

SUMMARY
Mammalian folate metabolism is comprised of cytosolic and mitochondrial pathways with nearly identical core reactions, yet the functional advantages of such an organization are not well understood. Using genome-editing and biochemical approaches, we find that ablating folate metabolism in the mitochondria of mammalian cell lines results in folate degradation in the cytosol. Mechanistically, we show that QDPR, an enzyme in tetrahydrobiopterin metabolism, moonlights to repair oxidative damage to tetrahydrofolate (THF). This repair capacity is overwhelmed when cytosolic THF hyperaccumulates in the absence of mitochondrially produced formate, leading to THF degradation. Unexpectedly, we also find that the classic antifolate methotrexate, by inhibiting its well-known target DHFR, causes even more extensive folate degradation in nearly all tested cancer cell lines. These findings shed light on design features of folate metabolism, provide a biochemical basis for clinically observed folate deficiency in QDPR-deficient patients, and reveal a hitherto unknown and unexplored cellular effect of methotrexate.
Graphical Abstract
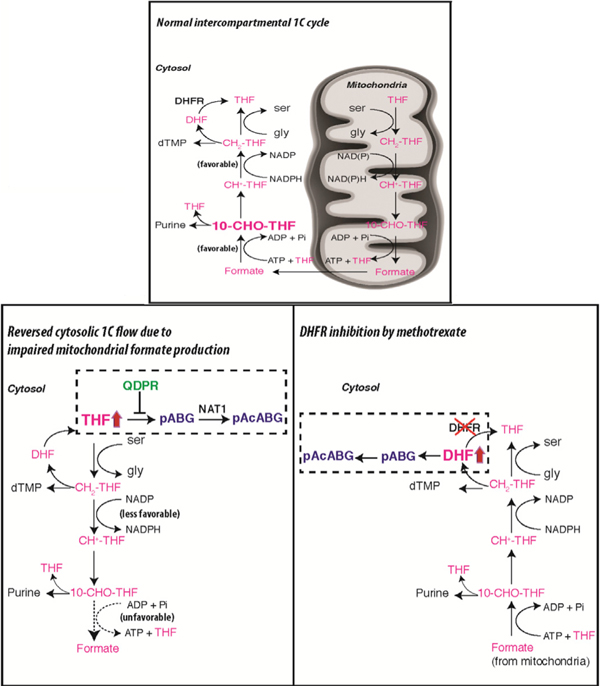
In Brief
Stratification of folate metabolism into distinct cytosolic and mitochondrial compartments enables repair of oxidative-stress-induced damage to folate metabolites while maintaining overall cellular pools of this nutrient.
INTRODUCTION
Folate is a vitamin cofactor that supports crucial biochemical processes, such as nucleotide, amino acid, and methyl group biosynthesis (Ducker and Rabinowitz, 2017; Locasale, 2013; Stover and Field, 2011; Tibbetts and Appling, 2010; Yang and Vousden, 2016). Reflecting the importance of folate metabolism in cell growth and proliferation, antifolates such as methotrexate are proven useful in treating cancers (Bertino, 2009; Chabner and Roberts, 2005; Goldman et al., 2010). An intriguing aspect of mammalian folate metabolism is the existence of parallel pathways with nearly identical core reactions in the cytosol and mitochondria (Figure 1A), but the functional advantages for such an organization are not well understood.
Figure 1. Formation of an Unusual Folate Metabolite in Mitochondrial-1C-Pathway-Deficient Cells.

(A) Compartmentation of mammalian 1C metabolism. SLC25A32 transports only the monoglutamate form of folates. In this intercompartmental cycle, 1C tends to flow clockwise, with serine oxidation in mitochondria and formate reduction in the cytosol.
(B) Folate-centered view of the mitochondrial 1C pathway.
(C) Western blot analysis of MDA-MB-468 cells stably expressing non-targeting or SHMT2-targeting short hairpin RNAs (shRNAs).
(D) Western blot analysis of MDA-MB-468 parental and MTHFD1L-knockout cells with or without ectopic expression of MTHFD1L.
(E) Western blot analysis of MDA-MB-468 parental and MTHFD1-knockout cells with or without ectopic expression of MTHFD1. The D125A mutation ablates the dehydrogenase and cyclohydrolase activity of MTHFD1.
(F–N) HPLC analysis of folates from isogenic MDA-MB-468 lines. ‘‘+ MTHFD1L’’ denotes ectopic MTHFD1L expression; ‘‘+ formate’’ denotes supplementing the labeling medium with 2 mM sodium formate; ‘‘+ hypoxanthine’’ denotes supplementing the labeling medium with 100 μM hypoxanthine, a necessity because the MTHFD1-knockout cells are purine auxotrophic. ‘‘+ MTHFD1’’ denotes ectopic MTHFD1 expression.
See also Figure S1.
A first clue indicating communication between the two folate pathways is that the mitochondrial pathway produces formate, which upon export into the cytosol, feeds into the cytosolic pathway for purine, dTMP, and methyl group biosynthesis (Tibbetts and Appling, 2010). This intercompartmental one-carbon (1C) cycle is predicted to be unidirectional with net formate flux out of mitochondria, on the grounds that high NAD(P):NAD(P)H ratios in mitochondria favor serine oxidation, whereas high NADPH:NADP (nicotinamide adenine dinucelotide phosphate, reduced and nicotinamide adenine dinucelotide phosphate, respectively) ratios in the cytosol favor formate reduction (Christensen and MacKenzie, 2006; Tibbetts and Appling, 2010). Importantly, functional operation of this intercompartmental 1C cycle was recently shown in vivo: mice lacking the final enzyme of the mitochondrial pathway, MTHFD1L, develop neural tube defects with 100% penetrance, and formate supplementation rescues the embryos beyond the stage of neural tube closure (Bryant et al., 2018; Momb et al., 2013).
Surprisingly, two recent studies reveal that a large proportion of mitochondria-derived formate is apparently of no use to the cell and consequently excreted (Ducker et al., 2016; Meiser et al., 2016). Likened to the Warburg effect in one study, these findings raise the important question of why formate is produced in excess of cellular demand for 1C.
Intracellular folates largely exist as polyglutamate derivatives and, as such, do not cross membranes (Lawrence et al., 2014). Within each compartment, the diverse reactions can be rationalized into three types: (1) those bringing 1C into the folate pool (e.g., SHMT2), (2) those removing 1C from the pool (e.g., MTHFD1L), and (3) those interconverting 1C between different oxidation states (e.g., MTHFD2). Since folate cofactors exist in catalytic amounts, recycling is key to maintaining function (Figure 1B); thus, 1C-supplying reactions are necessarily balanced by 1C-removing reactions, and 1C-interconverting reactions chemically bridge the two. Blocking folate recycling effectively ablates folate-dependent processes that require rapid turnovers in the respective compartment. Hence, deletion of SHMT2, MTHFD2, or MTHFD1L in cell lines results in a similar glycine-requiring growth phenotype (Ducker et al., 2016), although mitochondrial protein translation, for which the demand for 1C is quantitatively small, is affected differentially (Minton et al., 2018; Morscher et al., 2018).
We set out to unravel the evolutionary advantages of the dual-compartment organization of mammalian folate metabolism using genome-editing and biochemical approaches. We found that this organization affords flexibility to balance cellular demands for glycine and 1C. Unexpectedly, our results also revealed an unknown cellular effect induced by the clinically important drug methotrexate and a moonlighting function in folate metabolism for QDPR, an enzyme more widely known for its role in tetrahydrobiopterin metabolism.
RESULTS
An Unusual Folate Metabolite Induced by Mitochondrial 1C Pathway Disruption or Methotrexate Treatment
Using a high-performance liquid chromatography (HPLC) assay with radioactivity detection (Figures S1A–S1C), we examined the folate profiles of isogenic MDA-MB-468 cell lines in which the mitochondrial pathway was disrupted by knocking down SHMT2 using RNAi or knocking out MTHFD1L or the mitochondrial folate transporter SLC25A32 using CRISPR (Figures 1C, 1D, and S1D). Upon the mitochondrial 1C pathway disruption, we observed a dramatic decrease (4- to 5-fold) in 10-CHO-THF (10-formyl-tetrahydrofolate) and 5,10-CH+-THF (5,10-methenyl-tetrahydrofolate) abundance concomitant with an increase (50%–60%) in THF and 5,10-CH2-THF (5,10-methylene-tetrahydrofolate) abundance (Figures 1F–1I; Table S1), in agreement with a recent study (Ducker et al., 2016). Remarkably, an unusual metabolite also appeared; it is folate related as it contains the 3H label originating from [3′,5′,7,9-3H]-5-CHO-THF.
Reintroducing MTHFD1L into the MTHFD1L-knockout cells not only normalized the abundance of 10-CHO-THF and 5,10-CH+-THF but also eliminated the unusual metabolite (Figure 1J; Table S1), indicating that both effects are specific to loss of the mitochondrial pathway. Both effects were also rescued by including 2 mM formate in the labeling medium (Figure 1K; Table S1), consistent with the concept that formate is an important output of the mitochondrial 1C pathway.
We also knocked out the genes involved in the cytosolic 1C pathway, including SHMT1, MTHFR, TYMS, and MTHFD1, individually and in various combinations (Table S1). Only the MTHFD1-knockout cells showed a folate profile resembling those of the mitochondrial 1C pathway knockouts (Figure 1L; Table S1). MTHFD1 is a trifunctional enzyme consisting of an N-terminal dehydrogenase and cyclohydrolase domain and a C-terminal synthetase domain. Reconstituting the MTHFD1-knockout cells with either wild-type MTHFD1 or a point mutant (D125A) devoid of the dehydrogenase and cyclohydrolase activity (Sundararajan and MacKenzie, 2002) normalized the folate profile (Figures 1M and 1N; Table S1). Thus, both the mitochondrial 1C pathway that produces formate and the synthetase activity of MTHFD1 that accepts formate are required for sustaining a normal folate distribution.
Unexpectedly, treating wild-type MDA-MB-468 cells with methotrexate also led to the unusual metabolite (Figure 2A; Table S2). The substrate of the methotrexate target DHFR, DHF, also accumulated. A previous study reported methotrexate-induced formation of 10-CHO-DHF in MCF7 cells (Allegra et al., 1986). We confirmed this finding but noted that 10-CHO-DHF was a minor component appearing transiently at early time points (Figure S2A). Here, the unusual metabolite is not 10-CHO-DHF, because the two migrated at different positions on HPLC.
Figure 2. Formation of the Same Unusual Folate Metabolite upon Methotrexate Treatment.

(A) HPLC analysis of folates from MDA-MB-468 cells, untreated or treated with 1 μM methotrexate for indicated times.
(B) Western blot analysis of 293T parental, DHFR-knockout, DHFR-DHFRL1-double-knockout, and DHFR-TYMS-double-knockout cells.
(C) HPLC analysis of folates from 293T parental, DHFR-knockout, DHFR-DHFRL1-double-knockout cells, untreated or treated with 1 μM methotrexate for 24 hr. 100 μM hypoxanthine and 16 μM thymidine (HT) were included in the medium to support cell growth.
(D) HPLC analysis of folates from 293T DHFR-TYMS-double-knockout cells.
(E) HPLC analysis of folates from MDA-MB-468 TYMS-knockout cells, untreated or treated with 1 μM methotrexate for 6 hr. 16 μM thymidine was included in the medium to support cell growth.
See also Figure S2.
To determine whether this novel effect of methotrexate requires DHFR inhibition, we knocked out DHFR alone or together with its paralog DHFRL1 in 293T cells (Figure 2B). These cells are defective in folate-dependent processes and rely on exogenous purine and thymidine for growth. Remarkably, 293T DHFR-knockout cells displayed a folate profile closely resembling that of methotrexate-treated wild-type cells, with a prominent peak of the unusual metabolite; adding methotrexate or knocking out DHFRL1 produced no additional effects (Figure 2C; Table S2). Thus, formation of the unusual folate metabolite is an on-target effect of methotrexate.
To determine whether this effect of methotrexate requires DHF accumulation that accompanies DHFR inhibition, we knocked out the TYMS gene encoding thymidylate synthase, the only known source of DHF, in 293T DHFR-knockout cells and in MDA-MB-468 cells. Indeed, knocking out TYMS in the 293T DHFR-knockout background suppressed the accumulation of both DHF and the unusual folate metabolite (Figure 2D; Table S2). Similarly, methotrexate produced little effect in MDA-MB-468 cells with TYMS knockout or pretreatment with 5-fluoro-2′-deoxyuridine that blocks TYMS (Figures 2E and S2B; Table S2). In line with these biochemical observations and previous studies (Moran et al., 1979), MDA-MB-468 TYMS-knockout cells displayed resistance to methotrexate, and the TYMS-DHFR-double knockout cells displayed normal proliferation in the absence of exogenous purine (Figures S2C and S2D).
Identification of the Unusual Folate Metabolite Reveals It to Be a Folate Degradation Product, P-acetamidobenzoyl Polyglutamate
To better understand the rate of turnover of this folate metabolite, we performed a modified pulse-chase experiment. The unusual metabolite was allowed to form in the absence of formate in the labeling phase and then allowed to decay in the presence of formate in the chase phase (Figure 3A). The unusual metabolite exhibited a long half-life of ⁓40 hr (Figure 3B), suggesting a sluggish conversion to other species or excretion from the cell. Moreover, in acute settings of serine or methionine deprivation, while other folate species all readily interconverted, the unusual metabolite barely changed (Figures S3A and S4E). Together, these observations raised the possibility that the unusual metabolite is an end product rather than an intermediate of folate metabolism and that its formation is irreversible.
Figure 3. Identification of the Unusual Folate Metabolite.

(A) Schematic of the modified ‘‘pulse-chase’’ experimental design.
(B) HPLC quantification of folates at the indicated chase times. Data points are normalized to summed peak area of all known folates at 0.5 hr. Error bars depict SD.
(C) Schematic of pAcABGn hydrolysis facilitated by GGH, or acid plus heat. pABA, p-aminobenzoic acid; pABG, p-aminobenzoyl-glutamate.
(D) 3H-labeled, GGH-treated, HPLC-purified un-pAcABG usual metabolite was mixed with unlabeled pAcABG standard, untreated or treated with 1 N HCl at 99°C for 5 min or 1 hr, and analyzed by HPLC. The radioactive unusual metabolite (⁓5 Ci/mmol) displayed negligible UV absorption.
See also Figure S3.
Since a known end product of folate metabolism is the urinary folate catabolite p-acetamidobenzoylglutamate (pAcABG) (Murphy et al., 1976), we hypothesized that the unusual metabolite structurally resembles pAcABG yet retains the polyglutamate tail. HPLC analysis of extracts of 3H-folate-labeled cells that had not been treated with GGH revealed that the unknown peak no longer migrated at the expected position (Figures S3B and S3C), indicating that the parent compound is polyglutamated. Evidence that the unknown peak is pAcABG was provided by the observation that synthetic pAcABG (detected by UV absorbance) co-migrated with the radioactive peak and that both compounds were degraded by 1 N HCl (99°C), which hydrolyzes the amide bond, at similar rates and that the radioactive hydrolysis products co-migrated with the UV of the expected products, pABG and then pABA (Figures 3B and 3C). Thus, the unusual metabolite was identified as p-acetamidobenzoyl polyglutamate (pAcABGn). In other words, when formate is not generated in mitochondria, or when DHFR in the cytosol is inhibited by methotrexate, folates are degraded.
Folate Degradation Occurs through Nonenzymatic Oxidation and NAT1-Catalyzed Acetylation
Folate catabolism entails oxidative cleavage of the C9-N10 bond followed by enzymatic acetylation of the N10-amino group (Figure 4D) (Suh et al., 2001). Consistent with the oxidative nature of C9-N10 cleavage, H2O2 treatment not only exacerbated pAcABGn accumulation in MDA-MB-468 SHMT2-knockdown cells but also led to its formation in control cells with intact mitochondrial 1C metabolism (Figure 4A; Table S3).
Figure 4. Roles of Nonenzymatic Oxidation and NAT1 in Folate Catabolism.

(A) HPLC analysis of folates from MDA-MB-468 non-targeting control and SHMT2-knockdown cells, untreated or treated with 1 mM H2O2 for 5 hr. The asterisk denotes a 5-CH3-THF-derived metabolite, likely to be a 5-methyl-dihydro-pyrazino-s-trazine compound (Gregory, 1989).
(B) Western blot analysis of MDA-MB-468 parental and NAT1-knockout cells generated using two independent CRISPR guides.
(C) HPLC analysis of folates from the NAT1-knockout cells, untreated (top), treated with 1 μM methotrexate for 6 hr (middle), or treated with 1 μM methotrexate for 6 hr and then after cell lysis, further treated with recombinant DHFR and NADPH (bottom).
(D) Schematic of THF catabolism. ‘‘[O]’’ indicates the oxidative and presumably nonenzymatic nature of the cleavage step.
See also Figure S4.
Acetylation of the folate C9-N10 cleavage product has been ascribed to arylamine N-acetyltransferase 1 (NAT1), based on the substrate specificity of recombinant NAT1 (Minchin, 1995). A subsequent study found that mice deficient in Nat2 (the ortholog of human NAT1) lack pAcABG in urine (Wakefield et al., 2007). To confirm this function of NAT1, we knocked out NAT1 in MDA-MB-468 cells (Figure 4B). Subsequent SHMT2 knockdown or methotrexate treatment revealed the expected accumulation of the unacetylated precursor pABGn instead of pAcABGn (Figures 4C, S4A, and S4B). These results not only confirmed NAT1’s role in folate catabolism but also corroborated the identification of the folate breakdown product as pAcABGn.
Ablating Folate Metabolism in Mitochondria Results in Folate Degradation in the Cytosol
Figure 1H revealed folate degradation in MDA-MB-468 cells lacking the mitochondrial folate transporter (SLC25A32). This result implies that folates are degraded in the cytosol, assuming that the mitochondrial folate pool was indeed depleted, as originally demonstrated for Chinese hamster ovary mutant cells (Titus and Moran, 2000). This assumption was tested by taking advantage of a concept known as methyl-folate trap mediated by MTHFR, which is exclusively cytosolic (Figure S4C) (Shane and Stokstad, 1985). Whereas most folates readily interconvert, MTHFR-catalyzed 5-CH3-THF synthesis is irreversible and allosterically inhibited by downstream S-adenosylmethionine (SAM). Upon methionine deprivation, intracellular methionine and SAM concentrations drop precipitously, resulting in maximal MTHFR activation and trapping of most, if not all, cytosolic folates as 5-CH3-THF.
To ensure complete labeling in both cytosolic and mitochondrial compartments, cells were first cultured in 3H-5-CHO-THF for 2 weeks (Figure S4D). Upon subsequent, acute methionine deprivation, MDA-MB-468 SLC25A32-knockout cells showed nearly complete (>90%) conversion of 10-CHO-THF/5,10-CH+-THF and THF/5,10-CH2-THF into 5-CH3-THF, confirming the depletion of mitochondrial folates and thus locating folate degradation to the cytosol (Figures S4E and S4F).
QDPR Repairs Oxidative Damage to THF, and This Repair Capacity Is Overwhelmed When THF Hyperaccumulates in the Absence of Mitochondrially Produced Formate
Among folates, THF and 10-CHO-THF are the least stable, yet these two species are known to follow distinct chemical degradation pathways in vitro (Gregory, 1989). Whereas THF readily undergoes C9-N10 bond cleavage (Chippel and Scrimgeour, 1970), 10-CHO-THF is stable to cleavage and instead undergoes pteridine ring oxidation (Baggott and Johanning, 1999). Thus, the increased folate cleavage in the mitochondrial 1C pathway knockout cells may be explained by the observation that the proportion of THF in these cells is markedly increased (Figures 1G–1I). In support of this explanation, the formate rescue experiment (Figure 1K) indicates that once 10-CHO-THF is replenished from THF and formate, folate cleavage is prevented. Moreover, we found that hypoxanthine supplementation also boosted the proportion of 10-CHO-THF in the mitochondrial 1C pathway knockout cells, by feedback-inhibiting a de novo purine synthesis and thereby abolishing cytosolic 10-CHO-THF consumption (Figures S5A and S5B). Remarkably, hypoxanthine supplementation also largely prevented folate cleavage (Figure S5A). Together, these results suggest that unsubstituted THF is particularly vulnerable to oxidative cleavage in cells and that the mitochondrial 1C pathway plays a role in preventing its undue accumulation.
Two pioneering studies, both using in vitro enzymatic assays, have suggested that there exist cellular enzymes to repair oxidative damage to folates (Figure 5A) (Baggott and Johanning, 1999; Pollock and Kaufman, 1978). Oxidation of 10-CHO-THF to 10-CHO-DHF can be reversed by DHFR and ATIC (Baggott and Johanning, 1999), whereas cleavage of THF can be prevented by QDPR (Pollock and Kaufman, 1978), an enzyme more widely known for its role in recycling tetrahydrobiopterin, a cofactor that structurally resembles THF (Werner et al., 2011). (For a plausible chemical mechanism by which QDPR prevents THF cleavage, see Figure S5D). Using purified recombinant QDPR, we confirmed this remarkable activity (Figures 5B, 5C, and S5C). To test whether this activity of QDPR is relevant to folate homeostasis in cells, we knocked out or overexpressed QDPR in MDA-MB-468 parental and MTHFD1L- and MTHFD1-knockout cells (Figures 5D–5L and S5E–S5H; Table S3). For the parental cells, knocking out QDPR led to a moderate increase in pAcABG, whereas for the MTHFD1L- or MTHFD1-knockout cells where THF predominates, knocking out QDPR led to a staggering increase in pAcABG. Conversely, overexpressing QDPR fully prevented pAcABG accumulation in the MTHFD1L- and MTHFD1-knockout cells.
Figure 5. QDPR Repairs Oxidative Damage to Unsubstituted THF in Cells.

(A) Distinct chemical degradation and enzymatic repair pathways for THF and 10-CHO-THF.
(B) QDPR prevents 6(RS)-THF from decomposition at 37° C in vitro in an NADH-dependent manner.
(C) Unlike QDPR, DHFR does not prevent THF from decomposition at 37° C in vitro.
(D–L) HPLC analysis of folates from the indicated cells. 100 μM hypoxanthine was included in the medium to support the growth of the MTHFD1-knockout cells, which exhibit purine auxotrophy.
(M) Schematic of the serine-deprivation experiment (in [N]–[P]).
(N–P) Differential kinetics of THF decomposition into pAcABG in MDA-MB-468 parental, QDPR-knockout, and QDPR-overexpressing cells.
(Q) Differential degrees of pAcABG accumulation in MDA-MB-468 parental, QDPR-knockout, and QDPR-overexpressing cells, untreated or treated with 1 mM H2O2 for 3 hr. Error bars depict SD.
See also Figure S5.
Like QDPR, MTHFR shows a similar THF damage repair activity in vitro (Matthews and Kaufman, 1980). However, MTHFR is unlikely to play an analogous role in cells, because the MTHFR-knockout cells did not exhibit increased pAcABG formation, and the QDPR- and MTHFR-double-knockout cells showed the same moderate degree of pAcABG accumulation as the QDPR-knockout cells (Table S3).
To further test whether THF is the vulnerable species whose oxidative damage can be repaired by QDPR, we starved the cells of serine so as to convert most of cellular folates to THF and then compared the rates of THF decomposition into pAcABG in MDA-MB-468 parental, QDPR-knockout, and QDPR-overexpressing cells. Indeed, the parental cells showed a slow but visible rate of THF decomposition that was dramatically accelerated in the QDPR-knockout cells but fully obliterated in the QDPR-overexpressing cells (Figures 5M–5P). This slow rate of THF decomposition in the parental cells upon serine deprivation matched well with that of the MTHFD1L-knockout cells upon formate removal (Figure S5I), suggesting that in both cases, the repair capacity of QDPR is overwhelmed by excessive THF accumulation.
In addition, we compared the folate profiles of MDA-MB-468 parental, QDPR-knockout, and QDPR-overexpressing cells in response to H2O2 treatment. Again, QDPR knockout exacerbated H2O2-induced pAcABG formation, whereas QDPR over-expression abolished this response (Figure 5Q). Collectively, these results establish an unequivocal role for QDPR in repairing oxidative damage to THF in cells and indicate THF hyperaccumulation exceeding QDPR’s repair capacity as the basis for folate degradation seen in the mitochondrial 1C pathway knockout cells.
Lack of Evidence for QDPR Inhibition Accompanying Methotrexate Treatment or DHFR Knockout
We questioned whether QDPR is inhibited under conditions of DHFR inhibition, thereby explaining folate degradation seen in methotrexate-treated or DHFR-knockout cells. Surprisingly however, QDPR overexpression in MDA-MB-468 cells afforded no protection against methotrexate-induced folate degradation (Figures 6A and 6B), even though the same cells displayed no trace of serine deprivation or H2O2-induced folate degradation (Figures 5P and 5Q). Moreover, neither QDPR overexpression nor knockout altered the degree of folate degradation in MDA-MB-468 DHFR-knockout cells (Figures 6D–6F; Table S4). In addition, while the cellular thermal shift assay confirmed that DHFR was engaged and thus thermally stabilized by methotrexate (Huber et al., 2015), the thermal profile of QDPR remained unchanged upon methotrexate exposure (Figure 6C), consistent with the reported weak inhibition constant (⁓38 μM) of methotrexate for QDPR (Kaufman, 1987). These results thus point to a mechanism other than QDPR inhibition at work. A close inspection of the time course of cellular folates in response to methotrexate revealed a rapid phase of folate degradation that coincided with DHF accumulation followed by a slow phase of folate degradation that mirrored the slow DHF decay (Figures 6A and S6A). Since DHF is not a substrate of and cannot be protected by QDPR (Kaufman, 1987), the kinetics could be explained if a subpool of DHF is rapidly degraded, whereas the remainder is slowly degraded.
Figure 6. QDPR Genetically Interacts with DHFR, and ALDH1L1 Expression Alters the Folate Response to Methotrexate.

(A and B) Time course of folate interconversion and decomposition in MDA-MB-468 parental (A) and QDPR-overexpressing (B) cells upon 1 μM methotrexate treatment.
(C) Western blot analysis for the cellular thermal shift assay on MDA-MB-468 cells treated with 1 μM methotrexate for 30 min.
(D–L) HPLC analysis of folates from the indicated cells.
(M) ALDH1L1 protects 6(S)-THF from decomposition at 37°C in vitro.
(N) ALDH1L1 converts DHF into folic acid in aerobic buffers at 37°C.
(O and P) Time course of folate interconversion and decomposition in MDA-MB-468 parental and ALDH1L1-overexpressing cells upon 1 μM methotrexate treatment.
(Q and R) HPLC analysis of folates from the bladder cancer cell line RT4, untreated or treated with 1 μM methotrexate for 4 hr.
See also Figure S6.
Synthetic Interaction between DHFR and QDPR
Like in 293T cells (Figure 2D), the folate-profile phenotype of MDA-MB-468 DHFR-knockout cells was suppressed by TYMS knockout (Figures 6G and 6H; Table S4). Surprisingly however, MDA-MB-468 DHFR-TYMS-QDPR-triple-knockout cells displayed a folate profile resembling that of the DHFR-knockout cells, rather than that of the DHFR-TYMS-double-knockout cells, with substantial proportions of DHF (⁓15%) and pAcABG (31%–37%) (Figures 6I, 6J, and S6B; Table S4). Reconstituting the triple-knockout cells with either QDPR or DHFR eliminated the substantial accumulation of DHF and pAcABG (Figures 6K, 6L, and S6B; Table S4), thus ruling out off-target effects of the CRISPR knockout. These results indicate a synthetic interaction between DHFR and QDPR: knockout of both genes is required to expose a latent source of DHF that is distinct from TYMS. Presumably, when tetrahydrobiopterin recycling is blunted by QDPR knockout, THF and DHFR become the backup cofactor and recycling enzyme, respectively, for a biochemical pathway that normally uses tetrahydrobiopterin.
ALDH1L1 Expression Alters the Folate Response to Methotrexate
Folates are known to be stabilized through protein binding (Jones and Nixon, 2002). Therefore, we questioned whether overexpressing ALDH1L1, which binds THF polyglutamate tightly dissociation constant < 1 μM) (Min et al., 1988), would prevent THF degradation. This appeared to be the case, both in in vitro incubations of purified recombinant ALDH1L1 with THF (Figures 6M, S6D, and S6E) and in MDA-MB-468 ALDH1L1-overexpressing, MTHFD1L-knockout, or SLC25A32-knockout cells (Figure S6F). However, the effect of ALDH1L1 overexpression on the response of MDA-MB-468 cells to methotrexate was unexpected. The magnitude of folate degradation was indeed mitigated, but the majority (>60%) of folates were converted to folic acid, which has a fully oxidized pteridine ring (Figures 6O, 6P, and S6I; Table S5). This methotrexate-induced formation of folic acid required the folate-binding activity of ALDH1L1, but not its dehydrogenase activity, as shown with ALDH1L1 point mutants (Krupenko, 2009) impaired in one of the two activities (Figure S6G; Table S5). This effect was not an artifact of viral overexpression, since RT4, a bladder cancer line with endogenous expression of ALDH1L1 (Figure S6H), showed a similar folate response to methotrexate (Figures 6Q and 6R; Table S5).
A previous chemical study suggests that alkaline pH conditions can facilitate the oxidation of DHF to folic acid (Chippel and Scrimgeour, 1970). We therefore hypothesized that DHF initially formed upon methotrexate exposure is further converted to folic acid by ALDH1L1, perhaps using some general base in its folate-binding site. Indeed, this novel activity was confirmed in in vitro incubations of purified recombinant ALDH1L1 with DHF (Figures 6N, S6D, and S6E). Collectively, these results suggest that for cancer cells with high ALDH1L1 expression, methotrexate induces accumulation of folic acid, along with minor amounts of DHF and pAcABG.
Glycine Auxotrophy, a Growth Phenotype of Loss of the Mitochondrial 1C Pathway, Is Rescued by Overexpression of ALDH1L Enzymes
Glycine auxotrophy is a well-known growth phenotype of loss of the mitochondrial 1C pathway (Figures 7A–7C) (Chasin et al., 1974). An open question is why the cytosolic pathway cannot make sufficient glycine for cell growth. To allow continuous glycine synthesis by SHMTs, THF has to be continuously regenerated. Cytosolic THF is regenerated as 1C is consumed in biosynthesis, but this becomes inadequate when the demand for glycine exceeds the demand for 1C. The remaining route to regenerate cytosolic THF, via MTHFD1, is thought to be insufficient on the grounds that the dehydrogenase step runs against the high cytosolic NADPH:NADP gradients (Christensen and MacKenzie, 2006; Tibbetts and Appling, 2010).
Figure 7. Glycine Auxotrophy Is Rescued by Overexpression of ALDH1L Enzymes.

(A–I) Growth curves of MDA-MB-468 parental, MTHFD1L-knockout, and SLC25A32-knockout cells, with or without ALDH1L1 or ALDH1L2 overexpression, and with or without supplementation with 400 μM glycine or 100 μM hypoxanthine. Adding 16 μM thymidine on top of 100 μM hypoxanthine had no additional effects (data not shown). Error bars depict SD.
(J) Western blot analysis of 293T parental, MTHFD1L-knockout, and MTHFD1L-ALDH1L2-double-knockout cells. The double-knockout clones (A1) and (B1) were derived from the MTHFD1L-knockout clone using two independent CRISPR guides against ALDH1L2.
(K) Representative images of the cells in (J) plated in glycine-free medium. Scale bars: 400 μm.
See also Figure S7.
To overcome the putative block at the MTHFD1 dehydrogenase step, we attempted, in MDA-MB-468 MTHFD1L-knockout cells, overexpression of yeast MTD1, which uses NAD instead of NADP to drive the same reaction (West et al., 1996). However, yeast MTD1 overexpression did not rescue glycine auxotrophy, even though a substantial increase in 10-CHO-THF and 5,10-CH+-THF at the expense of THF and 5,10-CH2-THF was seen in these cells (Figures S7A and S7B). These results thus led us to a further hypothesis: the subsequent synthetase step cannot efficiently regenerate THF because it runs against cytosolic ATP:ADP gradients. Much higher ATP:ADP ratios are found in the cytosol than in mitochondria, due to abundant ADP and ATP translocases at the mitochondrial inner membrane (Maldonado and Lemasters, 2014; Nicholls and Ferguson, 2013).
ALDH1L1 bypasses the MTHFD1 synthetase step to regenerate THF irreversibly (Krupenko, 2009). Indeed, overexpression of ALDH1L1 in MDA-MB-468 MTHFD1L- and SLC25A32-knockout cells enabled growth in glycine-free medium, albeit at lower than wild-type rates (Figures 7D–7F). Addition of hypoxanthine dramatically stimulated cell growth, consistent with 10-CHO-THF and 5,10-CH+-THF depletion seen in these cells (Figure S6F) and with previous studies (Krupenko and Oleinik, 2002). Under such purine-replete conditions, adding glycine further promoted cell growth, indicating that although substantial cytosolic glycine synthesis is enabled by ALDH1L1, it is suboptimal for growth. Nevertheless, these results confirm that the MTHFD1 synthetase step is essentially irreversible and cannot make ATP and regenerate THF in cells.
The mitochondrial isoform ALDH1L2 is expected to facilitate THF regeneration in mitochondria for MDA-MB-468 MTHFD1L-knockout but not for SLC25A32-knockout cells. Indeed, ALDH1L2 overexpression rescued glycine auxotrophy of the former but not the latter (Figures 7G–7I). This led to the realization that the residual growth of 293T MTHFD1L-knockout cells in glycine-free medium (Ducker et al., 2016) could arise from the moderate ALDH1L2 expression in 293T. Indeed, knocking out ALDH1L2 in 293T MTHFD1L-knockout cells completely eliminated the residual growth (Figures 7J and 7K). Collectively, these results suggest that either SHMT1 or SHMT2 can make glycine if folate recycling (Figure 1B) in either compartment is restored. These results also point to a role of ALDH1Ls, when expressed, in balancing cellular demands for glycine and 1C, as will be discussed below, in connection to formate excretion observed in cells with intact mitochondrial folate metabolism (Ducker et al., 2016; Meiser et al., 2016).
How General Are These Findings across Cell Lines?
Finally, we note that loss of the mitochondrial 1C pathway led to evident folate degradation in 9 out of 15 tested mammalian cell lines (Table S6; Figures S7C–S7E). Presumably, the degree of unsubstituted THF hyperaccumulation depends on the level of MTHFR activity (Figure S7D), the flux of de novo purine synthesis, and the intracellular folate pool size, which in turn depends on extracellular folate availability, transport activities, and folyl-polyglutamate synthetase activity. Moreover, the THF damage repair capacity afforded by QDPR is likely to vary across cell lines. Remarkably, methotrexate treatment resulted in extensive folate degradation in all tested 10+ cell lines, except RT4 that shows high ALDH1L1 expression (Table S6).
DISCUSSION
Recently, targeting serine biosynthetic and mitochondrial folate pathways, or dietary serine and glycine restriction, has been proposed for cancer treatment (Maddocks et al., 2017; Mullarky et al., 2016; Nilsson et al., 2014; Pacold et al., 2016). The present study was initiated to delineate the metabolic consequences of ablating the mitochondrial folate pathway in cancer cell lines, revealing folate degradation as a previously unsuspected consequence in a subset of cancer cell lines. The subsequent investigation into how and why folate degradation occurs in cells yielded two additional, serendipitous findings. First, methotrexate, an antifolate widely used in treating cancers and arthritis, causes even more extensive folate degradation in nearly all tested cancer cell lines. Although folate degradation was not a known effect of methotrexate in human cells, it is worth noting that earlier studies on the effects of trimethoprim in bacteria (Kwon et al., 2008; Quinlivan et al., 2000), and of methotrexate on urinary folate catabolite excretion in rats (Saleh et al., 1981), have hinted at this possibility.
Second, QDPR, an enzyme in tetrahydrobiopterin metabolism, has a moonlighting function in preventing THF degradation in cells. This function of QDPR was first proposed in a classic paper in 1978, based on the clinical observation that systemic sand cerebral folate deficiency is commonly found in QDPR-deficient patients (Pollock and Kaufman, 1978). Here, we present unequivocal evidence for this function of QDPR in cells, using genome-editing approaches then unavailable. This finding represents a new example of metabolite damage repair, an important concept of metabolism that has been overlooked until recently (Linster et al., 2013).
Figure S7F depicts a model that integrates the above, seemingly disparate findings. Central to this model are the different chemical properties of distinct folate species, among which only unsubstituted THF and DHF are susceptible to irreversible, oxidative cleavage (Chippel and Scrimgeour, 1970; Gregory, 1989). For THF, the cell employs at least two mechanisms to preserve its integrity. First, the mitochondrially produced formate plays a crucial role in ‘‘charging’’ unsubstituted THF into the 10-CHO-THF state, thereby minimizing the irreversible loss of unsubstituted THF due to oxidative stress, an inevitable consequence of aerobic life (Figure S7F, left). In the second line of defense, when oxidative damage to unsubstituted THF does happen, QDPR immediately repairs this damage at a reversible stage, at the expense of reducing equivalents from NADH (Figure S7F, middle). For DHF, in contrast, the only cellular mechanism to prevent its cleavage appears to be keeping its level at bay using DHFR and NADPH. In this sense, methotrexate, by blocking DHFR, appears to target the Achilles’ heel in the ‘‘war against folate integrity’’ (Figure S7F, right).
Whether this novel effect of methotrexate contributes to its clinical efficacy awaits further study. Without folate degradation, converting nearly all of cellular folates into DHF by itself should be able to halt cell proliferation, since DHF cannot function as a 1C carrier. However, in light of the present study, if nearly all of DHF can be further channeled into the degradation pathway, the response might be more durable and less likely to succumb to low-level DHFR amplification, a commonly acquired resistance mechanism that probably works by converting a fraction of cellular DHF back to THF (Banerjee et al., 2002). It is also noteworthy that knocking out QDPR in a DHFR-knockout background led to a more severe depletion of the active folate (THF, 5,10-CH2-THF, 5,10-CH+-THF, and 10-CHO-THF) pools (compare Figure 6D with Figure 6F). Thus, dual inhibition of QDPR and DHFR might be more effective than DHFR inhibition alone for ablating cancer cell proliferation. The folate degradation product identified in the present study, pAcABGn, may act as a competitive inhibitor for folate enzymes. Folate decomposition is known to produce formaldehyde that can result in DNA damage (Burgos-Barragan et al., 2017), and it remains to be determined quantitatively how significant this source of formaldehyde is. Exploring these open questions and devising strategies to further enhance the degree of cellular folate depletion may provide insights into more effective use of methotrexate as a single agent or in combination with other drugs.
Our study has also provided new insights into the evolution of non-redundant 1C pathways in the cytosol and mitochondria that build on previous suggestions (Tibbetts and Appling, 2010). Compartmentation is a way to store free energy, which can then be tapped to drive otherwise less favorable processes; a classic example is chemiosmosis. Here, to drive a unidirectional, intercompartmental 1C cycle, free energy is tapped from the differential redox states and adenylate energy charges between the cytosol and mitochondria and presumably also from the proton gradient (which favors formate export into the cytosol). Such thermodynamic driving forces, realized by compartmentation and maintaining in each compartment distinct ratios of pyridine and adenine nucleotide pairs (Williamson et al., 1967; Maldonado and Lemasters, 2014), could be widespread and worth exploring.
Why is it advantageous to have a unidirectional, intercompartmental 1C cycle? We believe this design ensures that net synthesis of glycine in mitochondria and 10-CHO-THF and 5,10-CH2-THF in the cytosol are all thermodynamically favorable and can readily support biosynthesis whenever needed. Now consider situations where there is only the cytosolic pathway and choices can be made between NAD and NADP for the interconversion of 5,10-CH2-THF and 10-CHO-THF, and between ADP to ATP coupling and simple hydrolysis (or a reaction like ALDH1L1) for the interconversion of 10-CHO-THF and THF. Whichever the choices, net synthesis of glycine, 10-CHO-THF, and 5,10-CH2-THF cannot be simultaneously guaranteed to be favorable. In particular, the SHMT reaction is readily reversible, such that net 5,10-CH2-THF synthesis from serine would be unfavorable when cellular serine is low and glycine is high. Moreover, use of 10-CHO-THF hydrolase to drive net glycine synthesis, if left uncontrolled, would have the negative consequence of depleting both 5,10-CH2-THF and 10-CHO-THF. In E. coli, where there is only a single compartment, the solution appears to entail multiple complex regulatory mechanisms, including high glycine-inducible expression of the glycine cleavage system, allosteric inhibition of 10-CHO-THF hydrolase by glycine, and an additional glycinamide ribonucleotide formyltransferase that directly assimilates formate (Meedel and Pizer, 1974; Nagy et al., 1995).
Organizing folate metabolism into two pathways that can be uncoupled affords flexibility to balance cellular demands for glycine and 1C. Consider this simplified equation: serine = glycine + 1C. The demand for glycine does not necessarily match the demand for 1C. In fact, the former may often be higher, as glycine is needed in many processes including protein, purine, and glutathione synthesis. A surplus of 1C must be disposed of or else will trap all folates so as to prevent further glycine synthesis, thus providing a plausible explanation for the observed high rates of excretion of mitochondria-derived formate (Ducker et al., 2016; Meiser et al., 2016). Alternatively, ALDH1Ls can serve as an outlet for excess 1C and in fact will further tip the balance against 1C production, as proposed by H.A. Krebs in 1976 (Krebs et al., 1976) and corroborated by the present study.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Lewis C. Cantley (lcantley@med.cornell.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
MDA-MB-468, MCF7, HeLa, HCC38, T47D, BT474, and RT4 were purchased from ATCC. 293T, HCT116, HT29, NCI-H1299, NCI-H1975, LNCap, and DU145 were lab stocks. NCI-H3255 was a kind gift of the Pandolfi lab (BIDMC/Harvard Medical School). MDA-MB-468, 293T, MCF7, HeLa, T47D, BT474, HCT116, HT29, NCI-H1975, DU145, and MEFs were cultured in DMEM with 10% FBS. HCC38, NCI-H1299, NCI-H3255, and LNCap were cultured in RPMI with 10% FBS. Sf21 was purchased from Invitrogen, and cultured in SF900III for recombinant protein expression.
Microbe strains
One-shot E. coli BL21 (DE3) (Invitrogen) was grown in 2× YT medium at 37°C and then at 28°C after IPTG induction, for recombinant protein expression.
METHOD DETAILS
CRISPR/Cas9-mediated genome-editing in cell lines
Guide RNA sequence was cloned into PX459_V2.0 (Addgene) according to a published protocol (Ran et al., 2013). Cell lines with transfected with guide using Lipofectamine 3000 or Lipofectamine LTX (Invitrogen). 24 hr post transfection, puromycin (typically at 1 μg/ml) was added to enrich positively transfected cells. The puromycin selection typically lasts for 2 days, until mock-transfected, control cells were completely eliminated by puromycin. Single cell cloning was performed by serial dilution in 96-well plates. After 1.5–3 weeks, 15–20 clones were picked for knockout verification.
If antibodies were available, knockout clones were first screened by western blotting. The genotypes of selected clones were determined by PCR amplification of a genomic region encompassing the editing site, TOPO cloning of the individual alleles into PCR4-TOPO vector (Invitrogen), and Sanger sequencing. At least 20 bacterial colonies were picked for sequencing to ensure approximately equal representation of both alleles (and in the case of DHFRL1, all three alleles). Our attempts with online programs (e.g., CRISP-ID) that in theory obviate the need for vector cloning were not successful, presumably due to confounding sequencing background noises, and in some cases, relatively large deletions in one or both alleles.
The genotypes of the knockout clones that have been determined by sequencing of the individual alleles (above), are as follows.
MDA-MB-468_MTHFD1L-KO, MTHFD1L allele 1: c.202_212delCCCGCGGCGCG, MTHFD1L allele 2: c.184_209del AGCCCCGGCGGCCGAACGCCCGCGGC.
MDA-MB-468_SLC25A32-KO, SLC25A32 allele 1: c.60_61insAC, SLC25A32 allele 2: c.60_61insT.
293T-DHFR-DHFRL1- DKO: both DHFR alleles: c.31_59delGTGTCCCAGAACATGGGCATCGGCAAGAA, DHFRL1 allele 1: c. 40_41insA, DHFRL1 allele 2: c. 39_42delAA, DHFRL1 allele 3: c.40delinsGAC.
293T-DHFR-KO, both DHFR alleles: c.203_204insT.
MDA-MB-468_QDPR-KO sg2#2, QDPR allele 1: c.260_261_insC, QDPR allele 2: c.247_295_delATTCTTTGCGTTGCTGGAG GATGGGCCGGGGGCAATGCCAAATCCAAGT plus deletion of GTGAGT in the adjacent intron.
MDA-MB-468_QDPR-KO sg2#11: Both QDPR alleles: c.260_261_insA.
Molecular cloning of expression constructs
MGC cDNA clones of Rat GGH, human ALDH1L1, and mouse ALDH1L2, and human MTHFD1 were purchased from Open Biosystems. Human QDPR, DHFR, TYMS, and a codon-optimized version of yeast MTD1 cDNA and was synthesized by Integrated DNA Technologies.
Rat GGH cDNA was subcloned into an engineered pFASTBac vector with an N-terminal Honeybee secretion tag and a C-terminal His8 tag for expression in Sf21 through the Bac-to-Bac system, and into an engineered lentiviral gateway destination vector with a C-terminal FLAG-HA tandem affinity tag, pDEST-CTAP (Cantley lab stock) for expression in 293T. Human ALDH1L1 cDNA was subcloned into pDEST10 (Invitrogen) that has an N-terminal His6 tag for expression in Sf21 through the Bac-to-Bac system. Human ALDH1L1 cDNA was also subcloned into plenti6.3 (Invitrogen) for lentiviral expression in mammalian cells; the native stop codon of ALDH1L1 was included so that untagged, native ALDH1L1 was expressed. Mouse ALDH1L2, human QDPR, human MTHFD1, and human DHFR cDNAs were subcloned into pDEST-CTAP for lentiviral expression in mammalian cell lines; again, the native stop codon was included so that untagged, native form were expressed. Human QDPR and human DHFR cDNAs were also subcloned into pDEST17 for bacterial expression. Yeast MTD1 was subcloned into pLEX_305 (Addgene) for lentiviral expression in mammalian cells.
Recombinant protein purification
Recombinant rat GGH, and human ALDH1L1 were expressed in Sf21 cells (Invitrogen). Recombinant human QDPR and DHFR were expressed in E. coli BL21(DE3) (Invitrogen). All proteins were purified using the standard protocol for Ni-NTA purification under native conditions as described in ‘‘The QIAexpressionist Handbook’’ (QIAGEN). Recombinant rat GGH expressed in 293T was purified using anti-FLAG affinity gel (Sigma-Aldrich) and eluted using FLAG peptides (Sigma-Aldrich) according to manufacturer’s instructions. Because rat GGH is secreted into the culture medium, the protein was purified from the medium supernatant of Sf21 or 293T culture.
Generation of lentiviral stable cell lines
293T cells were co-transfected with the viral plasmid of interest with pVSVG and delta8.9 using Lipofectamine 2000 or 3000. Lentivirus-containing supernatants were collected at 48 hr after transfection. Virus from plenti6.3 or pDEST-CTAP was typically concentrated 5- or 10-fold using Lenti-X concentrator (Clontech), whereas virus from pLEX305 was directy used without concentration. Target cells were infected with 0.45 μm PVDF membrane-filtered viral supernatant in the presence of 8 mg/ml polybrene for 24 hr and then replaced in fresh media. At about 48–72 hr post-infection, puromycin (typically at 2 μg/ml) was added to select positively transduced cells for at least 3 days.
Western blot analysis
Cells were quickly washed twice with ice-cold PBS, and lysed in NP-40 lysis buffer (50 mM Tris- HCl, pH 8.0, 150 mM NaCl, 1.0% NP-40) freshly supplemented with protease inhibitors (Roche or Thermo). Lysates were cleared by centrifugation at 13,000 g at 4°C for 10 min. Protein concentration was determined by BCA assay. Proteins were denatured in NuPAGE LDS 4X sample buffer (Invitrogen) supplemented with β-mercaptoethanol. Typically, 20–50 μg of total protein was loaded per lane on a 4%–12% Bis-Tris gradient gel (Invitrogen) and analyzed by standard immunoblotting. Immunoblot signals were detected by enhanced chemiluminescence.
Cell proliferation assays
Cell proliferation was monitored by CellTiter-Glo Luminescent Assay (Promega). The base medium was composed of custom-made glycine-free DMEM (GIBCO) and 10% dialyzed FBS (GIBCO or Hyclone). To this base medium was added 400 μM glycine, and/or 100 μM hypoxanthine, and/or 16 μM thymidine, and/or 2 mM sodium formate, as specified in the figure legends. On day 0, 1000 cells were seeded on each well of a Corning 96-well plate. 5 replicate wells were used for each nutrient condition. Luminescence was typically measured on days 1, 3, 5, and 7. Results of CellTiter-Glo Luminescent Assay were further confirmed by crystal violet staining of 1–2 extra plates on the final day (typically day 7).
In vitro assay of QDPR or DHFR with THF
The reaction conditions were as follows: volume, 0.5 ml; buffer, 50 mM Tris-HCl pH 7.5; NADH, 0.5 mM; THF, 1.733 mM; QDPR or DHFR, 40 ug/ml; temperature, 37°C. The reaction was started by addition of freshly purified 6(R,S)-THF. At the time points indicated in Figures 5B–5C, 80-μl aliquots were removed, and quenched by 80 μl of stop buffer that contains 6% (w/v) sodium ascorbate and 400 mM β-mercaptoethanol in 50 mM Tris-HCl pH 7.5. The aliquots were kept on ice until the whole set was done, heated at 100°C for 2 min, centrifuged to remove denatured protein, and subjected to HPLC-UV analysis.
When the enzyme (QDPR or DHFR) was omitted, the dialysis buffer (against which the enzyme was dialyzed overnight, 50 mM NaPhos and 300 mM NaCl pH 8.0) was added instead. When NADH was omitted, 20 mM Tris-base (i.e., the buffer used to dissolve NADH) was added instead. 6(R,S)-THF was freshly purified from a DEAE-FF column a few minutes before the reaction, and contained no reducing agents.
In vitro assay of ALDH1L1 with THF or DHF
The conditions for the reaction with 6(S)-THF were as follows: volume, 0.5 ml; buffer, 42.5 mM sodium phosphate and 255 mM NaCl pH 8.0; 6(S)-THF, 20 μM or 40 μM; ALDH1L1, 35 μM; temperature, 37°C. For the enzyme-omitted controls, ALDH1L1 was replaced with the dialysis buffer against which ALDH1L1 was dialyzed overnight (50 mM NaPhos and 300 mM NaCl pH 8.0). The reaction was started by addition of freshly purified 6(S)-THF. At the time points indicated in Figure 6M, 70-μl aliquots were removed, and quenched by 35 μl of stop buffer that contains 9% (w/v) sodium ascorbate and 600 mM β-mercaptoethanol in 50 mM Tris-HCl pH 7.5. The aliquots were kept on ice until the whole set was done, heated at 100 C for 2 min, centrifuged to remove denatured protein, and subjected to HPLC-UV analysis.
6(S)-THF was enzymatically synthesized by reacting DHF with DHFR and NADPH, and then freshly purified from a DEAE-FF column a few minutes before the reaction. Purified 6(S)-THF contained no reducing agents.
The conditions for the reaction with DHF were as follows: volume, 0.5 ml; buffer, 42.5 mM sodium phosphate and 255 mM NaCl pH 8.0; DHF, 20 μM or 40 μM; ALDH1L1, 35 μM; temperature, 37°C. For the enzyme-omitted control, the dialysis buffer (against which ALDH1L1 was dialyzed overnight, 50 mM NaPhos and 300 mM NaCl pH 8.0) was added instead. The reaction was started by addition of freshly purified DHF. At the time points indicated in Figure 6N, 70-μl aliquots were removed, and quenched by 35 μl of stop buffer that contains 9% (w/v) sodium ascorbate and 600 mM β-mercaptoethanol in 50 mM Tris-HCl pH 7.5, followed by immediate boiling at 100°C for 2 min. The aliquots were centrifuged to remove denatured protein, and kept on ice until HPLC-UV analysis.
DHF was freshly purified from a DEAE-FF column a few minutes before the reaction, and contained no reducing agents. The folic acid impurity (⁓4%) was not separable from DHF using the DEAE-FF column.
Cellular Thermal Shift Assay
80%–90% confluent cells in a 60 mm plate were trypsinized and rinsed with DMEM/10% FBS. Cells were resuspended with 500 μl DMEM/10% FBS and distributed into two Eppendorf tubes. Each tube was treated with control vehicle (DMSO) or 1 μM methotrexate, respectively, and incubated in 37°C, 5% CO2 incubator for 30 min. Cell suspensions were distributed into PCR tubes (25 μl/tube, 8 tubes) and heated for 3 min using a gradient PCR condition (38, 39.7, 42.6, 47.3, 52.8, 57.3, 60.5, 62.5°C; T100 Thermal Cycler, Bio-rad). After a 3 min incubation at room temperature, cells were lysed by three repeated freeze-thaw cycles using liquid nitrogen and 37°C water bath. Cell lysates were centrifuged at 15,000 g for 20 min at 4°C. Soluble fractions were transferred into new tubes and the proteins were denatured by boiling for 10 min in an SDS sample buffer. Immunoblot analysis was performed using QDPR and DHFR antibodies and their corresponding secondary antibodies [anti-mouse IRDye 680RD (cat. #: P/N 925– 68070), anti-rabbit IRDye 800CW (cat. #: P/N 925–32211)]. The immunoblot signals were detected by Odyssey imaging system (LI-COR Biosciences).
LC-MS analysis of purine-related metabolites
Pre-cooled 80% methanol (1 mL) was added to each 6-cm dish of 70% confluent cells. The lysate was scraped into an Eppendorf tube and then centrifuged at 4°C for 10 minutes at 14,000 rpm. The supernatant was loaded onto a C18 Sep-pak SPE columns, and the flow-through was collected and then evaporated using a Speed-Vac. Targeted LC/MS analyses were performed on a Q Exactive Orbitrap mass spectrometer (Thermo Scientific) coupled to a Vanquish UPLC system (Thermo Scientific). The Q Exactive operated in polarity-switching mode. A Sequant ZIC-HILIC column (2.1 mm i.d. × 150 mm, Merck) was used for separation of metabolites. The flow rate was set at 150 μL/min. Mobile phases consisted of 100% acetonitrile for line A, and 0.1% NH4OH/20 mM CH3COONH4 in water for line B. Gradient ran from 85% to 30% A in 20 min followed by a wash with 30% A and re-equilibration at 85% A. Metabolites were identified on the basis of exact mass within 5 ppm and retention time. Relative metabolite quantitation was performed based on peak area for each metabolite. Data analysis was done using in-house written scripts. Purine-related metabolites were manually checked using Thermo X-Caliber. Normalization was based on protein content.
Preparation of folate and pAcABG standards
THF was prepared by NaBH4 reduction of folic acid followed by purification on a HiTrap DEAE FF column (1ml) eluted with a gradient program from 5 mM β-mercaptoethanol in water to 5 mM β-mercaptoethanol in 0.5 M Tris-HCl pH 7.5. A final concentration of 0.5 M β-mercaptoethanol was added to the fractions containing THF as detected by UV spectrophotometry. Note that for purification of THF to be used in the QDPR repair activity assay (Figures 5B and 5C), β-mercaptoethanol was omitted from the mobile phase, and such purified THF was immediately used for the QDPR repair activity assay. 10-CHO-THF was prepared from 5-CHO-THF (Sigma-Aldrich) via 5,10-CH+-THF and purified on the same HiTrap DEAE FF column by the same gradient program. Conversion of 5,10-CH+-THF to 10-CHO-THF was dramatically expedited by the Tris-HCl pH 8.0 buffer. Purified THF and 10-CHO-THF were flushed with nitrogen or argon and then stored in −80°C. pAcABG was prepared by reacting pABG (Sigma-Aldrich) with acetic anhydride (Sigma-Aldrich); the yield was nearly 100% and no column purification was necessary.
HPLC analysis for intracellular folates
The procedure was largely based on a published method (Wilson and Horne, 1986), in which cells were labeled with 3H-5-formyl-tetrahydrofolate (5-CHO-THF) or 3H-folic acid, and cellular folates liberated from proteins and protected from air oxidation by boiling in a neutral Tris-HCl buffer with ascorbate and β-mercaptoethanol. The polyglutamate tails were then cleaved by recombinant γ-glutamyl hydrolase (GGH), and the resulting folate monoglutamates resolved by HPLC using radioactivity detection.
Cells (1X105) seeded on a 6-cm Corning dish the day before were cultured in 4 mL of custom- made folate-free DMEM (GIBCO) supplemented with 10% dialyzed FBS and 2 μCi 5-CHO-THF (Moravek, specific activity: 10 Ci/mmol; final concentration: 50 nM) for 3 days. To prepare the folate extract, cells were quickly washed once with ice-cold PBS, scraped into 200 μl of extraction buffer (50 mM Tris-HCl pH 7.4, 200 mM β-mercaptoethanol, 3% (w/v) sodium ascorbate), and placed in a 100°C heating block rotating at 400 rpm for 2 min. A high concentration of ascorbate is critical for preventing air oxidation of reduced folates; 3% ascorbate was found to be much more effective than 0.3%. Lysate was cleared by centrifugation at 13,000 rpm at 4°C for 10 min, and then incubated with 15 ul of recombinant GGH (> 45 μg) at 37°C for 10 min. Reaction was stopped by heating at 100°C for 2 min, followed by centrifugation. Supernatant was stored in −80°C until HPLC analysis (typically within 3 days).
Radiolabeled folates were analyzed using an InfinityLab Poroshell 120 SB-C18 or EC-C18 column (4.6 X 150 mm, 2.7 μm) run with linear gradient elution of 15% B to 40% B over 30 min at a flow rate of 1 ml/min using an Agilent 1260 HPLC system coupled to Perkin Elmer 150TR Radiomatic Flow Scintillation Analyzer. HPLC line A is 25 mM sodium acetate pH 5.5 (for SB column) or pH 6.0 (for EC column) with 5 mM tetrabutylammonium bisulfate and 3 mM sodium ascorbate. HPLC line B is methanol.
Quantification was based on integrated areas of radioactive peaks. Note that pABG and pAcABG have a lower specific radioactivity than that of intact folates (see the chemical structures in Figure 4D). The percentage of 3H associated with pABG in commercial [3′,5′,7,9-3H]-5-CHO-THF differed from batch to batch (Shane, 1986). The percentages in the three batches we used were determined to be 57%, 39%, and 38%, using Shane’s method (Shane, 1986). Quantitation of pABG and pAcABG was based on these values, with the latter two rounded up to 40%.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data obtained from HPLC analysis were analyzed using Agilent ChemStation and Graphpad Prism. The supplemental tables show mean ± SD.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-MTHFD1L | Santa Cruz | H-106 |
| Rabbit polyclonal anti-SHMT2 | Cell Signaling | Cat#12762S |
| Rabbit monoclonal anti-SHMT1 | Cell Signaling | Cat#80715S |
| Mouse monoclonal anti-MTHFR | Sigma-Aldrich | Cat#SAB5300417 |
| Rabbit polyclonal anti-NAT1 | Abcam | Cat#ab175088 |
| Mouse monoclonal anti-ALDH1L1 | Abcam | Cat#ab56777; RRID: AB_940204 |
| Rabbit polyclonal anti-ALDH1L2 | Abcam | Cat#ab113496; RRID: AB_10864169 |
| Mouse monoclonal anti-QDPR | Santa Cruz | Cat#SC-376218; RRID: AB_10989783 |
| Mouse monoclonal anti-TYMS | Sigma-Aldrich | Cat#SAB1404503; RRID: AB_10739437 |
| Rabbit polyclonal anti-DHFR | Sigma-Aldrich | Cat#SAB1410298 |
| Mouse monoclonal anti-DHFR | Santa Cruz | Cat#SC-377091 |
| Mouse polyclonal anti-MTHFD1 | Abcam | Cat#Ab70203; RRID: AB_1269485 |
| Mouse monoclonal anti-beta actin | Abcam | Cat#ab6276; RRID: AB_2223210 |
| Rabbit polyclonal anti-beta tubulin | Abcam | Cat#ab6046; RRID: AB_2210370 |
| Bacterial and Virus Strains | ||
| BL21(DE3) | Invitrogen | Cat#C600003 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Folic acid | Sigma-Aldrich | Cat#F7876 |
| p-Aminobenzoyl glutamate (pABG) | Sigma-Aldrich | Cat#A0879 |
| p-Aminobenzoic acid (pABA) | Sigma-Aldrich | Cat#A9878 |
| Acetic anhydride | Sigma-Aldrich | Cat#320102 |
| 5-CHO-THF/folinic acid/leucovorin | Sigma-Aldrich | Cat#47612 |
| [3′,5′,7,9-3H]-folic acid | Moravek | Cat#MT783 |
| [3′,5′,7,9-3H]-5-CHO-THF | Moravek | Cat#MT521 |
| 5-Fluoro-2′-deoxyuridine | Sigma-Aldrich | Cat#F0503 |
| Crystal Violet | Sigma-Aldrich | Cat#C0775 |
| Recombinant DHFR | Sigma-Aldrich | Cat#SRP6119 |
| Sodium Borohydride | Sigma-Aldrich | Cat#8063730100 |
| Methotrexate | Sigma-Aldrich | Cat#A6770 |
| Sodium formate | Sigma-Aldrich | Cat#456020 |
| Critical Commercial Assays | ||
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | G7572 |
| Experimental Models: Cell Lines | ||
| MDA-MB-468 | ATCC | Cat#HTB-132 |
| RT4 | ATCC | Cat#HTB-2 |
| HeLa | ATCC | Cat#CCL-2 |
| MCF7 | ATCC | Cat#HTB-22 |
| 293T | Lab stocks | N/A |
| HCT116 | Lab stocks | N/A |
| Oligonucleotides | ||
| MTHFD1L CRISPR targeting sequence:GACGATGGAGTCCCGCGCCG CGG | This paper | N/A |
| SLC25A32 CRISPR targeting sequence:GCACGGTATTCCGCCACGTC CGG | This paper | N/A |
| QDPR CRISPR targeting sequence #2:GCAATTCTTTGCGTTGCTGG AGG | This paper | N/A |
| QDPR CRISPR targeting sequence #4:CCTGACCTTGGCTGGCGCAA AGG | This paper | N/A |
| NAT1 CRISPR targeting sequence #1:CAAAGGGAACAGCTCGGATC TGG | This paper | N/A |
| NAT1 CRISPR targeting sequence #2:TGCCATGGACTTAGGCTTAG AGG | This paper | N/A |
| DHFR/DHFRL1 CRISPR targeting sequence:CGTCGCTGTGTCCCAGAACA TGG | This paper | N/A |
| DHFR CRISPR targeting sequence:GAGAAGAATCGACCTTTAAA GGG | This paper | N/A |
| ALDH1L2 CRISPR targeting sequence #1:TGAACACCCCTACTACTCGG TGG | This paper | N/A |
| ALDH1L2 CRISPR targeting sequence #2:AAGAGGGCCACCGAGTAGTA GGG | This paper | N/A |
| SHMT2 sh-RNA targeting sequence | Harvard PlasmID Database | HsSH00253193 |
| CCGGAGAGTTGTGGACTTTAT | TRCN0000034807 | |
| Recombinant DNA | ||
| Gateway-pDEST17 | Invitrogen | Cat#11803012 |
| Gateway-pDEST10 | Invitrogen | Cat#11806–015 |
| Gateway-plenti6.3/V5-DEST | Invitrogen | Cat#V53306 |
| pLEX_305 | David Root Lab (Unpublished) | Addgene Plasmid#41390 |
| PX459_V2.0 | (Ran et al., 2013) | Addgene Plasmid#62988 |
| Software and Algorithms | ||
| Prism7 | Graphpad | Graphpad |
| Agilent OpenLAB CDS ChemStation | Agilent | Agilent |
Highlights.
Formate from mitochondrial serine metabolism protects cytosolic THF from degradation
Methotrexate causes degradation of cytosolic folate by a similar pathway
QDPR can reverse THF oxidation and preserve cytosolic folate levels
This role of QDPR can explain the folate deficiency in patients with QDPR mutations
ACKNOWLEDGMENTS
First and foremost, we thank Dean Appling (UT-Austin) for helpful advice throughout this study. We also thank Joshua Bryant and Dean Appling for providing wild-type and MTHFD1L-null MEFs. We thank Shana Sturla (ETH Zurich), I. David Goldman (Albert Einstein), Atsuo Sasaki (University of Cincinnati), Steven Gross (Weill Cornell), and all Cantley lab members, especially Cindy Hodakoski and Valbona Luga, for critical insights. This work was supported by a DoD BCRP postdoctoral fellowship W81XWH-13-1-0251 (Y.Z.); fellowships from the LAM Foundation LAM00100F01-14 (G.L.) and the Tuberous Sclerosis Alliance TSA-01-14 (G.L.); NIH grants 1F32HD074428-01 (J.M.), R01 CA046595 (J.B.), R01 HL121266 (J.B.), R01 GM051405 (J.B.), and R35 CA197588 (L.C.C.); a Lustgarten Foundation grant (L.C.C.); and a gift from the Mindy and Jon Gray family (L.C.C.).
DECLARATION OF INTERESTS
L.C.C. is a founder and member of the SAB of Agios Pharmaceuticals and Petra Pharmaceuticals, companies that are developing drugs to treat cancer. Petra Pharmaceuticals provides funds to support the laboratory of L.C.C.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and six tables and can be found with this article online at https://doi.org/10.1016/j.cell.2018.09.041.
REFERENCES
- Allegra CJ, Fine RL, Drake JC, and Chabner BA (1986). The effect of methotrexate on intracellular folate pools in human MCF-7 breast cancer cells. Evidence for direct inhibition of purine synthesis. J. Biol. Chem 261, 6478–6485. [PubMed] [Google Scholar]
- Baggott JE, and Johanning GL (1999). 10-Formyl-dihydrofolic acid is bioactive in human leukemia cells. J. Nutr 129, 1315–1318. [DOI] [PubMed] [Google Scholar]
- Banerjee D, Mayer-Kuckuk P, Capiaux G, Budak-Alpdogan T, Gorlick R, and Bertino JR (2002). Novel aspects of resistance to drugs targeted to dihydrofolate reductase and thymidylate synthase. Biochim. Biophys. Acta 1587, 164–173. [DOI] [PubMed] [Google Scholar]
- Bertino JR (2009). Cancer research: from folate antagonism to molecular targets. Best Pract. Res. Clin. Haematol 22, 577–582. [DOI] [PubMed] [Google Scholar]
- Bryant JD, Sweeney SR, Sentandreu E, Shin M, Ipas H, Xhemalce B, Momb J, Tiziani S, and Appling DR (2018). Deletion of the neural tube defect-associated geneMthfd1ldisrupts one-carbon and central energy metabolism in mouse embryos. J Biol Chem 293, 5821–5833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos-Barragan G, Wit N, Meiser J, Dingler FA, Pietzke M, Mulderrig L, Pontel LB, Rosado IV, Brewer TF, Cordell RL, et al. (2017). Mammals divert endogenous genotoxic formaldehyde into one-carbon metabolism. Nature 548, 549–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabner BA, and Roberts TG Jr. (2005). Timeline: Chemotherapy and the war on cancer. Nat. Rev. Cancer 5, 65–72. [DOI] [PubMed] [Google Scholar]
- Chasin LA, Feldman A, Konstam M, and Urlaub G (1974). Reversion of a Chinese hamster cell auxotrophic mutant. Proc. Natl. Acad. Sci. USA 71, 718–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chippel D, and Scrimgeour KG (1970). Oxidative degradation of dihydrofolate and tetrahydrofolate. Can. J. Biochem 48, 999–1009. [DOI] [PubMed] [Google Scholar]
- Christensen KE, and MacKenzie RE (2006). Mitochondrial one-carbon metabolism is adapted to the specific needs of yeast, plants and mammals. BioEssays 28, 595–605. [DOI] [PubMed] [Google Scholar]
- Ducker GS, and Rabinowitz JD (2017). One-Carbon Metabolism in Health and Disease. Cell Metab 25, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducker GS, Chen L, Morscher RJ, Ghergurovich JM, Esposito M, Teng X, Kang Y, and Rabinowitz JD (2016). Reversal of Cytosolic One-Carbon Flux Compensates for Loss of the Mitochondrial Folate Pathway. Cell Metab 23, 1140–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman ID, Chattopadhyay S, Zhao R, and Moran R (2010). The antifolates: evolution, new agents in the clinic, and how targeting delivery via specific membrane transporters is driving the development of a next generation of folate analogs. Curr. Opin. Investig. Drugs 11, 1409–1423. [PubMed] [Google Scholar]
- Gregory JF III. (1989). Chemical and Nutritional Aspects of Folate Research: Analytical Procedures, Methods of Folate Synthesis, Stability, and Bioavailability of Dietary Folates. In Advances in Food and Nutrition Research, Kinsella JE, ed. (Elsevier; ), pp. 1–101. [DOI] [PubMed] [Google Scholar]
- Huber KVM, Olek KM, Müller AC, Tan CSH, Bennett KL, Colinge J, and Superti-Furga G (2015). Proteome-wide drug and metabolite interaction mapping by thermal-stability profiling. Nat. Methods 12, 1055–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones ML, and Nixon PF (2002). Tetrahydrofolates are greatly stabilized by binding to bovine milk folate-binding protein. J. Nutr 132, 2690–2694. [DOI] [PubMed] [Google Scholar]
- Kaufman S (1987). Dihydropteridine reductase from sheep liver. Methods Enzymol 142, 97–103. [DOI] [PubMed] [Google Scholar]
- Krebs HA, Hems R, and Tyler B (1976). The regulation of folate and methionine metabolism. Biochem. J 158, 341–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupenko SA (2009). FDH: an aldehyde dehydrogenase fusion enzyme in folate metabolism. Chem. Biol. Interact 178, 84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupenko SA, and Oleinik NV (2002). 10-formyltetrahydrofolate dehydrogenase, one of the major folate enzymes, is down-regulated in tumor tissues and possesses suppressor effects on cancer cells. Cell Growth Differ 13, 227–236. [PubMed] [Google Scholar]
- Kwon YK, Lu W, Melamud E, Khanam N, Bognar A, and Rabinowitz JD (2008). A domino effect in antifolate drug action in Escherichia coli. Nat. Chem. Biol 4, 602–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence SA, Titus SA, Ferguson J, Heineman AL, Taylor SM, and Moran RG (2014). Mammalian mitochondrial and cytosolic folylpolyglutamate synthetase maintain the subcellular compartmentalization of folates. J. Biol. Chem 289, 29386–29396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linster CL, Van Schaftingen E, and Hanson AD (2013). Metabolite damage and its repair or pre-emption. Nat. Chem. Biol 9, 72–80. [DOI] [PubMed] [Google Scholar]
- Locasale JW (2013). Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat. Rev. Cancer 13, 572–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddocks ODK, Athineos D, Cheung EC, Lee P, Zhang T, van den Broek NJF, Mackay GM, Labuschagne CF, Gay D, Kruiswijk F, et al. (2017). Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 544, 372–376. [DOI] [PubMed] [Google Scholar]
- Maldonado EN, and Lemasters JJ (2014). ATP/ADP ratio, the missed connection between mitochondria and the Warburg effect. Mitochondrion 19 Pt A, 78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews RG, and Kaufman S (1980). Characterization of the dihydropterin reductase activity of pig liver methylenetetrahydrofolate reductase. J. Biol. Chem 255, 6014–6017. [PubMed] [Google Scholar]
- Meedel TH, and Pizer LI (1974). Regulation of one-carbon biosynthesis and utilization in Escherichia coli. J. Bacteriol 118, 905–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiser J, Tumanov S, Maddocks O, Labuschagne CF, Athineos D, Van Den Broek N, Mackay GM, Gottlieb E, Blyth K, Vousden K, et al. (2016). Serine one-carbon catabolism with formate overflow. Sci. Adv 2, e1601273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min H, Shane B, and Stokstad EL (1988). Identification of 10-formyltetra-hydrofolate dehydrogenase-hydrolase as a major folate binding protein in liver cytosol. Biochim. Biophys. Acta 967, 348–353. [DOI] [PubMed] [Google Scholar]
- Minchin RF (1995). Acetylation of p-aminobenzoylglutamate, a folic acid catabolite, by recombinant human arylamine N-acetyltransferase and U937 cells. Biochem. J 307, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minton DR, Nam M, McLaughlin DJ, Shin J, Bayraktar EC, Alvarez SW, Sviderskiy VO, Papagiannakopoulos T, Sabatini DM, Birsoy K, and Possemato R (2018). Serine Catabolism by SHMT2 Is Required for Proper Mitochondrial Translation Initiation and Maintenance of Formylmethionyl-tRNAs. Mol. Cell 69, 610–621.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momb J, Lewandowski JP, Bryant JD, Fitch R, Surman DR, Vokes SA, and Appling DR (2013). Deletion of Mthfd1l causes embryonic lethality and neural tube and craniofacial defects in mice. Proc. Natl. Acad. Sci. USA 110, 549–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran RG, Mulkins M, and Heidelberger C (1979). Role of thymidylate synthetase activity in development of methotrexate cytotoxicity. Proc. Natl. Acad. Sci. USA 76, 5924–5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morscher RJ, Ducker GS, Li SH-J, Mayer JA, Gitai Z, Sperl W, and Rabinowitz JD (2018). Mitochondrial translation requires folate-dependent tRNA methylation. Nature 554, 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullarky E, Lucki NC, Beheshti Zavareh R, Anglin JL, Gomes AP, Nicolay BN, Wong JCY, Christen S, Takahashi H, Singh PK, et al. (2016). Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc. Natl. Acad. Sci. USA 113, 1778–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M, Keating M, Boyle P, Weir DG, and Scott JM (1976). The elucidation of the mechanism of folate catabolism in the rat. Biochem. Biophys. Res. Commun 71, 1017–1024. [DOI] [PubMed] [Google Scholar]
- Nagy PL, Marolewski A, Benkovic SJ, and Zalkin H (1995). Formyltetra-hydrofolate hydrolase, a regulatory enzyme that functions to balance pools of tetrahydrofolate and one-carbon tetrahydrofolate adducts in Escherichia coli. J. Bacteriol 177, 1292–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, and Ferguson SJ (2013). Bioenergetics (Academic Press; ). [Google Scholar]
- Nilsson R, Jain M, Madhusudhan N, Sheppard NG, Strittmatter L, Kampf C, Huang J, Asplund A, and Mootha VK (2014). Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat. Commun 5, 3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacold ME, Brimacombe KR, Chan SH, Rohde JM, Lewis CA, Swier LJYM, Possemato R, Chen WW, Sullivan LB, Fiske BP, et al. (2016). A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat. Chem. Biol 12, 452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock RJ, and Kaufman S (1978). Dihydropteridine reductase may function in tetrahydrofolate metabolism. J. Neurochem 31, 115–123. [DOI] [PubMed] [Google Scholar]
- Quinlivan EP, McPartlin J, Weir DG, and Scott J (2000). Mechanism of the antimicrobial drug trimethoprim revisited. FASEB J 14, 2519–2524. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh AM, Pheasant AE, and Blair JA (1981). Folate catabolism in tumour-bearing rats and rats treated with methotrexate. Br. J. Cancer 44, 700–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shane B (1986). Identification of folylpoly(gamma-glutamate) chain length by cleavage to and separation of p-aminobenzoylpoly(gamma-glutamates). Methods Enzymol 122, 323–330. [DOI] [PubMed] [Google Scholar]
- Shane B, and Stokstad EL (1985). Vitamin B12-folate interrelationships. Annu. Rev. Nutr 5, 115–141. [DOI] [PubMed] [Google Scholar]
- Stover PJ, and Field MS (2011). Trafficking of intracellular folates. Adv. Nutr 2, 325–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh JR, Herbig AK, and Stover PJ (2001). New perspectives on folate catabolism. Annu. Rev. Nutr 21, 255–282. [DOI] [PubMed] [Google Scholar]
- Sundararajan S, and MacKenzie RE (2002). Residues involved in the mechanism of the bifunctional methylenetetrahydrofolate dehydrogenase-cyclohydrolase: the roles of glutamine 100 and aspartate 125. J. Biol. Chem 277, 18703–18709. [DOI] [PubMed] [Google Scholar]
- Tibbetts AS, and Appling DR (2010). Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr 30, 57–81. [DOI] [PubMed] [Google Scholar]
- Titus SA, and Moran RG (2000). Retrovirally mediated complementation of the glyB phenotype. Cloning of a human gene encoding the carrier for entry of folates into mitochondria. J. Biol. Chem 275, 36811–36817. [DOI] [PubMed] [Google Scholar]
- Wakefield L, Cornish V, Long H, Griffiths WJ, and Sim E (2007). Deletion of a xenobiotic metabolizing gene in mice affects folate metabolism. Biochem. Biophys. Res. Commun 364, 556–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner ER, Blau N, and Thöny B (2011). Tetrahydrobiopterin: biochemistry and pathophysiology. Biochem. J 438, 397–414. [DOI] [PubMed] [Google Scholar]
- West MG, Horne DW, and Appling DR (1996). Metabolic role of cytoplasmic isozymes of 5,10-methylenetetrahydrofolate dehydrogenase in Saccharomyces cerevisiae. Biochemistry 35, 3122–3132. [DOI] [PubMed] [Google Scholar]
- Williamson DH, Lund P, and Krebs HA (1967). The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem. J 103, 514–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SD, and Horne DW (1986). High-performance liquid chromatographic separation of the naturally occurring folic acid derivatives. Methods Enzymol 122, 269–273. [DOI] [PubMed] [Google Scholar]
- Yang M, and Vousden KH (2016). Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 16, 650–662. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.