

Abstract
The growing prevalence of multi-antibiotic-resistant bacteria necessitates looking at potential alternative approaches for attenuating infections by bacteria while reducing the rate of antibiotic resistance development. Enterococcus faecalis is responsible for a large percentage of clinical enterococci infections and its pathogenicity has been demonstrated to be influenced by quorum sensing (QS). In this study, we report the systematic study of the relationship between backbone hydrogens and the ability to activate the FsrC receptor. We demonstrate that N-methylation was particularly well-tolerated at one site (Phe7) and granted stability against protease digestion, increasing the peptide half-life relative to the native signal by more than 6-fold. The inclusion of the N-Me-Phe7 modification may be useful for improving the pharmacological properties of E faecalis QS inhibitors as part of the development of future therapeutic candidates.
Keywords: Quorum Sensing, Enterococcus faecalis, Gelatinase Biosynthesis-Activating Pheromone, Metabolic Stability, Peptidomimetics
Graphical Abstract
Enterococcus faecalis is an opportunistic pathogen present in the gut biome of humans, and accounts for more than half of all enterococci infections, including nosocomial endocarditis, neonatal infections, and urinary tract infections.1, 2 E. faecalis is intrinsically resistant to some antibiotics, including vancomycin, an antibiotic of last resort.1, 3 Moreover, E. faecalis can transfer its resistance genes to other bacteria through a pheromone-responsive gene transfer system.3, 4 Therefore, there is an urgent need to discover non-antibiotic therapeutics for E. faecalis infections. The evolutionary pressure to evolve against therapeutics can be lessened by targeting pathways that are not required for cell survival. Such a strategy will decrease the rate by which antibiotic resistance is acquired while still attenuating infections by reducing the organism’s pathogenicity.
In order to effectively attack their host and establish an infection, E. faecalis bacteria communicate with one another via quorum sensing (QS).5, 6 Gram-positive bacteria, like E. faecalis, release peptide signals into the environment at a constant rate.6 Once an individual cell detects that the environmental concentration of that peptide has met a species-dependent threshold concentration, the transcription of genes involved in group-beneficial traits, such as virulence factor production, antimicrobial agent production, digestive enzyme secretion, bioluminescence and biofilm formation, is activated.5–7 Because of the role QS plays in bacterial pathogenicity, this area has gained attention as a method to attenuate bacterial infections.8–11
The fsr QS circuit in E. faecalis relies on the production and response to an 11-amino-acid macrocyclic signaling peptide known as gelatinase biosynthesis-activating pheromone (GBAP).5, 12 GBAP contains an ester-linked macrocycle between the side chain of Ser3 and the C-terminus of Met11.12 GBAP activates a membrane-bound histidine kinase receptor, FsrC. Receptor activation leads to phosphorylation of the response regulator, FsrA. Phosphorylated FsrA acts as a transcription factor that induces the transcription of the fsrABDC, gelE, and sprE genes, directly resulting in autoinduction of the QS circuitry, gelatinase production, and serine protease production, respectively.1, 5, 13–15 Gelatinase directly affects E. faecalis adherence and biofilm formation, thus affecting processes such as translocation across intestinal epithelial cells, while SprE, a serine protease, has been implicated in similar processes.1, 14, 16–18
We have previously investigated the relative importance of each of GBAP’s residues on QS activation by modifying their functional groups (alanine scan) and chirality (D-amino acid scan).19 Information gained from this earlier work, combined with work done by others,20 enabled us to rationally design potent activators and inhibitors of the fsr QS circuit.21 Although these investigations have resulted in a greatly increased understanding of the structure-activity relationships (SARs) involved in QS activation, additional questions remained. For instance, the role of backbone interactions in GBAP activity had yet to be elucidated. In this study we utilized an N-methylation scan approach to systematically evaluate the importance of amide protons in GBAP activity.22–24 Through this analysis we identified two N-methyl modifications of interest in the design of potential therapeutics. The first produces an analog that is a weak antagonist, while the second produces an analog with significantly improved stability compared to the native signal molecule while retaining its potency. The activator modification improved the stability to protease degradation when incorporated into the parent scaffold, suggesting that its inclusion in future peptide designs could potentially lead to peptides with enhanced pharmacological properties.
Results and Discussion
Synthetic Strategies
Although we have demonstrated that tail modifications, i.e. modifications made to the two exocyclic residues, can be used to enhance the potency of GBAP analogs, we have also shown that the tail residues are not required to retain the activity of native GBAP.19 As such, all peptides investigated here have the tail residues removed and replaced with an acetyl capping group. Most peptides were produced using our previously published entirely on-resin synthetic approach19, 21 with the incorporation of Fmoc-protected N-methylated amino acids at the appropriate point in the synthesis. However, two analogs required modification to their protocols in order to be synthesized successfully.
The first of these two was the N-MeS3 analog (3). Our synthetic strategy relies on the selective deprotection of the Ser3 side chain in order to form the lactone linkage in the completed macrocycle. However, the Boc protecting group commonly utilized in commercial N-methylated amino acids cannot be removed selectively. Thus, in order to prepare this analog, we used Fmoc-Ser(Trt)-OH and methylated the serine on solid support after its addition to the peptide chain. This approach was successful with some modification of an existing protocol (see the SI for details).25
The second peptide that required an altered protocol was N-MeM11 (10). Because M11 participates in the lactone linkage in the final macrocycle, and because a cis conformation is favored following an N-methylated amino acid,26 the labile ester forming the macrocycle bridge between Ser3 and Met11 is well positioned for attack by the primary amine of the subsequent amino acid (tryptophan10). This attack is stabilized by a six-membered ring intermediate and leads to loss of the methionine-tryptophan dipeptide in the form of a diketopiperazine (DKP), causing a significant drop in yield as soon as the tryptophan’s Fmoc protecting group is removed. In order to address this issue, we prepared a dipeptide composed of tryptophan and the amino acid that follows it in the synthesis (glutamine9). By coupling this dipeptide to methionine following ester formation, we avoid the undesirable DKP formation. The details of the Fmoc-Gln(Trt)-Trp(Boc)-OH preparation on chlorotrityl resin and subsequent purification can be found in the SI. Table 1 shows the peptides prepared in this study and their EC50 values.
Table 1.
EC50 Values for N-methylated GBAP Analogsa
| Peptide # | Peptide Name | Sequence | EC50b [95% CI]c (nM) |
|---|---|---|---|
| 1 | GBAPd | QN(SPNIFGQWM) | 1.15 [0.825 – 1.59] |
| 2 | Ac-GBAP-Des(Q1N2)d | Ac-(SPNIFGQWM) | 1.01 [0.496 – 2.06] |
| 3 | Ac-GBAP-Des(Q1N2)[N-MeS3] | Ac-([NMeS3]PNIFGQWM) | -- |
| 4 | Ac-GBAP-Des(Q1N2)[N-MeN5] | Ac-(SP[NMeN5]IFGQWM) | > 1,000 |
| 5 | Ac-GBAP-Des(Q1N2)[N-MeI6] | Ac-(SPN[NMeI6]FGQWM) | > 1,000 |
| 6 | Ac-GBAP-Des(Q1N2)[N-MeF7] | Ac-(SPNI[NMeF7]GQWM) | 1.92 [1.46 – 2.54] |
| 7 | Ac-GBAP-Des(Q1N2)[N-MeG8] | Ac-(SPNIF[NMeG8]QWM) | -- |
| 8 | Ac-GBAP-Des(Q1N2)[N-MeQ9] | Ac-(SPNIFG[NMeQ9]WM) | -- |
| 9 | Ac-GBAP-Des(Q1N2)[N-MeW10] | Ac-(SPNIFGQ[NMeW10]M) | 322 [186 – 557] |
| 10 | Ac-GBAP-Des(Q1N2)[N-MeM11] | Ac-(SPNIFGQW[NMeM11]) | -- |
| 11 | GBAP-[N-MeF7] | QN(SPNI[NMeF7]GQWM) | 6.30 [2.33 – 17.1] |
| 12 | Linear GBAP | QNSPNIFGQWM | -- |
N-Methylation Structure-Activity Relationship (SAR) Results
The SAR data for the N-methylated analogs were collected using our β-galactosidase cell-based bacterial reporter assay. Briefly, quantification of QS activation is measured in an E. faecalis strain bearing a plasmid containing pGelE-lacZ. Upon activation of the fsr QS circuitry, FsrA will bind pGelE and transcribe lacZ. Thus, β-galactosidase activity can be used to monitor QS response (full experimental details can be found in the SI).19, 21 Overall, the majority of N-methyl analogs exhibited significant reduction in activity, emphasizing the important role backbone protons have in GBAP activity, either through hydrogen-bond interactions with the receptor or by allowing conformational flexibility compared to the bulky methyl group (Table 1). Starting with the N-terminus, N-MeS3 (3) was found to be inactive as an activator, and to have weak inhibitory activity (the signal of 50 nM competitor GBAP was reduced to around 60% at 10 μM of 3, the highest concentration tested). These results suggest that N-methylation at the Ser3 position could potentially enhance the inhibitory propensity of existing GBAP-based fsr inhibitors. Analogs 4 and 5 both had large drops in potency compared to native GBAP, with EC50 values greater than 1,000 nM. Similarly to 3, analogs 7, 8, and 10 lost all activity. However, neither of these three analogs exhibit any inhibitory activity. Analog 9 also exhibited a substantial decrease in activity, but had a measurable EC50 value of 322 nM (Table 1).
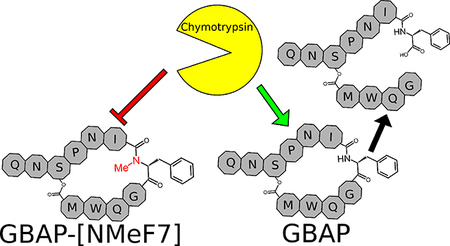
Interestingly, the N-MeF7 analog (6) was quite potent. With an EC50 value of 1.92 nM, it was comparably potent to native GBAP. This result reinforces the unique role the F7 position has in GBAP activity as this residue was found to be critical for activity (replacement with alanine resulted in a relatively inactive analog, GBAP-F7A, EC50 > 1000 nM),19 yet was also the site previously found to retain potency when its chirality was inverted or when a larger amino acid was substituted.19, 21 This is especially interesting since Phe7 is a potential cleavage site for degradation by chymotrypsin.27 Methylation at this site could therefore be applied to protect against degradation while simultaneously maintaining binding characteristics, allowing for the design of better drug leads. Due to the low solubility of the acetylated form of this analog (6), and to test that this modification can be successfully added to the native peptide (which would indicate potential effectiveness in other analogs as well), we prepared GBAP-N-MeF7 (11). This analog retains the native tail residues and lacks an acetyl capping group. As expected, it was found to have similar potency to the tail-removed analog (3-fold reduction), while having significantly improved solubility.
Stability Assays
In order to evaluate whether N-methylation at F7 makes the analog more resistant to degradation by proteases, we conducted stability assays to assess the length of time that the peptide remains intact when exposed to chymotrypsin or human plasma. We tested native GBAP (1) and linearized GBAP (12) as active and inactive controls whose stability can be compared with 11.
In order to get a baseline for the peptide stability, we tested native GBAP in phosphate-buffered saline (PBS) at pH 7.2 and ammonium carbonate at pH 8 without the addition of proteases at 25 °C. Since we have previously observed that GBAP’s terminal tail glutamine spontaneously degrades to pyroglutamate,19 we wanted to be able to differentiate between this product and any others that might be observed. These experiments also addressed the feasibility of conducting structural studies with GBAP with confidence that degradation would not occur during data collection. We found that native GBAP undergoes rapid chemical degradation in the pH 8 buffer at 25 °C, involving not only conversion of the N-terminal glutamine to pyroglutamate, but also slower opening of the macrocyclic ring due to hydrolysis of the ester linkage. Complete conversion to a ring-opened product containing the pyroglutamic residue was seen within 24 hours at 25 °C. At pH 7.2, GBAP degradation was much slower at 25 °C. Approximately 6% of the native GBAP had degraded after 24 hours. No ring opening was observed during those first 24 hours in the pH 7.2 conditions at 25 °C.
Due to the chemical instability of GBAP in the pH 8 buffer, we conducted the protease experiments in the pH 7.2 PBS buffer system at 37 °C. This pH (7.2) also mimics better physiologically relevant conditions such as serum and plasma. We tested and compared GBAP, 11, and 12 in the absence and presence of chymotrypsin (approximately 50 nM), an important protease of the digestive system where E. faecalis naturally resides, to assess their resilience against enzymatic degradation. Native GBAP chemically degraded much faster in the pH 7.2 buffer at the elevated temperature (37 °C vs. 25 °C) with a half-life of approximately 4 hours (at 24 hours, GBAP had completely degraded). When GBAP was treated with chymotrypsin, significantly different degradation product peaks were produced compared to the normal degradation, indicating that the peptide was indeed being cleaved by the protease (Figures 1 & 2). Analog 12 was hypothesized to have similar or better chemical stability to GBAP (due to the lack of the ester bond found in GBAP’s lactone ring), but was expected to have increased susceptibility to chymotrypsin compared to the other two macrocyclic peptides.
Figure 1.

Comparison of GBAP in PBS at 37 °C without (A and B) and with (C and D) treatment with 50 nM chymotrypsin. Full analytical HPLC spectra (A and C) are compared with zoomed views (B and D) of the same respective spectra. Solid blue lines indicate the GBAP traces prior to incubation. Dashed red lines indicate the GBAP traces after 8 hours of incubation. In both cases, the half-life for GBAP is approximately 4 hours, however the difference in products indicates that chymotrypsin can successfully digest GBAP.
Figure 2:

Degradation trends in PBS at 37 °C without (A) and with (B) treatment with 50 nM chymotrypsin. GBAP is shown in blue with circles, linear GBAP is shown in green with triangles, and GBAP-N-MeF7 is shown in orange with squares.
Indeed, 12 was chemically much more stable in the pH 7.2 buffer at 37 °C, with a half-life of more than 24 hours (approximately 70% remained after 24 hours). These results suggest that the conformation adopted by the macrocycle may help orient the tail residues to facilitate formation of the observed pyroglutamate degradation product. However, as predicted, 12 was much more susceptible to degradation by chymotrypsin with a half-life of only 2 hours (Figure 2). 11 was very stable both in PBS buffer alone or in the presence of chymotrypsin with a similar amount remaining after 24 hours in both cases. The results indicate that N-methylation at F7 induces remarkable stability against chymotrypsin in comparison to native GBAP. 11 had a half-life greater than 24 hours (with approx. 80–90% peptide remaining undigested at the 24-hour time point) when treated with chymotrypsin (Figure 2). These results further suggest that N-methylation at F7 results in conformational changes that make the formation of the pyroglutamate product less favored, while still allowing the peptide to retain its activity.
Lastly, we tested GBAP and 11 for their stability in human plasma. Overall, degradation in plasma was significantly faster for both analogs. However, a similar stability trend was observed, that is N-methylation at Phe7 resulted in an analog with higher resistance to degradation. GBAP exhibited very low stability in plasma with a half-life significantly lower than 30 min (first time point, < 4% peptide remaining) and complete degradation in 1 hour. GBAP-N-MeF7 (11) was more stable in plasma with a half-life just below 30 min (~30% peptide remaining) and complete degradation only after 2 hours. The plasma stability results again highlight the utility of incorporating N-methylation as a means to improve the pharmacological properties of lead peptide scaffolds.
Conclusions
The N-methyl scan revealed several amide protons (S3, G8, Q9, and M11) that are vital for GBAP activity, either by allowing the peptide to adopt its active conformation or through direct interactions with the receptor. In addition, N-methylation at several other sites (N5, I6, and W10) significantly reduced potency, although agonist activity remained. Only F7 tolerated N-methylation, with only a minimal loss in potency.
The tolerance to modification at F7 proved advantageous as F7 is a likely cleavage location if the peptide is recognized by chymotrypsin. N-methylation at F7 appeared to nearly eliminate cleavage by chymotrypsin, increasing the half-life of native GBAP from about 4 hours in the presence of the same concentration of chymotrypsin to well over 24 hours, as well as improve the peptide stability in human plasma. Combining the increased stability with the low impact that the modification has on the peptide activity has revealed a promising strategy for improving the enzymatic stability of future E. faecalis fsr QS modulators.
Experimental Section
General
All peptides were purified to ≥ 95% as confirmed by analytical reverse phase high-performance liquid chromatography (RP-HPLC). See Table S-1 for individual peptide purities.
Peptide Synthesis
Entirely on-resin fluorenylmethyloxycarbonyl (Fmoc)-based peptide synthesis28–30 was conducted as we previously described (see SI for full details). Due to the reduced nucleophilicity of N-methylated N-termini, longer coupling times and occasionally elevated temperature was necessary to attach the next amino acid. All amino acids were coupled for 4 h at room temperature. Synthesis continued if the chloranil test (see SI) was found to be negative, otherwise, the attachment was reattempted, but ran overnight at 50 °C.19
Serine was N-methylated on the growing peptide chain using an adapted procedure.25 The peptide was synthesized using our standard procedures up until after the removal of Fmoc from the Ser(Trt) residue. 4 equiv of ortho-nitrobenzenesulfonyl chloride (o-NBS chloride) was dissolved in 1 to 2 mL of DMF. 10 equiv of 2,4,6-Collidine was added and the mixture was shaken for 1 min. The resulting solution was then added to the resin and shaken for 15 min. This process was repeated once more before the resin was washed 5 times with DMF. This was sufficient to mono-protect the resin as indicated by a positive chloranil test result and a mostly-negative Kaiser test result (roughly 10% of the assayed resin beads still stained blue). 6 equiv of DBU was then dissolved in 1–2 mL of DMF, added to the resin, and shaken for 10 min. Approximately 20–25 equiv of dimethylsulfate (DMS) was added and the resulting mixture shaken for 1 hr. The resin was briefly washed and this process was repeated once more followed by washing with DMF 5 times shaking for 2 min each. After this second treatment, both the Kaiser and chloranil tests were negative indicating methylation of all available secondary and primary amines. To remove o-NBS, 10 equiv of β-mercaptoethanol was dissolved in 1–2 mL of DMF along with 5 equiv of DBU and added to the resin and shaken for 30 min. This process was repeated once, and the resin washed 5 times with DMF shaking for 2 min each. After these steps the Kaiser test was negative, but the chloranil test was positive, indicating successful mono-methylation and o-NBS removal.
The synthesis of the peptide containing the N-MeM11 modification required the synthesis of the dipeptide Fmoc-Gln(Trt)-Trp(Boc)-OH to avoid reaction of the N-terminal amine of tryptophan with the macrocycle ester to form the corresponding diketopiperazine and peptide truncation at the serine branching point. In order to retain the protecting groups, the dipeptide was prepared using chlorotrityl resin as the solid support. First, the chlorotrityl resin was dried under vacuum for 48 h. 1.1 eq (relative to the manufacturer’s reported loading for the resin) of Fmoc-Trp(Boc)-OH was dissolved in dry DCM and 2.5 eq of DIPEA was added. This solution was added to the resin in a 50 mL falcon tube (the screw-top cap prevents the cap popping off from increased pressure when shaking DCM solutions) and shaken for 1 h. 1 mL of methanol was added and the resin shaken for an additional 10 min. The resin slurry was decanted into a 20 mL solid phase reaction vessel and washed thoroughly three times each with DMF, DCM, and methanol. The resin was then dried under reduced pressure and the loading of the resin determined as describe above. The resin was allowed to stand in DCM for 30 min to swell. 3 equiv of Fmoc-Gln(Trt)-OH and 2.85 eq of HATU were dissolved in DMF and 4 eq of DIPEA was added. The resulting solution was added to the resin and shaken overnight (approx. 18 h). The dipeptide was cleaved from the resin by treating with 1% TFA and 5% TIPS in DCM 4–5 times for two min. The resin was then washed 3 times with the same cleavage solution and 5 times with DCM. The combined cleavage and wash fractions were then washed 1X with an equivalent volume of basic brine (approx. 250 mg/mL sodium bicarbonate saturated with sodium chloride), the DCM was dried over magnesium sulfate for approx. 2 hr. The dried solution was filtered, and the DCM removed under reduced pressure. The residue was then dissolved in acetonitrile and water and lyophilized for at least 18 hr. The resulting residue was then dissolved in the minimum amount of DCM and passed through a silica plug packed using 30% MeOH in DCM and eluted with the same solvent mixture. Fractions containing the product were verified with thin layer chromatography (TLC), combined, and the solvent removed under reduced pressure. The residue was dissolved in acetonitrile and water and lyophilized a second time. This process yielded the desired, still protected dipeptide in ≥ 80% yield. The dipeptide was then treated the same as any other amino acid for the coupling step following N-MeM11 attachment (see above).
Peptide Purification and Verification
Peptide purification by RP-HPLC and verification by mass spectrometry (MS) were conducted as previously described (see SI).19
β-Galactosidase Reporter Assay
The ability of the analogs to activate the QS circuit was evaluated as we previously described (see SI for full experimental details).19, 21
Chemical Stability Assays
Peptide solutions were prepared at 0.28 mM concentration in 1 mL of either PBS buffer (pH 7.2) or ammonium carbonate buffer (pH 8) and incubated at either 25 °C or 37 °C for 24 hours. Time points were taken at 0 min, 30 min, 1 hr, 2 hr, 4 hr, 8 hr, and 24 hr. At each time point, a 100 μL aliquot was taken and mixed with 20 μL of ACN. 90 μL was then injected via an autosampler and an analytical trace taken (see Purification by HPLC section above for details on the analytical method). Traces were overlaid for comparison and the percent of remaining peptide was calculated by comparing the integrated area under the undigested peak in each trace with that of the undigested peak in the 0 min time point trace (which was normalized to 100%). Note that due to the very low chemical stability of GBAP in the ammonium carbonate buffer, all other peptides were only tested in the PBS buffer conditions.
Chymotrypsin Stability Assays
Due to the low chemical stability of GBAP in ammonium carbonate buffer (pH 8), the chymotrypsin degradation assays were conducted in PBS buffer (pH 7.2). A 1 mL, 0.28 mM peptide solution was prepared in the buffer. 5 μL of 250 μg/mL chymotrypsin solution was added, giving a final chymotrypsin concentration of approximately 50 nM. The solution was incubated at 37 °C for 24 hours. Time points were taken at 0 min, 30 min, 1 hr, 2 hr, 4 hr, 8 hr, and 24 hr. At each time point, a 100 μL aliquot was taken and mixed with 20 μL of ACN. 90 μL was then injected via an autosampler and an analytical trace taken (see Purification by HPLC section above for details on the analytical method). Trace analysis was conducted similarly to the chemical stability assays.
Human Plasma Stability Assays
Stability assays were done with human plasma by adapting a reported protocol.31 A 500 μL, 0.56 mM peptide solution was prepared in PBS buffer (pH 7.2). This solution was combined with 500 μL of reconstituted human plasma to give a 0.28 mM final concentration of peptide. The resulting suspension was incubated at 37 °C for 24 hours. Time points were taken at 0 min, 30 min, 1 hr, 2 hr, 4 hr, 8hr, and 24 hr. At each time point, a 130 μL aliquot was taken and added to a 10 kDa micro-centrifuge-compatible filter and tube assembly. The assembly was centrifuged at 18,000 ×g for 15 min, giving approximately 100 μL of filtrate. 20 μL of ACN was added to the filtrate and 90 μL of the resulting solution was injected via an autosampler and an analytical trace taken (see Purification by HPLC section above for details on the analytical method). Trace analysis was conducted similarly to the chemical and chymotrypsin stability assays.
Supplementary Material
Acknowledgements
This work was supported by grants from the National Institutes of Health (R35GM128651), the National Science Foundation (CHE-1808370), the Cayman Biomedical Research Institute (CaBRI), and by the Nevada INBRE through a grant from the NIH (GM103440). The E. faecalis reporter strain, TX5274, was kindly provided by B. E. Murray (University of Texas Health Science Center, Houston).
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
Full details of experimental procedures, peptide characterization, and dose-response curves for all GBAP analogues. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- [1].Arias CA, and Murray BE (2012) The rise of the Enterococcus: beyond vancomycin resistance, Nat Rev Microbiol 10, 266–278 DOI 10.1038/nrmicro2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Huycke MM, Sahm DF, and Gilmore MS (1998) Multiple-drug resistant Enterococci: The nature of the problem and an agenda for the future, Emerg Infect Dis 4, 239–249 DOI 10.3201/eid0402.980211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Van Tyne D, and Gilmore MS (2014) Friend Turned Foe: Evolution of Enterococcal Virulence and Antibiotic Resistance, Annu Rev Microbiol 68, 337–356 DOI 10.1146/annurev-micro-091213-113003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Fisher K, and Phillips C (2009) The ecology, epidemiology and virulence of Enterococcus, Microbiology 155, 1749–1757 DOI 10.1099/mic.0.026385-0. [DOI] [PubMed] [Google Scholar]
- [5].Cook LC, and Federle MJ (2014) Peptide pheromone signaling in Streptococcus and Enterococcus, Fems Microbiol Rev 38, 473–492 DOI 10.1111/1574-6976.12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Abisado RG, Benomar S, Klaus JR, Dandekar AA, and Chandler JR (2018) Bacterial Quorum Sensing and Microbial Community Interactions, mBio 9 DOI 10.1128/mBio.02331-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Brown SP, and Johnstone RA (2001) Cooperation in the dark: signalling and collective action in quorum-sensing bacteria, Proc Biol Sci 268, 961–965 DOI 10.1098/rspb.2001.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nakayama J, Cao Y, Horii T, Sakuda S, and Nagasawa H (2001) Chemical synthesis and biological activity of the gelatinase biosynthesis-activating pheromone of Enterococcus faecalis and its analogs, Biosci Biotechnol Biochem 65, 2322–2325 DOI 10.1271/bbb.65.2322. [DOI] [PubMed] [Google Scholar]
- [9].Pattnaik SS, Ranganathan S, Ampasala DR, Syed A, Ameen F, and Busi S (2018) Attenuation of quorum sensing regulated virulence and biofilm development in Pseudomonas aeruginosa PAO1 by Diaporthe phaseolorum SSP12, Microb Pathog 118, 177–189 DOI 10.1016/j.micpath.2018.03.031. [DOI] [PubMed] [Google Scholar]
- [10].Yang Y, Koirala B, Sanchez LA, Phillips NR, Hamry SR, and Tal-Gan Y (2017) Structure-Activity Relationships of the Competence Stimulating Peptides (CSPs) in Streptococcus pneumoniae Reveal Motifs Critical for Intra-group and Cross-group ComD Receptor Activation, ACS Chem Biol 12, 1141–1151 DOI 10.1021/acschembio.7b00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rasko DA, and Sperandio V (2010) Anti-virulence strategies to combat bacteria-mediated disease, Nat Rev Drug Discov 9, 117–128 DOI 10.1038/nrd3013. [DOI] [PubMed] [Google Scholar]
- [12].Del Papa MF, and Perego M (2011) Enterococcus faecalis Virulence Regulator FsrA Binding to Target Promoters, J Bacteriol 193, 1527–1532 DOI 10.1128/jb.01522-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Qin X, Singh KV, Weinstock GM, and Murray BE (2001) Characterization of fsr, a Regulator Controlling Expression of Gelatinase and Serine Protease in Enterococcus faecalis OG1RF, J Bacteriol 183, 3372–3382 DOI 10.1128/JB.183.11.3372-3382.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Pinkston KL, Gao P, Diaz-Garcia D, Sillanpaa J, Nallapareddy SR, Murray BE, and Harvey BR (2011) The Fsr Quorum-Sensing System of Enterococcus faecalis Modulates surface Display of the Collagen-Binding MsCRAMM Ace through Regulation of gelE, J Bacteriol 193, 4317–4325 DOI 10.1128/JB.05026-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ali L, Goraya MU, Arafat Y, Ajmal M, Chen J-L, and Yu D (2017) Molecular Mechanism of Quorum-Sensing in Enterococcus faecalis: Its Role in Virulence and Therapeutic Approaches, Int J Mol Sci 18 DOI 10.3390/ijms18050960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zeng J, Teng F, and Murray BE (2005) Gelatinase Is Important for Translocation of Enterococcus faecalis across Polarized Human Enterocyte-Like T84 Cells, Infect Immun 73, 1606–1612 DOI 10.1128/IAI.73.3.1606-1612.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sifri CD, Mylonakis E, Singh KV, Qin X, Garsin DA, Murray BE, Ausubel FM, and Calderwood SB (2002) Virulence effect of Enterococcus faecalis protease genes and the quorum-sensing locus fsr in Caenorhabditis elegans and mice, Infect Immun 70, 5647–5650 DOI 10.1128/IAI.70.10.5647-5650.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Singh KV, Nallapareddy SR, Nannini EC, and Murray BE (2005) Fsr-independent production of protease(s) may explain the lack of attenuation of an Enterococcus faecalis fsr mutant versus a gelE-sprE mutant in induction of endocarditis, Infect Immun 73, 4888–4894 DOI 10.1128/IAI.73.8.4888-4894.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].McBrayer DN, Gantman BK, Cameron CD, and Tal-Gan Y (2017) An Entirely Solid Phase Peptide Synthesis-Based Strategy for Synthesis of Gelatinase Biosynthesis-Activating Pheromone (GBAP) Analogue Libraries: Investigating the Structure-Activity Relationships of the Enterococcus faecalis Quorum Sensing Signal, Org Lett 19, 3295–3298 DOI 10.1021/acs.orglett.7b01444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Nakayama J, Yokohata R, Sato M, Suzuki T, Matsufuji T, Nishiguchi K, Kawai T, Yamanaka Y, Nagata K, Tanokura M, and Sonomoto K (2013) Development of a Peptide Antagonist against fsr Quorum Sensing of Enterococcus faecalis, ACS Chem Biol 8, 804–811 DOI 10.1021/cb300717f. [DOI] [PubMed] [Google Scholar]
- [21].McBrayer DN, Cameron CD, Gantman BK, and Tal-Gan Y (2018) Rational Design of Potent Activators and Inhibitors of the Enterococcus faecalis Fsr Quorum Sensing Circuit, ACS Chem Biol 13, 2673–2681 DOI 10.1021/acschembio.8b00610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chatterjee J, Rechenmacher F, and Kessler H (2012) N-Methylation of Peptides and Proteins: An Important Element for Modulating Biological Functions, Angew Chem Int Ed Engl 52, 254–269 DOI 10.1002/anie.201205674. [DOI] [PubMed] [Google Scholar]
- [23].Chatterjee M, D’Morris S, Paul V, Warrier S, Vasudevan AK, Vanuopadath M, Nair SS, Paul-Prasanth B, Mohan CG, and Biswas R (2017) Mechanistic understanding of Phenyllactic acid mediated inhibition of quorum sensing and biofilm development in Pseudomonas aeruginosa, Appl Microbiol Biotechnol 101, 8223–8236 DOI 10.1007/s00253-017-8546-4. [DOI] [PubMed] [Google Scholar]
- [24].Chatterjee J, Gilon C, Hoffman A, and Kessler H (2008) N-Methylation of Peptides: A New Perspective in Medicinal Chemistry, Acc Chem Res 41, 1331–1342 DOI 10.1021/ar8000603. [DOI] [PubMed] [Google Scholar]
- [25].Tal-Gan Y, Freeman NS, Klein S, Levitzki A, and Gilon C (2010) Synthesis and structure-activity relationship studies of peptidomimetic PKB/Akt inhibitors: The significance of backbone interactions, Bioorg Med Chem 18, 2976–2985 DOI 10.1016/j.bmc.2010.02.031. [DOI] [PubMed] [Google Scholar]
- [26].Laufer B, Chatterjee J, Frank AO, and Kessler H (2009) Can N-methylated amino acids serve as substitutes for prolines in conformational design of cyclic pentapeptides?, J Pept Sci 15, 141–146 DOI 10.1002/psc.1076. [DOI] [PubMed] [Google Scholar]
- [27].Appel W (1986) Chymotrypsin: molecular and catalytic properties, Clin Biochem 19, 317–322 DOI 10.1016/S0009-9120(86)80002-9. [DOI] [PubMed] [Google Scholar]
- [28].Chan WC, and White PD (2000) Fmoc Solid Phase Peptide Synthesis: A Practical Approach, Vol. 222, Oxford University Press, Oxford. [Google Scholar]
- [29].Behrendt R, White P, and Offer J (2016) Advances in Fmoc solid-phase peptide synthesis, J Pept Sci 22, 4–27 DOI 10.1002/psc.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chantell CA, Onaiyekan MA, and Menakuru M (2012) Fast conventional Fmoc solid-phase peptide synthesis: a comparative study of different activators, J Pept Sci 18, 88–91 DOI 10.1002/psc.1419. [DOI] [PubMed] [Google Scholar]
- [31].Pakkala M, Hekim C, Soininen P, Leinonen J, Koistinen H, Weisell J, Stenman UH, Vepsalainen J, and Narvanen A (2007) Activity and stability of human kallikrein-2-specific linear and cyclic peptide inhibitors, J Pept Sci 13, 348–353 DOI 10.1002/psc.849. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.