

Abstract
Background
The honeybee (Apis mellifera) has been used with great success in a variety of behavioral studies. The lack of genomic tools in this species has, however, hampered efforts to provide genome-based explanations for behavioral data. We have combined the power of DNA arrays and the availability of distinct behavioral stages in honeybees to explore the dynamics of gene expression during adult development in this insect. In addition, we used caffeine treatment, a procedure that accelerates learning abilities in honeybees, to examine changes in gene expression underlying drug-induced behavioral modifications.
Results
Spotted microarrays containing several thousand cDNAs were interrogated with RNAs extracted from newly emerged worker bees, experienced foragers and caffeine-treated bees. Thirty-six differentially expressed cDNAs were verified by northern blot hybridization and characterized in silico by sequencing and database searches. Experienced foragers overexpressed royal jelly proteins, a putative imaginal disc growth factor, a transcriptional regulator (Stck) and several enzymes, including α-glucosidases, aminopeptidases and glucose dehydrogenase. Naive workers showed increased expression of members of the SPARC and lectin families, heat-shock cognate proteins and several proteins related to RNA translation and mitochondrial function. A number of novel genes overexpressed in both naive and experienced bees, and genes induced by caffeine, have also been identified.
Conclusions
We have shown the usefulness of this transcriptome-based approach for gene discovery, in particular in the context of the efficacy of drug treatment, in a model organism in which routine genetic techniques cannot be applied easily.
Background
Recent progress in genome sequencing has significantly widened a division within the major model organisms, some of which are genetically tractable whereas some of which are not but are valuable in other spheres of biology. In particular, the availability of the complete genome sequence in several species has allowed researchers to monitor gene transcription on a global scale for the first time, making possible an impressive leap from the study of individual genes or proteins to an integrated understanding of how gene networks enable complex functions to be carried out [1]. The promise of such methods for systems biologists, behavioralists and neuroethologists is enormous, as they help to integrate the different biological levels from genotype to phenotype.
The honeybee (Apis mellifera) has an exceptional track record as a behavioral model. It has contributed greatly to our understanding of insect navigation, social behavior and learning under natural conditions [2,3,4], and is now poised to become a valuable model in drug and pesticide evaluation because of the ease with which behavioral changes in this species can be monitored. In contrast, the recombinant-DNA-based genetic information available for the honeybee and other Hymenoptera is rudimentary and, consequently, little is known about the molecular and cellular mechanisms underlying the functioning of the brain and other vital systems in this social insect.
We have examined the practicability of using spotted microarrays representing several thousand cDNAs amplified from a standard unannotated library to generate a catalog of genes differentially expressed during behavioral development in the honeybee. The adult life of the honeybee worker is divided into two easily accessible but behaviorally quite different stages, which offer a unique opportunity to compare molecular processes in 'naive' individuals with those in experienced foragers. Young adult bees (nurses) perform 'simple' in-hive duties and largely depend on olfactory stimuli and colony context, whereas older bees (> 2-3 weeks) are engaged in complex, far-ranging foraging tasks and have strongly developed visual and olfactory perception. In contrast to experienced foragers, younger workers show no daily rhythms and, somewhat surprisingly, cannot be trained in laboratory olfactory tasks until they are 6-7 days old [5,6,7]. Interestingly, both colony status and artificial treatments can either accelerate or delay the rate of adult development [3,5,7,8].
The arrays were co-hybridized with RNAs extracted from naive, newborn individuals, experienced foragers, and drug-treated and untreated 3-day-old bees. We used caffeine treatment following our finding that it accelerates the development of associative olfactory learning in newborn workers (R.M., unpublished work). The candidate clones revealed by microarray analysis were verified by northern blot hybridization, sequenced and scored against the protein and DNA databases. This approach yielded a set of differentially expressed transcripts encoding conserved proteins with putative functions. Additionally, several genes of unknown function that may be involved in the control and execution of adult maturation were also identified. Our study shows that an analysis of a partial transcriptome can be successful in gaining insights into the dynamics of gene expression in a valuable model organism where conventional genetics is difficult to implement.
Results
Identifying changes in gene expression using cDNA microarrays
The expression pattern of approximately 2,500 unique honeybee transcripts, equivalent to approximately 20% of the estimated number of genes in the honeybee genome, was analyzed using cDNA probes reverse transcribed from mRNAs extracted from naive and experienced honeybees. Probes were prepared from the heads (minus appendages) and abdomens. Head RNA represents transcripts from the brain, cephalic glands and mandibular parts, whereas abdomen RNA comes largely from the digestive system, undeveloped reproductive system, the lymph-propelling organ, the sting and certain glands. Data from independent co-hybridizations were analyzed to identify spots (array elements) whose gene-expression ratio differed between the two conditions. Selected microarray data are shown in Table 1. By comparing transcript expression levels in the heads of naive and experienced individuals we selected 24 clones showing a more than twofold difference. An additional 11 clones were selected by comparing gene expression in the abdomens (Table 1).
Table 1.
Identity of genes differentially expressed between naive and experienced honeybees and in caffeine-treated honeybees
| Array data | ||||||
| bEST number | Gene | Head (foragers vs naive) | Abdomen (foragers vs naive) | Head (caffeine treated vs control) | Predicted function | Similarity to closest relative (%) |
| 5 | Hsc70 | -1.6 | +1.3 | +12.6 | Chaperone, heat shock response, neurotransmitter release | 95% |
| 22 | α-Gluc-1 | +2.5 | -1.1 | +2.2 | α-Glucosidase | 100% |
| 28 | Unknown | -3.2 | +2.3 | -1.5 | Unknown | N/A |
| 30 | Hsp20 | +1.3 | +2.1 | -1.1 | Chaperone | 65% |
| 31 | Hsp83 | -1.6 | +2.4 | +1.2 | Chaperone, heat shock response, protein folding | 92% |
| 36 | SPARC | -8.2 | -3.5 | +1.8 | Cell adhesion, calcium binding | 61% |
| 54 | GPDH 2 | -3.1 | +1.3 | -1.1 | Mitochondrial glycerol-3-phosphate dehydrogenase | 69% |
| 56 | Cox10 | -4.0 | -1.1 | -1.2 | Protoheme IX farnesyltransferase, cox assembly | 76% |
| 57 | Stck | -2.6 | -1.5 | +3.9 | LIM-domain protein, transcriptional regulation | 88% |
| 61 | Unknown | -5.2 | -1.3 | +1.4 | PDZ-domain protein, signal transduction | 50% |
| 82 | Aminopeptidase-1 | +1.6 | +1.0 | -1.2 | Membrane alanyl aminopeptidase | 51% |
| 92 | Trf | +1.2 | +3.0 | +2.8 | Iron transport, iron homeostasis, defense response | 63% |
| 97 | CG16857 | -3.3 | +2.1 | +1.5 | Ig-domain protein, cell adhesion | 63% |
| 99 | RpS19 | -4.7 | -1.7 | -1.6 | Structural protein of ribosome, protein biosynthesis | 83% |
| 102 | MRJP2 | +12.1 | +1.7 | -1.3 | Royal-jelly protein | 100% |
| 105 | Gld | +3.0 | -1.0 | +2.0 | Glucose dehydrogenase | 100% |
| 108 | PPIase | -4.0 | -1.1 | -1.5 | Peptidylprolyl isomerase | 92% |
| 109 | ATP synthase β | -5.9 | -1.4 | +1.6 | Hydrogen-transporting two-sector ATPase | 95% |
| 112 | Unknown | +5.5 | -1.1 | -1.3 | Unknown | N/A |
| 121 | IDGF | -1.4 | +1.9 | +2.4 | Imaginal disc growth factor, not chitinase | 54% |
| 122 | α-Glucosidase-2 | -2.3 | +9.8 | +1.2 | α-Glucosidase | 60% |
| 123 | Unknown | -3.3 | -1.1 | -1.7 | Unknown | N/A |
| 124 | Peritrophin | -5.6 | -4.3 | +1.4 | Structural protein of peritrophic membrane, chitin binding | 78% |
| 125 | Hymenoptaecin | +1.5 | +8.0 | +1.7 | Antibacterial protein, defense response | 100% |
| 127 | CG6112 | -7.1 | -3.8 | -1.3 | Ligand-gated ion channel subunit | 98% |
| 128 | Aminopeptidase-2 | -1.4 | -1.2 | +1.5 | Membrane alanyl aminopeptidase | 49% |
| 129 | Unknown | -1.9 | +1.2 | +5.6 | Unknown | N/A |
| 130 | Unknown | -7.2 | -1.7 | -1.1 | Unknown | N/A |
| 131 | Lectin | -16.0 | -5.9 | -1.3 | Ligand binding or carrier | 82% |
| 132 | CoxI | -4.9 | +1.0 | +1.1 | Respiratory-chain enzyme | 100% |
| 133 | Scp1 | -2.4 | -1.2 | -1.7 | Small chemosensory protein | 74% |
| 134 | ATPsynthase F0-6 | -14.1 | -1.4 | -1.0 | Proton pump, ATP synthesis | 100% |
| 136 | Unknown | N/A | +1.2 | +4.3 | Unknown | N/A |
| 138 | Gs2 | N/A | +1.8 | +3.5 | Glutamine synthase | 77% |
| 143 | Unknown | N/A | -1.0 | +3.4 | Unknown | N/A |
| 147 | CG5586 | N/A | -1.3 | -3.2 | WD40 and SOCS domain-containing protein | 38% |
| 148 | Aminotransferase | -2.6 | -1.4 | -3.5 | Ornithine-oxo-acid aminotransferase | 73% |
bEST number, cDNA number; gene, gene identifier; array data, fold change calculated from array hybridization data using pairwise comparisons for heads, abdomens and caffeine-treated bees; predicted function, putative function inferred from sequence similarity; Percentage similarity to the closest relative in GenBank. Accession numbers of bESTs reported in this paper: 5, BI946410; 22, BI946425; 28, BI946431; 30, BI946433; 31, BI946435; 36, BI946440; 54, BI946454; 56, BI946456; 57, BI946458; 61, BI946461; 82, BI946480; 92, BI946487; 97, BI946490; 99, BI946493; mrjp2 (102), af000632; gld (105), ab022907; 108, BI946499; 109, BI946500; 112, BI946503; 121, BI946511; 122, BI946512; 123, BI946513; 124, BI946514; hymenoptaecin (125), amu15956; 127, BI946517; 128, BI946519; 129, BI946520; 130, BI946522; 131, BI946524; 132, BI946525; 133, BI946526; 134, BI946527; 136, BI946528; 138, BI946532; 143, BI946537; 147, BI946542; 148, BI946543.
Whereas honeybee behavioral development is largely controlled by genetic factors, both colony status and artificial treatments have been shown to either accelerate or delay the maturation of adult workers [7,8]. We therefore examined gene expression in caffeine-treated bees following our finding that this drug accelerates the development of memory of olfactory associative learning in young workers (R.M., unpublished work). In this experiment we used cDNA probes reverse transcribed from mRNAs extracted only from the heads of caffeine-treated and untreated 3-day-old bees. We selected 11 clones showing a greater than twofold change in the level of expression in treated versus untreated individuals (Table 1).
Some of the differentially expressed cDNAs from the head and abdomen detected in these two experiments were found to represent the same transcripts; therefore, only 37 unique genes were revealed by this approach, both upregulated and downregulated, and are listed in Table 1.
Verification of microarray data by northern blot hybridization
Although spotted cDNA arrays can rapidly generate massive datasets, they are prone to methodological problems, such as the variability in absolute hybridization intensity owing to differences in the amount of DNA deposited on the chip for various genes, the existence of alternatively spliced variants and repetitive elements, and the lack of linearity between the quantified signals and the expression levels of the corresponding genes [9,10]. To ensure that the candidate transcripts identified by the microarray approach were indeed differentially expressed in naive and experienced bees, we conducted northern blot verification for 36 selected array data points. Figure 1 illustrates our array compared with northern blot analyses. We found good correlation between the array and northern blot data, and the differential expression patterns detected by microarray were reproduced in the northern blot analyses of the selected transcripts. One notable exception is clone bEST129, which microarrays showed to be upregulated in heads of caffeine-treated bees, but northern blots showed to be downregulated (Figure 1).
Figure 1.

Differential expression of 36 ESTs (indicated by red boxes) detected by microarrays and confirmed by northern blotting (lower panels for each EST). The three columns correspond to transcripts identified as differentially expressed in the heads and abdomens of newly born bees and experienced foragers, and in the heads of 3-day-old bees after caffeine treatment.
To determine whether a change in the level of a particular transcript in one compartment correlates with a similar change in other organs or tissues, we examined the expression patterns of 15 confirmed candidates in all three major body compartments (head, thorax and abdomen). By adding mRNA from the thorax, we expanded the range of tissues examined to include the wing and leg muscles. As shown in Figure 2, this experiment gave additional information on the spatio-temporal expression of our candidate genes. Some transcripts that were upregulated in one compartment showed decreased expression in another. For example, genes for the chaperonin Hsp20 and the transcriptional regulator Stck were both upregulated in the head but downregulated in the abdomen of foragers. The relative levels of mRNAs in different compartments were uneven, indicating an additional level of complexity of the regulatory mechanisms controlling the expression of these genes in time and space. Transcripts associated with 'functional' glands, including those for the royal-jelly proteins (RJPs) and certain enzymes, were restricted to the head, whereas α-glucosidase-2 was predominantly expressed in the abdomen. However, despite the fact that the cDNA library used in this study had been prepared from brain mRNAs, we found that the majority of the transcripts were expressed in other body compartments. This is consistent with previous studies in other species showing that the majority of genes are expressed with little spatial specificity [11].
Figure 2.
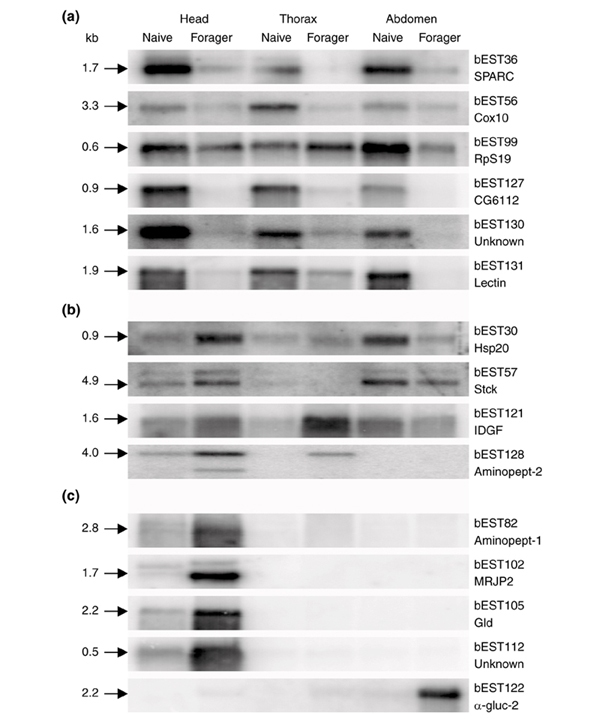
Northern blot hybridization showing the pattern of expression of selected genes in the head, thorax and abdomen. (a) Genes downregulated in foragers; (b) genes upregulated in foragers; (c) genes predominantly expressed in only one compartment.
In silico characterization of candidate clones
In-depth analysis of the 37 clones listed in Table 1 that were detected as differentially expressed by microarray analysis revealed both evolutionarily conserved and hitherto unknown genes. Details of the genes, including putative identifications and functional attributes, are shown in Table 1. The clones fall into three main groups. The first comprises genes encoding proteins whose functional attributes are based on large data sources from many organisms: ribosomal proteins (bEST: 99), metabolic enzymes (bEST: 22, 105, 138), developmentally regulated proteins and mitochondrial proteins (bEST: 54, 56), all sufficiently conserved in sequence and domain composition throughout many evolutionary lineages to be predicted with some confidence. This category also includes heat-shock cognate proteins (bEST: 5, 30, 31), which belong to a large, highly conserved family that includes molecular chaperones, stress proteins, signal transducers and developmental regulators [12]. The second group comprises transcripts encoding proteins that have at least one recognizable motif or domain, but whose functional attributes are generally unclear. For example, cell adhesion molecules are particularly heterogeneous between different organisms in terms of their domain combinations, making in silico comparisons particularly problematical (bEST: 97). The third group comprises 'orphan' genes that show no significant similarity to sequences deposited in GenBank and are likely to represent either honeybee-specific genes or genes that have not yet been sequenced in other species. Alternatively, these sequences might represent genes that encode highly divergent proteins that cannot be assigned to a known functional class without a three-dimensional structure [13]. One example of a novel gene that is significantly downregulated in experienced foragers, especially in the abdomen, is clone bEST123.
Discussion
Our rationale for comparing gene expression in naive, newly born bees and experienced foragers was based on previous findings suggesting that adult development in this insect is associated not only with behavioral maturation and division of labor, but also with significant molecular and cellular changes in the brain and other tissues. These changes include alterations in cholinergic and glutamatergic systems [14,15,16], increases in the levels of hormones and biogenic amines [17] and volumetric changes in some brain neuropils [18]. By comparing patterns of gene expression in naive versus experienced bees, we expected to identify transcripts that might control maturation of the brain and the innate immune system, activation of metabolic pathways, learning ability and other physiological functions. Furthermore, we were curious to determine if caffeine-induced improvements in memory consolidation and associative learning lead to changes in gene expression that can be analyzed by microarrays.
In this study we have not detected any differences in the level of expression of genes involved specifically in neuro-transmission, most probably as the result of experimental restrictions imposed by the use of heterogeneous body parts. It has been acknowledged that the marked cellular heterogeneity and low expression levels of many genes in the nervous system pose a serious challenge for microarray technologies [9,19]. One recent study of two different mouse strains has shown that of more than 7,000 expressed mouse genes detected on the array, only 24 were identified that were differentially expressed in all six brain regions in the two different strains [20]. In spite of its relatively small size (around 1 mm3), the honeybee brain contains 1 million neurons organized in functionally specialized and anatomically divergent neuropils [21]. Differences in the levels of mRNAs expressed in such a small amount of tissue are probably at the detection limit of current microarray experiments. Judging from their level of expression, the genes revealed in our present study are either moderately or highly expressed (Figure 2), and might therefore be more suitable for parallel comparisons than less abundant transcripts. Nonetheless, the identification of a number of differentially expressed transcripts in the honeybee encoding molecules from functionally defined gene-activity cascades, including some in the nervous system, underscores the utility of this approach as a screen for detecting changes that occur during complex biological processes.
As expected, a number of upregulated genes encoding metabolic enzymes and proteins expressed in 'functional' glands have been detected. These include α-glucosidase-1, glucose dehydrogenase and RJPs. The RJPs are related at the sequence level to the yellow protein family in Drosophila, but have diverse, poorly understood functions in the honeybee [22]. They are highly abundant in the hypopharyngeal gland of worker bees and are components of the so-called bee-milk, used to feed the queen larva. However, at least one member of the RJP family is also expressed in the brain [23]. One of the α-glucosidases described in this work (bEST122) seems to be a novel protein expressed predominantly in the abdomen (Figures 1,2), in contrast to the previously identified α-glucosidase-1 (bEST22), which is head-specific [24]. The expression levels of mitochondrial genes (bEST54, 56, 109, 134) were significantly reduced in foragers, suggesting that electron-transport system activity declines in older bees. It has been proposed that decreased levels of mitochondrial transcripts reflect changes in the electron-transport system and oxidative phosphorylation and contribute to the etiology of aging [25]. Similarly, the decreased levels of mRNAs encoding ribosomal proteins (bEST99) in foragers are likely to indicate the loss of homeostatic function in older individuals, as reported for other species [26].
Other genes known to be involved in cellular differentiation showed changes in the level of expression in newly born bees and experienced foragers. For example, sparc (bEST36), which encodes a calcium-binding glycoprotein expressed in extracellular matrix of various cell types undergoing morphogenesis, development and remodeling, is highly expressed in young bees, whereas a crystallin-like chaperonin, Hsp20, encoded by l(2)efl (lethal 2 essential for life, bEST30) is upregulated in the heads of foragers. Other genes upregulated in foragers encode proteins such as LIM (bEST57), which affects muscle adherens junction integrity and mechanosensory function, imaginal disc growth factor (IDGF; bEST121), and a novel protein (bEST112).
Among transcripts induced by caffeine treatment, we have identified those for a putative member of the LIM family (with a protein-protein interacting domain LIM; bEST57), IDGF (bEST121) and a glutamine synthetase (bEST138). The increased levels of mRNAs encoding two proteins implicated in immune responses, namely transferrin (bEST92) and hymenopteacin (bEST125), may indicate a reaction to physical injury and/or bacterial infection during injections. On the other hand, transferrin is also a key component in the control of iron homeostasis and it is conceivable that cellular changes induced by caffeine treatment, unrelated to injury, lead to the induction of its transcript. Such multifunctional roles for proteins are increasingly being found to be a rule rather than an exception, and even the best characterized of all proteins are increasingly being found to have bioinformatically unpredicted multifunctional characteristics [13]. For example, many ribosomal proteins have been found to be involved in extra-ribosomal activities such as iron binding, chromosomal condensation, DNA replication, transcription, RNA processing and DNA repair, and in protein-protein interactions in the p53 and Mdm2 pathways involved in malignant transformation [27]. The identification of caffeine-inducible transcripts suggests that the honeybee might be a convenient organism in which the efficacy of drug treatment could be rapidly tested before more costly and time-consuming experiments with vertebrates.
A number of the honeybee cDNAs, such as that for the putative PDZ-domain-containing protein (bEST61), show higher similarity to vertebrate sequences than to invertebrate sequences. This observation may indicate an under-representation of GenBank entries from insect species more closely related to the honeybee than is Drosophila melanogaster. One example of an unexpected evolutionary divergence of dipteran and hymenopteran sequences is the ligand-binding domain of the honeybee nuclear receptor Ultraspiracle (USP) (R.M., unpublished work; GenBank accession number AF263459), which clearly falls in the vertebrate-crab-tick-locust group rather than the dipteran-lepi-dopteran group. The reason for the divergence of USP sequences is unknown, but could result from a change in the type of ligand bound or the loss of ligand altogether [28].
Our study has revealed a collection of predicted proteins common to the honeybee and Drosophila as well as proteins that may be unique to the honeybee. Together with another study in the honeybee that compared the expression pattern of 288 'caste-related' genes [29], this dataset will be valuable for future comparative studies of gene-expression patterns in both insects. These comparisons will help to determine whether the behavioral differences between the 'simple' fly and the 'sophisticated' honeybee result from the invention of novel proteins, or are the output of changing patterns of expression of a basic set of genes common to both species. The haploid genome in the honeybee is similar in size to that in Drosophila (approximately 180 million base pairs) and is expected to contain a comparable number of genes, approximately 13,600 [30]. However, these two genomes produce radically different living systems, both structurally and functionally, supporting the view that there is essentially no correlation between the gene number and the phenotype of a given species [30].
Conclusions
We show here that array-based transcriptome analysis can be successfully used for basic cataloguing of gene expression in an organism for which genome and/or transcriptome information is virtually unavailable. Furthermore, in contrast to the most widely studied invertebrate model organisms - the fruit fly D. melanogaster and the nematode Caenorhabditis elegans - the honeybee has a range of complex individual and social behaviors, including a change from doing simpler activities when young to more complex activities when older. Thus, the availability of gene-expression data in the honeybee, combined with the ease with which honeybee brain chemistry and behavior can be perturbed, further strengthens the value of this insect in behavioral and pharmacological studies.
Materials and methods
Sample collection
Foraging honeybee workers were captured near the hive entrance and snap-frozen in liquid nitrogen (LN). To ensure that fully matured workers were collected, only those that carried pollen or nectar were selected. We estimate their age to vary from 20-35 days. To obtain newly emerged honeybees, a single brood frame was removed from the hive and incubated at 32°C (80% humidity). Individual insects were collected within 30-60 min after emergence and snap-frozen in LN. All dissections were done under permanent cooling (dry ice or LN). To obtain caffeine-treated bees, a colony of 30 newly born individuals in a small cage was fed for 3 days with honey containing 10 mM caffeine. A similar colony fed only with pure honey was used as control.
Array preparation
We used a standard, unannotated and non-normalized honeybee brain cDNA library made in lambda ZAP [31], kindly provided by G. Robinson, University of Illinois, Urbana-Champaign. Mass excision of cDNA-containing plasmids was carried out as recommended by the Stratagene uni-ZAP XR manual. Single colonies were picked manually and inoculated into 96-well microtiter plates containing 100 μl of LB medium per well. Following an overnight incubation, 5-μl bacterial aliquots from each well were taken for direct PCR amplification using 2 units of Taq DNA polymerase from Promega (plus 0.025 units of Stratagene Pfu) and 50 pmol of each primer (reverse and M13-20) per 100 μl reaction. Twenty-four PCR reactions from each of the 48 microtiter master plates were checked for the presence of amplified inserts. The PCR products were cleaned up by precipitation with iso-propanol and after several washes with 70% ethanol were resuspended in 3 × SSC, 0.01% sarcosyl. Array construction, labeling and hybridization were carried out according to protocols established by the Brown lab in Stanford [32] with minor modifications. Arrays were prepared by printing 4,608 samples in triplicate on polylysine-coated glass slides (Menzel) using a robotic device from Genetic Microsystems (model 418). These samples correspond to approximately 2,500 unique cDNAs. This estimate is based on random sequencing of 106 clones (aproximately 72% unique), the frequency of 'empty' clones resulting from abortive bacterial growth or failed plasmid purification (10%) or the success rate of insert amplification (85%).
RNA extraction, hybridization and data analysis
Total RNA was extracted from frozen tissues by homogenization in a guanidine-thiocyanate buffer [14] and sedimented overnight at 100,000g through a CsCl cushion (1.2 ml of 5.7 M CsCl overlaid with 4 ml RNA extract). We found that incorporation of fluorochromes was greatly improved in samples purified by CsCl sedimentation. Labeled probes were prepared by incorporating Cy3 and Cy5 dUTP (Amersham) during reverse transcription of total RNA (SuperScriopt II, Life Technologies). Hybridization was carried out for 2-5 h at 62°C in a small volume (60-120 μl) of ExpressHyb buffer (Clontech) under a plastic coverslip. Typically each run was repeated twice. Following a washing step, slides were scanned with the Affymetrix 428 scanner and analyzed using Affymetrix Pathways software v.1.0 and Microsoft Excel spreadsheets.
Sequencing and bioinformatics
High-throughput automated sequencing was done by the Australian Genome Research Facility (AGRF) in Brisbane using double-stranded plasmid DNA templates. Database searches were carried out at the National Center for Biotechnology Information (NCBI) using the BLAST server [33]. Additional searches were conducted at the Keck Center for Comparative and Functional Genomics, University of Illinois at Urbana-Champaign [34] to determine if our cDNAs are included in the recently established honeybee EST database. The following nine clones have not previously been found: bEST: 28, 54, 121, 123, 132, 134, 136, 143 and 147 (see Table 1 for GenBank accession numbers).
Northern blot analysis
Total RNA was isolated using Trizol reagent from Gibco BRL followed by mRNA purification on Oligo(dT)25 magnetic beads from Dynal (Oslo). RNA samples were denatured by mixing with an equal volume of formamide, containing 0.05% bromophenol blue and 0.01% SybrGreen II, incubated at 90°C for 5-7 min and immediately chilled on ice. Electrophoresis was carried out in small horizontal tanks (Hoeffer HE33) using 1.5% agarose gels submerged in TBE buffer (50 mM Tris-borate, 1 mM EDTA pH 8.2) at 20 V/cm. Alternatively, the glyoxal-based system (Ambion) was used for RNA separation, in particular to analyze larger (> 2 kb) transcripts (see the manufacturer's instruction manual (Ambion, catalogue number 1946) for details). Following electrophoretic resolution the gels were quantified with the Vistra FluoroImager and then soaked in 1 M ammonium acetate, 0.02 M NaOH and blotted onto Hybond N+ nylon membranes (Amersham) by capillary transfer. RNA was crosslinked to the membrane by UV irradiation, and after a brief wash in 2 × SSC the blots were prehybridized for 5-30 min. Hybridization was carried out either at 68°C (ExpressHyb solution, Clontech), or at 42°C (UltraHyb buffer, Ambion) for 16 h using 32P-labeled probes (RediPrime kit, Amersham). Blots were washed 3-4 times in 2 × SSC, 0.1% SDS at 50°C and exposed to a phosphorstorage screen (Molecular Dynamics) without drying. Computer generated images (MD Phosphor-Imager 400S) of individual gels were analyzed using ImageQuant software.
Acknowledgments
Acknowledgements
We thank Nadine McCarthy and Paul Helliwell for skilful assistance. This work was supported by Human Frontier Science Program grant RGO134/1999-B.
References
- Ferea TL, Brown PO. Observing the living genome. Curr Opin Genet Dev. 1999;9:715–722. doi: 10.1016/s0959-437x(99)00033-7. [DOI] [PubMed] [Google Scholar]
- Srinivasan MV, Zhang SW, Altwein M, Tautz J. Honeybee navigation: nature and calibration of the 'odometer'. Science. 2000;287:851–853. doi: 10.1126/science.287.5454.851. [DOI] [PubMed] [Google Scholar]
- Robinson GE, Fahrbach SE, Winston ML. Insect societies and the molecular biology of social behavior. BioEssays. 1997;19:1099–1108. doi: 10.1002/bies.950191209. [DOI] [PubMed] [Google Scholar]
- Menzel R, Müller U. Learning and memory in honeybees: from behavior to neural substrates. Annu Rev Neurosci. 1996;19:379–404. doi: 10.1146/annurev.ne.19.030196.002115. [DOI] [PubMed] [Google Scholar]
- Toma DP, Bloch G, Moore D, Robinson GE. Changes in period mRNA levels in the brain and division of labor in honey bee colonies. Proc Natl Acad Sci USA. 2000;97:6914–6919. doi: 10.1073/pnas.97.12.6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray S, Ferneyhough B. The effects of age on olfactory learning and memory in the honeybee Apis mellifera. NeuroReport. 1997;8:789–793. doi: 10.1097/00001756-199702100-00042. [DOI] [PubMed] [Google Scholar]
- Maleszka R, Helliwell P. Effect of juvenile hormone on short-term olfactory memory in young honeybees (Apis mellifera). Horm Behav. 2001;40:403–408. doi: 10.1006/hbeh.2001.1705. [DOI] [PubMed] [Google Scholar]
- Huang ZY, Robinson GE. Regulation of honey bee division of labor by colony age demography. Behav Ecol Sociobiol. 1996;39:147–158. [Google Scholar]
- Geschwind DH. Mice, micorarrays, and the genetic diversity of the brain. Proc Natl Acad Sci USA. 2000;97:10676–10678. doi: 10.1073/pnas.97.20.10676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramdas L, Coombes KR, Baggerly K, Abruzzo L, Highsmith WS, Krogmann T, Hamilton SR, Zhang W. Sources of nonlinearity in cDNA microarray expression measurements. Genome Biol. 2001;2:research00047.1–0047.7. doi: 10.1186/gb-2001-2-11-research0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han PL, Meller V, Davis RL. The Drosophila brain revisited by enhancer detection. J Neurobiol. 1996;31:88–102. doi: 10.1002/(SICI)1097-4695(199609)31:1<88::AID-NEU8>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Pirkkala L, Nykanen P, Sistonen L. Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. FASEB J. 2001;15:1118–1131. doi: 10.1096/fj00-0294rev. [DOI] [PubMed] [Google Scholar]
- Miklos GLG, Maleszka R. Protein functions and biological contexts. Proteomics. 2001;1:169–178. doi: 10.1002/1615-9861(200102)1:2<169::AID-PROT169>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Kucharski R, Ball EE, Hayward D, Maleszka R. Molecular cloning and expression analysis of a cDNA encoding a glutamate transporter in the honeybee brain. Gene. 2000;244:399–405. doi: 10.1016/s0378-1119(99)00503-x. [DOI] [PubMed] [Google Scholar]
- Guez D, Suchail S, Gauthier M, Maleszka R, Belzunces L. Contrasting effects of Imidacloprid on habituation in 7-day and 8-day old honeybees. Neurobiol Learn Mem. 2001;76:183–191. doi: 10.1006/nlme.2000.3995. [DOI] [PubMed] [Google Scholar]
- Maleszka R, Helliwell P, Kucharski R. Pharmacological interference with glutamate re-uptake impairs long-term memory in the honeybee, Apis mellifera. Behav Brain Res. 2000;115:49–53. doi: 10.1016/s0166-4328(00)00235-7. [DOI] [PubMed] [Google Scholar]
- Taylor DJ, Robinson GE, Logan J, Laverty R, Mercer AR. Changes in brain amine levels associated with the morphological and behavioral development of the worker honeybee. J Comp Physiol A. 1992;170:715–721. doi: 10.1007/BF00198982. [DOI] [PubMed] [Google Scholar]
- Withers GS, Fahrbach SE, Robinson GE. Selective neuroanatomical plasticity and division of labor in the honey bee colony. Nature. 1993;364:238–240. doi: 10.1038/364238a0. [DOI] [PubMed] [Google Scholar]
- Cao Y, Dulac C. Profiling brain transcription: neurons learn a lesson from yeast. Curr Opin Neurobiol. 2001;11:615–620. doi: 10.1016/s0959-4388(00)00258-0. [DOI] [PubMed] [Google Scholar]
- Sandberg R, Yasuda R, Pankratz DG, Carter TA, Del Rio JA, Wodicka L, Mayford M, Lockhart DJ, Barlow C. Regional and strain-specific gene expression mapping in the adult mouse brain. Proc Natl Acad Sci USA. 2000;97:11038–11043. doi: 10.1073/pnas.97.20.11038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strausfeld NJ, Homberg U, Kloppenburg P. Parallel organization in honey bee mushroom bodies by peptidergic Kenyon cells. J Comp Neurol. 2000;424:179–195. [PubMed] [Google Scholar]
- Maleszka R, Kucharski R. Analysis of Drosophila yellow-B cDNA reveals a new family of proteins related to the royal jelly proteins in the honeybee and to an orphan protein in an unusual bacterium Deinococcus radiodurans. Biochem Biophys Res Commun. 2000;270:773–776. doi: 10.1006/bbrc.2000.2506. [DOI] [PubMed] [Google Scholar]
- Kucharski R, Maleszka R, Hayward DC, Ball EE. A royal jelly protein is expressed in the subset of Kenyon cells in the mushroom bodies of the honey bee brain. Naturwiss. 1998;85:343–346. doi: 10.1007/s001140050512. [DOI] [PubMed] [Google Scholar]
- Ohashi K, Natori S, Kubo T. Expression of amylase and glucose oxidase in the hypopharyngeal gland with an age-dependent role change of the worker honeybee (Apis mellifera L.). Eur J Biochem. 1999;265:127–133. doi: 10.1046/j.1432-1327.1999.00696.x. [DOI] [PubMed] [Google Scholar]
- Schwarze SR, Weindruch R, Aiken JM. Decreased mitochondrial RNA levels without accumulation of mitochondrial DNA deletions in aging Drosophila melanogaster. Mutat Res Genomics. 1998;382:99–107. doi: 10.1016/s1383-5726(97)00013-7. [DOI] [PubMed] [Google Scholar]
- Shikama N, Brack C. Changes in the expression of genes involved in protein synthesis during Drosophila aging. Gerontology. 1996;42:123–136. doi: 10.1159/000213783. [DOI] [PubMed] [Google Scholar]
- Wool IG. Extraribosomal functions of ribosomal proteins. Trends Biochem Sci. 1996;21:164–165. [PubMed] [Google Scholar]
- Hayward DC, Bastiani MJ, Trueman JW, Truman JW, Riddiford LM, Ball EE. The sequence of Locusta RXR, homologous to Drosophila Ultraspiracle, and its evolutionary implications. Dev Genes Evol. 1999;209:564–571. doi: 10.1007/s004270050290. [DOI] [PubMed] [Google Scholar]
- Evans JD, Wheeler DE. Expression profiles during honeybee caste determination. Genome Biol. 2000;2:research0001.1–0001.6. doi: 10.1186/gb-2000-2-1-research0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miklos GLG, Maleszka R. Deus ex Genomix. Nat Neurosci. 2000;3:424–425. doi: 10.1038/74786. [DOI] [PubMed] [Google Scholar]
- Ebert PR, Rowland JE, Toma DP. Isolation of seven unique biogenic amine receptor clones from the honey bee by library scanning. Insect Mol Biol. 1998;7:151–162. doi: 10.1046/j.1365-2583.1998.72059.x. [DOI] [PubMed] [Google Scholar]
- Pat Brown's lab http://cmgm.stanford.edu/pbrown/
- National Center for Biotechnology Information http://www.ncbi.nlm.nih.gov/
- Honey bee brain EST project http://titan.biotec.uiuc.edu/bee/honeybee_project.htm