

Abstract
Transient, severe forebrain or global ischemia leads to delayed cell death of pyramidal neurons in the hippocampal CA1. The precise molecular mechanisms underlying neuronal cell death after global ischemia are as yet unknown. Glutamate receptor-mediated Ca2+ influx is thought to play a critical role in this cell death. In situ hybridization revealed that the expression of mRNA encoding GluR2 (the subunit that limits Ca2+ permeability of AMPA-type glutamate receptors) was markedly and specifically reduced in gerbil CA1 pyramidal neurons after global ischemia but before the onset of neurodegeneration. To determine whether the change in GluR2 expression is functionally significant, we examined the AMPA receptor-mediated rise in cytoplasmic free Ca2+ level ([Ca2+]i) in individual CA1 pyramidal neurons by optical imaging with the Ca2+indicator dye fura-2 and by intracellular recording. Seventy-two hours after ischemia, CA1 neurons that retained the ability to fire action potentials exhibited a greatly enhanced AMPA-elicited rise in [Ca2+]i. Basal [Ca2+]i in these neurons was unchanged. These findings provide evidence for Ca2+entry directly through AMPA receptors in pyramidal neurons destined to die. Downregulation of GluR2 gene expression and an increase in Ca2+ influx through AMPA receptors in response to endogenous glutamate are likely to contribute to the delayed neuronal death after global ischemia.
Keywords: global ischemia, glutamate receptor regulation, AMPA receptor, GluR2, GluRB, hippocampus, CA1, intracellular calcium, intracellular recording, neurodegeneration, delayed neurodegeneration, optical imaging, gerbil
Transient, severe global ischemia, occurring during cardiorespiratory arrest in patients or experimentally in animals, induces selective and delayed neuronal cell death (for review, see Schmidt-Kastner and Freund, 1991). Pyramidal neurons in the CA1 region of the hippocampus and some types of hilar neurons are particularly vulnerable and die after global ischemia (Kirino, 1982;Pulsinelli et al., 1982; Hsu and Buzsaki, 1993). In rats, cell loss is not prominent until 2–3 d after induction of ischemia (Pulsinelli et al., 1982). A rise in [Ca2+]i is thought to initiate a cascade of events leading to cell death, including activation of proteases and endonucleases, generation of free radicals that destroy cell membranes by lipid peroxidation, and induction of apoptosis (for review, see Rothman and Olney, 1986; Choi, 1990, 1995; Puttfarcken et al., 1993; Bredesen, 1995; Meldrum, 1995).
Although AMPA receptors were initially thought to be relatively impermeable to Ca2+, it is now clear that there are also AMPA receptors exhibiting considerable Ca2+permeability. AMPA receptors containing the GluR2 subunit exhibit low Ca2+ permeability, whereas AMPA receptors lacking GluR2 are much more Ca2+ permeable (Hollmann et al., 1991; Hume et al., 1991; Burnashev, 1996). AMPA receptors in most principal neurons of adult hippocampus are heteromeric, contain GluR2, and have a low permeability to Ca2+ (Bochet et al., 1994; Jonas et al., 1994; Geiger et al., 1995). GluR2-lacking, relatively Ca2+-permeable AMPA receptors have recently been implicated in the pathogenesis of neuronal degeneration after global ischemia. In rats, global ischemia leads to reduced expression of GluR2 mRNA in vulnerable pyramidal neurons of the hippocampal CA1 before the delayed cell death (Pellegrini-Giampietro et al., 1992a; Pollard et al., 1993); this reduction would lead to increased formation of Ca2+-permeable AMPA receptors. After ischemia, AMPA receptor-mediated EPSCs at the CA1–Schaffer collateral synapse are enhanced and increased in sensitivity to Joro spider toxin and 1-acetyl naphthyl spermine (Tsubokawa et al., 1995), channel blockers selective for GluR2-lacking, Ca2+-permeable AMPA receptors (Blaschke et al., 1993; Herlitze et al., 1993). Administration of the AMPA and kainate receptor antagonist 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo(f)quinoxaline (NBQX) protects CA1 neurons in animal models of global ischemia (Sheardown et al., 1990; Buchan et al., 1991); protection is observed even when NBQX is administered 16–24 hr after ischemia (Sheardown et al., 1993). Neuroprotection by NBQX implicates AMPA and/or kainate receptors in delayed neuronal death but does not distinguish between more and less of the Ca2+-permeable receptors.
Taken together, the data suggest that the “switching off” of GluR2 expression in CA1 after an ischemic insult is translated into the formation of new AMPA receptors lacking the GluR2 subunit. This change in receptor composition increases AMPA receptor-mediated Ca2+ entry in response to endogenous glutamate and enhances glutamate pathogenicity in this region (the GluR2 hypothesis; Pellegrini-Giampietro et al., 1997).
The present study was performed in gerbils to test whether the change in AMPA receptor expression enhances AMPA receptor-gated Ca2+ entry into CA1 pyramidal neurons. We first show by in situ hybridization that global ischemia in gerbils, as in rats, leads to a reduction in GluR2 mRNA in these neurons. We then show by intracellular recording and optical imaging of fura-2-injected CA1 neurons that Ca2+ influx through AMPA receptors is increased after global ischemia. The changes in GluR2 mRNA expression and AMPA receptor function precede neuronal death. These findings indicate that Ca2+ influx through AMPA receptors lacking the GluR2 subunit may be an important factor contributing to delayed neurodegeneration after global ischemia.
MATERIALS AND METHODS
Global ischemia in the gerbil
Global ischemia in the gerbil was produced by temporary bilateral occlusion of the carotid arteries. Adult male Mongolian gerbils (Tumblebrook Farms), weighing between 60 and 80 gm, were fasted overnight and anesthetized with intraperitoneal ketamine (100 mg/kg) and xylazine (6 mg/kg). The carotid arteries were occluded with nontraumatic aneurism clips and released after 5 min to allow cerebral reperfusion. Body temperature was maintained close to 37.5°C with a rectal thermistor and heat lamp until thermoregulation recovered. Control gerbils were sham-operated and sacrificed 48 or 72 hr later.
Histological analysis
Neuronal damage was monitored in the hippocampus by histological examination at various time points after ischemia. Animals were anesthetized and decapitated. The brains were then rapidly removed and placed into ice-cold PBS. Both hippocampi were dissected out and cut into 1-mm-thick transverse slices with a McIlwain tissue chopper. These slices were placed immediately into ice-cold fixative (2.5% glutaraldehyde and 4% formaldehyde in 0.1 m sodium cacodylate buffer, pH 7.4). After fixation at 4°C overnight, the tissue was osmicated (2% OsO4 in 0.1 m sodium cacodylate buffer, pH 7.4, for 2 hr), dehydrated, and embedded in Eponate 12 resin (Ted Pella Inc., Redding, CA). Two micrometer sections were cut and stained with toluidine blue. In addition, emulsion-dipped sections after hybridization were counterstained with hematoxylin and eosin, dehydrated in graded ethanols, cleared, and coverslipped.
In situ hybridization
Preparation of probes.35S-Uridine triphosphate (UTP)-labeled RNA probes were transcribed from GluR1, GluR2 (AMPA receptor), and NR1 (NMDA receptor) subunit cDNAs with a Stratagene (La Jolla, CA) transcription kit. cDNA templates were incubated (1 hr, 37°C) with the appropriate polymerase (T7 for GluR1 and GluR2, T3 for NR1) and with labeled and unlabeled nucleotides. RNA probes were purified by phenol and chloroform extraction.
Hybridization. Glutamate receptor mRNA expression was measured by in situ hybridization on brain sections of control and ischemic gerbils by a modification of the method ofPellegrini-Giampietro et al. (1991). In brief, coronal sections (20 μm) of brains from control (n = 6) and from 24 hr (n = 4), 48 hr (n = 8), and 72 hr (n = 4) ischemic gerbils were hybridized with35S-UTP-labeled RNA probes directed against the GluR1, GluR2, and NR1 subunit cDNAs. Before application of an RNA probe, sections were subjected to acetylation and were incubated (2 hr at 50°C) with 100 μl of prehybridization solution. For hybridization, slides were incubated overnight at 50°C with the35S-labeled RNA probe (106 cpm/section, 1 ng/μl). Sections were treated with RNase A (20 μg/ml) and dehydrated in ethanol. Slides were apposed to Kodak XAR-5 film (24–72 hr) or, for higher resolution studies, dipped in photographic emulsion (Kodak NTB-2) and exposed for 1–4 weeks. The anatomy of brain images from autoradiographs and from hematoxylin and eosin sections was assessed using the atlas of Paxinos and Watson (1991). Photomicrographs were obtained using a Nikon microscope and bright-field optics.
Signal specificity. Signal specificity was assessed by competition experiments in which radiolabeled probes were hybridized to sections in the presence of excess (100-fold) levels of the same unlabeled probe. This procedure resulted in virtually blank autoradiograms. In separate control studies, labeling by sense RNA probes or by antisense probes to sections pretreated with RNase A (100 μg/ml) showed no detectable labeling. Conditions were of sufficiently high stringency as to rule out cross-hybridization among GluR1, GluR2, and GluR3 (Pellegrini-Giampietro et al., 1991), and more distantly related glutamate receptor subunits (GluR5–GluR7, KA1, KA2). The GluR1 and GluR2 probes are “pan” probes (Sommer et al., 1990) in that they label both “flip” and “flop” splice variants. The NR1 probe is a pan probe in that it labels all eight splice variants; NR1 shares <20% sequence identity with NR2A–NR2C (Kutsuwada et al., 1992; Monyer et al., 1992) and is therefore unlikely to cross-react with mRNAs encoding NR2 subunits.
Quantitation and statistical analysis. For quantification of mRNA expression levels, autoradiograms were analyzed with a Molecular Dynamics (Sunnyvale, CA) 300A Computing Densitometer equipped with the National Institutes of Health IMAGE program. Film images were scanned at 2000 dpi resolution, and images of each section (∼1 × 106 pixels) were created. Mean optical densities in regions of maximal labeling of individual hippocampal subfields were averaged from a minimum of two sections from each animal for each probe, and film background was subtracted. Optical density values were expressed as grand means (±SEM) of individual means from three to eight gerbils per postischemic group. To enable comparisons between groups for any given probe, brain sections were cut from control and postischemic gerbils in the same experimental session, incubated the sections with the same solutions of RNA probes on the same day, and apposed the sections to the same sheet of film.
Mean optical density readings were statistically analyzed by Student’s unpaired t test. The percent change in optical density for postischemic gerbils was expressed relative to optical density values for the corresponding regions of the control gerbil hippocampus on the same film. The rationale of the quantitative analysis was based on the following considerations: (1) optical density readings taken from each region of interest varied little in different sections from the same animals; (2) the concentration of RNA probe used (106 cpm/section) produced saturating levels of hybridization and the maximal signal-to-noise ratio for the probes used (Pellegrini-Giampietro et al., 1991); and (3) use of35S-UTP-labeled brain paste standards indicated that exposure times were in the linear response range of the film (Pellegrini-Giampietro et al., 1991).
Ca2+ imaging and intracellular recording
Electrophysiology. For electrophysiological and Ca2+-imaging experiments, gerbils were decapitated 48 hr (n = 6) or 72 hr (n = 7) after ischemia. Control gerbils were sham-operated and decapitated 48 or 72 hr later (n = 6). Three unoperated animals did not differ from sham-operated animals, and data from these animals were pooled. Experiments were performed on transverse vibratome slices of gerbil dorsal hippocampus (300 μm). After preparation, slices were placed in a slice chamber and allowed to recover for at least 1 hr in an extracellular solution containing 124 mm NaCl, 2.0 mm KCl, 3 mm MgSO4, 3 mm CaCl2, 18 mmNaHCO3, 1.24 mmKH2PO4, 10 mmd-glucose, and 50 μm picrotoxin. Recordings were performed at room temperature in an interface recording configuration with a humidified atmosphere consisting of 95% O2/5% CO2. CA1 pyramidal neurons were impaled with sharp microelectrodes containing a solution of 10 mm fura-2 (potassium salt; Molecular Probes, Eugene, OR) in 1.5 m CsCl in the tip and a backfill solution of 3m CsCl. Electrode resistance was ∼200 MΩ initially and dropped to 60–80 MΩ over ∼20 min as the electrode solutions equilibrated. CA1 neurons were loaded with fura-2 by passing steady hyperpolarizing current (0.5–1.0 nA) and loaded with Cs+ by passing depolarizing pulses (100–200 msec, 0.2–0.7 nA, every 10 sec for 10 min). Final intracellular concentrations of fura-2 were 0.2–0.3 mm (Petrozzino et al., 1995). Cells were voltage-clamped in the discontinuous single electrode mode of an Axoclamp-2A (Axon Instruments, Burlingame, CA) with a switching frequency of 2–5 kHz. Details of the procedure are given in Results.
Imaging. Cells were imaged from the top surface of the slice by means of an upright microscope (Zeiss Axioskop) and a 20× dry objective (Zeiss). A charge-coupled device camera system (series 200; Photometrics Ltd., Tucson, AZ) was used to acquire digitized images of fura-2 fluorescence as described previously (Muller and Connor, 1991). The camera was operated in frame transfer mode. Image pairs were acquired using 350 and 380 nm excitation wavelengths. Exposure times for single frames were 100–200 msec. Image pairs were acquired every 30 sec. [Ca2+]i was calculated from image pairs by the ratio method (Grynkiewicz et al., 1985). Fluorescence data were background corrected before construction of ratio images. [Ca2+]i before, during, and after AMPA receptor stimulation compared using the Student’s unpaired t test.
RESULTS
Global ischemia induces selective, delayed neurodegeneration
To assess neuronal loss after induction of global ischemia in gerbils, we subjected brain sections of experimental and control animals to histological analysis. Toluidine blue-stained sections at the level of the hippocampus revealed no detectable cell loss 24 hr (data not shown) or 72 hr after induction of global ischemia (Fig.1A–D). In contrast, analysis of brain sections taken from animals 1 week after ischemia revealed virtually complete loss of neurons in the pyramidal cell layer of the hippocampal CA1 (Fig.1E,F). Only a few surviving neurons remained, and these may also have been deteriorating. The hippocampal CA3 and dentate gyrus exhibited no detectable cell loss (data not shown). These data confirm those of Kirino (1982) and Kirino and Sano (1984).
Fig. 1.

Global ischemia induces selective, delayed neuronal cell loss in the hippocampal CA1. Toluidine blue labeling of coronal brain sections at the level of the dorsal hippocampus from control (A, B) and experimental (C, D) gerbils revealed no detectable neuronal damage 72 hr after transient global ischemia. Global ischemia was induced by two-vessel occlusion as described in Materials and Methods. One week after ischemia, the pyramidal cell layer of the hippocampal CA1 exhibited virtually complete loss of neurons with only a few apparent survivors that may also have been deteriorating (E, F). A,C, E, lower magnification than inB, D, F. Scale bars:E, 100 μm; F, 50 μm.
GluR2 mRNA is decreased in CA1 after transient global ischemia
To examine patterns of glutamate receptor mRNA expression in adult gerbil brain, we performed in situ hybridization with riboprobes specific for GluR1, GluR2, and NR1 mRNAs. Autoradiograms of coronal sections of control gerbil brain at the level of the dorsal hippocampus showed that all three transcripts were expressed at high levels in the pyramidal cell layer of CA1 and CA3 and in the granule cell layer of the dentate gyrus (Fig. 2,left panel).
Fig. 2.
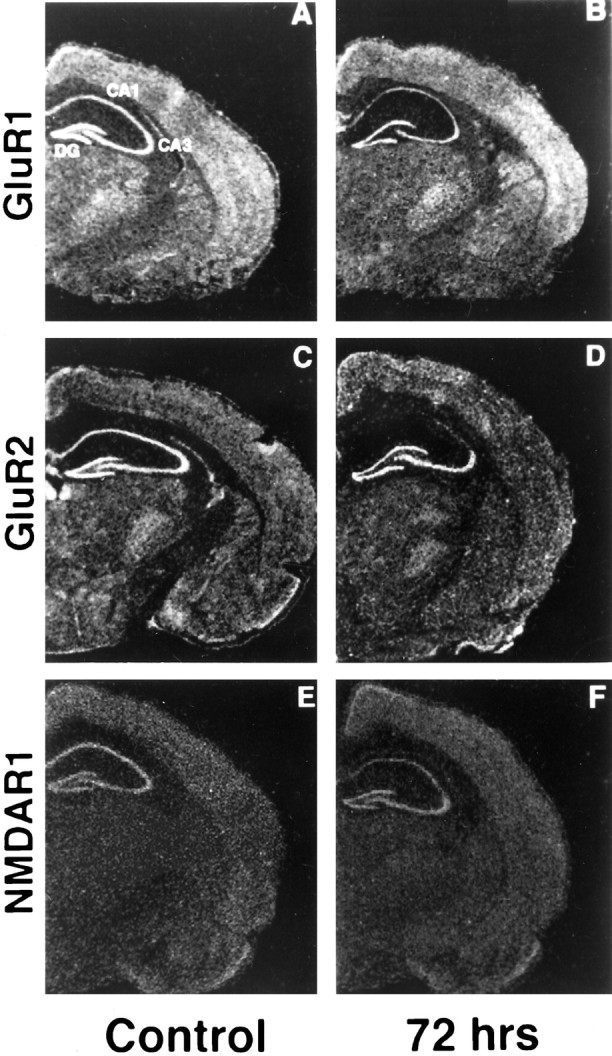
Expression of GluR2 mRNA is reduced specifically in CA1 after ischemia. Photomicrographs of autoradiograms of GluR1, GluR2, and NR1 mRNA in situ hybridization in coronal sections of gerbil brain at the level of the dorsal hippocampus from control animals (A, C, E) and from experimental animals 72 hr after ischemia (B,D, F) are shown. GluR2 mRNA was dramatically reduced in the pyramidal cell layer of the vulnerable CA1 but not in the pyramidal cell layer of CA3 or in the granule cell layer of the dentate gyrus, areas that do not undergo neurodegeneration. GluR1 and NR1 mRNAs were somewhat reduced in CA1 72 hr after ischemia.
Twenty-four hours after a 5 min period of global ischemia, GluR2 mRNA expression was significantly reduced in the CA1 pyramidal layer, to 85 ± 2% of control GluR2 expression. Forty-eight and 72 hr after ischemia, GluR2 mRNA expression was markedly reduced in CA1, to 48 ± 6 and 17 ± 10% of the control value, respectively (Figs. 3,4A). Expression of GluR1 and NR1 mRNAs was unchanged at 24 hr and exhibited only modest decreases, to 87 ± 5 and 93 ± 5% of the control value, respectively, at 48 hr in CA1 (Fig. 4A). Seventy-two hours after ischemia, the reduction in GluR1 and NR1 was to approximately half of the control value, to 45 ± 4 and 54 ± 3%, respectively (p < 0.01; Fig.4A). The ratio of GluR2 to GluR1 mRNA expression in a given region may be a predictor of the fraction of receptors that are Ca2+ permeable (for review, seePellegrini-Giampietro et al., 1992b; Burnashev, 1996). This ratio declined steadily to approximately one-third of the control value by 72 hr in CA1 (Fig. 4B). In other subfields of the hippocampus that do not undergo neurodegeneration, CA3 pyramidal cells and dentate gyrus granule cells, glutamate receptor mRNA expression showed little change. Seventy-two hours after ischemia, GluR1 and GluR2 mRNAs were both decreased by ∼15% in CA3 (p< 0.01) and were not significantly changed in dentate gyrus (decreased by ∼1.5%; p > 0.05). In the two regions, the ratio of GluR2 to GluR1 mRNAs was unchanged relative to control values (Fig.4B). No changes in expression of glutamate receptor mRNAs were noted in other areas of the brain.
Fig. 3.

Progressive decrease of GluR2 mRNA expression in the hippocampal CA1 after global ischemia. A–D, Expression of GluR2 mRNA in the pyramidal cell layer of the hippocampal CA1 decreased with time after ischemia. Expression of GluR2 mRNA was little changed in CA3 and dentate gyrus. Autoradiograms are described in Figure 2.
Fig. 4.

Time course of reduction in GluR2 mRNA expression in the CA1 pyramidal cell layer after ischemia. Glutamate receptor mRNA expression at the level of the hippocampus was measured as mean optical density within regions of interest in film autoradiograms from experimental and control animals. A, Values for GluR1, GluR2, and NR1 mRNA expression in CA1 of animals 24, 48, and 72 hr after ischemia are plotted as percent ± SEMs of the corresponding values for control animals. Twenty-four hours after ischemia, GluR2 mRNA expression was decreased relative to the corresponding control value; the reduction in GluR2 was significantly greater than the reduction in GluR1 (p < 0.01) and NR1 (p < 0.01) mRNA expression. At 48 hr GluR2 had decreased to 48 ± 6% of the corresponding control value; GluR1 and NR1 were only slightly decreased. Seventy-two hours after ischemia, GluR1 and NR1 were decreased to 45 ± 4 and 54 ± 3%, respectively, of control values; GluR2 was decreased to 17 ± 10% of control values (*p < 0.01 for each mRNA). Numbers of animals in each group are indicated inparentheses. B, Ratios of GluR2 to GluR1 mRNA expression in the hippocampal CA1, CA3, and dentate gyrus (DG) 24, 48, and 72 hr after ischemia are normalized to the control values. In CA1, the normalized ratio decreased progressively to 0.38 at 72 hr (asterisks indicate significance). In CA3 and dentate gyrus, the ratio remained constant.
Ischemia-induced changes in GluR2 mRNA expression are specific to CA1 pyramidal neurons
To examine AMPA receptor mRNA expression in individual pyramidal neurons, we performed microscopic examination of emulsion-dipped sections of postischemic and control gerbil brain. In control sections, labeling with an RNA probe directed to GluR2 mRNA revealed dense clusters of hybridization grains overlying individual neurons in CA1 (Fig. 5A), CA3, and dentate gyrus (data not shown). Seventy-two hours after ischemia, sections labeled with the GluR2 RNA probe revealed a reduced density of hybridization grains overlying individual CA1 pyramidal cells, indicative of decreased RNA expression per neuron (Fig.5B).
Fig. 5.

Changes in GluR2 mRNA expression are cell-specific. Emulsion-dipped coronal sections of the hippocampus from control and experimental animals 72 hr after ischemia show silver grains densely clustered over CA1 pyramidal neurons. A, Sections of control brain revealed dense clusters of silver grains overlying individual pyramidal neurons in the CA1; virtually all neurons in the field exhibited intense labeling. B, Seventy-two hours after ischemia, GluR2 labeling was dramatically reduced for all CA1 neurons. Sections were counterstained with hematoxylin and eosin. Arrows indicate representative pyramidal neurons. At this time, histological analysis showed no cell loss (see Fig. 1).
Global ischemia increases the AMPA receptor-mediated rise in [Ca2+]i in CA1 pyramidal neurons
To test whether transient global ischemia alters basal [Ca2+]i and AMPA-elicited Ca2+ influx, we performed intracellular recording and optical ratio imaging of individual fura-2-injected CA1 neurons in slices from control and postischemic gerbils. Neurons were selected for these measurements by three criteria: (1) having a resting membrane potential more negative than −60 mV at the time of penetration, (2) generating action potentials >65 mV in amplitude, and (3) requiring a hyperpolarizing current of <0.2 nA to maintain the membrane potential at −80 mV after Cs+ injection had blocked K conductances. Cells that failed these criteria increased in conductance, had their [Ca2+]i rise beyond where it was measurable with fura-2 (see below), and could not be studied further. The fraction of pyramidal cells that met these criteria was ∼80% in slices from control animals and animals 48 hr after ischemia and was ∼50% in slices from animals 72 hr after ischemia.
CA1 neurons selected by the above criteria were voltage clamped at −80 mV, dye-loaded by passing hyperpolarizing current for 10–20 min, and injected with Cs+ by application of suprathreshold depolarizing current pulses. Then the slices were perfused for at least 15 min with a solution containingd,l-2-aminophosphonovaleric acid (APV, 100 μm) and MK-801 (20 μm) to block Ca2+ influx through NMDA receptors, nimodipine (10 μm) and Cd2+ (100 μm) to block, at least partially, voltage-dependent Ca2+channels, and tetrodotoxin (3 μm) to block action potentials and synaptic release of glutamate by other neurons. The [Ca2+]i was determined ratiometrically from paired digitally acquired fluorescence images using excitation at 350 and 380 nm wavelengths. CA1 neurons, held at −80 mV, exhibited a low resting [Ca2+]i (between 50 and 100 nm) that did not differ significantly in control and in 48 and 72 hr postischemic slices (Figs.6B,7A). For this comparison, a basal [Ca2+]i was determined just before AMPA application, but the level generally had not changed from the time of fura-2 injection.
Fig. 6.

AMPA-elicited inward current and rise in [Ca2+]i in a CA1 pyramidal neuron after ischemia. A, Inward current elicited by AMPA [30 μm with 10 μm cyclothiazide (CTZ)] in a CA1 pyramidal neuron in a hippocampal slice from an animal 72 hr after ischemia. AMPA and CTZ were bath-applied for 30 sec (red bar). Then the AMPA was washed out with saline containing the NMDA receptor and the Ca2+ and Na+ channel blockers. After ∼5 min, CNQX (20 μm) was added to the other blockers to cause more rapid recovery. In the control neuron illustrated in B, the AMPA-elicited inward current in the presence of 30 μm CTZ was of somewhat lower amplitude but was similar in time course.B, Optical imaging (350 nm excitation images) of individual CA1 pyramidal neurons injected with fura-2 in hippocampal slices from a control animal (upper row) and an experimental animal 72 hr after ischemia (lower row, same cell as in A). a–b, Time of imaging indicated in current trace above. a, Image taken before bath application of agonist. b, Image taken at peak inward current after application of AMPA (100 μm with 10 μm CTZ). c, Image taken after recovery to near baseline current. Colorrepresents [Ca2+]i determined from the ratio of fluorescence obtained at two excitation wavelengths (350 and 380 nm); calibration is at right. AMPA elicited little change in [Ca2+]i in the control neuron. In contrast, AMPA elicited a rise in [Ca2+]i in the soma of the postischemic neuron and a smaller increase in its proximal dendrites.Red circles in the 350 nm excitation images indicate representative sites of Ca2+ measurement in cell somata and dendrites for data presented in Figures 7 and 8.
Fig. 7.
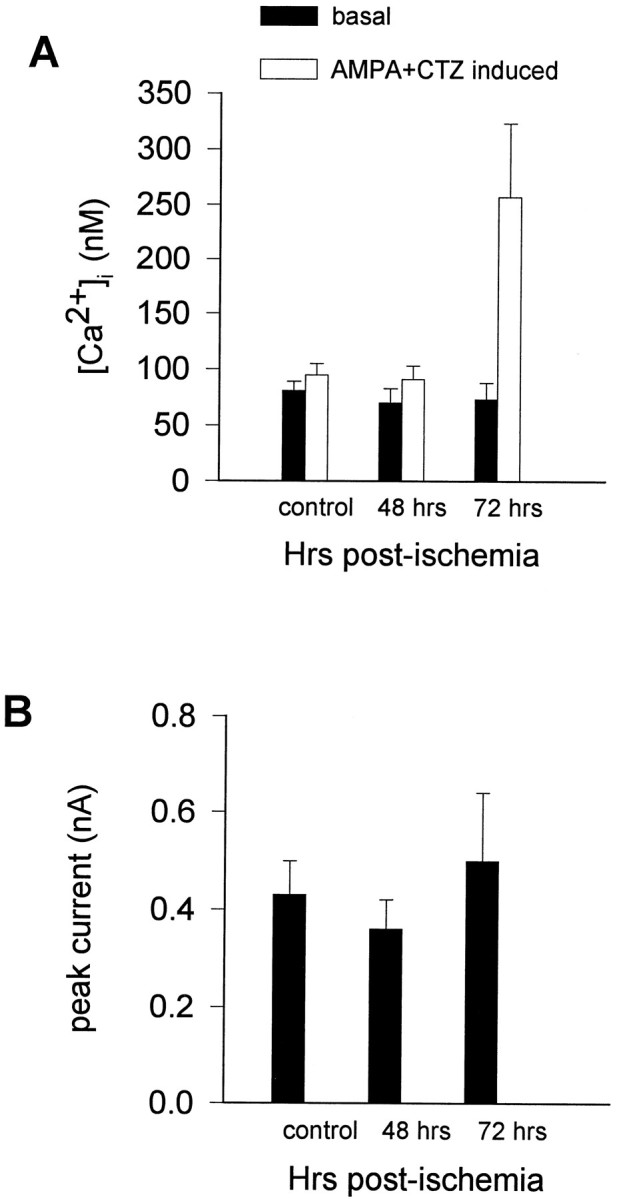
AMPA-elicited rises in [Ca2+]i in CA1 neurons are increased 72 hr after global ischemia. A, Basal and AMPA-elicited rise in [Ca2+]i in the somatic region of individual CA1 pyramidal neurons in hippocampal slices of control animals and ischemic animals 48 and 72 hr after reperfusion. Basal [Ca2+]i did not significantly differ in control and postischemic neurons (open bars). AMPA (30 μm with 10 μm CTZ to reduce desensitization) induced a slight rise in [Ca2+]i in control neurons and neurons from animals 48 hr after ischemia (filled bars). AMPA elicited a large rise in [Ca2+]iin neurons 72 hr after ischemia relative to that elicited in control neurons (p < 0.005) or to that in neurons 48 hr after ischemia (p < 0.025).B, The peak AMPA-induced current did not differ significantly in control versus postischemic neurons at 48 hr or at 72 hr.
After determination of basal [Ca2+]i, AMPA (100 μm) and cyclothiazide [CTZ, 10 μm; to reduce desensitization of AMPA receptors (Partin et al., 1994)] were applied for 30 sec in the bathing solution containing blockers of NMDA, Ca2+ and Na+ channels. In all CA1 neurons examined in both postischemic and control slices, AMPA elicited a slowly rising inward current (Fig. 6). After washout of AMPA for ∼5 min with the blocker solution and then with the blocker solution plus 20 μm 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) to speed recovery, the AMPA-elicited current returned to near the baseline value (Fig. 6). Finally, the slices were perfused with normal saline for at least 15 min, and the somata were voltage-clamped to a potential positive to 0 mV. Only cells that gave a Ca2+ signal well above background in response to this depolarization were included in the study. Only one neuron was examined per brain slice, and a neuron was subjected to only a single sequence of solutions.
In control slices, AMPA elicited a small rise in [Ca2+]i in 11 of 17 neurons (mean rise in [Ca2+]i for the 17 control neurons, 12.9 ± 3.5 nm). In slices from animals 48 hr after ischemia, AMPA elicited a small rise in [Ca2+]i in 8 of 12 CA1 neurons (mean rise in [Ca2+]i for the 12 neurons, 20.6 ± 8.1 nm, not significantly different from control). In slices from animals 72 hr after ischemia, the inward current was associated with a rise in [Ca2+]i that paralleled the slow rise in current in 8 of 9 CA1 neurons (mean rise in [Ca2+]i for the 9 neurons, 185 ± 72 nm) (Figs. 7, 8). The AMPA-elicited rise in [Ca2+]i was significantly greater in neurons 72 hr after ischemia than in control neurons (p < 0.005) or in neurons 48 hr after ischemia (p < 0.025). After washout of AMPA (with 20 μm CNQX in addition to the other blockers), [Ca2+]i, as well as the current, slowly returned toward initial values (Fig. 6).
Fig. 8.
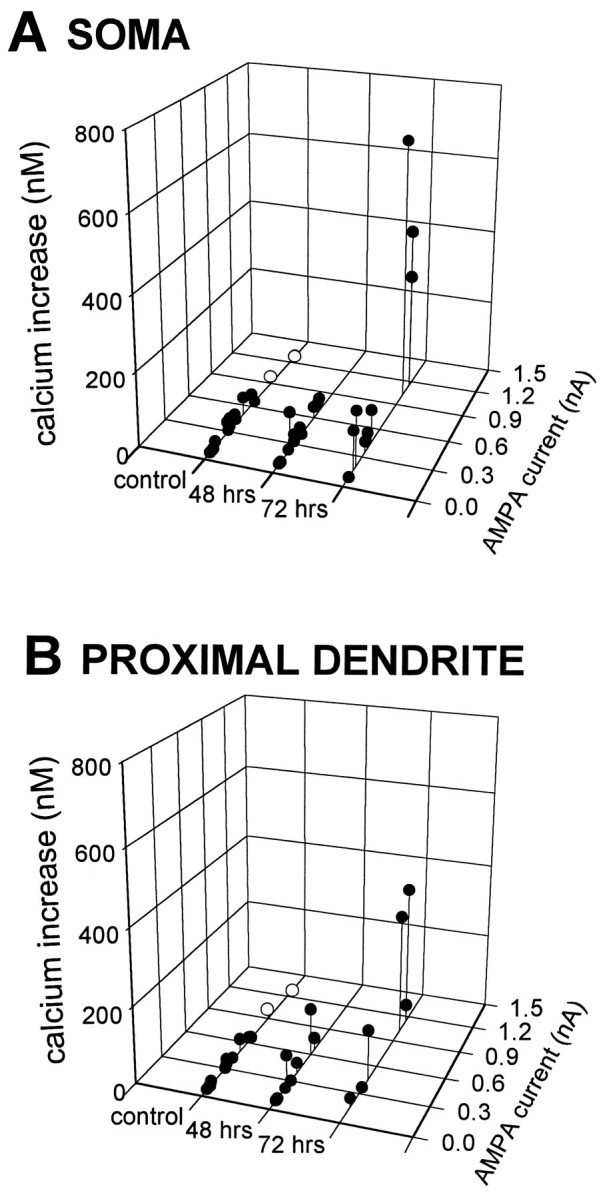
AMPA-elicited rises in [Ca2+]i in individual CA1 pyramidal neurons are increased 72 hr after ischemia. Peak inward currents and rises in [Ca2+]i elicited by AMPA (30 μm with 10 μm CTZ) are indicated for individual pyramidal neurons in slices from control animals and experimental animals 48 and 72 hr after ischemia. [In the two control neurons exhibiting the largest AMPA-elicited currents (open circles), the CTZ concentration was increased to 30 μm to elicit a current comparable with the large currents observed in three neurons 72 hr after ischemia.] AMPA-elicited rises in [Ca2+]i were greater in neurons from animals at 72 hr than in neurons from animals at 48 hr after ischemia or from control animals. A, Data from somata.B, Data from proximal dendrites.
In slices from animals 72 hr after ischemia, AMPA-induced currents tended to be greater than currents in control neurons, although the difference did not reach significance (0.05 < p< 0.1; Fig. 7B). Larger currents tended to be associated with larger changes in [Ca2+]i. To examine changes in [Ca2+]i in control neurons in which AMPA-induced currents were of the same magnitude as in the 72 hr postischemic neurons, we increased the concentration of CTZ from 10 μm to 30 μm for two additional neurons from control slices. Under these conditions, AMPA-elicited currents in the two neurons were in the range of the largest currents observed in cells 72 hr after ischemia (Fig. 8A); however, the change in [Ca2+]i was small in each cell (2 nm and 19 nm) and comparable with that measured in other control neurons at the lower concentration of CTZ. We also compared the AMPA receptor-mediated rise in [Ca2+]i in control neurons with that in 72 hr postischemic neurons after excluding the three postischemic cells with the largest AMPA currents; in this sample the AMPA-elicited rise in [Ca2+]i was still significantly larger in neurons 72 hr after ischemia than in control neurons (p < 0.005). Thus, the increase in the AMPA-elicited rise in [Ca2+]iin CA1 neurons 72 hr after ischemia is not simply caused by a bigger AMPA-elicited current.
A crucial aspect of these observations is the use of blockers of routes of Ca2+ entry into the cells other than Ca2+-permeable AMPA receptors. Inadequate voltage clamp, which might have permitted Ca2+ entry through voltage sensitive mechanisms, should have been the same in control and postischemic neurons. We conclude that the greater AMPA-induced rise in [Ca2+]i in CA1 neurons 72 hr after ischemia is caused by increased Ca2+ permeability of their AMPA receptors.
In addition to having greater AMPA-induced Ca2+signals, the postischemic neurons differed somewhat from control neurons in action potential generation. Recordings with K-acetate-filled electrodes showed that adaptation and broadening of spikes during a prolonged depolarization were reduced in postischemic neurons (J. A. Connor and J. J. Petrozzino, unpublished observations). These properties could not be well evaluated with Cs+-filled electrodes, which rapidly caused spike broadening and reduction of the afterhyperpolarization in control cells.
In slices from animals 72 hr after ischemia, the AMPA-elicited rise in [Ca2+]i was maximal in the cell somata and proximal dendrites (Fig. 8A,B) and decreased gradually with distance from the soma. AMPA-elicited rise in [Ca2+]i was undetectable in the distal dendrites (Fig. 6). In two of seven cells 72 hours after ischemia in which measurements were made in both soma and proximal dendrite, the rise in [Ca2+]i was somewhat greater in the dendrite. That the AMPA-elicited rise in [Ca2+]i was greater in pyramidal cell somata and proximal dendrites than in distal dendrites was somewhat unexpected in that the density of AMPA receptors and excitatory synapses is greater along dendritic processes (Eshhar et al., 1993;Baude et al., 1995; Siegel et al., 1995). This apparent discrepancy may be because of earlier insertion of newly synthesized AMPA receptors lacking GluR2 in the cell somata and proximal dendrites than in the distal dendrites. In addition, the Ca2+ handling capacity may be greater in the distal dendrites.
The slow application of AMPA in these experiments precluded measurement of a lag in the rise in [Ca2+]i that would be associated with accumulation of Ca2+ in the cell.
DISCUSSION
The present study examined AMPA receptor gene expression and AMPA receptor-mediated changes in [Ca2+]iin hippocampal CA1 neurons after transient global ischemia in gerbils. As in the rat, GluR2 mRNA expression was reduced in vulnerable CA1 neurons at times preceding ischemia-induced neurodegeneration. The reduction in GluR2 expression was specific to vulnerable CA1 pyramidal neurons; neurons in CA3 and dentate gyrus, which do not undergo neurodegeneration, exhibited little or no decrease in GluR2 mRNA expression. Expression of the other glutamate receptor mRNAs examined, GluR1 and NR1, was reduced in CA1 only modestly 48 hr after ischemia; the change observed in these mRNAs could reflect onset of neuronal damage, although neither morphological nor physiological deterioration was apparent at this time.
The novel observation of this study is that, 72 hr after induction of global ischemia but before cell death, AMPA elicited a pronounced rise in [Ca2+]i in CA1 pyramidal cells. The rise in [Ca2+]i was markedly greater than the rise seen in neurons from control animals or animals 48 hr after ischemia. Basal [Ca2+]i was unchanged in postischemic neurons, although our criteria for selecting cells in this study may have excluded cells in which [Ca2+]i was increased because of the onset of cell damage. Because Ca2+ entry through other Ca2+-permeable channels was blocked, these data indicate that the increased rise in [Ca2+]i was caused by Ca2+ influx through AMPA receptors, which provides functional evidence for the reduction in GluR2 protein synthesis predicted by the reduction in GluR2 mRNA. Moreover, the reduction in receptor mRNA preceded the increase in Ca2+ influx by at least 24 hr. In support of this mechanism, GluR2 protein expression in rats, detected by immunocytochemistry with a subunit-specific monoclonal antibody, is markedly reduced in CA1 (but not CA3) neurons by 36 hr after ischemia (Pulsinelli et al., 1995). Our findings do not rule out that sequestration and extrusion mechanisms had reduced capacity or that Ca2+-evoked Ca2+ release from cytoplasmic stores was increased after ischemia. However, if Ca2+ influx were the same in control and in postischemic neurons, the minimal levels of AMPA-induced rises in [Ca2+]i in control neurons would require very effective Ca2+regulation in control neurons or a dramatic increase in Ca2+-evoked Ca2+ release in postischemic neurons. We conclude that the observed elevations in [Ca2+]i were, in fact, predominantly if not exclusively caused by Ca2+ influx through Ca2+-permeable AMPA receptors.
A problem inherent in the study of postischemic neurons is that they are likely to die rapidly after expression of Ca2+-permeable AMPA receptors. Because neurons with resting and action potentials that failed to meet our criteria were eliminated from the present study, our sample is likely to be biased toward neurons relatively delayed in degeneration, although nonetheless fated to die (Fig. 1).
From studies of neuronal properties after global ischemia, we can summarize that neuroprotection is provided by non-NMDA glutamate antagonists (Sheardown et al., 1990, 1993; Buchan et al., 1991); that expression of GluR2 receptor mRNA and protein are reduced (Pellegrini-Giampietro et al., 1992b, 1994; Pulsinelli et al., 1995) (present study); and that AMPA receptor permeability to Ca2+ is increased, assessed pharmacologically and by Ca2+ imaging (Tsubokawa et al., 1995) (present study). The reduction in GluR2 expression, which the physiological data indicate is translated into new AMPA receptors lacking the GluR2 subunit, could allow AMPA receptor-mediated entry of Ca2+ into CA1 neurons to reach toxic levels. Neuroprotection by NBQX suggests that this Ca2+influx is necessary, if not sufficient, for the delayed cell death. This mechanism is supported by studies of glutamate-induced cell death in cultured neurons after oxygen and glucose deprivation (Ying et al., 1996). Insulted cultures exhibit increased AMPA- or kainate-induced Ca2+ accumulation sensitive to Joro spider toxin and increased vulnerability to AMPA receptor-mediated excitotoxicity, suggesting increased formation of GluR2-lacking, Ca2+-permeable AMPA receptors.
GluR2 expression, Ca2+ permeability, and cell death
Studies involving patch-clamp recording and reverse transcriptase-PCR demonstrate that abundance of GluR2 mRNA is inversely related to AMPA receptor Ca2+ permeability in a wide range of cell types (for review, see Burnashev, 1996). Excitatory principal neurons of the hippocampus (Bochet et al., 1994; Geiger et al., 1995) and neocortex (Jonas et al., 1994) exhibit low Ca2+ permeability and abundant GluR2 mRNA. AMPA receptors in these cells are primarily GluR1/GluR2 and GluR2/GluR3 assemblies, with homomeric GluR1 accounting for only ∼8% of AMPA receptor complexes (Wenthold et al., 1996). In contrast, GABAergic interneurons and dentate gyrus basket cells (Bochet et al., 1994; Jonas et al., 1994; Geiger et al., 1995; Racca et al., 1996) display higher Ca2+ permeability and less abundant GluR2 mRNA.
Available data indicate that the decrease in GluR2 expression in neurons in which GluR2 is normally abundant, rather than the absence of GluR2, is predictive of neuronal vulnerability. In normal hippocampus, GABAergic interneurons lacking GluR2 are viable and relatively resistant to ischemia-induced damage (Johansen et al., 1984). Moreover, transgenic mice with targeted disruption of the GluR2 gene survive, and their principal neurons, which express GluR2 in wild-type animals, are functional (Jia et al., 1996). Possible explanations for survival of neurons lacking GluR2 are (1) the presence of compensatory mechanisms for Ca2+ buffering and extrusion (for example, enhanced expression of Ca2+-binding proteins (Ribak et al., 1990; Kondo et al., 1997) and (2) a reduction in AMPA receptor currents, caused by reduced expression of GluR1 and GluR3 and/or expression of receptors with altered properties, such as enhanced desensitization (Geiger et al., 1995; Lambolez et al., 1996). Hence, the GluR2 hypothesis would apply primarily to neurons that normally express AMPA receptors with low Ca2+ permeability and that do not cope with significant Ca2+ influx via this class of receptor. In these cells, acute increases in Ca2+ permeability of AMPA receptors could account for cell death. Transgenic mice heterozygous for a Q/R editing-deficient GluR2 allele expressed AMPA receptors with increased Ca2+ permeability, particularly in hippocampal and neocortical principal neurons (Brusa et al., 1995). The mice developed recurrent seizures and died within the first 3 weeks of life, with cell loss in the hippocampus. Unedited GluR2 may have contributed to the formation in these animals of a greater number of Ca2+-permeable AMPA receptors than in the GluR2 knock-out mice.
Mechanisms of altered gene expression
The molecular mechanisms by which global ischemia alters GluR2 expression have not been determined. After ischemia, expression of transcription factors, including products of immediate early genes, stress proteins, and neurotrophic factors are also altered in CA1 (Aoki et al., 1993; Nowak et al., 1993; Takeda et al., 1993; Kindy et al., 1994; Kokaia et al., 1994; Kamme et al., 1995). These proteins are plausible candidates for the downregulation of GluR2 expression by reducing mRNA transcription and/or stability. Because some mRNAs and proteins are upregulated in CA1, the decreases in GluR2 mRNA and protein are not simply a result of a loss of transcriptional or translational capability but seem to result from a regulatory, although maladaptive, change. It is likely that the regulatory mechanism responsible for reduced expression of GluR2 is active in other physiological processes, such as development (Pellegrini-Giampietro et al., 1991).
A number of neuroprotective treatments in animal models of global ischemia prevent downregulation of GluR2 in CA1. Agonists of adenosine A1 receptors and activators of KATP channels are neuroprotective when given just before ischemia and block downregulation of GluR2 (Heurteaux et al., 1995). Aurintricarboxylic acid, a nonspecific endonuclease inhibitor that can prevent apoptosis, is neuroprotective when given intraventricularly at the time of ischemia and blocks downregulation of GluR2 (E. M. Aronica, J. A. Gorter, J. A. Kessler, M. V. L. Bennett, R. S. Zukin, and D. M. Rosenbaum, unpublished observations). Ischemic preconditioning, in which a period of ischemia sublethal for CA1 neurons is given several days before a period of ischemia that would otherwise be lethal to these neurons, also blocks downregulation of GluR2 (Heurteaux et al., 1995). In contrast to the foregoing examples, the AMPA/kainate antagonist NBQX is neuroprotective (Sheardown et al., 1990, 1993; Buchan et al., 1991) but does not block downregulation of GluR2 (Pellegrini-Giampietro et al., 1994), suggesting that NBQX affords neuroprotection by a direct block of GluR2-lacking, Ca2+-permeable AMPA receptors.
Another candidate mechanism for GluR2 regulation that could modify Ca2+ influx is RNA editing. The RNA encoding the GluR2 isoform that limits Ca2+ permeability is edited to result in a Q to R substitution in a channel-lining residue; the unedited subunit forms Ca2+-permeable channels in heteromeric or homomeric configurations (Sommer et al., 1991;Seeburg, 1996). However, editing of GluR2 mRNA at the Q/R site is virtually complete and invariant in hippocampus and neocortex under physiological conditions and unaltered after global ischemia (Kamphuis et al., 1995, Paschen et al., 1996).
Regulation of GluR2 expression in neurodegeneration
In adult rats, kainate-induced status epilepticus causes delayed and specific neuronal cell death; pyramidal neurons of the hippocampal CA3 and hilar interneurons are particularly vulnerable. GluR2 mRNA expression is dramatically reduced in the vulnerable CA3 after status epilepticus (Pollard et al., 1993; Friedman et al., 1994), and the reduction in GluR2 mRNA precedes the onset of neuronal cell death. GluR2 expression is unchanged in other regions resistant to seizure-induced damage, in particular CA1 and dentate gyrus. In pup rats, kainate induces status epilepticus but without the subsequent hippocampal cell death; GluR2 mRNA and protein expression are maintained in pup CA3 (L. F. Friedman, E. F. Sperber, S. L. Moshé, M. V. L. Bennett, and R. S. Zukin, unpublished observations). These and other findings (for review, seePellegrini-Giampietro et al., 1997) suggest that downregulation of GluR2 expression and entry of Ca2+ through AMPA receptors in response to endogenous glutamate may be involved in the pathogenesis of a number of neurodegenerative disorders in addition to delayed neuronal death after ischemia.
Footnotes
This work was supported by National Institutes of Health Grants NS 20752 and NS 31282 (R.S.Z.) and NS 07512 (M.V.L.B.), an Aaron Diamond postdoctoral fellowship award (E.M.A.), and a Human Frontiers Science Program award (J.A.G). M.V.L.B. is the Sylvia and Robert S. Olnick Professor of Neuroscience. We thank S. Rybak for technical assistance and C. Roy for excellent histological preparations.
Correspondence should be addressed to Dr. R. Suzanne Zukin, Department of Neuroscience, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461.
Dr. Gorter’s present address: University of Amsterdam, Department of Experimentele Dierkunde, Kruislaan 320, 1098 SM Amsterdam, The Netherlands.
Dr. Connor’s present address: The Lovelace Institute, 2425 Ridgecrest Drive, Albuquerque, NM 87108.
REFERENCES
- 1.Aoki M, Abe K, Kawagoe J, Nakamura S, Kogure K. Acceleration of HSP70 and HSC70 heat shock gene expression following transient ischemia in the preconditioned gerbil hippocampus. J Cereb Blood Flow Metab. 1993;13:781–888. doi: 10.1038/jcbfm.1993.99. [DOI] [PubMed] [Google Scholar]
- 2.Baude A, Nusser Z, McIlHinney RAJ, Molnar E, Somogyi P. High-resolution immunogold localization of AMPA type glutamate receptor subunits at synaptic and non-synaptic sites in rat hippocampus. Neuroscience. 1995;69:1031–1055. doi: 10.1016/0306-4522(95)00350-r. [DOI] [PubMed] [Google Scholar]
- 3.Blaschke M, Keller BU, Rivosecchi R, Hollmann M, Heinemann S, Konnerth A. A single amino acid determines the subunit-specific spider toxin block of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate/kainate receptor channels. Proc Natl Acad Sci USA. 1993;90:6528–6532. doi: 10.1073/pnas.90.14.6528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bochet P, Audinat E, Lambolez B, Crepel F, Rossier J, Iino M, Tsuzuki K, Ozawa S. Subunit composition at the single-cell level explains functional properties of a glutamate-gated channel. Neuron. 1994;12:383–388. doi: 10.1016/0896-6273(94)90279-8. [DOI] [PubMed] [Google Scholar]
- 5.Bredesen DE. Neural apoptosis. Ann Neurol. 1995;38:839–851. doi: 10.1002/ana.410380604. [DOI] [PubMed] [Google Scholar]
- 6.Brusa R, Zimmermann F, Koh DS, Feldmeyer D, Gass P, Seeburg PH, Sprengel R. Early onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science. 1995;270:1677–1680. doi: 10.1126/science.270.5242.1677. [DOI] [PubMed] [Google Scholar]
- 7.Buchan AM, Li H, Cho S, Pulsinelli WA. Blockade of the AMPA receptor prevents CA1 hippocampal injury following severe but transient forebrain ischemia in adult rats. Neurosci Lett. 1991;132:255–258. doi: 10.1016/0304-3940(91)90314-j. [DOI] [PubMed] [Google Scholar]
- 8.Burnashev N. Calcium permeability of glutamate-gated channels in the central nervous system. Curr Opin Neurobiol. 1996;6:311–317. doi: 10.1016/s0959-4388(96)80113-9. [DOI] [PubMed] [Google Scholar]
- 9.Choi DW. Cerebral hypoxia: some new approaches and unanswered questions. J Neurosci. 1990;10:2493–2501. doi: 10.1523/JNEUROSCI.10-08-02493.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi DW. Calcium: still center-stage in hypoxic–ischemic neuronal death. Trends Neurosci. 1995;18:58–60. [PubMed] [Google Scholar]
- 11.Eshhar N, Petralia RS, Winters CA, Niedzielski AS, Wenthold RJ. The segregation and expression of glutamate receptor subunits in cultured hippocampal neurons. Neuroscience. 1993;57:943–964. doi: 10.1016/0306-4522(93)90040-m. [DOI] [PubMed] [Google Scholar]
- 12.Friedman LK, Pellegrini-Giampietro DE, Sperber EF, Bennett MVL, Moshe SL, Zukin RS. Kainate-induced status epilepticus alters glutamate and GABAA receptor gene expression in adult rat hippocampus: an in situ hybridization study. J Neurosci. 1994;14:2697–2707. doi: 10.1523/JNEUROSCI.14-05-02697.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geiger JR, Melcher T, Koh DS, Sakmann B, Seeburg PH, Jonas P, Monyer H. Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron. 1995;15:293–204. doi: 10.1016/0896-6273(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 14.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 15.Herlitze S, Raditsch M, Ruppersberg JP, Jahn W, Monyer H, Schoepfer R, Witzemann V. Argiotoxin detects molecular differences in AMPA receptor channels. Neuron. 1993;10:1131–1140. doi: 10.1016/0896-6273(93)90061-u. [DOI] [PubMed] [Google Scholar]
- 16.Heurteaux C, Lauritzen I, Widmann C, Lazdunski M. Essential role of adenosine, adenosine A1 receptors, and ATP-sensitive K+ channels in cerebral ischemic preconditioning. Proc Natl Acad Sci USA. 1995;92:4666–4670. doi: 10.1073/pnas.92.10.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA-gated glutamate receptor channels depends on subunit composition. Science. 1991;252:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- 18.Hsu M, Buzsaki G. Vulnerability of mossy fiber targets in the rat hippocampus to forebrain ischemia. J Neurosci. 1993;13:3964–3979. doi: 10.1523/JNEUROSCI.13-09-03964.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hume R, Dingledine R, Heinemann S. Identification of a site in glutamate receptor subunits that controls calcium permeability. Science. 1991;253:1028–1031. doi: 10.1126/science.1653450. [DOI] [PubMed] [Google Scholar]
- 20.Jia ZP, Agopyan N, Miu P, Xiong ZG, Henderson J, Gerlai R, Tavrna FA, Velumian A, MacDonald J, Carlen P, Abramownewerly W, Roder J. Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron. 1996;17:945–956. doi: 10.1016/s0896-6273(00)80225-1. [DOI] [PubMed] [Google Scholar]
- 21.Johansen FF, Jorgensen MB, Ekstrom von Lubitz DK, Diemer NH. Selective dendrite damage in hippocampal CA1 stratum radiatum with unchanged axon ultrastructure and glutamate uptake after transient cerebral ischaemia in the rat. Brain Res. 1984;291:373–377. doi: 10.1016/0006-8993(84)91272-1. [DOI] [PubMed] [Google Scholar]
- 22.Jonas P, Racca C, Sakmann B, Seeburg PH, Monyer H. Differences in Ca2+-permeability of AMPA-type glutamate receptor channels in neocortical neurons caused by differential GluR-B subunit expression. Neuron. 1994;12:1281–1289. doi: 10.1016/0896-6273(94)90444-8. [DOI] [PubMed] [Google Scholar]
- 23.Kamme F, Campbell K, Wieloch T. Biphasic expression of the fos and jun families of transcription factors following transient forebrain ischaemia in the rat. Effect of hypothermia. Eur J Neurosci. 1995;7:2007–2016. doi: 10.1111/j.1460-9568.1995.tb00623.x. [DOI] [PubMed] [Google Scholar]
- 24.Kamphuis W, de Leeuw FE, Lopes da Silva FH. Ischaemia does not alter the editing status at the Q/R site of glutamate receptor-A, -B, -5 and -6 subunit mRNA. NeuroReport. 1995;6:1133–1136. doi: 10.1097/00001756-199505300-00015. [DOI] [PubMed] [Google Scholar]
- 25.Kindy MS, Hu Y, Dempsey RJ. Blockade of ornithine decarboxylase enzyme protects against ischemic brain damage. J Cereb Blood Flow Metab. 1994;14:1040–1045. doi: 10.1038/jcbfm.1994.136. [DOI] [PubMed] [Google Scholar]
- 26.Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- 27.Kirino T, Sano K. Selective vulnerability in the gerbil hippocampus following transient ischemia. Acta Neuropathol (Berl) 1984;62:201–208. doi: 10.1007/BF00691853. [DOI] [PubMed] [Google Scholar]
- 28.Kokaia Z, Metsis M, Kokaia M, Bengzon J, Elmer E, Smith ML, Timmusk T, Siesjo BK, Persson H, Lindvall O. Brain insults in rats induce increased expression of the BDNF gene through differential use of multiple promoters. Eur J Neurosci. 1994;6:587–596. doi: 10.1111/j.1460-9568.1994.tb00303.x. [DOI] [PubMed] [Google Scholar]
- 29.Kondo M, Sumino R, Okado H. Combinations of AMPA receptor subunit expression in individual cortical neurons correlate with expression of specific calcium-binding proteins. J Neurosci. 1997;17:1570–1581. doi: 10.1523/JNEUROSCI.17-05-01570.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kutsuwada T, Kashiwabuchi N, Mori H, Sakimura K, Kushiya E, Araki K, Meguro H, Masaki H, Kumanishi T, Arakawa M. Molecular diversity of the NMDA receptor channel. Nature. 1992;358:36–41. doi: 10.1038/358036a0. [DOI] [PubMed] [Google Scholar]
- 31.Lambolez B, Ropert N, Perrais D, Rossier J, Hestrin S. Correlation between kinetics and RNA splicing of α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors in neocortical neurons. Proc Natl Acad Sci USA. 1996;93:1797–1802. doi: 10.1073/pnas.93.5.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meldrum BS. Excitatory amino acid receptors and their role in epilepsy and cerebral ischemia. Ann NY Acad Sci. 1995;757:492–505. doi: 10.1111/j.1749-6632.1995.tb17509.x. [DOI] [PubMed] [Google Scholar]
- 33.Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, Burnashev N, Sakmann B, Seeburg PH. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science. 1992;256:1217–1221. doi: 10.1126/science.256.5060.1217. [DOI] [PubMed] [Google Scholar]
- 34.Muller W, Connor JA. Cholinergic input uncouples Ca2+ changes from K+ conductance activation and amplifies intradendritic Ca2+ changes in hippocampal neurons. Neuron. 1991;6:901–905. doi: 10.1016/0896-6273(91)90230-w. [DOI] [PubMed] [Google Scholar]
- 35.Nowak TS, Jr, Osborne OC, Suga S. Stress protein and proto-oncogene expression as indicators of neuronal pathophysiology after ischemia. Prog Brain Res. 1993;96:195–208. doi: 10.1016/s0079-6123(08)63267-7. [DOI] [PubMed] [Google Scholar]
- 36.Partin KM, Patneau DK, Mayer ML. Cyclothiazide differentially modulates desensitization of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor splice variants. Mol Pharmacol. 1994;46:129–138. [PubMed] [Google Scholar]
- 37.Paschen W, Schmitt J, Uto A. RNA editing of glutamate receptor subunits GluR2, GluR5 and GluR6 in transient cerebral ischemia in the rat. J Cereb Blood Flow Metab. 1996;16:548–556. doi: 10.1097/00004647-199607000-00004. [DOI] [PubMed] [Google Scholar]
- 38.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; New York: 1991. [DOI] [PubMed] [Google Scholar]
- 39.Pellegrini-Giampietro DE, Bennett MVL, Zukin RS. Differential expression of three glutamate receptor genes in developing rat brain: an in situ hybridization study. Proc Natl Acad Sci USA. 1991;88:4157–4161. doi: 10.1073/pnas.88.10.4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pellegrini-Giampietro DE, Zukin RS, Bennett MVL, Cho S, Pulsinelli WA. Switch in glutamate receptor subunit gene expression in CA1 subfield of hippocampus following global ischemia in rats. Proc Natl Acad Sci USA. 1992a;89:10499–10503. doi: 10.1073/pnas.89.21.10499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pellegrini-Giampietro DE, Bennett MVL, Zukin RS. Are Ca2+-permeable AMPA/kainate receptors more abundant in immature brain? Neurosci Lett. 1992b;144:65–69. doi: 10.1016/0304-3940(92)90717-l. [DOI] [PubMed] [Google Scholar]
- 42.Pellegrini-Giampietro DE, Pulsinelli WA, Zukin RS. NMDA and non-NMDA receptor gene expression following global brain ischemia in rats: effect of NMDA and non-NMDA receptor antagonists. J Neurochem. 1994;62:1067–1073. doi: 10.1046/j.1471-4159.1994.62031067.x. [DOI] [PubMed] [Google Scholar]
- 43.Pellegrini-Giampietro DE, Gorter JA, Bennett MVL, Zukin RS (1997) The GluR2 (GluR-B) hypothesis: Ca2+-permeable AMPA receptors in neurological disorders. Trends Neurosci, in press. [DOI] [PubMed]
- 44.Petrozzino JJ, Pozzo Miller LD, Connor JA. Micromolar Ca2+ transients in dendritic spines of hippocampal pyramidal neurons in brain slice. Neuron. 1995;14:1223–1231. doi: 10.1016/0896-6273(95)90269-4. [DOI] [PubMed] [Google Scholar]
- 45.Pollard H, Héron A, Moreau J, Ben-Ari Y, Khrestchatisky M. Alterations of the GluR-B AMPA receptor subunit flip/flop expression in kainate-induced epilepsy and ischemia. Neuroscience. 1993;57:545–554. doi: 10.1016/0306-4522(93)90004-y. [DOI] [PubMed] [Google Scholar]
- 46.Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- 47.Pulsinelli WA, Friedman LK, Nowak T, Pellegrini D, Morrison JM, Zukin RS. GluR2 receptor protein expression is reduced in hippocampal CA1 after global ischemia. Soc Neurosci Abstr. 1995;21:837. [Google Scholar]
- 48.Puttfarcken PS, Getz RL, Coyle JT. Kainic acid-induced lipid peroxidation: protection with butylated hydroxytoluene and U78517F in primary cultures of cerebellar granule cells. Brain Res. 1993;624:223–232. doi: 10.1016/0006-8993(93)90081-w. [DOI] [PubMed] [Google Scholar]
- 49.Racca C, Catania MV, Monyer H, Sakmann B. Expression of AMPA-glutamate receptor B subunit in rat hippocampal GABAergic neurons. Eur J Neurosci. 1996;8:1580–1590. doi: 10.1111/j.1460-9568.1996.tb01303.x. [DOI] [PubMed] [Google Scholar]
- 50.Ribak CE, Nitsch R, Seress L. Proportion of parvalbumin-positive basket cells in the GABAergic innervation of pyramidal and granule cells of the rat hippocampal formation. J Comp Neurol. 1990;300:449–461. doi: 10.1002/cne.903000402. [DOI] [PubMed] [Google Scholar]
- 51.Rothman SM, Olney JW. Glutamate and the pathophysiology of hypoxic–ischemic brain damage. Ann Neurol. 1986;19:105–111. doi: 10.1002/ana.410190202. [DOI] [PubMed] [Google Scholar]
- 52.Schmidt-Kastner R, Freund TF. Selective vulnerability of the hippocampus in brain ischemia. Neuroscience. 1991;40:599–636. doi: 10.1016/0306-4522(91)90001-5. [DOI] [PubMed] [Google Scholar]
- 53.Seeburg PH. The role of RNA editing in controlling glutamate receptor channel properties. J Neurochem. 1996;66:1–5. doi: 10.1046/j.1471-4159.1996.66010001.x. [DOI] [PubMed] [Google Scholar]
- 54.Sheardown MJ, Nielsen EO, Hansen AJ, Jacobsen P, Honore T. 2,3-Dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline: a neuroprotectant for cerebral ischemia. Science. 1990;247:571–574. doi: 10.1126/science.2154034. [DOI] [PubMed] [Google Scholar]
- 55.Sheardown MJ, Suzdak PD, Nordholm L. AMPA, but not NMDA, receptor antagonism is neuroprotective in gerbil global ischaemia, even when delayed 24 hr. Eur J Pharmacol. 1993;236:347–353. doi: 10.1016/0014-2999(93)90470-3. [DOI] [PubMed] [Google Scholar]
- 56.Siegel SJ, Janssen WG, Tullai JW, Rogers SW, Moran T, Heinemann SF, Morrison JH. Distribution of the excitatory amino acid receptor subunits GluR2(4) in monkey hippocampus and colocalization with subunits GluR5–7 and NMDAR1. J Neurosci. 1995;15:2707–2719. doi: 10.1523/JNEUROSCI.15-04-02707.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sommer B, Keinänen K, Verdoorn TA, Wisden W, Burnashev N, Herb A, Köhler M, Takagi T, Sakmann B, Seeburg PH. Flip and flop: a cell-specific functional switch in glutamate-operated channels of the CNS. Science. 1990;249:1580–1585. doi: 10.1126/science.1699275. [DOI] [PubMed] [Google Scholar]
- 58.Sommer B, Köhler M, Sprengel R, Seeburg PH. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell. 1991;67:11–19. doi: 10.1016/0092-8674(91)90568-j. [DOI] [PubMed] [Google Scholar]
- 59.Takeda A, Onodera H, Sugimoto A, Kogure K, Obinata M, Shibahara S. Coordinated expression of messenger RNAs for nerve growth factor, brain-derived neurotrophic factor and neurotrophin-3 in the rat hippocampus following transient forebrain ischemia. Neuroscience. 1993;55:23–31. doi: 10.1016/0306-4522(93)90451-k. [DOI] [PubMed] [Google Scholar]
- 60.Tsubokawa H, Oguro K, Masuzawa T, Nakaima T, Kawai N. Effects of a spider toxin and its analogue on glutamate-activated currents in the hippocampal CA1 neuron after ischemia. J Neurophysiol. 1995;74:218–225. doi: 10.1152/jn.1995.74.1.218. [DOI] [PubMed] [Google Scholar]
- 61.Wenthold RJ, Petralia RS, Blahos J, II, Niedzielski AS. Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J Neurosci. 1996;16:1982–1989. doi: 10.1523/JNEUROSCI.16-06-01982.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ying HS, Weishaupt J, Grabb M, Canzoniero LMT, Sensi SL, Monyer H, Choi DW. AMPA receptor expression in cultured rat hippocampal neurons following sublethal oxygen-glucose deprivation. Soc Neurosci Abstr. 1996;22:597. doi: 10.1523/JNEUROSCI.17-24-09536.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]