

Abstract
Flavin-dependent ‘ene’-reductases (EREDs) are known to stereoselectively reduce activated alkenes, but are inactive toward carbonyls. Herein we demonstrate that in the presence of photoredox catalysts, these enzymes will reduce aromatic ketones. Mechanistic experiments suggest this reaction proceeds via ketyl radical formation, a reaction pathway that is distinct from the native hydride transfer mechanism. Furthermore, this reactivity is accessible with no modification of the enzyme or cofactors, allowing both native and non-natural mechanisms to occur simultaneously. Based on control experiments, we hypothesize that binding to the enzyme active site attenuates the reduction potential of the substrate, enabling single electron reduction. This reactivity highlights opportunities to access new catalytic manifolds by merging photoredox catalysis with biocatalysis.
Keywords: biocatalysis, photoredox, reduction, hydrogen atom transfer, ‘ene’-reductase
Graphical abstract
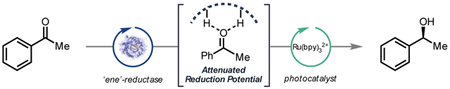
Flavin Misbehavin’: ‘Merging photocatalysis with biocatalysis leads to new chemical reactivity for flavin-dependent ‘ene’-reductases. Mechanistic studies support a mechanism in which enantioenriched alcohols are produced via hydrogen atom transfer from flavin to the ketyl radical.
The discovery of new catalytic reactions is a fundamental goal in organic synthesis. Small molecule catalysts, such as transition metal complexes or small organic molecules, have proven to be fertile scaffolds on which new chemical reactivity is developed.1 Enzymes are rarely used as general scaffolds for discovering new chemical reactions because of their perceived reaction specificity. The realization that substrate permissive enzymes can also exhibit catalytic promiscuity has opened the door to enzymes being used for reactions beyond their native function.2 From this perspective, devising new strategies for revealing non-natural reaction pathways in enzymes can significantly expand the potential applications of biocatalysis.
Synergistic catalysis is an attractive strategy for expanding the synthetic capabilities of a particular catalyst.3 By weaving together two catalytic cycles, it becomes possible to achieve chemical reactivity that would otherwise be inaccessible to either catalyst individually.4 For instance, the merger of enamine and transition metal catalysis enabled cross-coupling reactions that were previously unknown.5 Despite the growing trend of using biocatalytic approaches to organic synthesis, examples of synergy between small molecule catalysts and enzymes remain elusive.6 This is due, in part, to the challenge of achieving productive interactions between enzymatic and small molecule catalytic cycles.
Enzymes use a variety of strategies to enable kinetically challenging reactions. Oxidoreductases, for instance, use a series of hydrogen bonding interactions to render substrates more electrophilic, priming them for reduction by various cofactors. We recognized that if this mechanism of substrate activation could be merged with single electron reductants, it would be possible to use oxidoreductases that typically use ionic mechanisms to catalyze reactions that proceed via radical intermediates.7,8 To this end, we recently demonstrated that nicotinamide-dependent double bond reductases (DBRs) can catalyze a ‘new to nature’ enantioselective radical deacetoxylation of α-acetoxyketones when exposed to visible light and a xanthene dye photoredox catalyst.9 In this study, binding to the DBR activates the ketone for single electron reduction by the photocatalyst and orients the resulting α-acyl radical for selective hydrogen atom transfer from NADPH.
Building on this mechanistic understanding, we questioned whether a similar mode of activation could be achieved within the active site of flavin-dependent ‘ene’-reductases (EREDs). These substrate-promiscuous enzymes are widely used for the asymmetric reduction of activated olefins, which occurs via stereoselective delivery of hydride from flavin hydroquinone (FMNhq).10 Two hydrogen bond donors (typically Histidine/Histidine, or Histidine/Asparagine) are used to activate and orient the substrate for reduction. We envisioned that these residues could similarly activate ketones for single electron reduction. Furthermore, recent studies have shown that EREDs are highly compatible with photoredox catalysis, although no examples currently exist of using photocatalysis to achieve new reactivity with these enzymes.11,17 As a model system, we targeted the reduction of ketones to alcohols, a reaction pathway that is not observed in EREDs, or any other flavoenzyme known in nature. We imagined that this reaction would be attractive for streamlining the biocatalytic reduction of enones to saturated alcohols, a process that currently requires two enzymes using natural reaction mechanisms.12 Furthermore, mechanistically novel biocatalytic carbonyl reductions may offer advantages, such as altered selectivity profiles and irreversibility.
From a design perspective, we imagined that this non-natural carbonyl reaction could proceed via the mechanism indicated in Figure 2. Initial reduction of the ERED with NADH affords reduced flavin (FMNhq). The enzyme binds an equivalent of the ketone, activating it for single electron reduction (E1/2 = −2.1 V in MeCN).13 Concurrently, visible light photoexcitation of a Ru(II) photocatalyst produces the excited state Ru(II)* that is known to be readily quenched by NADH analogues to afford a more strongly reducing Ru(I) species.14 Subsequently, Ru(I) (E1/2II/I = −1.33 V) reduces the ERED-bound ketone to the corresponding ketyl radical.15 We hypothesize that the ketyl radical is quenched via hydrogen atom transfer from flavin and electron transfer from FMNsq to Ru(II)* regenerates FMNox .16 As active sites of EREDs are partially exposed to the bulky solution, we anticipate that electron transfer to substrates bound within the active site can occur without requiring a bioconjugation event.
Figure 2.

Proposed Reaction Mechanism
We initiated our investigation by examining the reduction of acetophenone with eight structurally diverse EREDs. While no reaction was observed in the absence of photocatalyst, addition of Ru(bpy)3Cl2 and irradiation with blue light enabled all nine EREDs to reduce acetophenone to the corresponding alcohol (SI Table 2). The most active enzyme was morphinone reductase from P. putida (MorB), which provided product in quantitative yield as a scalemic mixture favoring the (R)-enantiomer (80:20 e.r.) (Table 1, entry 1). Comparable yields can be achieved with 0.5 mol % enzyme, although extended reaction times are required to reach full conversion. Interestingly, pH had a negligible effect on reaction efficiency (SI Table 6). Preferential formation of the opposite enantiomer of the alcohol is achieved by 12-oxophytodienoate reductase (OPR-1) (82:18 e.r.) and YersER (86:14 e.r.), albeit with diminished yield by comparison to MorB (Table 1, entries 2 and 3). A brief survey of photocatalysts revealed Ru(bpy)3Cl2 and Ru(bpz)3Cl2 to be most effective (SI Table 4).
Table 1.
Reaction Optimization
 | |||
|---|---|---|---|
| entrya | change from standard conditions | yield (%) | e.r. |
| 1 | no change | 99 | 80:20 |
| 2 | OPR1 instead of MorB | 38 | 18:82 |
| 3 | YersER instead of MorB | 20 | 14:86 |
| 4 | no MorB | N.R. | - |
| 5 | d1-glucose and no Ru(bpy)3CI2 | N.R. | - |
| 6 | no irradiation | N.R | - |
| 7 | no GDH-105 and glucose | N.R | - |
Standard Reaction Conditions: 10 Dmol substrate, 1 mol% MorB, 1 mol% Ru(bpy)3CI2, 2 mol% NAD(P)+, 9 mg glucose, 0.25 mg GDH-105, 250 DL reaction volume.
A series of control experiments were conducted with MorB to confirm the importance of each component of the system (Table 1, entries 4–7). Both NAD+ and GDH-105 were required for reactivity, suggesting that reduced flavin (FMNhq) is required for ketone reduction (Table 1, entry 7 and SI Table 1, entry 2). Importantly, these studies suggest the reaction is not occurring via hydride transfer from NADPH to acetophenone (Table 1, entry 5). Surprisingly, replacing NAD(P)+ and GDH-105 with a photochemical reducing system involving TEOA and Ru(bpy)3Cl2 provided only low conversion, presumably because cofactor turnover is faster with the GDH/glucose system.17 (SI Table 5).
To probe the formation of a ketyl radical, an aromatic ketone containing a pendant cyclopropane was prepared and subjected to the reaction conditions. Two products were identified as the ring-opened ketone and the corresponding ring-opened alcohol, with the remainder of the mass balance being unreacted starting material (Figure 3A and SI Figure 1). Importantly, in the absence of the ERED only starting material was observed, supporting the hypothesis that enzyme-mediated substrate activation is required to achieve single electron reduction and ketyl radical formation (SI Figure 1).
Figure 3.

Mechanistic studies to determine the presence of a radical intermediate and identification of the H-atom source.
We next focused on determining which residues are responsible for substrate binding and activation (Figure 3B). Like other EREDs, MorB is understood to bind its natural substrate via hydrogen bonding from a His/Asn pair, making these side chains the most likely candidates. To probe this hypothesis, we used site-directed mutagenesis to mutate each residue to alanine and tested the reduction of acetophenone. When asparagine was mutated to alanine (N189A), the product was observed in quantitative yield, albeit with significantly diminished e.r. (57:43). Scrutton previously observed that this mutation results in cyclohexanone binding in two different conformations, potentially accounting for the poor selectivity observed with this variant.18 Mutation of the histidine to alanine (H186A), however, only formed the product in 15% yield. This mutation is known to inhibit the oxidative half-reaction with cyclohexanone, presumably due to diminished substrate binding. Collectively, these results suggest that the canonical binding residues are responsible for orienting the substrate and activating it for single electron reduction.
To gain insight into the radical termination event, we investigated the source of the hydrogen atom. While our previous studies indicated that reduced FMNhq could function as a hydrogen atom source, we recognized that a cysteine or tyrosine located within the enzyme active site could also serve a similar role (Figure 3c).19 To probe this possibility, isotope incorporation experiments were performed with acetophenone and wild type MorB. When the reaction was run using KPi buffered D2O as the solvent, no deuterium was incorporated into the alcohol product, as evidenced by 13C NMR. This strongly suggests that tyrosines in the active site are not serving as H-atom donors. Interestingly, when the standard reaction conditions were repeated using d1-glucose as an in situ deuterium source for labeling FMN, no deuterium incorporation was observed in the product. In both cases, the reaction goes to full conversion within 24 hours with no change in enantioselectivity from the standard reaction conditions.
Surprised by these unusual isotope incorporation results, we hypothesized that a kinetic isotope effect could prevent deuterated flavin from quenching the ketyl radical. Instead, the deuterium at the flavin N-5 position could exchange with solvent, followed by hydrogen atom transfer to the ketyl radical. (SI Figure 3). Indeed, when a rate study was performed, the reaction became significantly slower in the presence of d1-glucose. (SI Figure 4). This effect was further magnified when both glucose and the solvent were deuterated. Due to the pKa of the anionic FMNhq (pKa > 20) and lack of solvent exchange observed under standard olefin reduction experiments, we postulate that this exchange is mediated by oxidation of FMNhq by photoexcited Ru(II)* to generate the neutral flavin semiquinone.20 Due to its increased acidity (pKa = 8.3), flavin semiquinone could exchange rapidly with solvent under the reaction conditions (SI Figure 3).
To better understand the scope and limitations of this reaction, a panel of aromatic ketone substrates were tested for reduction with MorB and Ru(bpy)3Cl2. Para- and meta- substituents are tolerated, forming the product in good yields and with promising levels of enantioselectivity (Figure 4, 14–18). Interestingly, electron withdrawing substituents proved to be more reactivity, presumably due to their increased reduction potentials. An ortho-substituent is also tolerated and provides product in low yield with poor enantioselectivity (Table 4, 19). A more sterically napthyl ketone is also reactive, although extended reaction times are required to reach high conversions (Table 4, 20). Increasing the steric bulk of the other substituent on the ketone provided products with poor levels of enantioselectivity (Table 4, 21,22). This limitation can be overcome by using FlOYE as a catalyst (98:2 er), although this reaction occurs with diminished yields (Supplemental Table 2). Alternatively, we imagine protein engineering could be applied to increase the enantioselectivity of a particular transformation. Finally, non-acetophenone derivatives were unreactive, presumably due to their low reduction potentials.
Figure 4.

Substrate Scope
Finally, we envisioned that a single enzyme with ene-reductase and ketoreductase catalytic activity could streamline biocatalytic synthesis. When ketone 23 was subjected to the reaction conditions, we observed global reduction of the substrate, presumably with the ERED conducting its native reactivity, followed by a non-natural ketone reduction to provide the desired product in 89% yield and 83:17 er (Figure 5, 24). In contrast, in the absence of light only alkene reduction is observed in 98% yield (Figure 5, 25).
Figure 5.

Change in Product Selectivity
One of the current limitations of biocatalysis in organic synthesis is the perception that enzymes are limited to their natural catalytic activity, suggesting that novel reactivity in biocatalysis can only be obtained by screening nature for new transformations, or with significant engineering of the biocatalyst. This perception is in stark contrast to small molecule chiral catalysts, which are often used to impart selectivity in a wide range of chemical transformations. Herein, we demonstrate that a new mechanistic pathway can be accessed to generate chiral products that are unexpected for this enzyme class without biocatalyst modification. Furthermore, as significant efforts are often made to engineer enzymes to tolerate conditions typically used in organic synthesis, finding more examples of catalytic promiscuity expands their synthetic utility and maximizes the return on investment for such engineering projects.
Experimental Section
General Reaction Procedure.
In an anaerobic chamber (Coy, Grass Lake, MI), a stock solution of turnover mix containing GDH-105 (5 mg) and glucose (180 mg) in 1 mL of degassed KPi (100 mM, pH 8) was prepared. A catalyst mix solution containing NAD+ (14.3 mg) and Ru(bpy)3Cl2 (8 mg) in 1 mL of degassed KPi (100 mM, pH 8) was prepared. To a dram vial containing 150 μL of degassed KPi (100 mM, pH 8) was added 50 μL of turnover mix, followed by 10 μL of catalyst mix and enzyme (volume of enzyme varies with concentration). Finally, 10 μL of ketone (1.0 M in DMSO) was added. The vial was sealed with a cap and removed from the anaerobic chamber. The reaction was placed on a shaker and illuminated with 460 nm light for 24 hours.
Supplementary Material
Figure 1.

Merging photoredox catalysis with biocatalysis
Acknowledgements
T.K.H thanks the NIH-NIGMS (R01 GM127703), Searle Scholars Award (SSP-2017–1741), Sloan Research Fellowship, the Princeton Catalysis Initiative, and Princeton University for support. B.A.S. and S.I.K. thank the Edward C. Taylor Fellowship for support.
Footnotes
Supporting information for this article is given via a link at the end of the document
References
- [1].Yoon TP, Jacobsen EN, Science. 2003, 299, 1691–1693. [DOI] [PubMed] [Google Scholar]
- [2].a) Bornscheur UT; Kazlauskas RJ, Angew. Chem. Int. Ed 2004, 43, 6032–6040. [DOI] [PubMed] [Google Scholar]; b) Renata H, Wang ZJ, Arnold FH, Angew. Chem. Int. Ed 2015, 54, 3351–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Arnold FH, Angew. Chem. Int. Ed 2017, 57, 4143–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Allen AE, MacMillan DWC, Chem. Sci 2012, 3, 633–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Nicewicz DA, MacMillan DWC, Science, 2008, 322, 5898, 77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Du J, Skubi K, Schultz DM, Yoon TP 2014, 344, 392. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gil A, Albericio F, Alvarez Chem M. Rev. 2017, 117, 8420. [DOI] [PubMed] [Google Scholar]
- [5].a) Krautwald S, Sarlah D, Schafroth MA, Carreira EM, Science 2013, 340, 1065. [DOI] [PubMed] [Google Scholar]; (b) Ibrahem I, Córdova A, Angew. Chem. Int. Ed 2006, 45, 1952–1956. [DOI] [PubMed] [Google Scholar]
- [6].Hönig M, Sondermann P, Turner NJ, Carreira EM, Angew. Chem. Int. Ed 2017, 56, 8942–8973. [DOI] [PubMed] [Google Scholar]
- [7].Zipse H. Org. Biomol. Chem 2003, 1, 692–699. [DOI] [PubMed] [Google Scholar]
- [8].a) Turek AK, Hardee DJ, Ullman AM, Nocera DG, Jacobsen EN, Angew. Chem. Int. Ed 2016, 55, 539–544. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zipse H. Org. Biomol. Chem 2003, 1, 692–699. [DOI] [PubMed] [Google Scholar]
- [9].Biegasiewicz KF, Cooper SJ, Emmanuel MA, Miller DC, Hyster TK, Nat. Chem 2018, 10, 770–775. [DOI] [PubMed] [Google Scholar]
- [10].a) Kohli RM, Massey V, J. Biol. Chem 1998, 273(98), 32763–32770. [DOI] [PubMed] [Google Scholar]; b) Winkler CK, Tasnádi G, Clay D, Hall M, K Faber J Biotechnol 2012, 162, 381– 389. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Winkler CK, Faber K, Hall M, Curr. Opin. Chem. Biol 2018, 43, 97–105. [DOI] [PubMed] [Google Scholar]
- [11].a) Lee SH, Choi DS, Pesic M, Lee YW, Paul CE, Hollmann F, Park CB, Angew. Chem. Int. Ed 2017, 56, 8681–8685. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Litman ZC, Wang Y, Zhao H, Hartwig JF, Nature, 2018, 560, 355–359. [DOI] [PubMed] [Google Scholar]
- [12].a) Brenna E, Gatti FG, Monti D, Parmeggiani F, Sacchetti A. ChemCatChem 2012, 4, 653–659. [Google Scholar]; b) Toogood HS, Scrutton NS, ACS Catal. 2018, 8, 3532–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fukuzumi S, Ishikawa K, Hironaka K, Tanaka T, J. Chem. Soc. Perkin Trans II 1987, 751–760. [Google Scholar]
- [14].a) Maidan R, Willner I, J. Am. Chem. Soc 1986, 108, 1080.-. [Google Scholar]; b) Pac C, Ihama M, Yasuda M, Miyauchi Y, Sakurai H, J. Am. Chem. Soc 1981, 103, 6496–1497. [Google Scholar]
- [15].Prier CK, Rankic DA, MacMillan DWC, Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sandoval BA, Meichan AJ, Hyster TK, J. Am. Chem. Soc 2017, 139, 11313–11316. [DOI] [PubMed] [Google Scholar]
- [17].Peers MK, Toogood HS, Heyes DJ, Mansell D, Coe JB, Scrutton NS, Catal. Sci. Technol, 2016, 6, 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Messiha HL, Munro AW, Bruce NC, Barsukov I, Scrutton NS, J. Biol. Chem 2005, 280, 10695–10709. [DOI] [PubMed] [Google Scholar]
- [19].a) Stubbe J, Nocera DG, Yee CS, Chang MCY, Chem. Rev 2003, 103(6), 2167–2202. [DOI] [PubMed] [Google Scholar]; b) Morinaka BI, Vagstad AL, Helf MJ, Gugger M, Kegler C, Freeman MF, Bode HB, Piel J, Angew. Chem. Int. Ed 2014, 53, 8503–8507. [DOI] [PubMed] [Google Scholar]
- [20].Macheroux P, Ghisla S, Sanner C, Rüterjans H, Müller F. BMC Biochemistry, 2005, 6, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.