

Abstract
Histone demethylases play a critical role in mammalian gene expression by removing methyl groups from lysine residues in degree- and site-specific manner. To specifically interrogate members and isoforms of this class of enzymes, we have developed demethylase variants with an expanded active site. The mutant enzymes are capable of performing lysine demethylation with wild type proficiency, but are sensitive to inhibition by cofactor-competitive molecules embellished with a complementary steric ‘bump’. The selected inhibitors show more than 20-fold selectivity over the wild type demethylase, thus overcoming issues typical to pharmacological and genetic approaches. The mutant-inhibitor pairs are shown to act on a physiologically relevant full-length substrate. By engineering a conserved amino acid to achieve member-specific perturbation, this study provides a general approach for studying histone demethylases in diverse cellular processes.
Keywords: Histone demethylase, Small molecule inhibition, Bump-and-hole, Protein engineering, N-oxalylglycine
Graphical Abstract
Give me some space: Bulky residues in histone demethylase KDM4A are replaced with amino acids with smaller side chain. The extra space created in the active site can accommodate inhibitors with a steric ‘bump’ resulting in member-specific perturbation of the KDM family.
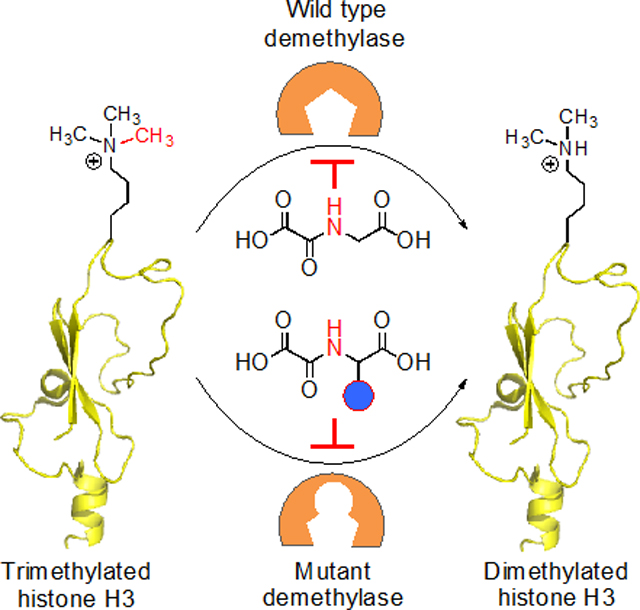
2-ketoglutarate (2KG) and iron-dependent lysine demethylases (KDMs) oxidize C-H bonds of a methyl group to demethylate cellular proteins (Figure 1).[1] Of particular importance is histone demethylation that regulates chromatin-dependent processes by modulating interactions between nucleosomes and transcriptional machinery.[2] Cells employ an array of KDMs (>30 in humans) to demethylate histones, a key step in the regulation of gene expression, in a wide range of biological pathways, including early embryonic development.[3] Lysine demethylation occurs in a site and degree-specific manner (mono-, di- and tri-demethylation) and can have opposing transcriptional outcomes. For example, demethylation of trimethylated K4 and K9 residues in histone H3 results in gene activation and repression, respectively, and two distinct sets of KDMs participate in these site-specific demethylation events: KDM5A-D act on H3K4me3 while KDM4A-E for H3K9me3.[4] Lysine demethylation of non-histone proteins is emerging as a key signaling mechanism for controlling cellular processes such as the cell cycle.[5] Small-molecule inhibitors carrying drug-like or peptide scaffolds have been developed to target KDMs and other 2KG-dependent enzymes.[6] Majority of the cofactor-competitive, metal-chelating inhibitors target a wide range of 2KG-dependent enzymes. Even for the substrate-competitive macrocyclic inhibitors,[7] achieving subtype specificity has been challenging. Such limitations resulting from off-target activity and occasional lack of cell-permeability call for new molecular tools to interrogate specific members of the KDM family.
Figure 1.
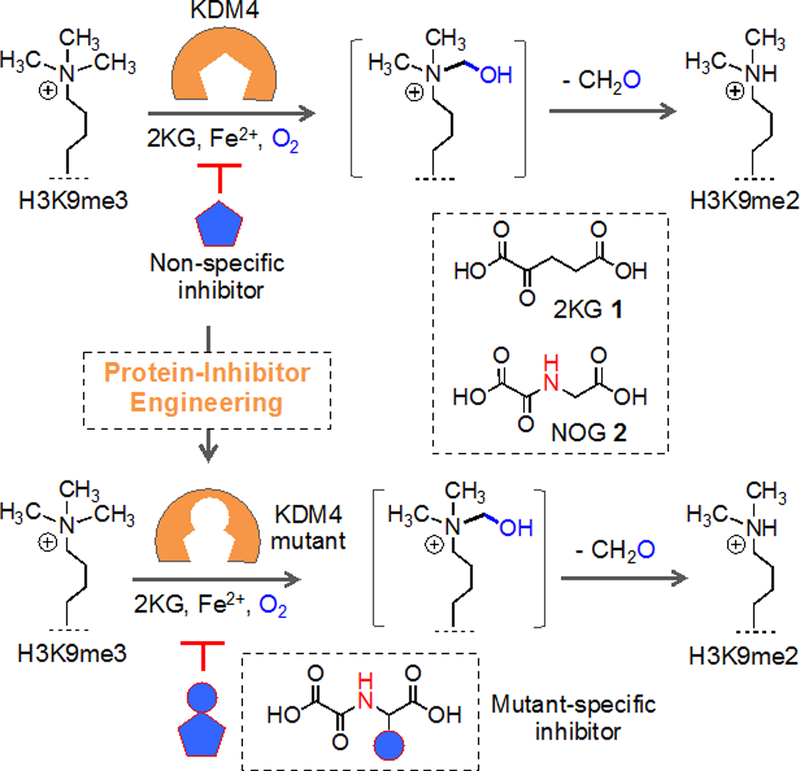
Schematic representation of analogue-sensitive histone demethylase approach. KDM4A, a representative member of the family, demethylates H3K9me3 to H3K9me2 using cofactor 2KG 1 to activate transcription. KDM4A is mutated to carry a ‘hole’ in the pocket while maintaining the catalytic activity. Cofactor-competitive inhibitor carrying complementary steric ‘bump’ specifically inhibits a mutant enzyme. N-oxalylglycine (NOG) 2 serving as inhibitor scaffold because of its structural similarity to 1.
Analogue-sensitive chemical genetics presents a distinctive solution employing orthogonal enzyme-cofactor or enzyme-inhibitor pairs that typically do not interfere with wild type proteins.[8] In this approach, a steric or electronic component (or both) of an enzyme-cofactor interface is engineered in a complementary manner to achieve target specificity while retaining the important attributes of small-molecule modulators (ability to provide conditional, dose-dependent and temporal perturbation). We recently developed KDM4 variants with expanded active sites that are capable of accepting 2KG analogues carrying a hydrophobic appendage for active removal of methyl groups from H3.[9] The ‘bumped’ 2KG analogues did not activate a series of wild type KDMs, thus ensuring specificity.
Given that the members of the KDM family play pivotal roles in mammalian development, single and combined knockouts are often lethal.[10] To elucidate biological functions of a specific isoform within a KDM subfamily, we surmised that KDM variants that act much like a wild type enzyme but are selectively inhibited by a designed molecule could constitute a powerful approach (Figure 1). Akin to analogue-sensitive kinase mutants,[11] the engineered KDMs are active with the natural cofactor, thus potentially circumventing the issues related to dominant negative effect typically observed for catalytically inactive variants. During the course of our engineering of KDM4A for mutant-specific activation, we identified N198A and S288A mutations with demethylase activity towards H3K9me3 peptide using 2KG as cofactor.[9] This initial observation provided us with the impetus to develop analogue-sensitive histone demethylases using KDM4A as a model enzyme.
Analysis of the crystal structure of KDM4A bound with 2-hydroxyglutarate (2HG), a 2KG-competitive inhibitor, revealed that N198 and S288 reside within a few angstroms from 2HG (Figure 2A).[12] Therefore, we generated additional mutants (N198G and S288G) to expand the active site further to test against a panel of inhibitors with varying degrees of steric appendages. We examined the ability of the mutants to demethylate the H3K9me3 substrate peptide using MALDI mass spectrometry. All four mutants exhibited robust demethylase activity towards the substrate peptide only in the presence of cofactor 2KG (Figure 2C–F). In these single time-point experiments, mutants performed a second-degree demethylation (removal of two methyl groups) as opposed to wild type KDM4A which typically removes only one methyl group within the allocated timeframe (Figure 2B). This is likely due to the expanded active site that allows the substrate peptide to adopt a catalytically competent orientation during each demethylation step. Furthermore, we observed that N198G is more active than it’s alanine counterpart, and an opposite trend was seen with the S288A and G mutants (Figure 2C–F). These results suggest that overall demethylase activity of the ‘hole-modified’ mutants is a manifestation of the nature of the stereoelectronic engineering during mutation (e.g. loss of an amide and a hydroxymethylene side chains in N198 and S288, respectively) and not entirely due to the larger active site space.
Figure 2.
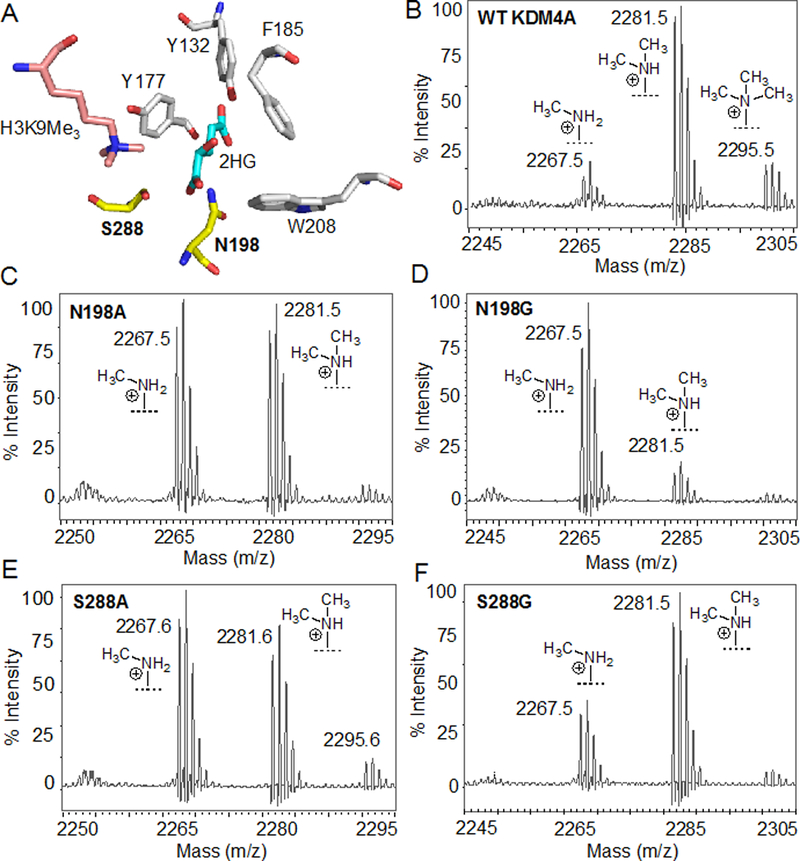
Engineering of KDM4A. (A) Crystal structure (PDB 2YBP) showing KDM4A active site bound with 2-hydroxyglutarate (2HG) and H3K9me3. N198 and S288 residues are engineered in the current study. (B-F) MALDI spectra for demethylase activity of wild type KDM4A (B), and N198A (C), N198G (D), S288A (E) and S288G (D) mutants towards H3K9me3 substrate peptide.
To gain further insight into catalytic activity of the mutants, we employed a coupled fluorescence assay to measure the amount of formaldehyde generated via KDM-mediated C-H hydroxylation of the methyl groups (Figure 3A).[12–13] In this assay, formaldehyde dehydrogenase converts the formaldehyde to formic acid using NAD+ as an oxidant that in turn is reduced to NADH. Time-dependent accumulation of NADH was measured during the demethylase assay with increasing concentration of 2KG (Figure S1). The assay determines the overall catalytic efficiency instead of individual mono- and di-demethylation (Figure 3B, S1). We observed a similar magnitude of KM values for all the enzymes with S288G being the tightest binder (KM ~6.9 μM) and N198G being the lowest (KM ~16 μM) in the series (Figure 3C). Interestingly, among the mutants, N198G had the maximal turnover (kcat) and the S288G mutant with the weakest, reflecting a complex interplay of substrate binding, transition-state stabilization, product release and potential product inhibition.
Figure 3.
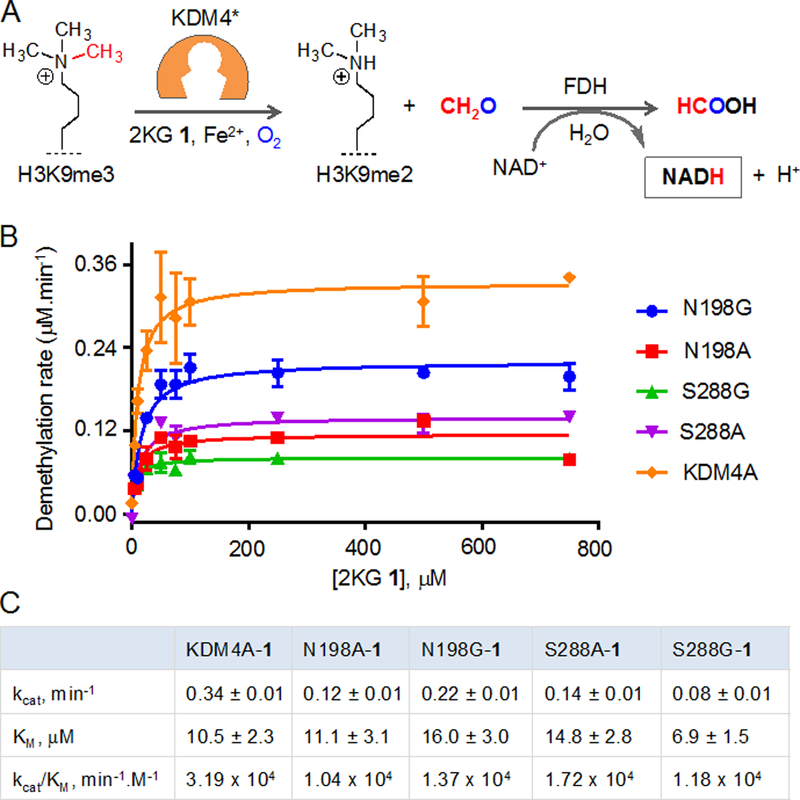
Catalytic efficiencies of the wild type and mutant KDM4A enzymes. (A) Schematic showing coupled fluorescence assay. Formaldehyde generated during demethylation of H3K9me3 peptide is oxidized to formic acid by FDH which in turn reduces NAD+ to NADH. (B) Michaelis-Menten plots for KDM4A, and its alanine and glycine mutants at N198 and S288 with increasing concentration of cofactor 2KG. (C) Steady-state kinetic parameters of the enzymes obtained from plots shown in B.
The overall catalytic proficiency (kcat/KM) of N198G mutant was higher than that of N198A, and this trend was reversed for the S288A and S288G mutants, thus corroborating well with our initial MALDI screening results. With respect to wild type system (KDM4A-2KG), the mutant-2KG pairs showed comparable kinetic proficiency. kcat/KM values for S288A and N198G are only 1.8 and 2.3 folds lower than that of wild type enzyme, respectively (Figure 3C). The catalytic prowess of these demethylase variants is remarkable and significantly higher than many reported engineered enzymes.[14] Collectively, kinetic analysis suggests that the selected KDM4A mutants are capable of performing rapid histone demethylation to support dynamic chromatin-templated processes such as transcription.
These results set the stage for identification of ‘bumped’ inhibitors for the ‘hole-modified’ mutants. In our initial MALDI assay, we used 1mM 2KG to detect the activity of wild type and mutant KDM4A. We realized that finding an effective inhibitor would be challenging at such a high concentration of cofactor due to the competitive nature of inhibition. Towards this end, we performed a series of MALDI demethylase assays with varied concentrations of 2KG (Figure S2–6). The results led us to select 50 μM of 2KG for subsequent inhibitory experiments. At this cofactor concentration, which is about 3–4 folds higher than the KM values for the five enzymes, we recorded greater than 50% substrate demethylation. It is noteworthy that the typical concentration of 2KG in human cells is in the range of ten to hundred micromolar.[15] Thus, screening compounds against the cofactor at the physiologically relevant concentration is likely to yield relevant inhibitors.
Upon optimizing the assay condition, we examined two cofactor-competitive KDM4A inhibitors: 5-carboxy-8-hydroxyquinone (IOX1) and N-oxalylglycine (NOG).[16] Both IOX1 and NOG are broad-spectrum inhibitors of 2KG-dependent enzymes with IC50 in the micromolar range (Figure 1, S7). IOX1 engages with KDM4A through the phenolic and carboxylic moieties by mimicking the carboxylic groups in 2KG. NOG binds to KDM4A due to its structural similarity to 2KG, but presence of the amido-acid moiety prevents its catalytic processing, resulting in cofactor-competitive inhibition. We tested wild type KDM4A and the four ‘hole-modified’ mutants against IOX1 and NOG under the optimized assay conditions. Both compounds inhibited the wild type as well as mutant proteins, albeit to different degrees, mirroring the ability of the engineered enzymes to bind 2KG (Figure S7).
These results prompted us to develop ‘bumped’ analogues of NOG to selectively inhibit the mutant enzymes. We focused on NOG-based scaffold mainly because of their structural similarity to the cofactor and straightforward synthetic access. Furthermore, we have recently demonstrated success in developing ‘bumped’ inhibitors for engineered DNA demethylases using bulky NOG derivatives.[17] We synthesized compounds 3-10 carrying variegated steric bulk at C4 and examined their inhibitory activity as described above (Figure 4A, Scheme S1). Although unmodified NOG 2 significantly inhibited all the enzymes tested, the ‘bumped’ compounds displayed a differential profile. For example, none of the analogues inhibited wild type KDM4A at 100 μM concentration, suggesting a lack of binding, likely due to a potential steric clash in the active site (Figure 4A, S8). N198A mutant was significantly inhibited by ethyl-NOG 3 and propyl-NOG 5, demonstrating that mutants with expanded active sites could accommodate bulky inhibitors (Figure 4A, S9). Consistently, N198G with a larger pocket was inhibited by leucine-NOG 7 carrying a bulky isobutyl group (Figure 4A, S10). Interestingly, we did not observe any significant inhibition of S288A and S288G mutants by the current set of NOG analogues (Figure 4A, S11–12). Given that the N198 and S288 occupy spatially different sites in the enzyme pocket (Figure 2A), it is likely that the NOG analogues could target the ‘hole’ generated by N198A/G mutations only. It would be of future interest to develop compounds based on other scaffolds such as IOX1 to inhibit mutations at S288 site. Nonetheless, screening of a small panel of compounds led us to identify three potential ‘bumped’ inhibitors for N198A and G mutants.
Figure 4.
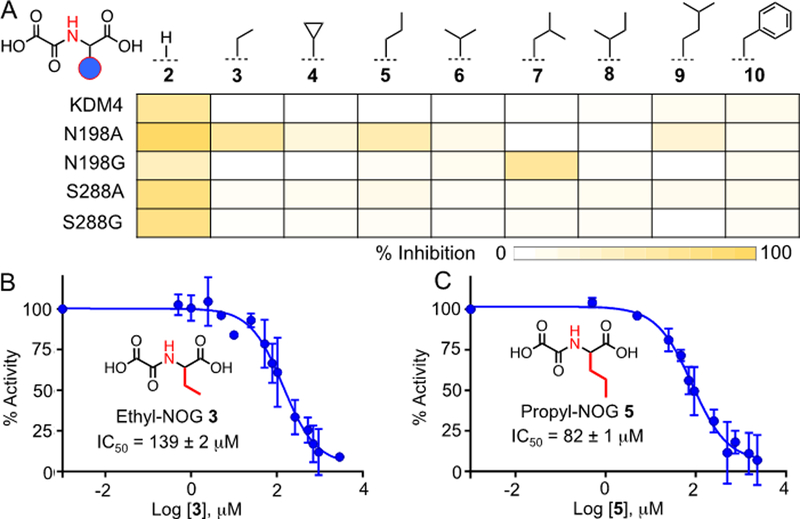
Inhibition of KDM4A and its mutants by NOG analogues. (A) Heat-map diagram of % inhibition of demethylase activity by 100 μM of NOG and its analogues as determined by MALDI-MS. (B-C) IC50 of ethyl-NOG 3 and propyl-NOG 5 towards N198A mutant.
To further validate the inhibition of N198A and N198G mutants by NOG analogues 3, 5 and 7, we measured their IC50 values using MALDI demethylase assay. In a dose-dependent experiment, ethyl-NOG 3 inhibited N198A mutant with an IC50 of 139 μM (Figure 4B, S13). The Hill slope value of −1.06 resembled the theoretical value of −1.0 for competitive inhibition, suggesting that the decrease in activity was not due to compound-mediated aggregation of the protein.[18] The inhibitory activity further improved with propyl-NOG 5 that showed an IC50 of 80 μM against the same mutant (Figure 4C, S14), suggesting a better fit for the inhibitor in the expanded pocket. N198G mutant was moderately inhibited by the leucine-NOG 7 with an IC50 of 288 μM. We did not observe any significant inhibition of wild type KDM4A by these ‘bumped’ NOG analogues at 300 μM concentrations (Figure S17–21). About 50% inhibition of KDM4A was observed at 3 mM concentration of ethyl-NOG 3, displaying at least 20-fold selectivity over the wild type enzyme (Figure S17, S19). Compound 5 demonstrated a significantly higher level of selectivity with no more than 30% inhibition of wild type KDM4A at 3 mM concentration (Figure S17, S20). In contrast, unmodified NOG 2 inhibited the wild type protein with an IC50 of 22 μM, closely agreeing with the reported value (Figure S17, S18).[12, 19] The results therefore validate the role of structure-guided protein and small molecule engineering to identify specific inhibitors for the engineered histone demethylases.
Finally, to examine whether the mutant proteins are active on full-length histone H3, we synthesized H3Kc9me3, a trimethylated thialysine analogue at position 9 of H3 via alkylation of the H3C9 mutant with 2-bromoethyltrimethyl ammonium salt (Figure 5A).[20] The reaction product was analyzed by ESI-HRMS (Figure 5B). Such chemically prepared histone analogues carrying site- and degree-specific lysine methylation pattern have been extensively used to test activity of chromatin-modifying proteins in biochemical assays.[21] The N198A mutant showed noticeable demethylase activity on the chemically methylated histone in the presence of 2KG 1 as confirmed by western blot with the appropriate antibody (Figure 5C,D). We observed that NOG analogue 3 significantly inhibited the activity of the mutant demethylase (Figure 5C,D). These results collectively demonstrate that the ‘hole-modified’ mutant and its cognate bumped’ inhibitor are capable of controlling lysine demethylation on full-length histone substrate.
Figure 5.
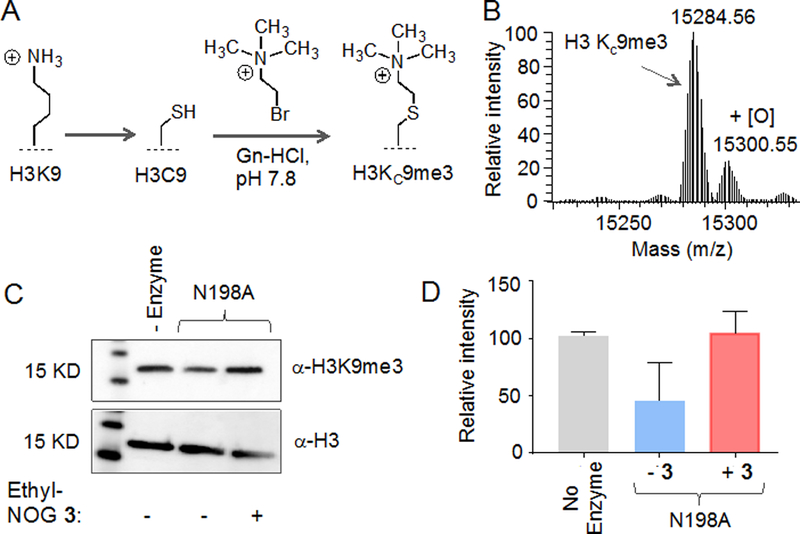
Activity of mutant enzymes on full-length histone H3. (A) Schematic showing chemically methylated histone H3 using thialysine approach. (B) ESI-HRMS spectrum of H3KC9me3. (C) Demethylation of full-length H3KC9me3 by N198A mutant as determined by western blot using H3K9me3 antibody (top panel); Histone H3 as loading control (bottom panel). Demethylation was significantly inhibited by the ethyl-NOG 3. (D) Quantitative representation of the western blot data shown in C. Error bars are based on two independent biological replicates.
In this piece of work, we have developed histone demethylase variants carrying an expanded active site via structure-guided protein engineering with the purpose of perturbing a specific mutant by a designed molecule. We showed that engineered enzymes are capable of demethylating histone peptide using the natural cofactor 2KG. Steady-state kinetic analysis employing a coupled fluorescence assay revealed that the mutants are catalytically competent similar to their wild type counterpart. This is particularly important given that loci-specific histone demethylation occurs within minutes upon appropriate external stimuli to effect gene expression.[22] Catalytically efficient engineered KDM4A is expected to perform rapid demethylation events in response to cellular and developmental cues. To specifically interrogate the mutant demethylases, we screened two cofactor-competitive inhibitor scaffolds and found both to be effective. In a subsequent structure-activity study with a panel of eight NOG analogues, we identified NOG analogues 3 and 5 carrying an ethyl and a propyl ‘bump’, respectively, as selective inhibitors for N198A mutant of KDM4A. Although the inhibitory potency of the compounds is moderate, it displayed more than 20-fold selectivity over wild type demethylase. These results underscore the importance of the complementary steric engineering at the protein-ligand boundary to achieve selectivity. We also confirmed the potential of the orthogonal enzyme-inhibitor pair to conditionally modulate methylation level on full-length histone.
Looking to the future, we will expand the collection of NOG-based molecules to identify inhibitors with improved potency. We envision expanding the active site further via a second site mutagenesis to accommodate bulkier analogues for higher selectivity. Given that N198 and S288 residues are highly conserved among KDM4 members, we expect the NOG analogues developed here to find general application as inhibitors of the engineered members in the subfamily. More importantly, majority of the lysine demethylases (>30 in humans) rely on 2KG for catalysis, thus suggesting a potential broad applicability of our engineering approach. We anticipate future experiments involving knock-in of the analogue-sensitive KDM4A mutant into the endogenous loci using CRISPR-cas9 based genome-editing techniques.[23] Cell-permeable ‘bumped’ NOG analogues in combination with ‘hole-modified’ KDMs would allow temporal manipulation of histone methylation and subsequent gene expression in a manner not attainable by currently available pharmacological and genetic tools.
Experimental Section
Synthetic procedure and characterization of all the relevant compounds; methods for protein expression and purification, and biochemical assays; supplementary figures, tables and NMR spectra are provided in Supporting Information.
Supplementary Material
Acknowledgements
We thank the University of Pittsburgh, the National Science Foundation (MCB-1817692) and the National Institutes of Health (R01GM123234) for financial support; Profs. A. Bhagwat, D. Fujimori, M. Luo, R. Trievel for the expression constructs used in the current study; Dr. D. Chakraborty and members of our laboratory for critical reading and editing of the manuscript. S. Arora is Andrew Mellon fellow. Support for MALDI-TOF MS instrumentation was provided by a grant from the National Science Foundation (CHE-1625002).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Loenarz C, Schofield CJ, Nat. Chem. Biol. 2008, 4, 152–156. [DOI] [PubMed] [Google Scholar]
- [2].Klose RJ, Kallin EM, Zhang Y, Nat. Rev. Genet. 2006, 7, 715–727. [DOI] [PubMed] [Google Scholar]
- [3].(a) Gerace EL, Moazed D, Mol. Cell 2010, 40, 683–684; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Das PP, Shao Z, Beyaz S, Apostolou E, Pinello L, De Los Angeles A, O’Brien K, Atsma JM, Fujiwara Y, Nguyen M, Ljuboja D, Guo G, Woo A, Yuan GC, Onder T, Daley G, Hochedlinger K, Kim J, Orkin SH, Mol. Cell 2014, 53, 32–48; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Black JC, Allen A, Van Rechem C, Forbes E, Longworth M, Tschop K, Rinehart C, Quiton J, Walsh R, Smallwood A, Dyson NJ, Whetstine JR, Mol. Cell 2010, 40, 736–748. [DOI] [PubMed] [Google Scholar]
- [4].(a) Torres IO, Kuchenbecker KM, Nnadi CI, Fletterick RJ, Kelly MJ, Fujimori DG, Nat. Commun. 2015, 6, 6204; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, Shi Y, Cell 2006, 125, 467–481. [DOI] [PubMed] [Google Scholar]
- [5].Cho HS, Suzuki T, Dohmae N, Hayami S, Unoki M, Yoshimatsu M, Toyokawa G, Takawa M, Chen T, Kurash JK, Field HI, Ponder BA, Nakamura Y, Hamamoto R, Cancer Res. 2011, 71, 655–660. [DOI] [PubMed] [Google Scholar]
- [6].Thinnes CC, England KS, Kawamura A, Chowdhury R, Schofield CJ, Hopkinson RJ, Biochim. Biophys. Acta 2014, 1839, 1416–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kawamura A, Munzel M, Kojima T, Yapp C, Bhushan B, Goto Y, Tumber A, Katoh T, King ON, Passioura T, Walport LJ, Hatch SB, Madden S, Muller S, Brennan PE, Chowdhury R, Hopkinson RJ, Suga H, Schofield CJ, Nat. Commun. 2017, 8, 14773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].(a) Islam K, ACS Chem. Biol. 2015, 10, 343–363; [DOI] [PubMed] [Google Scholar]; (b) Lopez MS, Kliegman JI, Shokat KM, Methods Enzymol. 2014, 548, 189–213. [DOI] [PubMed] [Google Scholar]
- [9].Breski M, Dey D, Obringer S, Sudhamalla B, Islam K, J. Am. Chem. Soc. 2016, 138, 13505–13508. [DOI] [PubMed] [Google Scholar]
- [10].Pedersen MT, Kooistra SM, Radzisheuskaya A, Laugesen A, Johansen JV, Hayward DG, Nilsson J, Agger K, Helin K, EMBO J. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].(a) Bishop AC, Buzko O, Shokat KM, Trends Cell Biol. 2001, 11, 167–172; [DOI] [PubMed] [Google Scholar]; (b) Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, Wood JL, Morgan DO, Shokat KM, Nature 2000, 407, 395–401. [DOI] [PubMed] [Google Scholar]
- [12].Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, Leung IK, Li XS, Woon EC, Yang M, McDonough MA, King ON, Clifton IJ, Klose RJ, Claridge TD, Ratcliffe PJ, Schofield CJ, Kawamura A, EMBO Reports 2011, 12, 463–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].(a) Roy TW, Bhagwat AS, Nucleic Acids Res. 2007, 35, e147; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Krishnan S, Collazo E, Ortiz-Tello PA, Trievel RC, Anal. Biochem. 2012, 420, 48–53. [DOI] [PubMed] [Google Scholar]
- [14].Shah K, Liu Y, Deirmengian C, Shokat KM, Proc. Natl. Acad. Sci. USA 1997, 94, 3565–3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pritchard JB, J. Pharm. Exp. Therap. 1995, 274, 1278–1284. [PubMed] [Google Scholar]
- [16].(a) Hopkinson RJ, Tumber A, Yapp C, Chowdhury R, Aik W, Che KH, Li XS, Kristensen JB, King ON, Chan MC, Yeoh KK, Choi H, Walport LJ, Thinnes CC, Bush JT, Lejeune C, Rydzik AM, Rose NR, Bagg EA, McDonough MA, Krojer T, Yue WW, Ng SS, Olsen L, Brennan PE, Oppermann U, Muller-Knapp S, Klose RJ, Ratcliffe PJ, Schofield CJ, Kawamura A, Chem. Sci. 2013, 4, 3110–3117; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McDonough MA, McNeill LA, Tilliet M, Papamicael CA, Chen QY, Banerji B, Hewitson KS, Schofield CJ, J. Am. Chem. Soc. 2005, 127, 7680–7681. [DOI] [PubMed] [Google Scholar]
- [17].Sudhamalla B, Wang S, Snyder V, Kavoosi S, Arora S, Islam K, J. Am. Chem. Soc. 2018, 140, 10263–10269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fallahi-Sichani M, Honarnejad S, Heiser LM, Gray JW, Sorger PK, Nat. Chem. Biol. 2013, 9, 708–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rose NR, Ng SS, Mecinovic J, Lienard BM, Bello SH, Sun Z, McDonough MA, Oppermann U, Schofield CJ, J. Med. Chem. 2008, 51, 7053–7056. [DOI] [PubMed] [Google Scholar]
- [20].Simon MD, Chu F, Racki LR, de la Cruz CC, Burlingame AL, Panning B, Narlikar GJ, Shokat KM, Cell 2007, 128, 1003–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Shiau C, Trnka MJ, Bozicevic A, Ortiz Torres I, Al-Sady B, Burlingame AL, Narlikar GJ, Fujimori DG, Chem. Biol. 2013, 20, 494–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].(a) Ingvarsdottir K, Edwards C, Lee MG, Lee JS, Schultz DC, Shilatifard A, Shiekhattar R, Berger SL, Mol. Cell. Biol. 2007, 27, 7856–7864; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Radman-Livaja M, Liu CL, Friedman N, Schreiber SL, Rando OJ, PLoS Genet. 2010, 6, e1000837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].(a) Bartkowiak B; Yan C; Greenleaf AL Biochim. Biophys. Acta 2015, 1849, 1179 Moyer, T. C.; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Holland AJ Methods Cell Biol. 2015, 129, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.