect peptide-dependent “background” binding to the beads. Extreme, unreproducible enrichments in these negative controls may indicate contamination of the phage library with host bacterial cells. Replicate IPs should be quantitatively and visually compared for high concordance.
Figure 5 illustrates analysis of sample data obtained by screening the 90-mer human peptidome library against two Sjögren’s Syndrome patients (in duplicate). Data normalization and background bias removal using the Generalized Poisson model provide antibody-dependent p-values of enrichment for each peptide (Step 58). These enrichments are reproducible and largely patient specific. However, peptides from the Ro52 antigen are strongly enriched by both patients.
Figure 5.
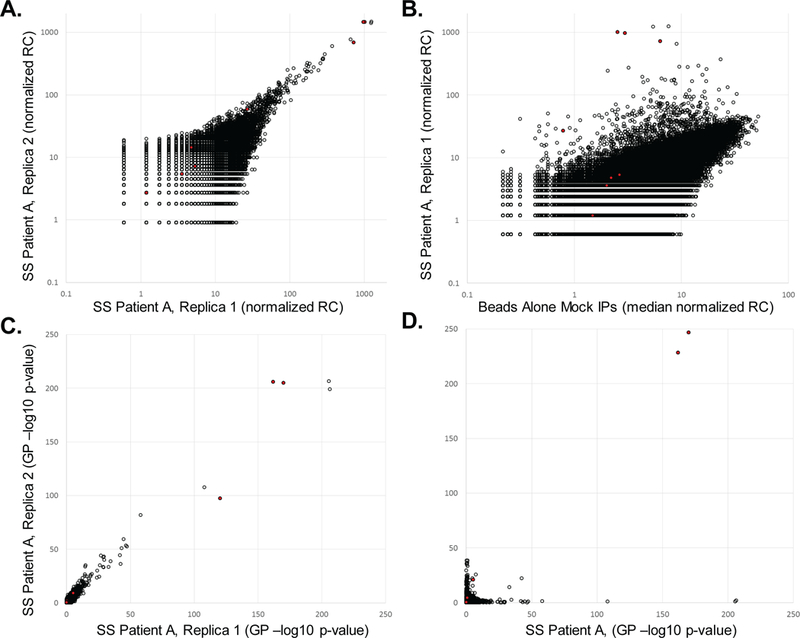
Output from the sequencing data analysis pipeline. (A) Sjögren’s Syndrome (SS) patient A’s serum sample was screened against the human peptidome library and analyzed in duplicate. Read counts were divided by the total reads, multiplied by 1×106 and then plotted. The scatter plot illustrates the reproducibility of the post-immunoprecipitation clonal distributions between the two replicas. Red filled circles highlight peptides from the Ro52 (TRIM21) protein, to which this patient was known to have autoantibodies. (B) Comparison of patient A’s immunoprecipitated clonal distribution to that of a set of mock IPs (no sample input), which illustrates (i) the bias in the starting library and (ii) antibody-dependent enrichment of specific phage clones (including strong enrichment of three Ro52 peptides). (C) Generalized Poisson (GP) based p-values calculated using the data in (A) as input. Background bias has been removed from this distribution, which illustrates reproducible antibody dependent enrichments. (D) Comparing the enrichment scores (-log10 p-values) of two different individuals illustrates their largely non-overlapping enrichment profiles. However, three peptides from Ro52 are among the shared enrichments. De-identified serum samples were analyzed in accordance with JHU Human Subject Research exemption IRB00049327.
Supplementary Material
Design of 96 ad_min_BCX_P7 PCR2 i7 indexing primers.
Acknowledgements
The development of the PhIP-seq technology platform has been an ongoing collaboration with Professor Stephen J. Elledge of the Harvard Medical School Genetics Department and the Howard Hughes Medical Institute. Special thanks to Tianxing Mary Shi (Department of Art as Applied to Medicine, Johns Hopkins School of Medicine) for creating artwork used in Figure 1. Recent improvements in the PhIP-seq methodology have been supported under a U24 Resource-Related Research Projects Cooperative Agreement awarded by the NIH (5U24AI118633–02; to HBL and SJE), NIH grant DE-12354–15A1 (to ANB), a grant from the Jerome L. Greene Foundation (to HBL and ANB), and a grant from the Sjögren’s Syndrome Foundation (to HBL and ANB).
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare that they have no competing financial interests.
References
- 1.Larman HB et al. Autoantigen discovery with a synthetic human peptidome. Nature biotechnology 29, 535–541 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Larman HB et al. PhIP-Seq characterization of autoantibodies from patients with multiple sclerosis, type 1 diabetes and rheumatoid arthritis. Journal of autoimmunity 43, 1–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Larman HB et al. Cytosolic 5’-nucleotidase 1A autoimmunity in sporadic inclusion body myositis. Annals of neurology 73, 408–418 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Finton KA et al. Ontogeny of recognition specificity and functionality for the broadly neutralizing anti-HIV antibody 4E10. PLoS pathogens 10, e1004403 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu GJ et al. Viral immunology. Comprehensive serological profiling of human populations using a synthetic human virome. Science 348, aaa0698 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kosuri S & Church GM Large-scale de novo DNA synthesis: technologies and applications. Nat Methods 11, 499–507 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Atak A et al. Protein microarray applications: Autoantibody detection and posttranslational modification. Proteomics 16, 2557–2569 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Yu X et al. Multiplexed Nucleic Acid Programmable Protein Arrays. Theranostics 7, 4057–4070 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Henkel S, Wellhausen R, Woitalla D, Marcus K & May C Epitope Mapping Using Peptide Microarray in Autoantibody Profiling. Methods Mol Biol 1368, 209–224 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Finton KA et al. Autoreactivity and exceptional CDR plasticity (but not unusual polyspecificity) hinder elicitation of the anti-HIV antibody 4E10. PLoS pathogens 9, e1003639 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu GJ et al. Systematic autoantigen analysis identifies a distinct subtype of scleroderma with coincident cancer. Proceedings of the National Academy of Sciences of the United States of America (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu H, Luo H, Yan M, Zuo X & Li QZ Autoantigen Microarray for High-throughput Autoantibody Profiling in Systemic Lupus Erythematosus. Genomics Proteomics Bioinformatics 13, 210–218 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miersch S & LaBaer J Nucleic Acid programmable protein arrays: versatile tools for array-based functional protein studies. Curr Protoc Protein Sci Chapter 27, Unit27 22 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Zhu J et al. Protein interaction discovery using parallel analysis of translated ORFs (PLATO). Nature biotechnology 31, 331–334 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Larman HB, Liang AC, Elledge SJ & Zhu J Discovery of protein interactions using parallel analysis of translated ORFs (PLATO). Nature protocols 9, 90–103 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jhaveri DT et al. Using Quantitative Seroproteomics to Identify Antibody Biomarkers in Pancreatic Cancer. Cancer Immunol Res 4, 225–233 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MacConaill LE et al. Unique, dual-indexed sequencing adapters with UMIs effectively eliminate index cross-talk and significantly improve sensitivity of massively parallel sequencing. BMC Genomics 19, 30 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Design of 96 ad_min_BCX_P7 PCR2 i7 indexing primers.