

Introduction
Over the past two decades, several new agents have been approved by regulatory agencies for the treatment of different types of lymphoid malignancies.[1] Within non-Hodgkin’s lymphoma (NHL), most new agents where approved for follicular B cell lymphoma. In contrast, only two drugs have been approved for relapsed diffuse large B cell lymphoma in more than three decades. In many cases, approved drugs targeted unique molecular mechanism, in other cases several agents where approved for targeting the same oncogenic pathway such as the PI3 kinase pathway and BTK. Many other investigational agents targeting intra-cellular mechanisms are under investigation. A summary of single agent activity of several new agents in patients with diffuse large B cell lymphoma, follicular lymphoma, and mantle cell lymphoma is shown in Figure 1. In this review, we will focus on the current status of drug development aiming at new and unique targets.
Figure 1.
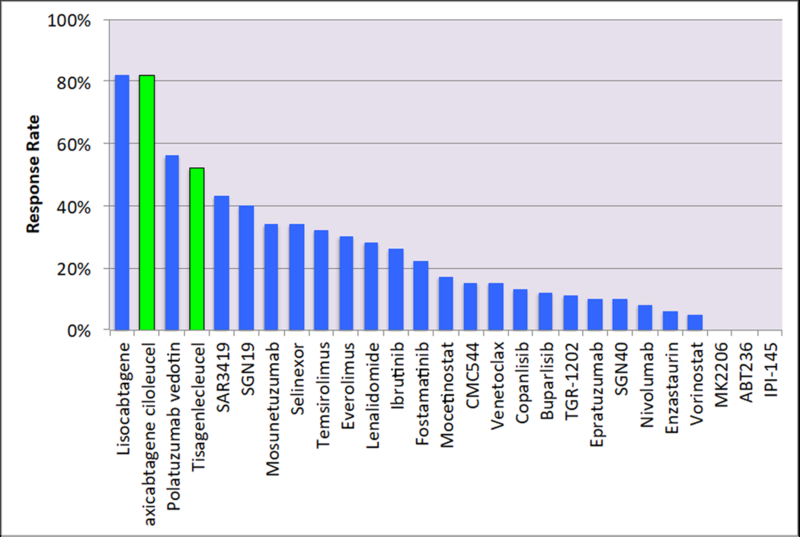
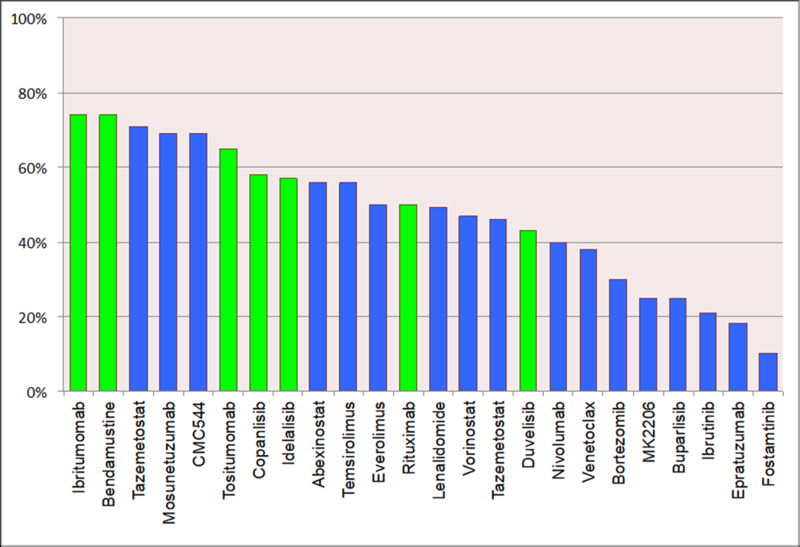
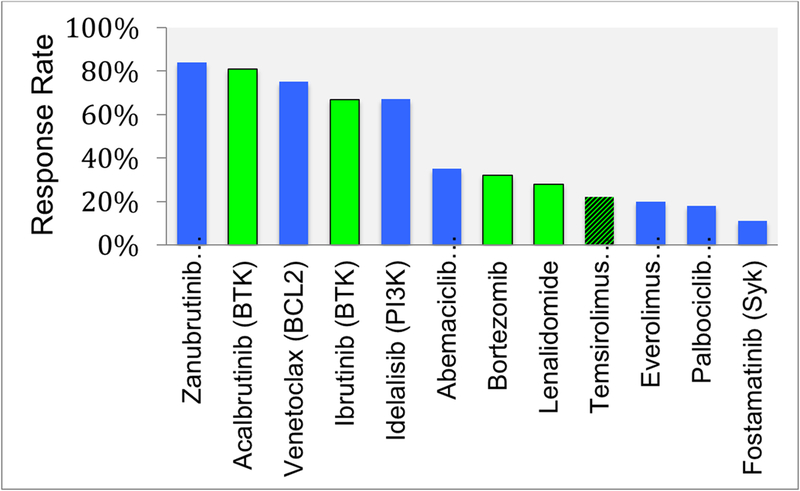
Single agent activity in common types of NHL. A) Diffuse large B cell lymphoma, B) Follicular lymphoma, and 3) Mantle cell lymphoma. Green color bars denote agents that are approved by the U.S. Food and Drug Administration (FDA). Temsirolimus in figure 1 c is shown in hashed green color indicating the drug is approved by the European Medicine Agency (EMA) but not by the FDA.
EZH2 inhibitors
Enhancer of zeste homolog 2 (or EZH2) is the enzymatic subunit that catalyzes the lysine Lys27 methylation of histone H3 (H3K27). EZH2 plays an essential role in germinal center biology.[2, 3] EZH2 genetic deletion in mice completely abrogated germinal centers formation. Heterozygous point mutations affecting tyrosine 641 (Y641) within the C-terminal catalytic SET domain of EZH2 have been identified in follicular lymphoma and GCB-DLBCL, with an incidence of approximately 15–20% in both tumor types.[4, 5] Y641 is a gain-of-function activating mutation, leading to increased levels of H3K27me3 and resulting in suppression of gene expression. In vivo, expression of the gain-of-function mutant allele in GC B cells synergizes with BCL2 protein overexpression, and accelerates the development of lymphomas, providing a rationale for the development of drugs aimed at inhibiting EZH2 activity alone or in combination with BCL2 inhibitors. In contrast, EZH2 genetic inactivation (deletion, frameshift, nonsense and missense mutations) have been identified in myelodysplastic syndromes (MDS), myeloproliferative neoplasms (MPN), and T-cell acute lymphocytic leukemia. These observations raised the possibility of implicating EZH2 loss-of-function in the development of malignancy. Furthermore, this raised concerns regarding the potential toxicity of long term EZH2 inhibitors.
To date, there are two EZH inhibitors in clinical development, one selectively targets EZH2 (Tazemetostat, EPZ-6438), and a second inhibits both EZH1 and EZH2 (DS-3201b). In a first-in-man, phase I study of patients with relapsed lymphoma or solid tumors, tazemetostat demonstrated a reasonable safety profile and a promising clinical activity.[6] Eight of 21 patients (38%) with B-cell NHL had a major clinical response, including 3 complete responses. The maximum tolerated dose was not reached, and the recommended phase II dose was determined to be 800 mg twice daily. Objective tumor responses were observed only in solid tumor patients with INI1- or SMARCA4-negative tumors. Tazemetostat was also evaluated in a phase II clinical trial in 156 patients with FL or DLBCL. Patients were stratified based on the presence of EZH2 mutations in their tumors. The highest response rate (71%) was observed in patients with FL that harbored EZH2 mutations. However, clinical responses were also observed in FL and GCB-DLBCL that did not carry the mutations (Figure 1). The most common toxicities, regardless of grade, included fatigue, nausea, cough, diarrhea, and thrombocytopenia.
Most recently, a dual EZH 1/2 inhibitor (DS-3201b) was recently evaluated in a phase I clinical trial. DS-3201b demonstrated clinical activity across a range of B- and T- cell NHL subtypes, with an overall response rate of 53%. Remarkably, in a small subset of patients with T cell lymphoma, 80% had a major response.
New B cell receptor (BCR) signaling pathway targets
Aberrant activation of BCR signaling pathway is implicated in the pathogenesis and progression of a variety of B-cell malignancies. Fostamatinib disodium (R788) was the first agent to target BCR signaling pathway by inhibiting SYK protein. However, initial clinical results were not very encouraging. Subsequently, several agents were developed to target the downstream protein Bruton’s tyrosine kinase (BTK). To-date, two BTK inhibitors (Ibrutinib and Acalabrutinib) have been approved for the treatment of lymphoid malignancies. In NHL, these agents are currently approved only for the treatment of patients with relapsed MCL (Figure 1).
The success of BTK inhibitors for the treatment of B cell lymphoid malignancies has generated enthusiasm about the development of other agents that can disrupt different nodes that enhance B cell receptor signaling. To-date, the leading compounds are aimed at targeting IRAK4 and MALT1 (Figure 2).
Figure 2.
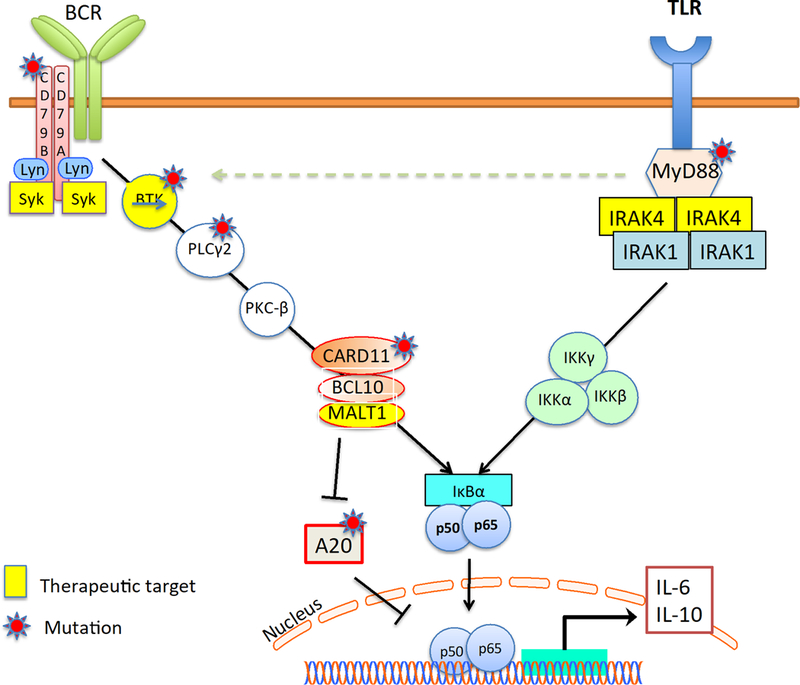
Interaction between the B cell receptor (BCR) signaling and the Toll-like receptor (TLR) signaling pathways. Several proteins in these pathways are currently being targeted for the treatment of lymphoma.
IRAK4.
Toll-like receptor (TLR) signaling adaptor protein MYD88, is frequently mutated in a variety of lymphoid malignancies, including Waldenstrom’s macroglobulinemia, marginal zone lymphoma, and DLBCL.[7] In most of these cases, tumors harboring the L265P mutations are prone to be more sensitive to BTK inhibitors. Interleukin-1 receptor-associated kinase 4 (IRAK4), accounts for almost all of the biological functions of MYD88, making it an attractive therapeutic target for MYD88-driven lymphoid malignancies (Figure 2).[8] In one study, small molecule inhibitors of IRAK4 was shown to be toxic against DLBCL lines harboring MYD88 L265P. Mechanistically, IRAK4 inhibition attenuated NF-κB and IL-6/IL-10-JAK-STAT3 signaling pathways. Furthermore, BTK or BCL2 inhibition enhanced the anti-proliferative activity of IRAK4 inhibitors DLBCL xenograft models.[9] CA-4948 is a reversible oral kinase inhibitor of IRAK4. CA-4948 demonstrated a dose-dependent efficacy in ABC-DLBCL MYD88 -L265P xenograft tumor models and patient-derived tumors, and it synergized with the anti Bcl-2 inhibitor, venetoclax. A phase I study of the IRAK4 inhibitor CA-4948 is currently being evaluated in patients with relapsed and refractory lymphoma (NCT03328078).
MALT1.
In 1999, the MALT1 gene was discovered after a new chromosomal translocation t(11;18)(q21;q21) was identified in cases of MALT (Mucosa associated lymphoid tissue) lymphoma. The C-terminal part of the fusion protein contained a domain homologous to the caspases, and hence defined MALT1 as a “paracaspase”.
MALT1 plays a key role in activating B and T lymphocytes, through regulating NF-kB activation.[10, 11] The activation of NF-kB pathway is achieved through several mechanisms, including recruiting TRAF6, and cleavage of the negative regulator TNFAIP3 (A20) (Figure 2). Accordingly, MALT1 is an attractive target for the treatment of NF-kB driven lymphomas, especially DLBCL of the ABC subtype.[12, 13] In preclinical testing, MI-2, a covalent inhibitor of MALT1 and, mepazine and thioridazine, which are reversible MALT1 inhibitors, demonstrated promising clinical activity against ABC-DLBCL in vitro and in mouse models. Other MALT1 inhibitors are also under development. Although no MALT1 inhibitor is currently in clinical testing, both Janssen and Abbvie plan to conduct clinical trials of MALT1 inhibitors in patients with relapsed lymphoma.
MCL1
The B-cell lymphoma 2 (BCL2) protein family, a key regulator of cell survival and death, is comprised of three functionally distinct subgroups: 1) the anti-apoptotic proteins (BCL2, BCL-XL, BCL-W, MCL1, A1/BFL-1), 2) the pro-apoptotic effector proteins (BAX and BAK), and 3) the pro-apoptotic BH3-only proteins (BIM, PUMA, BID, BAD, BIK, BMF, NOXA, HRK). Pharmacologic inhibition of the anti-apoptotic proteins can be achieved through the use of small molecules that mimic BH3 domain. These BH3-mimetics, bind to BCL2 or MCL1, leading to the release of pro-apoptotic proteins and triggering apoptosis. The first BH3 mimetic drug to be approved by regulatory agencies is venetoclax, a BCL2 selective oral inhibitor.
The MCL1 gene is frequently amplified in human cancers, and its protein overexpression is observed in a variety of tumors, including multiple myeloma, acute myeloid leukemia, chronic myeloid leukemia, B-cell acute lymphoblastic leukemia, among others. Mcl-1 can mediate resistance to several cytotoxic agents and to the Bcl-2 selective inhibitor venetoclax.[14] Several Mcl-1 selective inhibitors are currently under clinical development for the treatment of cancer. AZD5991,a rationally designed macrocyclic molecule with high selectivity and affinity for Mcl-1 currently in clinical development.[15] AZD5991 binds directly to Mcl-1 and induces rapid apoptosis in cancer cells, by activating the Bak-dependent mitochondrial apoptotic pathway. of AZD5991 is currently being evaluated in a Phase I clinical trial in patients with hematological malignancies (NCT03218683).
S63845 is a selective, BH3 mimetic MCL1 inhibitor. with no appreciable binding to BCL-2 or BCL-XL.[16] S63845 binds to the BH3-binding groove of human MCL1 with a Kd of 0.19 nM. S63845 potently killed MCL1-dependent cell lines in vitro and in xenograft mouse models. Interestingly, S63845 showed potent cytotoxic activity against c-MYC-driven human Burkitt lymphoma cell lines. A phase I clinical trial is currently evaluating the safety and efficacy of S63845 in patients with leukemia (NCT02979366). A third compound, AMG 176, is a potent, selective, and orally bioavailable MCL1 inhibitor that induces a rapid apoptosis in vitro in a variety of hematologic malignancies.[17] A phase I study is currently evaluating AMG 176 in patients with relapsed multiple myeloma and leukemia (NCT02675452)
PRMT5
Arginine methylation is a posttranslational modification catalyzed by protein arginine methyltransferases (PRMTs). PRMTs catalyze the mono or dimethylation of arginine residues on histone and non-histone proteins. Aberrant expression or functional dysregulation of PRMTs is associated with various disease states, including cancer.[18]
Currently, nine members of PRMTs have been identified in human cells. PRMT5 is a type II PRMT that specifically catalyzes symmetric dimethylation of arginine residues on H3 or H4. PRMT5 regulates transcriptional repression, RNA splicing, and signal transduction. PRMT5 is upregulated in a variety of cancers, including lymphoma. PRMT5 has been shown to play an important role in lymphomagenesis, controlling the growth of transformed B cells. The precise mechanisms involved in PRMT5 oncogenic activity are poorly described. However, some mechanisms have been recently identified, including arginine methylation of the tumor suppressor p53, that inhibits p53-mediated apoptosis.[19] Furthermore, PRMT5 supports constitutive CYCLIN D1/CDK4/6 activity leading to inactivation of the RBL2/E2F tumor suppressor pathway (8). Collectively, these data suggested that targeting PRMT5 may have a therapeutic value in a variety of cancers, including lymphoma.
PRMT5 overexpression has been observed in mantle cell lymphoma (MCL). In turn, PRMT5 inhibition has been shown to inhibit proliferation and/or induce cell death in MCL cell lines.[20] In most cases, wild type P53 was required for optimal anti-lymphoma activity of PRMT5 inhibitors. To-date, at least one PRMT5 inhibitor (GSK3326595) is currently being investigated in patients with relapsed solid tumors and NHL (NCT02783300).
Acknowledgement
This work is partially supported by NIH P50 CA192937 (MSK Lymphoma SPORE), the Leukemia and Lymphoma Society of America SCOR grant 7014–17, and by the NCI Cancer Center Support Grant (P30 CA008748),
Selected References
- 1.Younes A, Ansell S, Fowler N et al. The landscape of new drugs in lymphoma. Nat Rev Clin Oncol 2016. [DOI] [PMC free article] [PubMed]
- 2.Heyn H, Esteller M. EZH2: an epigenetic gatekeeper promoting lymphomagenesis. Cancer Cell 2013; 23: 563–565. [DOI] [PubMed] [Google Scholar]
- 3.Souroullas GP, Jeck WR, Parker JS et al. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat Med 2016; 22: 632–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bodor C, Grossmann V, Popov N et al. EZH2 mutations are frequent and represent an early event in follicular lymphoma. Blood 2013; 122: 3165–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Intlekofer AM, Joffe E, Batlevi CL et al. Integrated DNA/RNA targeted genomic profiling of diffuse large B-cell lymphoma using a clinical assay. Blood Cancer J 2018; 8: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Italiano A, Soria JC, Toulmonde M et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol 2018; 19: 649–659. [DOI] [PubMed] [Google Scholar]
- 7.Kuppers R IRAK4 inhibition to shut down TLR signaling in autoimmunity and MyD88-dependent lymphomas. J Exp Med 2015; 212: 2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ngo VN, Young RM, Schmitz R et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011; 470: 115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelly PN, Romero DL, Yang Y et al. Selective interleukin-1 receptor-associated kinase 4 inhibitors for the treatment of autoimmune disorders and lymphoid malignancy. J Exp Med 2015; 212: 2189–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hailfinger S, Lenz G, Ngo V et al. Essential role of MALT1 protease activity in activated B cell-like diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A 2009; 106: 19946–19951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dufner A, Schamel WW. B cell antigen receptor-induced activation of an IRAK4-dependent signaling pathway revealed by a MALT1-IRAK4 double knockout mouse model. Cell Commun Signal 2011; 9: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferch U, Kloo B, Gewies A et al. Inhibition of MALT1 protease activity is selectively toxic for activated B cell-like diffuse large B cell lymphoma cells. J Exp Med 2009; 206: 2313–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xin BT, Schimmack G, Du Y et al. Development of new Malt1 inhibitors and probes. Bioorg Med Chem 2016; 24: 3312–3329. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y, Mondello P, Erazo T et al. NOXA genetic amplification or pharmacologic induction primes lymphoma cells to BCL2 inhibitor-induced cell death. Proc Natl Acad Sci U S A 2018; 115: 12034–12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tron AE, Belmonte MA, Adam A et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat Commun 2018; 9: 5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kotschy A, Szlavik Z, Murray J et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016; 538: 477–482. [DOI] [PubMed] [Google Scholar]
- 17.Caenepeel S, Brown SP, Belmontes B et al. AMG 176, a Selective MCL1 Inhibitor, Is Effective in Hematologic Cancer Models Alone and in Combination with Established Therapies. Cancer Discov 2018; 8: 1582–1597. [DOI] [PubMed] [Google Scholar]
- 18.Li Y, Chitnis N, Nakagawa H et al. PRMT5 is required for lymphomagenesis triggered by multiple oncogenic drivers. Cancer Discov 2015; 5: 288–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berger SL. Out of the jaws of death: PRMT5 steers p53. Nat Cell Biol 2008; 10: 1389–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan-Penebre E, Kuplast KG, Majer CR et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol 2015; 11: 432–437. [DOI] [PubMed] [Google Scholar]