

SUMMARY
Diffuse intrinsic pontine gliomas (DIPGs) are incurable childhood brainstem tumors with frequent histone H3 K27M mutations and recurrent alterations in PDGFRA and TP53. We generated genetically engineered inducible mice and showed that H3.3 K27M enhanced neural stem cell self-renewal while preserving regional identity. Neonatal induction of H3.3 K27M cooperated with activating PDGFRα mutant and Trp53 loss to accelerate development of diffuse brainstem gliomas that recapitulated human DIPG gene expression signatures and showed global changes in H3K27 post-translational modifications, but relatively restricted gene expression changes. Genes upregulated in H3.3 K27M tumors were enriched for those associated with neural development where H3K27me3 loss released the poised state of apparently bivalent promoters whereas downregulated genes were enriched for those encoding homeodomain transcription factors.
Graphical Abstract
INTRODUCTION
Diffuse intrinsic pontine gliomas (DIPGs) are incurable brainstem tumors arising almost exclusively in children, with peak incidence between 6–8 years. These devastating tumors comprise approximately half of all pediatric high-grade gliomas (HGGs) (Jones and Baker, 2014). Recurrent, somatic mutation in histone H3 is a molecular hallmark distinguishing pathogenesis of HGG in children and adults (Schwartzentruber et al., 2012; Wu et al., 2014). Histone H3 K27M mutations occur in ~80% of DIPGs, and other HGGs arising in midline brain structures such as the thalamus. Diffuse midline glioma, H3 K27M-mutant is now recognized as a distinct entity by the World Health Organization classification system (Louis et al., 2016). In contrast, histone H3 G34R/V mutations are mutually exclusive with H3 K27M and occur in ~15% of cortical HGGs in older adolescents and young adults (Jones and Baker, 2014).
The genetic configuration of H3 K27M mutations implies a strongly dominant mode of action. 75% of K27M mutations occur in H3F3A, one of 15 genes encoding histone H3 variants. H3 K27M expression in the context of primary tumors or heterologous cell types, confers a dominant and profound decrease in H3K27me3, a posttranslational modification (PTM) associated with transcriptional repression (Bender et al., 2013; Chan et al., 2013; Herz et al., 2014; Lewis et al., 2013; Venneti et al., 2013). Although the K27M-mediated loss of H3K27me3 is context-independent, the high frequency association of H3 K27M with diffuse midline gliomas of childhood indicates that it only confers a selective advantage in specific developmental settings.
In addition to H3 mutations, DIPGs also contain alterations targeting canonical cancer signaling pathways, most frequently p53 loss of function and PDGFRα activation through gene amplification and/or mutation. Numerous other lower frequency mutations contribute to significant inter- and intratumoral DIPG heterogeneity (Mackay et al., 2017). Diffuse midline gliomas with K27M mutation, including DIPGs, also show distinct DNA methylation patterns when compared with other HGGs (Bender et al., 2013). This highlights the unique biology of K27M mutant gliomas, however, it is difficult to disentangle the effects of H3 K27M mutation from the signatures of midline developmental origin.
Experimental systems to study H3 K27M have relied on exogenous overexpression of H3.3 K27M along with different combinations of cooperating mutations to induce knockdown or deletion of Trp53 and/or activation of PDGFR signaling, and most did not target brainstem or midline brain structures. Mutants were virally transduced into neural progenitor cells (NPCs) induced from human embryonic stem (ES) cells or neural stem cells (NSCs) isolated from embryonic mouse forebrain and generated low or high-grade glioma, respectively, when implanted into brain (Funato et al., 2014; Mohammad et al., 2017). In utero electroporation of constructs encoding various mutants into mouse embryos evaluated cooperating effects of H3.3 K27M with other mutations in a limited number of hindbrain tumors, but predominantly targeted the cortex due to technical challenges with hindbrain delivery (Pathania et al., 2017). In an alternate approach, overexpressed H3.3 K27M in combination with Trp53 deletion and overexpression of PDGF-B, a PDGFRα ligand not typically mutated in human tumors, were introduced by in vivo retroviral transduction into neonatal brainstem using RCAS-tVA (Cordero et al., 2017). The varying levels of expression that can result from viral transductions or electroporation of different constructs contributes heterogeneity to each of these systems. This can be significant, as overexpression of the wild-type (WT) H3.3 was associated with altered neurogenic properties, showing important functional consequences of histone dosage, independent of K27M mutation (Pathania et al., 2017). Despite this limitation, all of these models showed that H3.3 K27M was insufficient to drive oncogenic transformation in the absence of other mutations but cooperated with other mutations to drive tumors. However, none of the models evaluated genomic occupancy of H3K27me3 in tumors or cells from brainstem or midbrain and minimal information is available about changes in genomic occupancy of other H3 PTMs in response to H3.3 K27M.
In this study, we set out to determine the consequence of H3.3 K27M mutations in different brain regions, how H3.3 K27M-mediated depletion of H3K27me3 impacts other aspects of epigenetic regulation, and how this connects with changes in transcription and oncogenic activity.
RESULTS
H3.3 K27M Promotes Self-Renewal and Mediates Global H3K27me3 Depletion but Discrete Transcription Changes
To study H3.3 K27M in the context of developing brain, we generated conditional knock-in mice, H3f3aLSL-K27M-Tag/+, in which H3.3 K27M is expressed from the endogenous H3f3a locus following Cre recombinase (Cre)-mediated excision of a loxP-flanked transcriptional STOP cassette (LSL). We included a C-terminal FLAG-HA tandem epitope tag immediately upstream of the termination codon to allow detection of the mutant protein (Figure S1A,B). A second mouse line, H3f3aLSL-WT-Tag/+, with identical construction but without the K27M mutation, was generated for controlled comparisons.
To investigate the role of H3.3 K27M in NSCs, we bred H3f3aLSL-K27M-Tag/+ or H3f3aLSL-WT-Tag/+ mice to Nestin-Cre mice (Tronche et al., 1999), which constitutively express Cre in neural stem and progenitor cells throughout the central nervous system beginning at ~E10.5. NSCs were isolated from hindbrain or forebrain (H- or F-NSC, respectively) of the resulting embryos at E15.5. Analysis of these NSCs over multiple passages in neurosphere growth conditions showed that H3.3 K27M promoted increased cell growth compared with H3.3 WT, regardless of origin location (Figure S2A,B). Clonogenic growth assays with H-NSCs showed H3.3 K27M significantly enhanced self-renewal capacity compared to H3.3 WT, and generated larger spheres, reflecting a modest increase in proliferation (Figure 1A,B). Interestingly, while the renewal capacity for H3.3 WT was similar from passages 3 through 9, H3.3 K27M cells displayed progressively enhanced clonogenic growth. Both genotypes began losing self-renewal by passage 11 (Figure 1A).
Figure 1. H3.3 K27M Promotes Self-Renewal and Mediates Global H3K27me3 Depletion but Discrete Transcription Changes That Do Not Disrupt Regional Signatures.

(A,B) Self-renewal of H3.3 K27M and H3.3 WT H-NSCs was assessed by clonogenic growth in methylcellulose at subsequent passages measuring number (A) and size (B) of spheres. *p < 0.05, **p < 0.01, ***p < 0.001, n.s., not significant. Error bars show +/− SEM. n = 3 per genotype (C) Scatterplot comparing expression in H3.3 K27M and H3.3 WT H-NSCs (RNA-seq; log2(FPKM+1)). (D) Plot of H3.3 K27M/H3.3 WT log2 ratio for RNA-seq versus H3K27me3 in H-NSCs. Colored dots depict genes up (red) and downregulated (blue) in H3.3 K27M compared to WT, with p < 0.05 and log2 fold change greater than 0.75 or less than −0.75, respectively, compared to the gene loci bulk (gray). (E) PCA of H3.3 WT and H3.3 K27M F- and H- NSCs. (F,G) Regional specific expression of Foxg1 (F) and Irx2 (G) shown by in situ hybridization (Allen Brain Atlas, E18.5) and average IGV tracks in H3.3 K27M F- and H-NSCs. *indicates Gm20554 locus near Irx2. Scale bar = 1 mm. (H) Average H-NSC tracks for three H3.3 K27M upregulated genes, Lin28b, Igf2bp2 and Plag1. *indicates Chchd7 locus near Plag1. In (F-H), tracks show H3K27me3, H3K27ac and H3K4me3 enrichment and RNA-seq in H3.3 WT or H3.3 K27M expressing NSCs. For each pair of tracks, n = 3 per genotype, scale is the same for both genotypes. See also Figures S1, S2, S3 and Tables S1–S3.
Consistent with reported effects of H3.3 K27M, chromatin immunoprecipitation with high throughput sequencing (ChIP-seq) combined with spike-in normalization revealed a profound global H3K27me3 reduction in H3.3 K27M compared to H3.3 WT NSCs (Figure S2C,D). However, RNA-seq analysis showed differences in the transcriptomes of H3.3 K27M and WT NSCs were relatively modest and selective rather than global (Figure 1C, Figure S2E, Table S1). To assess whether global loss of the transcriptional repression-associated PTM H3K27me3 could result in a global increase in all transcription, we also normalized the RNA-seq to a spike-in control, which confirmed the absence of an overall gain in transcription. Integrated analysis showed that the bulk of both up and downregulated genes have a similar decrease in H3K27me3 at their promoters (Figure 1D, S2F).
H3.3 K27M Does Not Disrupt Regional Expression Signatures
NSC expression signatures differentiating hindbrain and forebrain origin were not dramatically changed by loss of H3K27me3 (Figure 1E), and the majority of genes that were differentially upregulated in H3.3 WT H- or F-NSCs were also differentially upregulated in H3.3 K27M NSCs (Figure S3A). Gene Ontology (GO) analysis of these expression signatures showed similar enrichment in H3.3 WT or H3.3 K27M NSCs for regional development, including telencephalon regionalization in forebrain (p = 1.6E-08), and nervous system development and homeodomain transcription factors in hindbrain (p = 1.3E-07 and 3.7E-07, respectively, Table S2). For example, in situ hybridization data from the Allen Brain Atlas (Thompson et al., 2014) showed expression of Foxg1, which is not expressed in hindbrain, or Irx2, which is not expressed in forebrain, remains silenced in H3.3 K27M H- and F-NSCs respectively, even with substantial loss of H3K27me3 at these loci (Figure 1F,G). Although global transcription was mostly unaffected by loss of H3K27me3, we detected specific H3.3 K27M-mediated gene expression changes including upregulation of genes involved in neural development and proliferation (Figure S3B, Table S1, S3). The most significant H3.3 K27M induced changes included increased expression of genes important for regulating NPC proliferation and differentiation such as Lin28b, Igf2bp2 and Plag1 (Figure 1H, Table S1). These data indicate that H3.3 K27M contributes to programming enhanced self-renewal and a proliferative, progenitor cell state, while driving only selective changes in the transcriptome.
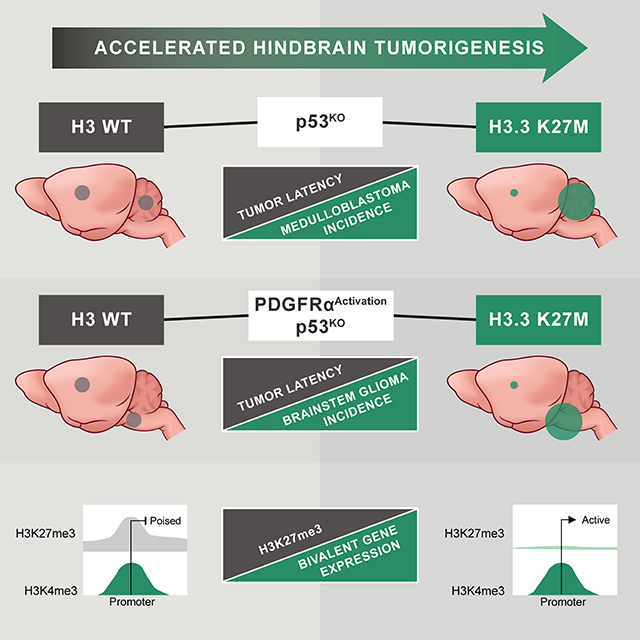
H3.3 K27M Accelerates Formation and Penetrance of Medulloblastoma Induced by Trp53-Deficiency
To model the contribution of H3.3 K27M to gliomagenesis in children, we bred H3f3aLSL-K27M-Tag/+ or H3f3aLSL-WT-Tag/+ mice with tamoxifen-inducible Nestin-CreERT2 mice (Zhu et al., 2012). The resulting Nestin-CreERT2;H3f3aLSL-K27M-Tag/+ or Nestin-CreERT2;H3f3aLSL-WT-Tag/+ mice (hereafter H3.3 K27M or H3.3 WT) were induced postnatal day 0 and 1 to activate the knock-in alleles in neonatal NSCs/NPCs. H3.3 K27M alone failed to cause brain tumor formation, and induction of either H3.3 K27M or H3.3 WT alleles did not cause obvious abnormalities or premature death within 1 year of age (Figure 2A,B). To evaluate cooperative oncogenic activity, H3.3 K27M or H3.3 WT mice were bred with Trp53flox mice (Jonkers et al., 2001) (hereafter p53cKO). Neonatal deletion of Trp53 induced highly penetrant brain tumors, with mice developing macroscopically visible cerebellar medulloblastomas in 59%, supratentorial HGG in 27%, and both concurrently in the remainder. H3.3 WT expression combined with p53cKO did not significantly alter tumor location, histopathology or latency (Figure 2A,C). In contrast, H3.3 K27M combined with p53cKO significantly accelerated brain tumor development (Figure 2B) and increased medulloblastoma frequency (Figure 2D, p = 0.0004). The histopathology of all evaluated supratentorial tumors was HGG and most developed as large masses with extensive infiltration of adjacent cerebral tissues. Pleomorphic cells occasionally showing astrocytic or oligodendroglial differentiation were associated with brisk mitotic activity and, in rare cases, areas of necrosis (Figure 2E). All histologically assessed tumors arising in the cerebellum appeared embryonal and were classified as medulloblastoma, with classic (Figure 2F) or large cell anaplastic morphologies at similar frequencies in all genotypes. Expression of the epitope-tagged knock-in H3.3 WT or H3.3 K27M was detected by nuclear FLAG expression, with H3.3 K27M;p53cKO tumors also showing the expected loss of H3K27me3, regardless of tumor histology (Figure 2G,H).
Figure 2. H3.3 K27M Accelerates Medulloblastoma Formation Caused by Trp53-Deficiency.

(A) Kaplan-Meier survival analysis in mice with induced H3.3 WT (n = 4), p53cKO (n = 37; gray), or H3.3 WT;p53cKO (n = 42), ns, p = 0.195. (B) Kaplan-Meier survival analysis with induced H3.3 K27M (n = 5), p53cKO (n = 46), or H3.3 K27M;p53cKO (n = 51), *p < 0.0001. Cohorts in A and B bred separately and used littermate controls to compare survival and tumor spectrum. (C,D) Location of macroscopic brain tumors in cohorts shown in the panel A (C) and the panel B (D). Supra, Supratentorial; CB, Cerebellar; Spinal, Spinal cord; BS, Brainstem. (E,F) H&E stain of representative supratentorial HGG (E) or medulloblastoma (F) observed in all genotypes. *in upper images indicates tumor. Scale bar = 1 mm (top images), 50 μm (bottom images). (G,H) Expression of FLAG-tagged H3.3 (upper images) and H3K27me3 (lower images) is shown by IHC on sections of representative supratentorial HGG (G) or medulloblastoma (H) for the indicated genotypes. Scale bar = 50 μm.
Activating PDGFRα Mutant Cooperates with Trp53 Deficiency to Preferentially Accelerate Spontaneous High-Grade Gliomas
PDGFRα is the most frequently mutated receptor tyrosine kinase in pediatric HGG (Mackay et al., 2017). Therefore, to model cooperative effects of pediatric HGG mutations, we generated LSL-PDGFRAV544ins transgenic mice with Cre-inducible expression of a mutant human PDGFRα containing a 15 amino acid duplication in the transmembrane domain (PDGFRAV544ins, Figure S4A,B). This alteration occurred as a heterozygous mutation in DIPG resulting in ligand-independent activation of PDGFRα (Paugh et al., 2013). Nestin-CreERT2;LSL-PDGFRAV544ins (hereafter PDGFRA) mice induced at P0 and P1 to express PDGFRAV544ins in neonatal NSCs/NPCs and their subsequent progeny, did not exhibit obvious abnormality, or reduced lifespan when observed for over 1 year (Figure 3A). In contrast, PDGFRA;p53cKO mice developed brain tumors faster than p53cKO mice, and substantially shifted the tumor spectrum to HGG (96%) including a significant increase of tumors involving the brainstem (52%, p < 0.0001) (Figure 3A,B). The cooperative effects of PDGFRA show that Trp53 loss alone is not sufficient for efficient gliomagenesis from brainstem neonatal NSCs/NPCs compared to the supratentorial compartment. Brainstem HGGs showed moderate nuclear pleomorphism, variable astrocytic differentiation, mitotic activity, extensive infiltration and strong nuclear Olig2, consistent with the pathology of human DIPG. Tumor cells also expressed robust cytoplasmic PDGFRα. However, consistent detection of H3K27me3 shows that these tumors did not acquire a somatic H3 K27M mutation or employ another genetic or epigenetic mechanism to suppress levels of this PTM (Figure 3C).
Figure 3. Active PDGFRα Mutant Cooperates with Trp53 Deficiency to Accelerate High-Grade Glioma Formation.

(A) Kaplan-Meier survival analysis in mice with induced mutant PDGFRA (n = 10), p53cKO (n = 46), or PDGFRA;p53cKO (n = 46), *p < 0.0001. (B) Location of macroscopic brain tumors for cohorts shown in (A): Supra, Supratentorial; CB, Cerebellar; Spinal, Spinal cord; BS, Brainstem. p53cKO cohort same as in Figure 2B. (C) HGG in PDGFRA;p53cKO mice. Sagittal section immunostained with anti-human PDGFRα (top image), and higher magnification of the pons for H&E stain and IHC of PDGFRα, Olig2 and H3K27me3 in representative HGG. Scale bar = 1 mm (whole brain image), 50 μm (higher magnification images). See also Figure S4.
H3.3 K27M Accelerates Spontaneous DIPG from Postnatal Neural Progenitors
H3F3A, TP53 and PDGFRA are the most commonly mutated genes in human DIPGs, and can occur in varying combinations (Buczkowicz et al., 2014; Fontebasso et al., 2014; Taylor et al., 2014; Wu et al., 2014). To assess the cooperative oncogenic effect of these mutations, we bred the respective engineered mice to Nestin-CreERT2 to generate H3.3 K27M;PDGFRA;p53cKO and H3.3 WT;PDGFRA;p53cKO mice. Mutations were induced at P0 and P1, resulting in highly penetrant brainstem and supratentorial HGGs in H3.3 WT;PDGFRA;p53cKO. Importantly, H3.3 K27M accelerated HGG development and significantly increased the proportion of HGGs arising in the brainstem from 59% to 95% (p < 0.0001) (Figure 4A,B). Tumors were diffusely infiltrative HGGs with similar histopathology to human DIPG for both H3.3 WT and K27M. Tumor cells showed robust expression of cytoplasmic PDGFRα, and also expressed Olig2. Expression of the knock-in H3.3 WT or H3.3 K27M was detected by FLAG in all tumors tested. While H3.3 WT tumors contained strong H3K27me3 expression, H3.3 K27M tumors consistently displayed marked loss of H3K27me3 (Figure 4C). Interestingly, H3K27me3 levels in H3.3 WT mouse DIPGs were noticeably higher than in H3.3 WT mouse NSCs and comparable to the levels found in human H3 WT HGG xenografts (Figure 4D), likely reflecting differences in H3K27me3 associated with developmental context.
Figure 4. H3.3 K27M Accelerates DIPG Formation from Postnatal Neural Progenitors.

(A) Kaplan-Meier survival analysis of cohorts with induced PDGFRA;p53cKO combined with H3.3 WT (n = 44) or H3.3 K27M (n = 43; green), p < 0.0001. (B) Location of macroscopic brain tumors for cohorts shown in (A): Supra, Supratentorial; CB, Cerebellar; Spinal, Spinal cord; BS, Brainstem. (C) DIPG in H3.3 WT;PDGFRA;p53cKO and H3.3 K27M;PDGFRA;p53cKO mice. Sagittal sections immunostained with anti-human PDGFRα. Boxed areas in brainstem are shown at higher magnification for H&E, and IHC for PDGFRα, Olig2, FLAG-tagged H3.3, or H3K27me3 in representative HGG. Scale bar = 1 mm (whole brain images), 50 μm (higher magnification images). (D) Western blot of acid extracted mouse hindbrain NSCs, mouse DIPGs and xenografted human HGGs that express WT H3 (H3 WT) or the H3.3 K27M mutant from the endogenous H3f3a/H3F3A promoter. A H3.3 K27M-specific antibody is used to confirm mutation status. Epitope tagged mouse H3.3 K27M protein is slightly larger than human H3.3 K27M protein. Xenografted human HGG H3 WT is a cerebellar tumor and H3.3 K27M is a DIPG.
Gene Expression in Spontaneous Mouse H3.3 K27M DIPGs Significantly Resembles That in Human H3.3 K27M DIPG
Gene expression signatures used to identify molecular subclasses of human HGG reflect the heterogeneity of this disease in adult and pediatric supratentorial HGGs and DIPGs (Huse et al., 2011; Paugh et al., 2011; Paugh et al., 2010; Puget et al., 2012). We used single sample gene set enrichment analysis (ssGSEA) to compare the gene expression signatures of the mouse brainstem gliomas with signature gene sets for human HGG subclasses (Proneural, Proliferative and Mesenchymal) (Phillips et al., 2006), and signature gene sets from normal mouse neural cell types (Zhang et al., 2014) (Table S4). Both mouse DIPGs and human DIPGs, with or without H3.3 K27M mutation, reflected intertumoral heterogeneity with varying enrichment for the different HGG subgroups and normal neural cell types (Figure 5A), and intermixing of H3.3 K27M and H3 WT tumors when viewed by PCA (Figure 5B). Two main subgroups of mouse DIPG showed predominantly Proneural or Proliferative signatures from the human HGG subclasses, and GO analysis of the most differentially expressed genes between tumors in these subgroups similarly identified signatures in synaptic transmission (p = 1.9E-26) or cell cycle (p = 1.75E-68), consistent with the HGG subgroup categories (Table S4).
Figure 5. Gene Expression in Mouse H3.3 K27M DIPGs Significantly Resembles That in Human H3.3 K27M DIPG.

(A) Heatmaps of ssGSEA scores comparing signatures for human HGG subgroups (PN, proneural; Pro, Proliferative; Mes, Mesenchymal) and normal murine neural cell types (N, Neurons; Astro, Astrocytes; MO, Myelinating oligodendrocytes; NFO, Newly formed oligodendrocytes; OPC, Oligodendrocyte precursor cells) between spontaneous mouse DIPG expressing H3.3 K27M (n = 20) or H3.3 WT (n = 9) or primary human DIPGs with H3.3 K27M (n = 20) or H3 WT (n = 3). For each panel, tumors were first separated by genotype then ordered by hierarchical clustering of gene signatures from human HGG subgroups. (B) PCAs of mouse and human DIPGs in (A). (C) GSEA showing significant enrichment in H3.3 K27M mouse DIPGs of genes upregulated in H3.3 K27M compared with H3.3 WT human DIPGs. Running enrichment score plots (left) and gene expression heatmaps in mouse H3.3 K27M or H3.3 WT DIPGs showing top leading edge genes (right). (D) Expression of leading edge upregulated genes Pbx3, Eya1 and Plag1. Boxplots depict log2-scale RNA-seq CPM values for primary human and mouse DIPGs, and mouse hindbrain (H-NSC) and forebrain (F-NSC) NSCs expressing H3.3 K27M or H3.3 WT. Box plots show the interquartile range (IQR). Median is shown as a horizontal line, highest and lowest values up to 1.5 times the IQR are shown with dotted lines outside box, and outliers greater than 1.5 times the IQR are shown as black squares. See also Figures S5 and S6 and Tables S4 and S5.
DIPGs, regardless of H3.3 K27M status, have distinct expression patterns from cortical pediatric HGGs, as shown by PCA (Figure S5A). We previously reported that these expression differences are significantly associated with transcription factors and developmental processes (Paugh et al., 2011). To assess transcription effects of H3.3 K27M without the confounding influence of regional expression differences in tumors arising from multiple anatomic locations, we compared DIPGs in mouse and human. The human H3 WT DIPGs used in our comparison were of similar age to the H3.3 K27M DIPGs (median age 8.9 versus 6, respectively, p = 0.9), had expression signatures that group with other DIPGs, and MRI images consistent with typical DIPG (Figure 5B, S5A–D). Gene set enrichment analysis (GSEA) demonstrated that human H3.3 K27M signatures were significantly enriched in mouse H3.3 K27M DIPGs (Figure 5C, Table S5). Pbx3, Eya1 and Plag1 are among the most significant (Figure 5C) and show a clear expression shift by H3.3 K27M in human DIPG, mouse DIPG and mouse embryonic NSCs (Figure 5D). H3.3 K27M downregulated genes in human DIPGs were not strongly enriched in mouse H3.3 WT compared to H3.3 K27M DIPG. However, a number of transcription factors associated with neural development were consistently downregulated by H3.3 K27M in human and mouse DIPGs as well as mouse hindbrain neural stem cells (Figure S6A). Several differentially expressed genes also show regional differences in NSCs, such as higher basal expression of Pbx3 and En1 in hindbrain, marked H3.3 K27M-dependent differential expression of Six1, En1 and Hoxd8 in hindbrain versus forebrain, and downregulation of Cdkn2a in forebrain (Figure 5D and S6A,B).
Spontaneous H3.3 K27M DIPGs Exhibit Global Changes in H3K27 Epigenetic State and Selective Expression Changes in PRC1 and PRC2 Targets
To better understand the epigenetic effects of H3.3 K27M in spontaneous DIPG, we compared the global occupancy of PTMs associated with gene repression (H3K27me3) and activation (H3K27ac and H3K4me3) in H3.3 K27M;PDGFRA;p53cKO and H3.3 WT;PDGFRA;p53cKO DIPGs. H3.3 K27M facilitated a genome-wide reduction of H3K27me3 and a reciprocal increase in H3K27ac, with minimal global changes in H3K4me3 (Figure S7A–C). Promoter regions recapitulated the global changes in H3K27me3 and H3K27ac (Figure S7D–F). Notably, while the levels of the H3K27 modifications were dramatically changed by H3.3 K27M, the positioning of the H3K27 PTMs across the promoter region was unaltered (Figure 6A,B).
Figure 6. Spontaneous H3.3 K27M DIPGs Exhibit Global Changes in H3K27 Epigenetic State and Selective Expression Changes in PRC1 and PRC2 Targets.

(A,B) Promoter based histograms representing counts within 40 bp bins across 4 kb region centered at TSS for H3K27me3 (A) or H3K27ac (B) in H3.3 K27M (n = 6) and H3.3 WT (n = 5) mouse DIPGs. (C) Plot of H3.3 K27M/H3.3 WT log2 ratio in mouse DIPGs for promoter regions comparing RNA-seq versus H3K27me3. Colored data points depict genes up (red) and downregulated (blue and purple) in H3.3 K27M tumors, with p < 0.05 and a log2 fold change greater than 0.75 or less than −0.75, respectively, compared to the gene loci bulk (gray). Purple data points show downregulated genes with H3K27me3 log2 fold change of −0.75 or greater (relative H3K27me3 retention). RNA-seq: H3.3 WT, n = 9; H3.3 K27M, n = 20. (D) Gene ontology and Enrichr ChEA2016 analysis of H3.3 K27M upregulated genes. Length of bar indicates p value. (E,F) Plots of H3.3 K27M/H3.3 WT log2 ratio in mouse DIPGs for promoter regions comparing H3K27ac versus H3K27me3 (E) and H3K4me3 versus H3K27me3 (F). H3K4me3 (H3.3 WT, n = 3; H3.3 K27M, n = 2). Shaded density histograms illustrate relative overlap of PTM changes in promoters of up (red) and downregulated (blue) genes compared to the gene loci bulk (gray). (G) Average tracks (identical scale for each genotype pair) showing H3K27me3, H3K27ac and H3K4me3 enrichment in H3.3 WT or H3.3 K27M expressing mouse DIPGs and average RNA-seq tracks for Usp44, Lif and Six1. See also Figures S7 and S8 and Table S6.
We next integrated analysis of changes in the epigenome and transcriptome to evaluate the mechanisms driving aberrant gene regulation in H3.3 K27M tumors. H3K27me3 is normally associated with transcriptionally silent genes (Margueron and Reinberg, 2011), but as observed in H3.3 K27M NSCs, the dramatic genome-wide decrease of H3K27me3 in DIPGs produces relatively modest transcriptome changes. Overall, there were more genes upregulated than downregulated in H3.3 K27M DIPGs (Figure 6C; 299 up, (red) vs. 155 down, (blue or purple)) and H3.3 K27M upregulated genes display marked H3K27me3 decrease agreeing with their expression change. Consistent with a role for H3K27me3 in regulation of genes involved in development, cell fate and differentiation, genes upregulated in mouse H3.3 K27M DIPGs are significantly enriched for association with neural development and differentiation (GO), and with PRC1 and PRC2 targets (Enrichr) (Figure 6D and Table S6). While decrease in H3K27me3 at H3.3 K27M upregulated gene promoters is similar to genes with unchanged expression (Figure 6E, red (H3.3 K27M up) versus gray (unchanged) histograms on top of plot), both activation-associated H3K27ac and H3K4me3 show an average gain consistent with increased gene expression (Figure 6E,F, right marginal plots; peaks of red histograms are shifted up versus both blue (H3.3 K27M down) and gray). Usp44 is an example of a K27M upregulated gene showing clear loss of H3K27me3 and gain of H3K27ac (Figure 6G). The average epigenetic signature for H3.3 K27M downregulated genes is discordant with their expression and reflects the global loss of H3K27me3 and gain of H3K27ac. However, these genes show less H3K27ac and H3K4me3 compared to genes with H3.3 K27M upregulated or unchanged expression (Figure 6E,F, right marginal plots; peaks of blue histograms are shifted down versus both red and gray). Lif highlights a locus where loss of H3K27me3 and relatively unchanged H3K27ac is nonetheless accompanied by reduced gene expression (Figure 6G).
Interestingly, a small number of downregulated genes (n = 38) in H3.3 K27M tumors do not follow global trends for H3K27me3 and H3K27ac. The promoters of this group of genes retain H3K27me3, and show reduced H3K27ac and H3K4me3 (Figure 6C,E,F, purple) as exemplified by the Six1 locus (Figure 6G). Strikingly, this group of genes is significantly enriched for targets of BMI1, a core component of the PRC1 complex involved in gene silencing, as well as targets of the PRC2 components, JARID2 and EZH2 (Enrichr), highly associated with development and gene regulation (GO) (Table S6).
Globally, H3.3 K27M-dependent changes in PTMs at enhancers resemble those at promoters. Enhancers also show reciprocal shifts of H3K27me3 and -ac, minimal change in H3K4me3 and lack evidence of redistribution of any of these PTMs (Figure S8A–C). Enhancers associated with H3.3 K27M upregulated genes (Figure S8D, red) also demonstrate a H3.3 K27M-dependent increase in H3K27ac consistent with increased activation, compared to downregulated (blue) or unchanged (gray) gene-associated enhancers. As with promoters, most enhancers show a similar H3.3 K27M-dependent loss of H3K27me3 regardless of the H3.3 K27M-dependent expression status of the nearest gene (Figure S8D). However, while the promoters of H3.3 K27M upregulated genes usually have a true H3K27me3 peak in H3.3 WT DIPG that is lost in H3.3 K27M DIPG, active enhancers in H3.3 K27M DIPG that are associated with H3.3 K27M upregulated genes usually lack discrete H3K27me3 peaks in H3.3 WT DIPG, suggesting the change in enhancer activity is an indirect effect of H3.3 K27M-mediated H3K27me3 loss (Figure S8E,F).
Epigenetic Release at Bivalent Promoters is Associated with Upregulated Genes in H3.3 K27M DIPG Oncogenic Signature
Epigenetic signatures are associated with important effects on gene expression, especially during development. Under normal conditions, H3K27me3 is found primarily at repressed loci, H3K4me3 associates with active promoters and the combination of H3K27me3 and H3K4me3 marks poised or bivalent promoters. Loss of H3K27me3 from bivalent promoters is associated with increased gene expression in normal developmental transitions (Zhou et al., 2011). To determine the effect of H3.3 K27M-mediated H3K27me3 loss on expression of genes with bivalent promoters, we identified all promoters marked with both H3K27me3 and H3K4me3 in H3.3 WT mouse DIPGs (Figure 7A, 12%, gold, left bar). There was significant increase in the proportion of apparently bivalent promoters in H3.3 WT tumors among promoters of genes with H3.3 K27M-dependent differential expression, including 57% of upregulated genes (p = 0.031) and 39% of downregulated genes (Figure 7A, gold, center and right bars). Notably, genes that are differentially expressed in H3.3 K27M DIPGs and have H3K27me3+H3K4me3+ at their promoters in H3.3 WT tumors are significant PRC1 and PRC2 targets (Enrichr, p < E-27) highly associated with development and neurogenesis (GO, p < E-10, and E-7 respectively) (Table S7), and include genes we identified as differentially upregulated in hindbrain NSCs such as Lgr5, Irx1 and Irx2.
Figure 7. Epigenetic Release at Bivalent Promoters Associated with Differentially Expressed Genes in H3.3 K27M DIPG Oncogenic Signature.

(A) Stacked bar graphs showing peak call status for H3K27me3 and H3K4me3 in the 2 kb surrounding the transcriptional start site (TSS) of all genes, H3.3 K27M up or downregulated genes in H3.3 WT DIPGs. Differential genes defined as in Figure 6. Gold represents proportion of potential bivalent gene targets (H3K27me3+ H3K4me3+). (B) Violin plot showing the average RNA-seq signal for all genes in H3.3 WT mouse DIPGs for each promoter status. The width of the violin shows how common expression levels are, with the widest part of the violin corresponding to the mode average. (C) Average tracks (identical scale for each genotype pair) showing H3K27me3, H3K27ac and H3K4me3 enrichment in H3.3 WT or H3.3 K27M expressing mouse DIPGs and average RNA-seq tracks for three potentially bivalent genes, Pbx3, Eya1 and Meis2. n = 6 (H3.3 K27M) and n = 5 (H3.3 WT) for H3K27me3 and H3K27ac; n = 2 (H3.3 K27M) and n = 3 (H3.3 WT) for H3K4me3; n = 20 (H3.3 K27M) and n = 9 (H3.3 WT) for RNA-seq. *portion of G630016G05 gene near Meis2. Red arrows, primer locations used for qPCR in (D). (D) ReChIP experiment from Pbx3, Eya1 and Meis2 promoters. Signal for primary ChIPs is shown as percent of the starting ChIP input (top row). The material pulled down with the H3K27me3 primary IP was used for ReChIP with indicated antibodies. ReChIP signal is shown as percent of original chromatin input for primary ChIP (bottom row). n = 2 for each genotype. Error bars show standard deviation. See also Table S7.
Although these apparently bivalent promoters are marked with H3K27me3 and H3K4me3 in H3.3 WT tumors, some genes were clearly expressed in contrast to the expected silent bivalent promoter (Figure 7B; gold violin, potentially bivalent genes and 7C; black track, RNA-seq in H3.3 WT for three H3K27me3+H3K4me3+ genes). These could represent bivalent promoters associated with variable expression levels, or a mixed cell or allele population in which the same promoters are marked with H3K27me3 in one subpopulation and H3K4me3 in another. To address this possibility, we performed co-occupancy analysis by sequential ChIP (ReChIP). At Pbx3, Eya1 and Meis2 promoters, ChIP analysis for each PTM showed recruitment of both H3K27me3 and H3K4me3 in H3.3 WT DIPGs, while all three gene loci showed substantial loss of H3K27me3 recruitment in H3.3 K27M DIPGs (red arrows in Figure 7C, Figure 7D, upper graphs). ReChIP of H3K4me3 from the chromatin pulled down by an H3K27me3 ChIP showed considerable enrichment compared to IgG control at all three loci, confirming that H3K4me3 and H3K27me3 co-occupy the same fragment of DNA (Figure 7D, lower graphs). While the expression of these H3K27me3, H3K4me3 marked genes suggests that they are actively transcribed in a portion of the H3.3 WT tumor cell population, ReChIP indicates that a true bivalent population exists. Together, these data indicate that bivalency release through loss of H3K27me3 is a plausible mechanism for H3.3 K27M-mediated differential gene expression signatures important in DIPG development.
DISCUSSION
H3 K27M mutations represent a unifying feature of incurable childhood brain tumors that are otherwise molecularly heterogeneous. The selective association of these mutations with pediatric midline diffuse gliomas, especially DIPG, indicates a critical connection between epigenetic dysregulation and developmental context. Elucidating the mechanisms through which these mutations contribute to cancer is essential to improve outcome for DIPG patients. Our results provide a number of insights into the consequences of H3.3 K27M that contribute to DIPG pathogenesis.
H3.3 K27M, in the absence of other mutations, caused a transient increase in self-renewal of hindbrain NSCs in vitro without inducing immortality or delaying senescence. This would be predicted to increase the pool of cells with greatest propensity for transformation, but only for a limited duration. A role for this mutation in early stages of tumor initiation is consistent with the clonal incidence of H3.3 K27M mutations in DIPGs and the restricted developmental window of susceptibility during childhood in which DIPGs arise. Strikingly, the genes most differentially induced by H3.3 K27M in NSCs included Lin28b, Plag1 and Igf2bp2; heterochronic genes associated with regulating developmental differences in fetal and adult NSCs (Fujii et al., 2013; Nishino et al., 2013; Yang et al., 2015). Upregulation of LIN28B and PLAG1 was also seen with overexpression of H3.3 K27M in NPCs derived from human ES cells, however, increased neurosphere formation in vitro was only seen with the combination of three alterations; H3.3 K27M, mutant PDGFRα and p53 knockdown (Funato et al., 2014). It is possible that the H3.3 K27M-dependent enhancement of self-renewal that we detected in NSCs acutely isolated from embryos was not readily detected in the human NPCs, which require prolonged culturing for in vitro induction and may be less developmentally synchronized.
H3.3 K27M accelerated hindbrain tumorigenesis from neonatal progenitors. Unexpectedly, combined H3.3 K27M expression and Trp53 deletion accelerated medulloblastoma formation. Multiple lines of evidence highlight an emerging role for H3K27me3 loss in pediatric hindbrain tumorigenesis including pediatric posterior fossa group A (PFA-1) ependymoma (Bayliss et al., 2016; Pajtler et al., 2018) and Group 3 medulloblastoma (Vo et al., 2017). Thus, acceleration of medulloblastoma formation by H3.3 K27M may reflect an increased potency for H3K27me3 loss to contribute to hindbrain tumor development. Combining PDGFRα activation with Trp53 deletion in neonatal NSCs/NPCs shifted the spectrum of tumors to HGGs, including a high proportion involving brainstem. Consistent with the hypothesis that developing hindbrain may have an increased vulnerability to transformation associated with H3K27me3 depletion, H3.3 K27M significantly increased the incidence of diffuse brainstem gliomas driven by combined PDGFRα activation and Trp53 deletion, and further accelerated tumor development.
We induced genetically engineered mutations in Nestin-positive cells in neonatal mice to coincide with the developmental period when most gliogenesis occurs, including the period of greatest postnatal growth in the pons, from P0 to P4 (Lindquist et al., 2016). A recent study using neonatal in vivo electroporation to introduce H3.3 K27M combined with p53 knockdown failed to induce tumors, while in utero electroporation to overexpress H3.3 K27M with CRISPR/Cas9-mediated Trp53 deletion induced gliomas (Pathania et al., 2017). In contrast, our results show cooperative effects of H3.3 K27M in generating both medulloblastomas and high-grade gliomas when induced in neonatal mice. This may reflect technical differences such as expression from endogenous loci and different targeted cell populations.
The H3.3 K27M;PDGFRA;p53cKO, and H3.3 WT;PDGFRA;p53cKO models reported here investigate the most common human DIPG mutation targets, recapitulate the spectrum of gene expression subgroups in human HGGs, and show significant similarity in gene expression signatures to primary human DIPGs with and without H3 K27M mutation, respectively. Many comparisons between primary pediatric gliomas with and without H3.3 K27M mutation include H3 WT cortical gliomas and are confounded by regional developmental epigenetic and expression signatures along with variations in other oncogenic mutations. Our experimental system provides a robust setting to evaluate the direct effects of H3.3 K27M in the context of DIPGs induced at the same developmental time point and with the same oncogenic drivers.
Integrated analysis of the epigenome and transcriptome of H3.3 K27M;PDGFRA;p53cKO compared to H3.3 WT;PDGFRA;p53cKO DIPGs showed global changes in H3K27 PTMs, but selective changes in gene expression significantly associated with signatures of neural development. Genes with apparently bivalent promoters were significantly enriched among those upregulated with H3.3 K27M mutation. Thus, promoters marked by both H3K27me3 and H3K4me3 in H3.3 WT tumors would be poised for expression, while loss of H3K27me3 at these promoters in H3.3 K27M tumors would release the bivalent state, resulting in upregulation (Figure 8). This outcome is consistent with a direct effect of depleted H3K27me3 on selective changes in gene expression, and is reminiscent of altered expression of bivalent genes associated with differentiation or development in response to deletion of Ezh2 or Eed encoding PRC2 components (Lu et al., 2018). This key selectivity has not been previously demonstrated for H3.3 K27M. Funato et al. found that H3K4me3 in promoters remained stable, but H3K27me3 decreased in gene bodies, not promoters, of genes upregulated in human NPCs overexpressing H3.3 K27M. Overexpression of H3.3 K27M or WT H3 in mouse forebrain NSCs showed that the majority of differentially expressed genes were not associated with H3K27me3 in H3 WT NSCs, leading to the suggestion that these expression changes were indirect effects of the mutation (Mohammad et al., 2017). The clear association of upregulated genes with H3.3 K27M-mediated release of bivalent promoters identified in our mouse model may be attributed to both the regulation of H3.3 K27M at physiological levels from its endogenous promoter and our direct analysis of DIPG tumors rather than NSC cultures in vitro. The low levels of H3K27me3 in NSCs compared to tumors (Figure 4D) may explain why the role of bivalency in H3.3 K27M-dependent changes in gene expression was not previously identified in comparisons with NSCs.
Figure 8. H3.3 K27M Impact on Poised Promoters in DIPGs.

In H3 WT DIPGs (top), poised promoters, primed for expression, bear both H3K27me3 (purple) and H3K4me3 (orange) PTMs on the same or nearby nucleosomes (left side). Some genes appear to have both poised and active promoter states represented in different H3 WT cells (or on different alleles within the same cell), as the genes are expressed and bulk analyses show both H3K27me3 and H3K27ac (not shown) present at the promoter (right side). In H3.3 K27M cells (bottom), H3K27me3 is diminished globally and bivalent gene promoters can be converted from poised to active, resulting in increased expression compared to the H3 WT state.
While our mouse DIPGs are genetically engineered, the spontaneous development of DIPGs recapitulates the human process and may involve initiation from slightly different developmental states or acquisition of other mutations that could introduce intertumoral heterogeneity. Importantly, an independent study to identify the consequences of H3.3 K27M depletion in DIPG patient derived xenografts also demonstrated significant enrichment in upregulation of genes with K27M-dependent release of bivalent promoter regulation (Silveira et al, unpublished).
A small collection of downregulated genes that were very significantly enriched for BMI1 targets and homeobox transcription factors retained H3K27me3 in H3.3 K27M;PDGFRA;p53cKO DIPGs despite global reduction of this PTM. The selective retention, or suggestion of increased deposition of H3K27me3 in the context of H3 K27M-mediated depletion, has been demonstrated in primary human tumors, cell lines and model systems (Bender et al., 2013; Chan et al., 2013; Funato et al., 2014; Pathania et al., 2017). CDKN2A was previously reported as a target for residual PRC2 activity associated with selective downregulation in H3 K27M model systems (Chan et al., 2013; Cordero et al., 2017; Mohammad et al., 2017), although this was not consistent in all DIPG cell lines (Piunti et al., 2017). Cdkn2a was modestly downregulated in H3.3 K27M;PDGFRA;p53cKO compared to much more significant decreases in homeodomain transcription factors that were already heavily marked with H3K27me3 in H3.3 WT tumors, suggesting tight regulation by strong transcriptional repressors that overcome H3.3 K27M-dependent effects by effectively recruiting residual PRC2 activity. Additional genes that were downregulated despite substantial loss of H3K27me3 may represent indirect H3.3 K27M-independent effects.
Our results demonstrate that H3.3 K27M enhances self-renewal of NSCs without inducing immortalization, and accelerates hindbrain tumorigenesis, of either medulloblastoma or high-grade glioma from neonatal stem/progenitor cells. Upregulation of genes normally restrained by bivalent promoter PTMs results in transcriptional changes in genes relevant for both development and tumorigenesis, perhaps creating an expanded pool of cells susceptible to transformation that may progress to DIPG if they acquire other critical mutations during a narrow window of development. Because epigenetic state is strongly interconnected with development and differentiation, it is likely that the specific collection of H3.3 K27M-dependent target genes may vary depending on the age and precise cellular state from which DIPG initiates, contributing to heterogeneity in tumor expression signatures and highlighting the power of the inducible genetically engineered approach for controlled comparisons. Furthermore, these experimental systems provide immune competent, physiologically relevant spontaneous models of DIPG that will be useful for future mechanistic and preclinical studies of DIPG pathogenesis and therapeutic response.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Suzanne Baker (Suzanne.Baker@stjude.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Generating Genetically Engineered Mutant Mice
Procedures for all mouse experiments were approved by the Institutional Animal Care and Use Committee at St. Jude Children’s Research Hospital and are in compliance with national and institutional guidelines.
To generate the H3f3aLSL-K27M-Tag knock-in allele, we performed an optimized strategy for construct development as described (Larson and Baker, 2019). In brief, BAC-isolated H3f3a genomic sequence was engineered by recombineering (Liu et al., 2003) for targeting vector assembly into the pBR322-DT backbone with a Diphtheria toxin (DT) negative selection cassette. The targeting vector contains a 16.2 kb sequence containing all H3f3a exons and partial flanking sequence. In order to make this allele Cre-inducible, a STOP sequence cassette capable of expressing Puromycin (Puro) and flanked by loxP sites (LSL) (a gift from Tyler Jacks, Addgene plasmid 11584) was inserted by recombineering into intron 1 upstream of exon 2. Exon 2 was subcloned and subjected to site-directed mutagenesis (QuickChange II SDM Kit, Agilent Technologies, 200523) to create the K27M point-mutation. For accurate tracking mutant protein expression, a previously reported tandem FLAG/HA epitope (Lewis et al., 2013) was inserted by recombineering just upstream of the canonical H3f3a STOP codon in exon 4. Since this epitope is very distal to the engineered point mutation, a Frt-flanked Neomycin (Neo) cassette was simultaneously inserted into intron 3 just upstream of exon 4. The completed vector was electroporated into mouse ES cells for homologous recombination, and clones positively selected with Puromycin (Sigma, P8833) and Geneticin (Life Technologies, 10131–027). Correctly targeted ES clones were identified by Southern blot analysis of EcoRI/BamHI-digested genomic DNA probed with a 5’ external probe, or SacI-digested genomic DNA probed with a 3’ external probe. Correctly targeted ES clones were blastocyst injected to generate chimeras and subsequently establish founder knock-in mice. To eliminate potential unwanted hypomorphic activity in vivo, we deleted the Neo cassette by intercrossing founder knock-in mice with a mouse strain ubiquitously expressing Flippase (The Jackson Laboratory, 012930). To generate the H3f3aLSL-WT-Tag knock-in allele, the same protocol was followed as described above without subcloning and engineering a point mutation in exon 2. H3f3a knock-in mice were intercrossed with C57BL/6J mice (The Jackson Laboratory, 000664) for three generations prior to breeding with other targeted alleles.
To generate Cre-inducible LSL-PDGFRAV544ins transgenic mice, standard subcloning techniques were used to construct the transgene which contains a ubiquitous chicken beta-actin gene promoter with human cytomegalovirus enhancer (CMV/CAG; plasmid with CMV/CAG sequence was a gift from R. Greenberg, University of Pennsylvania) upstream of the LSL cassette. This is followed by cDNA encoding human PDGFRAV544ins mutant (Paugh et al., 2013), an internal ribosomal entry sequence (IRES) and the cDNA encoding human placental alkaline phosphatase to facilitate detection of transgene expression. The construct was terminated by an SV40 intronic sequence and polyadenylation signal. Following pronuclear injection into fertilized FVB/NJ oocytes and implantation into foster mothers, the transgenic founder was identified by polymerase chain reaction (PCR) with primers recognizing the transgene-specific sequence (5’-TCTGCTAACCATGTTCATGCCTTC-3’ and 5’-GACCGAAAGGAGCGCACGAC-3’). Single integration site was confirmed by FISH using a probe specifically recognizing the whole transgene sequence.
Fluorescence in situ Hybridization (FISH)
To determine LSL-PDGFRAV544ins transgene localization in the mouse genome, purified plasmid DNA containing the whole transgene was labeled with AlexaFluor 488–5-dUTP (Molecular Probes, C11397) by nick translation to generate transgene probe. Chromosome 4 control probe (RP23–335E10) was labeled with AlexaFluor 594–5-dUTP (Molecular Probes, C11400). The labeled probes were combined with sheared mouse DNA and hybridized to metaphase chromosomes derived from the derived transgenic mouse lung fibroblast culture in a solution containing 50% formamide, 10% dextran sulfate, and 2X SSC. The chromosomes were then stained with 4,6-diamidino-2-phenylindole (DAPI) and analyzed.
Neurosphere in vitro Culture
Brains were dissected from E15.5 mouse embryos and separated into forebrain and hindbrain. Cells were dissociated by mechanical force and seeded in ultra-low attachment T25 flasks (Corning, 3815) in Mouse NeuroCult Proliferation Medium (Stem Cell Technologies, 05702) + human EGF (20 ng/mL) (Miltenyi Biotech, 130–097-751). Neurospheres were passaged weekly using Accutase (Millipore, SCR005) dissociation and reseeded at 0.5 × 106 cells per T25 flask. All molecular experiments were carried out at passage 3.
Induction of Cre Recombinase Activity
Tamoxifen (Sigma, T5648) was dissolved in corn oil (Sigma, C8267) at 5 mg/mL at 37°C, 0.22 μm filter sterilized and stored for up to 7 days at 4°C in the dark. Cre activity was induced by intraperitoneal injection of 3 mg tamoxifen solution/40 g body weight using a 30 gauge insulin syringe (Becton Dickinson, 309301). Mice were induced at postnatal day 0 and 1 with daily injections separated by 24 hr.
In vivo Tumor Models
Cohorts of mice were aged and monitored for brain tumor symptoms. When moribund, mice were anesthetized with avertin (10 mg/25 g) and transcardially perfused with PBS. The brain was carefully dissected, a portion of the tumor was snap frozen and the remaining tissue was fixed with 4% paraformaldehyde in PBS overnight at 4°C. Since our experimental mice harbor a mixed strain background and different cohorts may produce slightly different results, we carefully compared littermate controls derived separately from H3f3aLSL-WT-Tag and H3f3aLSL-K27M-Tag breeding cohorts to properly analyze tumor survival and overall tumor burden frequencies. Overall with a Trp53-deficient background, our model directs highly penetrant lethal brain tumors, and less than 10% of mice generated succumb to peripheral tumors or die of unknown causes.
Human DIPG data
Gene expression data from human DIPGs are from (Wu et al., 2014). Informed consent for specimen analysis was obtained under protocols approved by the St. Jude Children’s Research Hospital Institutional Review Boards.
METHOD DETAILS
Neural Stem Cell Self-Renewal
Neural stem cell self-renewal was evaluated using clonogenic growth of single cells plated in methyl cellulose-containing medium as described (Gritti et al., 1999) with the following modifications: Single cells were sorted by FACS (FACSAria Fusion Sorter) to seed 400 cells/well into black-walled 96 well plates (Corning, 3904), and resuspended in NeuroCult basal medium containing proliferation supplement (Stem Cell Technologies, 05702), human EGF (Miltenyi Biotech, 130–097-751), and 0.6% methyl cellulose (Sigma, M0512). Clonogenic growth was scored 9 days later after staining viable cells with 2 μM SYTO 9 (ThermoFisher Scientific, S34854). Plates were imaged on a TE2000E2 microscope (Nikon) equipped with a Plan Apo 4X 0.2 NA lens and a standard GFP filter cube (excitation 480/30 nm; emission 535/40 nm). Plates were maintained at 37°C and 5% CO2 during imaging. Tiled images of the entire well (5×6 fields) were acquired using a motorized stage (Prior Instruments), a DS-Qi1 camera (Nikon) and the well plate template in the JOBS module of NIS Elements version 4.30.02. For analysis, images were automatically segmented by intensity using standard object count tools in NIS Elements. Images of colonies with diameter greater than 50 μm were scored by a researcher blinded to genotype.
Histology and Immunohistochemistry
Fixed tissue was processed, embedded in formalin, and cut into 5 μm sections. Hematoxylin and Eosin (H&E) staining (ThermoFisher Scientific, 7221, 7111), was performed according to manufacturer’s instructions. Immunohistochemistry (IHC) was performed using heat-induced antigen retrieval with citric acid-based buffer followed by primary antibodies at 1:1000 dilution: FLAG (Sigma, mouse monoclonal, F1804), H3K27me3 (Cell Signaling, rabbit monoclonal, 9733), PDGFRα (Cell Signaling, rabbit monoclonal, 5241), Olig2 (Millipore, rabbit polyclonal, AB9610). Anti-mouse or anti-rabbit biotinylated secondary antibodies (Vector Laboratories, BA-2000 or BA-1000, respectively) were used at 1:1000 dilution with horseradish peroxidase-conjugated streptavidin (VECTASTAIN Elite ABC Kit, Vector Laboratories, PK-6100). Staining was developed with DAB substrates (Vector Laboratories, SK-4100), and sections were counterstained with hematoxylin (Vector Laboratories, H-3401).
RNA Extraction and ERCC Spike-In
For in vitro cultures, 1 mL of Trizol (Invitrogen, 15596–018) was added to 2×106 dissociated neural stem cells from neurospheres. 2 μL of 1:20 diluted ERCC spike in (Life Technologies, 4456740, Lot 1412017) was added directly to the Trizol and total RNA was extracted following the standard Trizol protocol except the final 70% EtOH wash was repeated three times. RNA was resuspended in 20 μL of nuclease-free water. For snap frozen brain tumors, 1 mL of Trizol was added to tissue and homogenized by expelling through a series of small gauge needle syringes. Standard Trizol protocol was performed with three chloroform extractions and three 70% EtOH RNA pellet washes. RNA was resuspended in 20–40 μL of nuclease-free water.
Chromatin Immunoprecipitation (ChIP)
Snap frozen tumor portions were ground into a powder before fixation. Single cell suspensions were generated from cultured neurospheres before fixation. Samples were fixed for 5 min with 1% paraformaldehyde in PBS at room temperature (RT). Fixation was quenched with 250 mM Glycine, pelleted and washed 2x with PBS + protease inhibitors (PI) (Sigma, P8340). Pellets were resuspended in lysis buffer + PI (50 mM HEPES pH 7.9, 140 mM NaCl, 1mM EDTA pH 8.0, 10% Glycerol, 0.5% Nonidet P-40, 0.25% Triton X-100) for 10 min on ice for cell suspensions or for 20 min with rotation at 4°C for tumors. Bare nuclei were washed 2x with wash buffer + PI (10 mM Tris HCl pH 8.0, 0.2 M NaCl, 1 mM EDTA pH 8.0, 0.5 mM EGTA pH 8.0) 2x with shearing buffer + PI (10 mM Tris HCL pH 8.0, 1 mM EDTA pH 8.0, 0.1% SDS), then resuspended in shearing buffer + PI. Chromatin was sheared on a Covaris M220 ultrasonicator (Microtube, 75W, 5% duty cycle, 200 cycles/burst, 5 min or Millitube, 75W, 10% duty cycle, 200 cycles/burst, 15 min), then centrifuged for 10 min at 16000 g to remove cellular debris. Sonication size was verified on an Agilent Bioanalyzer using a High Sensitivity DNA Assay. For ChIP-seq, sheared chromatin from Drosophila melanogaster S2 cells (ATCC, CRL-1963) was added to the mammalian chromatin prior to ChIP for sample normalization. Chromatin containing spike-in was kept as an input control. ChIP reactions were performed using a modified Upstate Biotechnology protocol. Briefly, sheared chromatin was diluted 1:10 with dilution buffer + protease inhibitors (21 mM Tris HCL pH 8.0, 1 mM EDTA pH 8.0, 167 mM NaCl, 1.1% Triton X-100, 0.1% SDS), precleared Protein A sepharose beads (GE Healthcare, 17–5280-04) and 50 μg bovine serum albumin (BSA) with rotation at 4°C for 1–2 hr. The precleared chromatin was combined with the antibody of interest, Protein A sepharose beads and BSA and rotated overnight at 4°C. The bead bound chromatin was washed once each with low salt buffer (20 mM Tris HCl pH 8.0, 2 mM EDTA pH 8.0, 150 mM NaCl, 1% Triton X-100, 0.1% SDS), high salt buffer (20 mM Tris HCl pH 8.0, 2 mM EDTA pH 8.0, 0.5 M NaCl, 1% Triton X-100, 0.1% SDS), LiCl buffer (10 mM Tris HCl pH 8.0, 1 mM EDTA pH 8.0, 250 mM LiCl, 1% Nonidet P-40, 1% Deoxycholate) and twice with TE (10 mM Tris HCl pH 8.0, 1 mM EDTA pH 8.0). Chromatin was eluted off the beads with 0.1 M NaHCO3/1% SDS by rotating 30 min at RT, then the supernatant was incubated overnight with 0.2 M NaCl at 65°C (input controls were processed along with the ChIP samples from this point on). Samples were incubated for 2 hr at 37°C with 10 μg Proteinase K, then cleaned up using a QIAquick PCR Purification Kit (Qiagen, 28104) and eluted in 50 μL 10 mM Tris HCl pH 8.5. Antibodies used were (μL antibody per IP shown): H3K27me3 (Cell Signaling, 9733, lot 8; 4 μL), H3K27ac (Cell Signaling, 8173, lot 1; 4 μL) and H3K4me3 (Cell Signaling, 9751, lot 8; 5 μL).
Library Preparation and Sequencing
All library preparation and sequencing was carried out by the Hartwell Center at St Jude Children’s Research Hospital. For RNA-seq, RNA quality was checked by 2100 Bioanalyzer RNA 6000 Nano assay (Agilent) or LabChip RNA Pico Sensitivity assay (Perkin Elmer) before library generation. Libraries were prepared from total RNA with the TruSeq Stranded Total RNA Library Prep Kit (Illumina). For ChIP-seq, libraries were prepared from 5–10 ng of DNA using the NEBNext ChIP-Seq Library Prep Reagent Set for Illumina with NEBNext Q5 Hot Start HiFi PCR Master Mix according to the manufacturer’s instructions (New England Biolabs) with the following modifications: a second 1:1 Ampure cleanup was added after adaptor ligation. The Ampure size selection step prior to PCR was eliminated. Completed ChIP-seq libraries were analyzed for insert size distribution on a 2100 BioAnalyzer High Sensitivity kit (Agilent) or Caliper LabChip GX DNA High Sensitivity Reagent Kit (Perkin Elmer). All libraries were quantified using the Quant-iT PicoGreen dsDNA assay (Life Technologies), Kapa Library Quantification kit (Kapa Biosystems) or low pass sequencing on a MiSeq Nano v2 run (Illumina). One hundred cycle paired end sequencing (RNA-seq) or fifty cycle single end sequencing (ChIP-seq) was performed on an Illumina HiSeq 2500 or HiSeq 4000.
ReChIP
The primary ChIP reaction was carried out with 1600ng of chromatin for each sample in a total volume of 500 μL (20 mM Tris HCL pH 8.0, 1 mM EDTA pH 8.0, 150 mM NaCl, 1% Triton X-100, 0.1% SDS) with 100 μL Protein A sepharose beads, 100 μg BSA and 20 μL H3K27me3 antibody and rotated overnight at 4°C. The bead bound chromatin was washed as in the standard ChIP protocol, but was eluted with 300 μL of 0.1 mg/mL H3K27me3 peptide (AnaSpec, AS-64378, Lot 1457617) in low salt ChIP wash buffer for 3 hr on ice with occasional gentle vortexing. Samples were spun for 1 min at 16000 g and eluate was removed to a fresh tube. 10% of the volume was kept to verify the primary IP result, and the remaining material was divided for two secondary IPs: 5 μL H3K4me3 or 5 μL Normal Rabbit IgG (Cell Signaling, 2729S), 50 μL Protein A sepharose beads and 50 μg BSA and incubated at 4°C overnight with rotation. Secondary IPs were washed and eluted as in standard ChIP protocol and input, primary and secondary IPs had cross links reversed and were cleaned up as in standard ChIP protocol. Loci were analyzed by qPCR using Quantitect SYBR Master Mix (Qiagen, 204145) on a CFX96 Real Time PCR System (BioRad, 1855195). Primers were designed using PrimerBLAST (NCBI). Pbx3: Forward primer = 5’-CCTCTAGAGAACTTGGCGCT-3’, Reverse primer = 5’GGAAGTGCAACTTTCTCCGC-3’; Eya1: Forward primer = 5’-CCTGCACACTCGCTACCT-3’, Reverse primer = 5’-CTCAGATGCTATCTGCCGCT-3’; Meis2: Forward primer = 5’-AGCCGAGACTTCTGAGTTGT-3’, Reverse primer = 5’AGTGGGGATCGTTGTTGGTA-3’.
Quantitative Reverse Transcriptase PCR (qRT-PCR)
cDNA was generated using the standard SuperScript III (ThermoFisher Scientific, 18080–051) protocol with 100 ng of total RNA. 1 μL of each 20 μL cDNA reaction was used per qRT-PCR reaction. qPCR reactions used Quantitect SYBR Master Mix (Qiagen) and were run on a CFX96 Real Time PCR System (BioRad).
Western Blot Analysis
Histone protein was purified by acid extraction using previously described techniques (Shechter et al., 2007). Protein was resolved using standard SDS-PAGE techniques and transferred to a 0.45 μm nitrocellulose membrane (GE Healthcare, RPN2020D). 1μg of protein per lane was probed for H3.3 K27M using 1:1000 dilution primary antibody (Millipore, rabbit polyclonal, ABE419) followed by 1:2500 dilution anti-rabbit HRP-conjugated secondary antibody (GE Healthcare Life Sciences, NA934) and detected by chemiluminescence (SuperSignal West Dura Extended Duration Substrate, ThermoFisher Scientific, 34076). This membrane was re-probed for H3K27me3 using 1:1000 dilution primary antibody (Cell Signaling, rabbit monoclonal, 9733) followed by 1:10000 dilution anti-rabbit IRDye800CW-conjugated secondary antibody (LI-COR, 926–32213). On a separate blot, 0.2 μg from the same protein aliquot was probed for total H3 using 1:1000 dilution primary antibody (Abcam, rabbit polyclonal, ab1791) followed by 1:2500 dilution anti-rabbit HRP-conjugated secondary antibody (GE Healthcare Life Sciences, NA934) and detected by chemiluminescence (SuperSignal West Dura Extended Duration Substrate, ThermoFisher Scientific, 34076). All images were developed using the LI-COR Odyssey Fc.
ChIP-Seq Analysis
Mapping reads and visualizing data
We used BWA (version 0.7.12; default parameter) (Li and Durbin, 2010) to align the reads to the mouse and Drosophila melanogaster hybrid reference genome (mm9+dm3) and then marked duplicated reads with Picard (version 1.65), with only nonduplicated reads kept by samtools (version 0.1.18, parameter “-q 1 -F 1024”, (Li et al., 2009)). Mapped reads were then split to two bam files (mapped to mm9 and dm3 respectively). To control the quality of the data and estimate the fragment size, the nonduplicated version of SPP (version 1.11) was used to draw cross-correlation and calculate relative strand correlation value with support of R (version 2.14). Upon manually inspecting the cross-correlation plot generated by SPP, the smallest fragment size estimated by SPP was used to extend each read and to generate bigwig file for visualization on integrated genome viewer (IGV) (version 2.3.82) (Thorvaldsdottir et al., 2013). We scaled coverage according to spike-in normalization factor (Orlando et al., 2014) to generate bigwig track for each sample. To show average of several replicates as a single track in the browser, the bigwig files were merged to a single average bigwig file using UCSC tools bigWigtoBedGraph, bigWigMerge and bedGraphToBigWig.
Peak calling, promoter and enhancer characterization
MACS2 (version 2.1.1 20160309) (Zhang et al., 2008) was used to call narrow peaks (H3K27ac and H3K4me3) with option “nomodel” and “extsize” defined as fragment size estimated above, FDR corrected p value cutoff 0.01. For broad peaks (H3K27me3), SICER (Zang et al., 2009) (version 1.1, with parameters of redundancy threshold 1, window size 200 bp, effective genome fraction 0.86, gap size 600 bp, FDR 0.00001 with fragment size defined above) was used for domain calling. Enriched regions were identified by comparing the IP library file to input library file. Peak regions were defined as the union of peak intervals in both H3.3 WT and H3.3 K27M samples. To avoid gender bias, regions in chromosome X and Y were excluded for all subsequent analyses. Promoters were defined as mouse RefSeq TSS±1000 bp regions. Enhancers were identified by H3K27ac MACS peaks merged from both H3.3 K27M and H3.3 WT samples but excluding those loci overlapping within TSS±1000bp regions.
Spike-in normalization, differential analysis and peak annotation
ChIP-seq raw read counts were reported for each region/each sample using BEDtools 2.25.0 (Quinlan and Hall, 2010). The spike-in normalization was performed by counting Drosophila melanogaster reads and mouse reads in each IP sample and corresponding Input sample and using those counts to generate spike-in normalization factor for each sample, which was calculated as (IP_dm3.reads/IP_mm9.reads)/(INPUT_dm3.reads/INPUT_mm9.reads). Raw read counts were voom normalized and statistically contrasted using the pipeline limma in R (version 3.30.13). Normalization factor defined above was used to modify mouse library size in edgeR (version 3.16.5) for counts per million (CPM) calculation and differential analysis. An empirical Bayes fit was applied to contrast H3.3 K27M samples to H3.3 WT samples and to generate log fold changes, p values and false discovery rates for each peak region. For signal visualizations of ChIP-seq peaks, promoters or enhancers, CPM values were log2 transformed. Genomic feature annotation of peaks and histograms showing average ChIP-seq intensity around peak center (±2000 bp) were generated using annotatePeaks.pl, (a program from HOMER suite; version 4.8.3, http://homer.salk.edu/homer/) (Heinz et al., 2010) which was modified to allow library sizes to be adjusted according to their respective spike-in normalization factors.
Analysis of Apparent Bivalency
Individual H3.3 WT tumors that had both H3K27me3 and H3K4me3 ChIP-seq data (n = 3) were scored for the presence of MACS called peaks overlapping the 2kb surrounding gene TSS and promoters were binned into four categories (H3K27me3− H3K4me3−, H3K27me3+ H3K4me3−, H3K27me3− H3K4me3+, and H3K27me3+ H3K4me3+ (apparent bivalency). To be binned as H3K27me3+ H3K4me3+, both H3K27me3 and H3K4me3 peaks at a promoter had to co-occur in a single tumor.
Transcriptome sequencing and analysis
Total stranded RNA sequencing data were generated and mapped against mouse genome assembly NCBIM37.67 using the StrongArm pipeline described previously (Wu et al., 2016). The gene level quantification values were obtained with HTSeq (v4.8.3) (Anders et al., 2015) based on GENCODE annotation and normalized by TMM method with ‘edgeR’ package. Analysis of normalization by ERCC spike in showed strong correlation with TMM normalization (R2 = 0.98) indicating that the K27M mutation did not cause a global shift in transcription. Differential expression analysis was performed with ‘voom’ method in R ‘limma’ package. To define a H3.3 K27M responsive gene set, we performed differential expression analysis using the RNA-seq data from human DIPG tumors (Wu et al., 2014) with H3F3A K27M mutations and WT H3F3A. Significantly up and downregulated genes were defined by at least 2 fold changes with p < 0.05. Gene set enrichment analysis (GSEA) was carried out using GSEA (Subramanian et al., 2005) with above-defined human H3.3 K27M DIPG gene sets. Single sample gene set enrichment analysis (ssGSEA) was used to demonstrate heterogeneity among mouse DIPG and human DIPG samples as previously described in Chow et al. (Chow et al., 2011). Cell-specific markers were derived from Zhang et al. (Zhang et al., 2014), and human HGG expression subtypes were derived from Phillips et al.(Phillips et al., 2006). Gene expression FPKM (fragment per kb per million mapped reads) values were used for both GSEA and ssGSEA. The single sample enrichment scores were Z-score normalized for heatmaps (R package pheatmap). Unsupervised scaled Principle Component Analysis (PCA) was performed on TMM normalized log2 CPM counts for top 1000 most variable genes with FactoMineR package (Le et al., 2008).
Gene ontology signatures were evaluated with the Gene Ontology Consortium resource, www.geneontology.org (The Gene Ontology, 2017).
Epigenetic enrichment of H3.3 K27M-mediated differentially expressed gene targets was evaluated using the transcription factor ChIP-seq database (ChEA 2016) from Enrichr, http://amp.pharm.mssm.edu/Enrichr (Kuleshov et al., 2016).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical comparisons between H3.3 WT and H3.3 K27M NSCs for growth analysis were performed using a longitudinal random effects model. Statistical comparisons between H3.3 WT and H3.3 K27M NSCs for self-renewal analysis were performed by multiple unpaired t-test using GraphPad Prism 7.0d software. Kaplan-Meier survival curve comparisons were performed by log-rank (Mantel-Cox) test using GraphPad Prism 7.0d software. Statistical comparisons between brain tumor spectrum incidence were performed using Fisher’s exact tests. The sample sizes (n) are indicated in the figure legends and represent biological replicates. For statistical analysis of enrichment of upregulated bivalent targets in H3.3 K27M DIPG, differential expression analysis was performed by computing an empirical Bayes t-statistic as implemented in the Voom and Limma methods (Limma R package). Significantly upregulated genes in H3.3 K27M vs H3 WT DIPGs were defined by p < 0.05 and a log2 fold change greater than 0.75. A permutation procedure was used to evaluate enrichment of promoter bivalency status among differentially expressed genes. The Kruskal-Wallis test statistic was used to characterize differences in the distribution of the genes empirical Bayes t-statistics (Limma R package) according to their promoter bivalency status. The statistical significance of the Kruskal-Wallis test statistic (p value) was determined by repeating the differential expression analysis described above with permuted assignments of the treatment labels to expression profiles. In each analysis, the smaller of all possible or 1000 randomly selected permuted assignments were evaluated to compute the p value.
DATA AND SOFTWARE AVAILABILITY
The RNA-seq and ChIP-seq data reported in this paper are deposited at NCBI Gene Expression Omnibus (GEO), accession GSE108364, and can be reached through this link: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE108364.
Supplementary Material
Figure S1 (related to Figure 1). Design of Epitope-Tagged Conditional H3f3aLSL-K27MTag Knock-In Allele.
(A) Schematic of endogenous H3f3a allele, targeting vector, targeted allele before and after Flippase-mediated excision of the Neo cassette, and Cre-mediated excision of STOP cassette. H3f3a allele: Exons are indicated as numbered boxes. Restriction sites are indicated: E, EcoRI; S, SacI; B, BamHI. External 5' probe detects an endogenous 14.6 kb EcoRI/BamHI-digested genomic DNA fragment. External 3' probe detects an endogenous 11.8 kb SacI-digested genomic DNA fragment. Targeting Vector: STOP cassette equipped with Puromycin resistance (Puro) and STOP sequences flanked by loxP sites (turquois arrowheads) positioned in intron 1. Red star indicates K27M point mutation on exon 2. Neomycin resistance cassette (Neo) flanked by Frt sites (striped yellow arrowheads) positioned in intron 3. Epitope tag positioned in exon 4 (tan tag). Vector backbone Diphtheria toxin negative selection cassette (DT) positioned downstream of H3f3a exon 4. H3f3aLSL-K27M-Neo-Tag allele: Correctly targeted allele detected by 5' external probe hybridizing to 7.1 kb EcoRI/BamHI-digested genomic DNA fragment, and a 3' external probe hybridizing to 7.0 kb SacI-digested genomic DNA fragment. H3f3aLSL-K27M-Tag allele: Successful Flippase-mediated excision of the Neo cassette. H3f3aK27M-Tag allele: Knock-in allele after Cre-mediated excision of the STOP cassette. (B) Representative Southern blot screening of DNA from ES cell clones. Here, four clones with correctly targeted H3f3a allele were identified by both 5’ (left) or 3’ (right) probes.
Figure S2 (related to Figure 1). Region-Specific Signatures Maintained Despite H3.3 K27M Effects.
NSCs were isolated from forebrain and hindbrain (F- and H-NSC) of E15.5 embryos and grown as neurospheres. (A, B) Growth of F- (A) or H- (B) NSCs expressing H3.3 K27M (n = 4) or H3.3 WT (n = 2) were assayed over sequential passages by calculating mean cumulative cell numbers +/− SEM. p < 0.0001 for both (A) and (B). (C,D) Scatterplot comparing H3K27me3 ChIP-seq peak signal intensity (log2(CPM+1)) in H3.3 K27M versus H3.3 WT F- (C) and H- (D) NSCs. n = 3 in each genotype group for each region. (E) Scatterplot comparing expression levels in H3.3 K27M and H3.3 WT F-NSCs (RNA-seq plotted as log2(FPKM+1)). n = 3 in each group. (F) RNA-seq versus H3K27me3 in F-NSCs. Colored data points depict genes upregulated (red) and downregulated (blue) in H3.3 K27M, with p < 0.05 and a log2 fold change greater than 0.75 or less than −0.75, respectively, compared to the gene loci bulk (gray). n = 3 in each group. For all NSC experiments, n represents NSC cultures isolated from independent embryos.
Figure S3 (related to Figure 1). Regional Gene Signatures and H3.3 K27M-Mediated Gene Expression Changes in Hindbrain and Forebrain NSCs.
(A) Venn diagrams showing the overlap of regional up-regulated gene signatures (defined as at least log2 two-fold expression increase, p < 0.05) in forebrain (versus hindbrain) or hindbrain (versus forebrain) within each genotype (H3.3 WT or H3.3 K27M). Intersection shows the number and percentage of forebrain or hindbrain genes regionally regulated in H3.3 WT that are also regionally regulated in H3.3 K27M NSCs. (B) Venn diagrams showing the overlap between the genes up-regulated and down-regulated by H3.3 K27M versus H3.3 WT (log2 fold change greater than 0.75 or less than −0.75, p < 0.05), respectively in either forebrain or hindbrain NSCs. Intersection shows shared genes upregulated by H3.3 K27M or downregulated by H3.3 K27M in both hindbrain and forebrain. (A,B) Number of genes in each area of the Venn diagrams is shown.
Figure S4 (related to Figure 3). Design of Conditional LSL-PDGFRAV544ins Transgenic Allele.
(A) Schematic of conditional PDGFRAV544ins transgene construct, containing a CMV/CAG promoter, STOP cassette equipped with Puromycin resistance (Puro) and STOP sequences flanked by loxP sites (turquois arrowheads), cDNA of mutant human PDGFRA (hPDGFRA V544ins), internal ribosome entry site (IRES), human placental alkaline phosphatase reporter gene (hPLAP), and SV40 splicing and polyadenylation signal (SV40 intron+polyA). (B) Fluorescence in situ hybridization (FISH) for the transgene (green), or control chromosome 4 probe (red), showing a single transgene integration site on chromosome 4. Scale bar = 10 μm.
Figure S5 (related to Figure 5). Typical DIPGs Cluster Primarily By Brain Location, Not H3 Mutation Status.
(A) PCA (top 1000 most variable genes) of human HGGs including typical (n = 3) or atypical (n = 1) H3 WT DIPGs, H3.3 K27M DIPGs (n = 20), H3 WT cortical HGGs (n = 25) and H3.3 G34R cortical HGGs (n = 4). Percentage of variance captured by each component is reported on axes. Small arrows indicate tumors featured in panels below. (B-D) Magnetic Resonance Imaging (MRI) was available for three of the four H3 WT DIPGs. Axial view (Ax); Sagittal view (Sag). (B) Typical DIPG MRI: Axial T2-weighted fluid-attenuated inversion recovery (FLAIR) and sagittal postcontrast T1-weighted magnetization-prepared rapid gradient-echo (MPRAGE) images of the brain in a 5 year old patient. The tumor is non-enhancing and is centered on the ventral-inferior pons. In transverse images it involves about 85% of the cross-sectional area of the brainstem and engulfs the basilar artery ventrally but shows some sparing of the tegmentum dorsally. (C) Typical DIPG MRI: Axial postcontrast T2-weighted FLAIR and sagittal postcontrast T1-weighted images of the brain in an 8 year old patient. The minimally enhancing tumor involves almost the entire pons and extends to the upper medulla oblongata. Anterior exophytic growth results in engulfment of the basilar artery. These features are similar to those described in (B) and are considered typical for DIPG. (D) Atypical DIPG MRI: Axial postcontrast T2-weighted FLAIR, T1-weighted postcontrast and sagittal postcontrast T1-weighted MPRAGE images of the brain in a 6 year old patient. The location of the lesion would be characteristic of DIPG and the T2 signal abnormalities also involve almost the entire cross-sectional area of the brainstem. There is however a very well-defined, avidly enhancing central component suggesting a focal tumor and the surrounding, non-enhancing T2 signal changes could correspond to perilesional edema only. There is no engulfment of the basilar artery. Although some degree of enhancement within the tumor lesion is seen in up to 60–70% of patients with DIPG, it is usually less prominent, less well-defined and sometimes multifocal. Atypical DIPG shown in D was not included in molecular analyses with H3 WT DIPGs in this study.
Figure S6 (related to Figure 5). H3.3 K27M Downregulated Genes in Mouse DIPGs Show Similar Regulation in Human DIPGs.
Expression of H3.3 K27M-dependent down-regulated genes Six1, En1, Hoxd8, and Cdkn2a. Boxplots depict log2-scale RNA-seq CPM values for primary human and mouse DIPGs, and mouse hindbrain (H-NSC) and forebrain (F-NSC) NSCs expressing H3.3 K27M or H3.3 WT. Box plots show the interquartile range (IQR). Median is shown as a horizontal line, highest and lowest values up to 1.5 times the IQR are shown with dotted lines outside box, and outliers greater than 1.5 times the IQR are shown as black squares. (B) Average tracks for Cdkn2a locus showing H3K27me3, H3K27ac and H3K4me3 enrichment and RNA-seq in H3.3 WT or H3.3 K27M expressing mouse DIPGs (identical scale for each genotype pair). * indicates Gm12610 locus upstream of Cdkn2a. n = 6 (H3.3 K27M) and n = 5 (H3.3 WT) for H3K27me3 and H3K27ac; n = 2 (H3.3 K27M) and n = 3 (H3.3 WT) for H3K4me3; n = 20 (H3.3 K27M) and n = 9 (H3.3 WT) for RNA-seq. Cdkn2a encodes two tumor suppressor genes through expression of different first exons. Locations of exons 1α (p16Ink4a) and 1β (p19Arf) are marked with red arrows.
Figure S7 (related to Figure 6). H3.3 K27M Drives Extensive Shifts in H3K27 Methylation and Acetylation Occupancy Across the DIPG Epigenome in H3.3 K27M versus H3.3 WT DIPGs.
(A-C) Peak-based plots depicting average ChIP signal (x axis) relative to log2 fold change (K27M/WT) of ChIP signal (y axis) and demonstrating genome-wide (A) H3K27me3, (B) H3K27ac, and (C) H3K4me3 marks. Corresponding histograms show average signal for H3.3 WT and H3.3 K27M for +/− 10 kb surrounding peak center. (D-F) Promoter-based plots depicting average ChIP signal (x axis) relative to log2 fold change of ChIP signal (y axis) for 2 kb promoter region centered on TSS for (D) H3K27me3, (E) H3K27ac, and (F) H3K4me3 marks, n = 6 (H3.3 K27M) and n = 5 (H3.3 WT) for H3K27me3 and H3K27ac; n = 2 (H3.3 K27M) and n = 3 (H3.3 WT) for H3K4me3.
Figure S8 (related to Figure 6). Enhancers Reflect Similar Global H3.3 K27M-Dependent Epigenetic Trends as Promoters, but Do Not Appear to be Direct Drivers of H3.3 K27M-Dependent Gene Expression.
(A-C) Active enhancer-based plots depicting average ChIP signal (x axis) relative to log2 fold change (H3.3 K27M/H3.3 WT) of ChIP signal (y axis) for (A) H3K27me3, (B) H3K27ac, and (C) H3K4me3 marks. Corresponding histograms show average signal for H3.3 WT and H3.3 K27M centered on enhancers. (D) Plot of H3.3 K27M/H3.3 WT log2 ratio in mouse DIPGs for active enhancer regions comparing log2 fold change of H3K27ac versus H3K27me3. Colored data points depict genes upregulated (red) and downregulated (blue) in H3.3 K27M tumors with p < 0.05 and a log2 fold change greater than 0.75 or less than −0.75, respectively, compared to the enhancer loci bulk (gray). Shaded density histograms illustrate relative overlap of PTM changes at enhancers of up and downregulated genes compared to the gene enhancer of bulk loci that are not significantly changed (gray). Active enhancers were associated with nearest expressed gene. (E,F) Average tracks (identical scale for each genotype pair, promoters on the left) showing H3K27me3, H3K27ac and H3K4me3 enrichment and RNA-seq in H3.3 WT or H3.3 K27M expressing mouse DIPGs for two representative upregulated genes with potentially bivalent promoters, Epcam (E) and Npy (F). n = 6 (H3.3 K27M) and n = 5 (H3.3 WT) for H3K27me3 and H3K27ac; n = 2 (H3.3 K27M) and n = 3 (H3.3 WT) for H3K4me3; n = 20 (H3.3 K27M) and n = 9 (H3.3 WT) for RNA-seq. Active enhancers are marked with blue bars (blue arrows) below RNA-seq. (A-F) Active enhancers were called based on presence of a H3K27ac peak call in either H3.3 WT or H3.3 K27M samples and being at least 1 kb away from a known promoter.
Table S1 (related to Figure 1). H3.3 K27M-Mediated Differential Expression in Embryo-Derived Neural Stem Cells
Table S2 (related to Figure 1). Analysis of Hindbrain and Forebrain Neural Stem Cell Regional Expression Patterns
Table S3 (related to Figure 1). H3.3 K27M-Mediated Exclusive and Overlapping Differential Expression Between Forebrain and Hindbrain Neural Stem Cells
Table S4 (related to Figure 5). ssGSEA Analysis
Table S5 (related to Figure 5). GSEA Leading Edge H3.3 K27M Gene Targets
Table S6 (related to Figure 6). H3.3 K27M-Mediated Differential Expression In Mouse DIPG
Table S7 (related to Figure 7). Gene Ontology and Enrichr Analysis of H3.3 K27M-Upregulated Bivalent Genes
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-FLAG (clone M2) | Sigma | Cat# F1804, Lot SLBS353OV (RRID:AB_262044) |
| Rabbit monoclonal anti H3K27me3 (clone C36B11) | Cell Signaling Technology | Cat# 9733, Lot 8 (RRID:AB_2616029) |
| Rabbit monoclonal anti-PDGFRα (clone D13C6) | Cell Signaling Technology | Cat# 5241, Lot 3 (RRID:AB_10692773) |
| Rabbit polyclonal anti-Olig2 | Millipore | Cat# AB9610, Lot 2060464 (RRID:AB_570666) |
| Rabbit monoclonal anti-H3K27ac (clone D5E4) | Cell Signaling Technology | Cat# 8173, Lot 1 (RRID:AB_10949887) |
| Rabbit monoclonal anti-H3K4me3 (clone C42D8) | Cell Signaling Technology | Cat# 9751, Lot 8 (RRID:AB_2616028) |
| Normal rabbit IgG | Cell Signaling Technology | Cat# 2729S (RRID:AB_1031062) |
| Biotinylated Horse anti-mouse IgG | Vector Laboratories | Cat# BA-2000, (RRID:AB_2313581) |
| Biotinylated Goat anti-rabbit IgG | Vector Laboratories | Cat# BA-1000 (RRID:AB_2313606) |
| Rabbit polyclonal anti-H3.3 K27M | Millipore | Cat# ABE419 (RRID:AB_2728728) |
| Rabbit polyclonal anti-H3 | Abcam | Cat# ab1791, Lot GR153323–6 (RRID:AB_302613) |
| HRP-conjugated Donkey anti-rabbit IgG | GE Healthcare Life Sciences | Cat# NA934, Lot 9495175 (RRID:AB_772206) |
| IRDye 800CW Goat anti rabbit IgG | LI-COR | Cat# 926–32213, Lot C70918–03 (RRID:AB_621848) |
| Biological Samples | ||
| Human primary DIPG tumor samples | (Wu et al., 2014) | NA |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Geneticin (G418) | Life Technologies | Cat# 10131–027 |
| Puromycin | Sigma | Cat# P8833 |
| AlexaFluor 488–5-dUTP | ThermoFisher | Cat# C11397 |
| AlexaFluor 594–5-dUTP | ThermoFisher | Cat# C11400 |
| Mouse NeuroCult Proliferation Medium | Stem Cell Technologies | Cat# 05702 |
| Human EGF | Miltenyi Biotech | Cat# 130–097-751 |
| Accutase | Millipore | Cat# SCR005 |
| Methyl cellulose | Sigma | Cat# M0512 |
| SYTO 9 Green Fluorescent Nucleic Acid Stain | ThermoFisher | Cat# S34854 |
| Tamoxifen | Sigma | Cat# T5648 |
| Corn oil | Sigma | Cat# C8267 |
| Hematoxylin | ThermoFisher | Cat# 7221 |
| Eosin | ThermoFisher | Cat# 7111 |
| Trizol | Invitrogen | Cat# 15596–018 |
| ERCC RNA Spike-In Mix | Life Technologies | Cat# 4456740, Lot 1412017 |
| Protease inhibitors | Sigma | Cat# P8340 |
| Protein A sepharose beads | GE Healthcare Life Sciences | Cat# 17–5280-04 |
| H3K27me3 peptide | AnaSpec | Cat# AS-64378, Lot 1457617 |
| Quantitect SYBR Master Mix | Qiagen | Cat# 204145 |
| SuperSignal West Dura Extended Duration Substrate | ThermoFisher | Cat# 34076 |
| Critical Commercial Assays | ||
| QuickChange II SDM Kit | Agilent Technologies | Cat# 200523 |
| Vectastain Universal Elite ABC Kit | Vector Laboratories | Cat# PK-6100 |
| DAB Substrate Kit | Vector Laboratories | Cat# SK-4100 |
| SuperScript III First-Strand Synthesis System | ThermoFisher | Cat# 18080051 |
| QIAquick PCR Purification Kit | Qiagen | Cat# 28104 |
| Deposited Data | ||
| RNA sequencing mapped raw data | This paper | GEO: GSE108364 |
| ChIP sequencing mapped raw data | This paper | GEO: GSE108364 |
| Experimental Models: Cell Lines | ||
| Drosophila melanogaster D.MEL S2 | ATCC | CRL-1963 (RRID:CVCL_Z232) |
| Experimental Models: Mouse Strains | ||
| B6.129S4-Gt(ROSA)26Sortm2(FLP*)Sor/J | The Jackson Laboratory | Stock# 012930 (RRID:IMSR_JAX:012930) |
| C57BL/6J | The Jackson Laboratory | Stock# 000664 (RRID:IMSR_JAX:000664) |
| H3f3aLSL-K27M-Tag | This paper | NA |
| H3f3aLSL-WT-Tag | This paper | NA |
| Nestin-Cre | The Jackson Laboratory (Tronche et al., 1999) | Stock# 003771 (RRID:IMSR_JAX:003771) |
| LSL-PDGFRAV544ins | This paper | NA |
| Nestin-CreERT2 | (Zhu et al., 2012) | NA |
| Trp53flox | The Jackson Laboratory (Jonkers et al., 2001) | Stock# 008462 (RRID:IMSR_JAX:008462) |
| Oligonucleotides | ||
| LSL-PDGFRAV544ins Forward | Integrated DNA Technologies | 5’-TCTGCTAACCATGTTCATGCCTTC-3’ |
| LSL-PDGFRAV544ins Reverse | Integrated DNA Technologies | 5’-GACCGAAAGGAGCGCACGAC-3’ |
| Pbx3 Forward | Integrated DNA Technologies | 5’-CCTCTAGAGAACTTGGCGCT-3’ |
| Pbx3 Reverse | Integrated DNA Technologies | 5’-GGAAGTGCAACTTTCTCCGC-3’ |
| Eya1 Forward | Integrated DNA Technologies | 5’-CCTGCACACTCGCTACCT-3’ |
| Eya1 Reverse | Integrated DNA Technologies | 5’-CTCAGATGCTATCTGCCGCT-3’ |
| Meis2 Forward | Integrated DNA Technologies | 5’-AGCCGAGACTTCTGAGTTGT-3’ |
| Meis2 Reverse | Integrated DNA Technologies | 5’-AGTGGGGATCGTTGTTGGTA-3’ |
| Recombinant DNA | ||
| Plasmid: Lox-STOP-lox (LSL) | Addgene | Addgene Plasmid #11584 |
| Software and Algorithms | ||
| Gene Ontology Consortium | (The Gene Ontology, 2017) | http://www.geneontology.org |
| Enrichr | (Kuleshov et al., 2016) | http://amp.pharm.mssm.edu/Enrichr/ |
| Samtools v0.1.18 | (Li et al., 2009) | https://github.com/samtools/samtools |
| BEDtools v2.25.0 | (Quinlan and Hall, 2010) | http://bedtools.readthedocs.io/ |
| UCSC tools | NA | https://genome.ucsc.edu/util.html |
| BWA v0.7.12 | (Li and Durbin, 2010) | http://bio-bwa.sourceforge.net |
| R | NA | https://www.r-project.org |
| Bioconductor | NA | https://www.bioconductor.org/ |
| IGV v2.3.82 | (Thorvaldsdottir et al., 2013) | http://software.broadinstitute.org/software/igv/ |
| HOMER v4.8.3 | (Heinz et al., 2010) | http://homer.ucsd.edu/ |
| HTSeq v0.6.1p1 | (Anders et al., 2015) | https://htseq.readthedocs.io/ |
| MACS2 V2.1.1 | (Zhang et al., 2008) | https://github.com/taoliu/MACS |
| SICER v1.1 | (Zang et al., 2009) | https://home.gwu.edu/~wpeng/Software.htm |
| FactoMineR | (Le et al., 2008) | https://CRAN.R-project.org/package=FactoMineR |
| GSEA | (Subramanian et al., 2005) | http://software.broadinstitute.org/gsea/index.jsp |
| Prism 7.0d | GraphPad | NA |
| PrimerBLAST | NCBI | https://www.ncbi.nlm.nih.gov/tools/primer-blast/ |
| Other | ||
| Ultra-low attachment T25 cell culture flasks | Corning | Cat# 3815 |
| 96-well, black walled cell culture plates | Corning | Cat# 3904 |
| 30-gauge insulin syringe | Becton Dickenson | Cat# 309301 |
| 0.45μm nitrocellulose membrane | GE Healthcare Life Sciences | Cat# RPN2020D |
HIGHLIGHTS.
H3.3 K27M mutation enhances neural stem cell self-renewal
Neonatal PDGFRα activation and Trp53 loss induces supratentorial and brainstem glioma
H3.3 K27M preferentially accelerates hindbrain tumorigenesis
H3.3 K27M drives bivalent gene activation associated with neurodevelopment in DIPG
SIGNIFICANCE.
Histone H3 K27M mutations occur in 80% of DIPGs and exert a dominant effect driving global loss of H3K27me3. It is unclear how this dramatic change in epigenetic regulation contributes to oncogenic transformation. We used genetically engineered mouse models to show that H3.3 K27M alone enhanced neural stem cell self-renewal. Neonatal induction of H3.3 K27M cooperated with active PDGFRα mutant and p53 inactivation to accelerate DIPG formation. H3.3 K27M and the resulting H3K27me3 loss drove selective regulation of bivalent promoters in tumors, dysregulating neural development genes. These genetically engineered models of spontaneous DIPG recapitulate the most common mutations from human tumors, reveal insights into disease pathogenesis, and provide physiologically relevant immunocompetent models for future mechanistic and preclinical studies.
ACKNOWLEDGEMENTS
We thank Kristen Cox and Chanrika Williams for genotyping, Qun Liu for blastocyst injections, Kristen Correia for pronuclear injections, Jim Houston for FACS, the St. Jude Transgenic/Gene Knockout Core, Cytogenetic Core, Center for In Vivo Imaging and Therapeutics (CIVIT), Cell and Tissue Imaging Core and Hartwell Center. We thank Tyler Jacks for the LSL cassette.
This work was supported by NIH grants CA096832 (SJB, PJM, DWE, JZ), CA188516 (SJB), CA211481 (CIVIT), Swim Across America (SJB), the NCI Cancer Center Support Grant CA21765, Musicians Against Childhood Cancer and ALSAC.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayliss J, Mukherjee P, Lu C, Jain SU, Chung C, Martinez D, Sabari B, Margol AS, Panwalkar P, Parolia A, et al. (2016). Lowered H3K27me3 and DNA hypomethylation define poorly prognostic pediatric posterior fossa ependymomas. Sci Transl Med 8, 366ra161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W, et al. (2013). Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 24, 660–672. [DOI] [PubMed] [Google Scholar]
- Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, Morrison A, Lewis P, Bouffet E, Bartels U, et al. (2014). Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet 46, 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KM, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, Gupta N, Mueller S, James CD, Jenkins R, et al. (2013). The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev 27, 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow LM, Endersby R, Zhu X, Rankin S, Qu C, Zhang J, Broniscer A, Ellison DW, and Baker SJ (2011). Cooperativity within and among Pten, p53, and Rb pathways induces high-grade astrocytoma in adult brain. Cancer Cell 19, 305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero FJ, Huang Z, Grenier C, He X, Hu G, McLendon RE, Murphy SK, Hashizume R, and Becher OJ (2017). Histone H3.3K27M Represses p16 to Accelerate Gliomagenesis in a Murine Model of DIPG. Mol Cancer Res 15, 1243–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset PO, Bechet D, Faury D, De Jay N, Ramkissoon LA, et al. (2014). Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat Genet 46, 462–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii Y, Kishi Y, and Gotoh Y (2013). IMP2 regulates differentiation potentials of mouse neocortical neural precursor cells. Genes Cells 18, 79–89. [DOI] [PubMed] [Google Scholar]
- Funato K, Major T, Lewis PW, Allis CD, and Tabar V (2014). Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 346, 1529–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritti A, Frolichsthal-Schoeller P, Galli R, Parati EA, Cova L, Pagano SF, Bjornson CR, and Vescovi AL (1999). Epidermal and fibroblast growth factors behave as mitogenic regulators for a single multipotent stem cell-like population from the subventricular region of the adult mouse forebrain. J Neurosci 19, 3287–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz HM, Morgan M, Gao X, Jackson J, Rickels R, Swanson SK, Florens L, Washburn MP, Eissenberg JC, and Shilatifard A (2014). Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science 345, 1065–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse JT, Phillips HS, and Brennan CW (2011). Molecular subclassification of diffuse gliomas: seeing order in the chaos. Glia 59, 1190–1199. [DOI] [PubMed] [Google Scholar]
- Jones C, and Baker SJ (2014). Unique genetic and epigenetic mechanisms driving paediatric diffuse high-grade glioma. Nat Rev Cancer 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, and Berns A (2001). Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet 29, 418–425. [DOI] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson JD, and Baker SJ (2019). Engineering Inducible Knock-In Mice to Model Oncogenic Brain Tumor Mutations from Endogenous Loci. Methods Mol Biol 1869, 207–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le S, Josse J, and Husson F (2008). FactoMineR: An R package for multivariate analysis. J Stat Softw 25, 1–18. [Google Scholar]
- Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, and Allis CD (2013). Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist RA, Guinto CD, Rodas-Rodriguez JL, Fuentealba LC, Tate MC, Rowitch DH, and Alvarez-Buylla A (2016). Identification of proliferative progenitors associated with prominent postnatal growth of the pons. Nat Commun 7, 11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Jenkins NA, and Copeland NG (2003). A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res 13, 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, and Ellison DW (2016). The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131, 803–820. [DOI] [PubMed] [Google Scholar]
- Lu TT, Heyne S, Dror E, Casas E, Leonhardt L, Boenke T, Yang CH, Sagar, Arrigoni L, Dalgaard K, et al. (2018). The Polycomb-Dependent Epigenome Controls beta Cell Dysfunction, Dedifferentiation, and Diabetes. Cell Metab 27, 1294–1308 e1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, Bjerke L, Clarke M, Vinci M, Nandhabalan M, et al. (2017). Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 32, 520–537 e525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, and Reinberg D (2011). The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad F, Weissmann S, Leblanc B, Pandey DP, Hojfeldt JW, Comet I, Zheng C, Johansen JV, Rapin N, Porse BT, et al. (2017). EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med 23, 483–492. [DOI] [PubMed] [Google Scholar]
- Nishino J, Kim S, Zhu Y, Zhu H, and Morrison SJ (2013). A network of heterochronic genes including Imp1 regulates temporal changes in stem cell properties. Elife 2, e00924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando DA, Chen MW, Brown VE, Solanki S, Choi YJ, Olson ER, Fritz CC, Bradner JE, and Guenther MG (2014). Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Rep 9, 1163–1170. [DOI] [PubMed] [Google Scholar]
- Pajtler KW, Wen J, Sill M, Lin T, Orisme W, Tang B, Hubner JM, Ramaswamy V, Jia S, Dalton JD, et al. (2018). Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania M, De Jay N, Maestro N, Harutyunyan AS, Nitarska J, Pahlavan P, Henderson S, Mikael LG, Richard-Londt A, Zhang Y, et al. (2017). H3.3(K27M) Cooperates with Trp53 Loss and PDGFRA Gain in Mouse Embryonic Neural Progenitor Cells to Induce Invasive High-Grade Gliomas. Cancer Cell 32, 684–700 e689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paugh BS, Broniscer A, Qu C, Miller CP, Zhang J, Tatevossian RG, Olson JM, Geyer JR, Chi SN, da Silva NS, et al. (2011). Genome-wide analyses identify recurrent amplifications of receptor tyrosine kinases and cell-cycle regulatory genes in diffuse intrinsic pontine glioma. J Clin Oncol 29, 3999–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paugh BS, Qu C, Jones C, Liu Z, Adamowicz-Brice M, Zhang J, Bax DA, Coyle B, Barrow J, Hargrave D, et al. (2010). Integrated molecular genetic profiling of pediatric highgrade gliomas reveals key differences with the adult disease. J Clin Oncol 28, 3061–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paugh BS, Zhu X, Qu C, Endersby R, Diaz AK, Zhang J, Bax DA, Carvalho D, Reis RM, Onar-Thomas A, et al. (2013). Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res 73, 6219–6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, et al. (2006). Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9, 157–173. [DOI] [PubMed] [Google Scholar]
- Piunti A, Hashizume R, Morgan MA, Bartom ET, Horbinski CM, Marshall SA, Rendleman EJ, Ma Q, Takahashi YH, Woodfin AR, et al. (2017). Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat Med 23, 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puget S, Philippe C, Bax DA, Job B, Varlet P, Junier MP, Andreiuolo F, Carvalho D, Reis R, Guerrini-Rousseau L, et al. (2012). Mesenchymal transition and PDGFRA amplification/mutation are key distinct oncogenic events in pediatric diffuse intrinsic pontine gliomas. PLoS One 7, e30313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, et al. (2012). Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231. [DOI] [PubMed] [Google Scholar]
- Shechter D, Dormann HL, Allis CD, and Hake SB (2007). Extraction, purification and analysis of histones. Nat Protoc 2, 1445–1457. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor KR, Mackay A, Truffaux N, Butterfield Y, Morozova O, Philippe C, Castel D, Grasso CS, Vinci M, Carvalho D, et al. (2014). Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet 46, 457–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Gene Ontology, C. (2017). Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res 45, D331–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CL, Ng L, Menon V, Martinez S, Lee CK, Glattfelder K, Sunkin SM, Henry A, Lau C, Dang C, et al. (2014). A high-resolution spatiotemporal atlas of gene expression of the developing mouse brain. Neuron 83, 309–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, and Mesirov JP (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14, 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, and Schutz G (1999). Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet 23, 99–103. [DOI] [PubMed] [Google Scholar]
- Venneti S, Garimella MT, Sullivan LM, Martinez D, Huse JT, Heguy A, Santi M, Thompson CB, and Judkins AR (2013). Evaluation of histone 3 lysine 27 trimethylation (H3K27me3) and enhancer of Zest 2 (EZH2) in pediatric glial and glioneuronal tumors shows decreased H3K27me3 in H3F3A K27M mutant glioblastomas. Brain Pathol 23, 558–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo BT, Li C, Morgan MA, Theurillat I, Finkelstein D, Wright S, Hyle J, Smith SMC, Fan Y, Wang YD, et al. (2017). Inactivation of Ezh2 Upregulates Gfi1 and Drives Aggressive Myc-Driven Group 3 Medulloblastoma. Cell Rep 18, 2907–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Barnhill RL, Lee S, Li Y, Shao Y, Easton J, Dalton J, Zhang J, Pappo A, and Bahrami A (2016). The landscape of fusion transcripts in spitzoid melanoma and biologically indeterminate spitzoid tumors by RNA sequencing. Mod Pathol 29, 359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, Zhu X, Qu C, Chen X, Zhang J, et al. (2014). The genomic landscape of diffuse intrinsic pontine glioma and pediatric nonbrainstem high-grade glioma. Nat Genet 46, 444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Yang SL, Herrlinger S, Liang C, Dzieciatkowska M, Hansen KC, Desai R, Nagy A, Niswander L, Moss EG, and Chen JF (2015). Lin28 promotes the proliferative capacity of neural progenitor cells in brain development. Development 142, 1616–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang C, Schones DE, Zeng C, Cui K, Zhao K, and Peng W (2009). A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 25, 1952–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou VW, Goren A, and Bernstein BE (2011). Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet 12, 7–18. [DOI] [PubMed] [Google Scholar]
- Zhu G, Chow LM, Bayazitov IT, Tong Y, Gilbertson RJ, Zakharenko SS, Solecki DJ, and Baker SJ (2012). Pten deletion causes mTorc1-dependent ectopic neuroblast differentiation without causing uniform migration defects. Development 139, 3422–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (related to Figure 1). Design of Epitope-Tagged Conditional H3f3aLSL-K27MTag Knock-In Allele.
(A) Schematic of endogenous H3f3a allele, targeting vector, targeted allele before and after Flippase-mediated excision of the Neo cassette, and Cre-mediated excision of STOP cassette. H3f3a allele: Exons are indicated as numbered boxes. Restriction sites are indicated: E, EcoRI; S, SacI; B, BamHI. External 5' probe detects an endogenous 14.6 kb EcoRI/BamHI-digested genomic DNA fragment. External 3' probe detects an endogenous 11.8 kb SacI-digested genomic DNA fragment. Targeting Vector: STOP cassette equipped with Puromycin resistance (Puro) and STOP sequences flanked by loxP sites (turquois arrowheads) positioned in intron 1. Red star indicates K27M point mutation on exon 2. Neomycin resistance cassette (Neo) flanked by Frt sites (striped yellow arrowheads) positioned in intron 3. Epitope tag positioned in exon 4 (tan tag). Vector backbone Diphtheria toxin negative selection cassette (DT) positioned downstream of H3f3a exon 4. H3f3aLSL-K27M-Neo-Tag allele: Correctly targeted allele detected by 5' external probe hybridizing to 7.1 kb EcoRI/BamHI-digested genomic DNA fragment, and a 3' external probe hybridizing to 7.0 kb SacI-digested genomic DNA fragment. H3f3aLSL-K27M-Tag allele: Successful Flippase-mediated excision of the Neo cassette. H3f3aK27M-Tag allele: Knock-in allele after Cre-mediated excision of the STOP cassette. (B) Representative Southern blot screening of DNA from ES cell clones. Here, four clones with correctly targeted H3f3a allele were identified by both 5’ (left) or 3’ (right) probes.
Figure S2 (related to Figure 1). Region-Specific Signatures Maintained Despite H3.3 K27M Effects.
NSCs were isolated from forebrain and hindbrain (F- and H-NSC) of E15.5 embryos and grown as neurospheres. (A, B) Growth of F- (A) or H- (B) NSCs expressing H3.3 K27M (n = 4) or H3.3 WT (n = 2) were assayed over sequential passages by calculating mean cumulative cell numbers +/− SEM. p < 0.0001 for both (A) and (B). (C,D) Scatterplot comparing H3K27me3 ChIP-seq peak signal intensity (log2(CPM+1)) in H3.3 K27M versus H3.3 WT F- (C) and H- (D) NSCs. n = 3 in each genotype group for each region. (E) Scatterplot comparing expression levels in H3.3 K27M and H3.3 WT F-NSCs (RNA-seq plotted as log2(FPKM+1)). n = 3 in each group. (F) RNA-seq versus H3K27me3 in F-NSCs. Colored data points depict genes upregulated (red) and downregulated (blue) in H3.3 K27M, with p < 0.05 and a log2 fold change greater than 0.75 or less than −0.75, respectively, compared to the gene loci bulk (gray). n = 3 in each group. For all NSC experiments, n represents NSC cultures isolated from independent embryos.
Figure S3 (related to Figure 1). Regional Gene Signatures and H3.3 K27M-Mediated Gene Expression Changes in Hindbrain and Forebrain NSCs.
(A) Venn diagrams showing the overlap of regional up-regulated gene signatures (defined as at least log2 two-fold expression increase, p < 0.05) in forebrain (versus hindbrain) or hindbrain (versus forebrain) within each genotype (H3.3 WT or H3.3 K27M). Intersection shows the number and percentage of forebrain or hindbrain genes regionally regulated in H3.3 WT that are also regionally regulated in H3.3 K27M NSCs. (B) Venn diagrams showing the overlap between the genes up-regulated and down-regulated by H3.3 K27M versus H3.3 WT (log2 fold change greater than 0.75 or less than −0.75, p < 0.05), respectively in either forebrain or hindbrain NSCs. Intersection shows shared genes upregulated by H3.3 K27M or downregulated by H3.3 K27M in both hindbrain and forebrain. (A,B) Number of genes in each area of the Venn diagrams is shown.
Figure S4 (related to Figure 3). Design of Conditional LSL-PDGFRAV544ins Transgenic Allele.
(A) Schematic of conditional PDGFRAV544ins transgene construct, containing a CMV/CAG promoter, STOP cassette equipped with Puromycin resistance (Puro) and STOP sequences flanked by loxP sites (turquois arrowheads), cDNA of mutant human PDGFRA (hPDGFRA V544ins), internal ribosome entry site (IRES), human placental alkaline phosphatase reporter gene (hPLAP), and SV40 splicing and polyadenylation signal (SV40 intron+polyA). (B) Fluorescence in situ hybridization (FISH) for the transgene (green), or control chromosome 4 probe (red), showing a single transgene integration site on chromosome 4. Scale bar = 10 μm.
Figure S5 (related to Figure 5). Typical DIPGs Cluster Primarily By Brain Location, Not H3 Mutation Status.
(A) PCA (top 1000 most variable genes) of human HGGs including typical (n = 3) or atypical (n = 1) H3 WT DIPGs, H3.3 K27M DIPGs (n = 20), H3 WT cortical HGGs (n = 25) and H3.3 G34R cortical HGGs (n = 4). Percentage of variance captured by each component is reported on axes. Small arrows indicate tumors featured in panels below. (B-D) Magnetic Resonance Imaging (MRI) was available for three of the four H3 WT DIPGs. Axial view (Ax); Sagittal view (Sag). (B) Typical DIPG MRI: Axial T2-weighted fluid-attenuated inversion recovery (FLAIR) and sagittal postcontrast T1-weighted magnetization-prepared rapid gradient-echo (MPRAGE) images of the brain in a 5 year old patient. The tumor is non-enhancing and is centered on the ventral-inferior pons. In transverse images it involves about 85% of the cross-sectional area of the brainstem and engulfs the basilar artery ventrally but shows some sparing of the tegmentum dorsally. (C) Typical DIPG MRI: Axial postcontrast T2-weighted FLAIR and sagittal postcontrast T1-weighted images of the brain in an 8 year old patient. The minimally enhancing tumor involves almost the entire pons and extends to the upper medulla oblongata. Anterior exophytic growth results in engulfment of the basilar artery. These features are similar to those described in (B) and are considered typical for DIPG. (D) Atypical DIPG MRI: Axial postcontrast T2-weighted FLAIR, T1-weighted postcontrast and sagittal postcontrast T1-weighted MPRAGE images of the brain in a 6 year old patient. The location of the lesion would be characteristic of DIPG and the T2 signal abnormalities also involve almost the entire cross-sectional area of the brainstem. There is however a very well-defined, avidly enhancing central component suggesting a focal tumor and the surrounding, non-enhancing T2 signal changes could correspond to perilesional edema only. There is no engulfment of the basilar artery. Although some degree of enhancement within the tumor lesion is seen in up to 60–70% of patients with DIPG, it is usually less prominent, less well-defined and sometimes multifocal. Atypical DIPG shown in D was not included in molecular analyses with H3 WT DIPGs in this study.
Figure S6 (related to Figure 5). H3.3 K27M Downregulated Genes in Mouse DIPGs Show Similar Regulation in Human DIPGs.
Expression of H3.3 K27M-dependent down-regulated genes Six1, En1, Hoxd8, and Cdkn2a. Boxplots depict log2-scale RNA-seq CPM values for primary human and mouse DIPGs, and mouse hindbrain (H-NSC) and forebrain (F-NSC) NSCs expressing H3.3 K27M or H3.3 WT. Box plots show the interquartile range (IQR). Median is shown as a horizontal line, highest and lowest values up to 1.5 times the IQR are shown with dotted lines outside box, and outliers greater than 1.5 times the IQR are shown as black squares. (B) Average tracks for Cdkn2a locus showing H3K27me3, H3K27ac and H3K4me3 enrichment and RNA-seq in H3.3 WT or H3.3 K27M expressing mouse DIPGs (identical scale for each genotype pair). * indicates Gm12610 locus upstream of Cdkn2a. n = 6 (H3.3 K27M) and n = 5 (H3.3 WT) for H3K27me3 and H3K27ac; n = 2 (H3.3 K27M) and n = 3 (H3.3 WT) for H3K4me3; n = 20 (H3.3 K27M) and n = 9 (H3.3 WT) for RNA-seq. Cdkn2a encodes two tumor suppressor genes through expression of different first exons. Locations of exons 1α (p16Ink4a) and 1β (p19Arf) are marked with red arrows.
Figure S7 (related to Figure 6). H3.3 K27M Drives Extensive Shifts in H3K27 Methylation and Acetylation Occupancy Across the DIPG Epigenome in H3.3 K27M versus H3.3 WT DIPGs.
(A-C) Peak-based plots depicting average ChIP signal (x axis) relative to log2 fold change (K27M/WT) of ChIP signal (y axis) and demonstrating genome-wide (A) H3K27me3, (B) H3K27ac, and (C) H3K4me3 marks. Corresponding histograms show average signal for H3.3 WT and H3.3 K27M for +/− 10 kb surrounding peak center. (D-F) Promoter-based plots depicting average ChIP signal (x axis) relative to log2 fold change of ChIP signal (y axis) for 2 kb promoter region centered on TSS for (D) H3K27me3, (E) H3K27ac, and (F) H3K4me3 marks, n = 6 (H3.3 K27M) and n = 5 (H3.3 WT) for H3K27me3 and H3K27ac; n = 2 (H3.3 K27M) and n = 3 (H3.3 WT) for H3K4me3.
Figure S8 (related to Figure 6). Enhancers Reflect Similar Global H3.3 K27M-Dependent Epigenetic Trends as Promoters, but Do Not Appear to be Direct Drivers of H3.3 K27M-Dependent Gene Expression.
(A-C) Active enhancer-based plots depicting average ChIP signal (x axis) relative to log2 fold change (H3.3 K27M/H3.3 WT) of ChIP signal (y axis) for (A) H3K27me3, (B) H3K27ac, and (C) H3K4me3 marks. Corresponding histograms show average signal for H3.3 WT and H3.3 K27M centered on enhancers. (D) Plot of H3.3 K27M/H3.3 WT log2 ratio in mouse DIPGs for active enhancer regions comparing log2 fold change of H3K27ac versus H3K27me3. Colored data points depict genes upregulated (red) and downregulated (blue) in H3.3 K27M tumors with p < 0.05 and a log2 fold change greater than 0.75 or less than −0.75, respectively, compared to the enhancer loci bulk (gray). Shaded density histograms illustrate relative overlap of PTM changes at enhancers of up and downregulated genes compared to the gene enhancer of bulk loci that are not significantly changed (gray). Active enhancers were associated with nearest expressed gene. (E,F) Average tracks (identical scale for each genotype pair, promoters on the left) showing H3K27me3, H3K27ac and H3K4me3 enrichment and RNA-seq in H3.3 WT or H3.3 K27M expressing mouse DIPGs for two representative upregulated genes with potentially bivalent promoters, Epcam (E) and Npy (F). n = 6 (H3.3 K27M) and n = 5 (H3.3 WT) for H3K27me3 and H3K27ac; n = 2 (H3.3 K27M) and n = 3 (H3.3 WT) for H3K4me3; n = 20 (H3.3 K27M) and n = 9 (H3.3 WT) for RNA-seq. Active enhancers are marked with blue bars (blue arrows) below RNA-seq. (A-F) Active enhancers were called based on presence of a H3K27ac peak call in either H3.3 WT or H3.3 K27M samples and being at least 1 kb away from a known promoter.
Table S1 (related to Figure 1). H3.3 K27M-Mediated Differential Expression in Embryo-Derived Neural Stem Cells
Table S2 (related to Figure 1). Analysis of Hindbrain and Forebrain Neural Stem Cell Regional Expression Patterns
Table S3 (related to Figure 1). H3.3 K27M-Mediated Exclusive and Overlapping Differential Expression Between Forebrain and Hindbrain Neural Stem Cells
Table S4 (related to Figure 5). ssGSEA Analysis
Table S5 (related to Figure 5). GSEA Leading Edge H3.3 K27M Gene Targets
Table S6 (related to Figure 6). H3.3 K27M-Mediated Differential Expression In Mouse DIPG
Table S7 (related to Figure 7). Gene Ontology and Enrichr Analysis of H3.3 K27M-Upregulated Bivalent Genes
Data Availability Statement
The RNA-seq and ChIP-seq data reported in this paper are deposited at NCBI Gene Expression Omnibus (GEO), accession GSE108364, and can be reached through this link: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE108364.