

Summary
DNA N6-adenine methylation (6mA) has recently been described in diverse eukaryotes, spanning unicellular organisms to metazoa. Here, we report a DNA 6mA methyltransferase complex in ciliates, termed MTA1c. It consists of two MT-A70 proteins and two homeobox-like DNA-binding proteins, and specifically methylates dsDNA. Disruption of the catalytic subunit, MTA1, in the ciliate Oxytricha leads to genome-wide loss of 6mA and abolishment of the consensus ApT dimethylated motif. Mutants fail to complete the sexual cycle, which normally coincides with peak MTA1 expression. We investigate the impact of 6mA on nucleosome occupancy in vitro by reconstructing complete, full-length Oxytricha chromosomes harboring 6mA in native or ectopic positions. We show that 6mA directly disfavors nucleosomes in vitro in a local, quantitative manner, independent of DNA sequence. Furthermore, the chromatin remodeler ACF can overcome this effect. Our study identifies a diverged DNA N6-adenine methyltransferase, and defines the role of 6mA in chromatin organization.
Keywords: DNA methylation, N6-methyladenine, nucleosome positioning, synthetic chromosomes, chromatin remodeling
Graphical Abstract
Introduction
Covalent modifications on DNA have long been recognized as a hallmark of epigenetic regulation. DNA N6-methyladenine (6mA) has recently come under scrutiny in eukaryotic systems, with proposed roles in retrotransposon or gene regulation, transgenerational epigenetic inheritance, and chromatin organization (Luo et al., 2015). 6mA exists at low levels in Arabidopsis thaliana (0.006%- 0.138% 6mA / dA), rice (0.2%), C. elegans (0.01–0.4%), Drosophila (0.001-0.07%), Xenopus laevis (0.00009%), mouse ES cells (0.0006 – 0.007%), human cells (Greer et al., 2015; Koziol et al., 2015; Liang et al., 2018; Wu et al., 2016; Xiao et al., 2018; Zhang et al., 2015; Zhou et al., 2018) and the mouse brain (Yao et al., 2017), although it accumulates in abundance (0.1–0.2%) during vertebrate embryogenesis (Liu et al., 2016). Disruption of DMAD, a 6mA demethylase, in the Drosophila brain leads to the accumulation of 6mA and Polycomb-mediated silencing (Yao et al., 2018). The existence of 6mA in mammals remains a subject of debate. Quantitative LC-MS analysis of HeLa and mouse ES cells failed to detect 6mA above background levels (Schiffers et al., 2017). A recent study, however, reported that loss of 6mA in human cells promotes tumor formation (Xiao et al., 2018), suggesting that 6mA is a biologically relevant epigenetic mark.
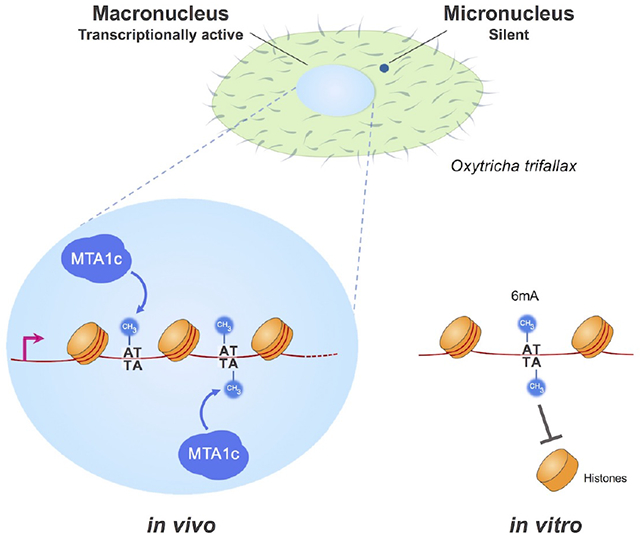
In contrast to metazoa, 6mA is abundant in various unicellular eukaryotes, including ciliates (0.18 – 2.5%) (Ammermann et al., 1981; Cummings et al., 1974; Gorovsky et al., 1973; Rae and Spear, 1978), and the green algae Chlamydomonas (0.3 – 0.5%) (Fu et al., 2015; Hattman et al., 1978). High levels of 6mA (up to 2.8%) were also recently reported in basal fungi (Mondo et al., 2017). Ciliates have long served as powerful models for the study of chromatin modifications (Brownell et al., 1996; Liu et al., 2007; Strahl et al., 1999; Taverna et al., 2002; Wei et al., 1998). They possess two structurally and functionally distinct nuclei within each cell (Bracht et al., 2013; Yerlici and Landweber, 2014). In the ciliate Oxytricha trifallax, the germline micronucleus is transcriptionally silent and contains ~100 megabase-sized chromosomes (Chen et al., 2014). In contrast, the somatic macronucleus is transcriptionally active, being the sole locus of Pol II-dependent RNA production in non-developing cells (Khurana et al., 2014). The Oxytricha macronuclear genome is extraordinarily fragmented, consisting of ~16,000 unique chromosomes with a mean length of ~3.2kb, most encoding a single gene. Macronuclear chromatin yields a characteristic ~200bp ladder upon digestion with micrococcal nuclease, indicative of regularly spaced nucleosomes (Gottschling and Cech, 1984; Lawn et al., 1978; Wada and Spear, 1980). Yet it remains unknown how and where nucleosomes are organized within these miniature chromosomes, and if this in turn regulates (or is regulated by) 6mA deposition.
Intriguingly, in green algae, basal yeast, and ciliates, 6mA is enriched in ApT dinucleotide motifs within nucleosome linker regions near promoters (Fu et al., 2015; Hattman et al., 1978; Karrer and VanNuland, 1999; Mondo et al., 2017; Pratt and Hattman, 1981; Wang et al., 2017). Here we identify four ciliate proteins – named MTA1, MTA9, p1, and p2 – necessary for 6mA methylation. MTA1 and MTA9 contain divergent MT-A70 domains, while p1 and p2 are homeobox-like proteins that likely function in DNA binding. We delineate key biochemical properties of this methyltransferase and dissect the function of 6mA in vitro and in vivo.
Results
Epigenomic profiles of chromatin and transcription in Oxytricha
We generated genome-wide in vivo maps of nucleosome positioning, transcription, and 6mA in the macronuclei of asexually growing (vegetative) Oxytricha trifallax cells using MNase-seq, poly(A)+ RNA-seq, TSS-seq, and single molecule real time (SMRT) sequencing (Figure 1). The smallest Oxytricha chromosome is only 430bp in length, with a single well-positioned nucleosome. Strikingly, 6mA is enriched in three consecutive nucleosome depleted regions directly downstream of transcription start sites (TSSs; Figure 1A). Each region contains varying levels of 6mA (Figure 1B), with the +1/+2 nucleosome linker being most densely methylated (Table S1). In general, highly transcribed chromosomes tend to bear more 6mA, suggesting a positive role of this DNA modification in gene regulation (Figure 1C). The majority of methylation marks are located within an ApT motif (Figures 1D and 1E). 6mA occurs on sense and antisense strands with approximately equal frequency, indicating that the underlying methylation machinery does not function strand-specifically. Quantitative LC-MS analysis confirmed the presence of 6mA in Oxytricha (Figure S1; see Methods).
Figure 1. Epigenomic profiles of Oxytricha chromosomes.
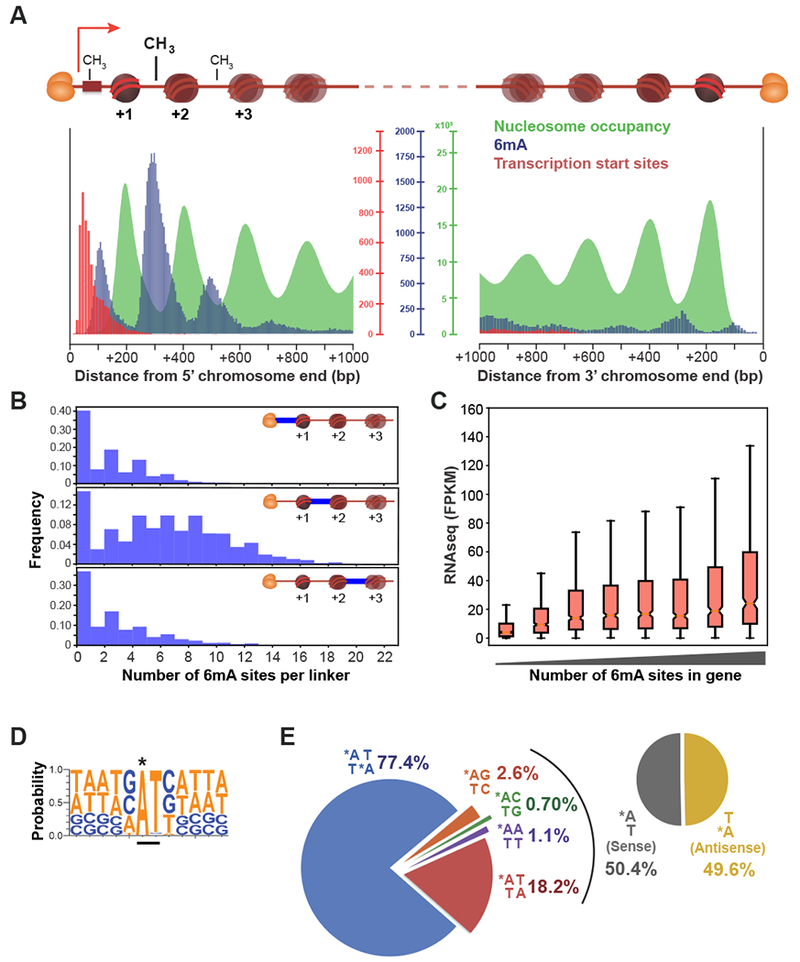
(A) Meta-chromosome plots of chromatin organization at Oxytricha macronuclear chromosome ends. Heterodimeric telomere end-binding protein complexes (orange ovals) protect each end in vivo. Horizontal red bar: promoter. The 5’ chromosome end is proximal to TSSs. “Nucleosome occupancy”: normalized MNase-seq coverage; “6mA”: total 6mA number; “Transcription start sites”: total number of called TSSs.
(B) Histograms of the total number of 6mA marks within each linker in Oxytricha chromosomes. Distinct linkers are depicted as horizontal blue lines.
(C) poly(A)-enriched RNAseq levels positively correlate with 6mA. Genes are sorted according to the total number of 6mA marks 0-800 bp downstream of the TSS. FPKM = Fragments per Kilobase of transcript per Million mapped RNAseq reads. Notch in the boxplot denotes median, ends of boxplot denote first and third quartiles, upper whisker denotes third quantile + 1.5 × interquartile range (IQR), and lower whisker denotes data quartile 1 – 1.5 × IQR.
(D) Composite analysis of 65,107 methylation sites reveals that 6mA (marked with *) occurs within an 5’-ApT-3’ dinucleotide motif.
(E) Distribution of various 6mA dinucleotide motifs across the genome. Asterisk: 6mA.
Purification and reconstitution of the ciliate 6mA methyltransferase, MTA1c
To uncover the functions of 6mA in vivo, we set out to identify and disrupt putative 6mA methytransferases (MTases). The Oxytricha genome encodes a large number of candidate methyltransferases (Table S2), rendering it impractical to test gene function, one at a time or in combination. To identify the ciliate 6mA MTase, we undertook a biochemical approach by fractionating nuclear extracts and identifying candidate proteins that co-purified with DNA methylase activity. The organism of choice for this experiment was Tetrahymena thermophila, a ciliate that divides significantly faster than Oxytricha (~2 hours vs 18 hours); (Cassidy-Hanley, 2012; Laughlin et al., 1983). This faster growth time rendered it feasible to culture large amounts of Tetrahymena cells for nuclear extract preparation. Tetrahymena and Oxytricha exhibit similar genomic localization and 6mA abundance (Figures S1 and S2A–F). We thus reasoned that the enzymatic machinery responsible for 6mA deposition is conserved between Tetrahymena and Oxytricha, and that Tetrahymena could serve as a tractable biochemical system for identifying the ciliate 6mA MTase.
We prepared nuclear extracts from log-phase Tetrahymena cells, since 6mA could be readily detected at this developmental stage through quantitative mass spectrometry and PacBio sequencing (Figures S1 and S2A–F). Nuclear extracts were incubated with radiolabeled S-adenosyl-L-methionine and PCR-amplified DNA substrate to assay for DNA methylase activity. Passage of the nuclear extract through an anion exchange column resulted in the elution of two distinct peaks of DNA methylase activity, both of which were heat sensitive (Figure 2C, S3A, S3B). Western blot analysis confirmed that both peaks of activity mediate methylation on 6mA (Figure S3C). The resulting fractions were further purified and subjected to mass spectrometry. Only four proteins – termed MTA1, MTA9, p1 and p2 – were detected at higher abundance in fractions with high DNA methylase activity (Figures 2C and 2D). p1 and p2 contain homeobox-like domains, suggesting a DNA binding function for an undetermined process (Figure S3D). On the other hand, MTA1 and MTA9 are both MT-A70 proteins. Such domains are widely known to mediate m6A RNA methylation in eukaryotes (Liu et al., 2014). MTA1 and MTA9 received the large majority of peptide matches, relative to all other MT-A70 genes encoded by the Tetrahymena genome (Figure 2D and Table S3). Curiously, although poly(A)-selected RNA transcripts were present from all MT-A70 genes (Figure 2D), almost all peptides in fractions with high DNA methylase activity corresponded to MTA1 and MTA9. The poly(A)+ RNA expression profiles of MTA1, MTA9, p1, and p2 are remarkably similar (Figure S2K), peaking early in the sexual cycle. This coincides with a sharp increase in nuclear 6mA, as evidenced from immunostaining (Wang et al., 2017). Accumulation of MTA1, MTA9, p1, and p2 therefore correlates with the presence of 6mA in vivo.
Figure 2. Purification and characterization of the ciliate 6mA methyltransferase.
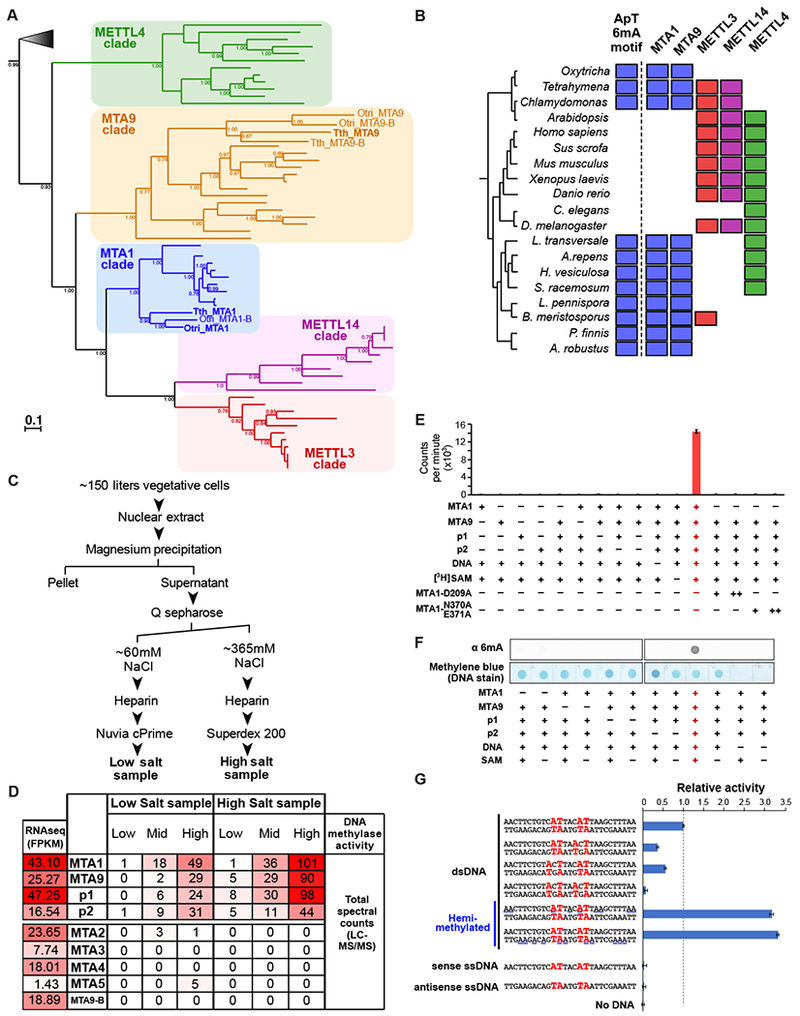
(A) Phylogenetic analysis of MT-A70 proteins. Bold MTA1 and MTA9 genes are experimentally characterized in this study. Paralogs of MTA1 and MTA9 are labeled as “-B”. Posterior probabilities > 0.65 are shown. Gray triangle represents outgroup of bacterial sequences. The complete phylogenetic tree is shown in Figure S2G. Gene names are in Table S2. Tth: Tetrahymena thermophila; Otri: Oxytricha trifallax.
(B) Phylogenetic distribution of the occurrence of ApT 6mA motifs and MT-A70 protein families. Filled square denotes its presence in a taxon. The basal yeast clade is comprised of L. transversale, A.repens, H. vesiculosa, S. racemosum, L. pennispora, B. meristosporus, P. finnis, and A. robustus.
(C) Experimental scheme depicting the partial purification of DNA methyltransferase activity from Tetrahymena nuclear extracts.
(D) Gene expression and protein abundance of candidate genes in partially purified Tetrahymena nuclear extracts. UniProt IDs are listed in Table S2. RNAseq data are from (Xiong et al., 2012). FPKM = Fragments per Kilobase of transcript per Million mapped RNAseq reads. “Low”, “Mid”, and “High” DNA methylase activity correspond to fractions eluting from the Nuvia cPrime and Superdex 200 columns in panel C. “Total spectrum counts”: total number of LC-MS/MS fragmentation spectra that match peptides from a target protein.
(E) DNA methyltransferase assay using [3H]SAM. Vertical axis represents scintillation counts. Error bars represent s.e.m. (n = 3).
(F) Dot blot assay using cold SAM.
(G) DNA methyltransferase assay performed on different nucleic acid substrates in the presence of MTA1, MTA9, p1, and p2. Sense ssDNA are 5’→3’; antisense are 3’→5’. ApT dinucleotides are labeled in bold red. Horizontal blue lines in hemimethylated dsDNA substrates denote possible locations where 6mA may be installed by EcoGII (prior to this assay). “Relative activity” denotes scintillation counts normalized against the unmethylated 27bp dsDNA substrate with two ApT motifs (top-most dsDNA substrate). An enlarged bar plot of relative activity on 27bp unmethylated dsDNA substrates is included in Figure S3K. Error bars represent s.e.m. (n = 3).
We next investigated the phylogenetic relationship of MTA1 and MTA9 to other eukaryotic MT-A70 domain-containing proteins. Two widely studied mammalian MT-A70 proteins – METTL3 and METTL14 (Ime4 and Kar4 in yeast) – form a heterodimeric complex that is responsible for m6A methylation on mRNA. METTL3 is the catalytically active subunit, while METTL14 functions as an RNA-binding scaffold protein (Śledź and Jinek, 2016; Wang et al., 2016a, 2016b). MTA1 and MTA9 derive from distinct monophyletic clades, outside of those that contain mammalian METTL3, METTL14, and C. elegans DAMT-1 (METTL4) (Figure 2A). Thus, MTA1 and MTA9 are divergent MT-A70 family members that are phylogenetically distinct from all previously studied RNA and DNA N6-methyladenine MTases. We then asked whether MTA1 and MTA9 are also present in other eukaryotes with a similar occurrence of 6mA in ApT motifs as Tetrahymena. We queried the genomes of Oxytricha, green algae, and eight basal yeast species, all of which exhibit this distinct methylation pattern (as evidenced from Figure 1, Figure S2A–E, (Fu et al., 2015), and (Mondo et al., 2017)). For all of these taxa, we can identify MT-A70 homologs that are monophyletic with MTA1 and MTA9 (Figure 2B). On the other hand, MT-A70 homologs from multicellular eukaryotes, including Arabidopsis, C. elegans, Drosophila, and mammals, grouped exclusively with METTL3, METTL14, and METTL4 lineages, but not MTA1 or MTA9. None of these latter genomes exhibit a consensus ApT dinucleotide methylation motif for 6mA (Greer et al., 2015; Koziol et al., 2015; Liang et al., 2018; Liu et al., 2016; Wu et al., 2016; Xiao et al., 2018; Zhang et al., 2015). We note that the absence of an ApT dinucleotide motif is based on data from a limited number of cell types, developmental stages, and culture conditions tested in these studies. Nonetheless, within the scope of currently available data, the presence of MTA1 and MTA9 correlates with the distinctive genomic localization of 6mA within ApT motifs.
We then sought to determine whether MTA1 and/or MTA9 are bona fide 6mA methyltransferases. MTA1, but not MTA9, contains a catalytic DPPW motif (Figure S3E)—a hallmark of N6-adenosine methyltransferases (Iyer et al., 2016). Surprisingly, recombinant full length Tetrahymena MTA1 and MTA9 (Figure S3G) showed no detectable DNA methyltransferase activity, individually or together (Figure 2E). Examination of the MTA1 and MTA9 sequences revealed that neither protein possesses a predicted nucleic acid binding domain (Figure S3D). In contrast, METTL3, which catalyzes m6A methylation on RNA, contains two tandem CCCH-type zinc finger motifs, necessary for RNA binding (Huang et al., 2018; Wang et al., 2016a). Additional co-factors may thus be necessary for MTA1/7 to engage DNA substrates. Indeed, the p1 and p2 proteins that co-elute with MTA1/7 in nuclear extracts possess homeobox-like domains predicted to bind DNA. We then tested whether these accessory factors, in addition to MTA1/7, are necessary for 6mA methylation. Strikingly, mixing recombinant, full length p1, p2, MTA1, and MTA9 resulted in robust 6mA methylation in vitro (Fig 2E and 2F). This activity was abolished when each protein was omitted, indicated that all four are necessary for 6mA methylation. Furthermore, MTA1 harboring a D209A mutation in the catalytic DPPW motif showed no activity, even in the presence of MTA9, p1 and p2 (Figure 2E). We also created double mutations in MTA1 (N370A, E371A), which lie in the conserved region that interacts with the 2’ and 3’-hydroxyl groups of the ribose moiety in the SAM cofactor (Figure 2E). This mutant protein also exhibited no 6mA methylase activity. Taken together, we find that four proteins – MTA1, MTA9, p1, and p2 – are necessary for 6mA methylation in vitro, with MTA1 the likely catalytic subunit. Henceforth, we refer to these four proteins as the putative MTA1 complex (MTA1c).
Purification of the MTA1c proteins from an E. coli over-expression system raises the possibility of methyltransferase activity arising from contaminating Dam methylase; however, we exclude this possibility for three reasons: 1) the DNA substrate used in this assay does not contain 5’-NATC-3’ sites, which are recognized and methylated by Dam methylase (Horton et al., 2006). 2) Methyltransferase activity was only observed when all four recombinant proteins were incubated with DNA. If contaminating Dam methylase were present in one or more of these protein preparations, then background activity should be observed when subsets of these proteins are used in the assay. 3) Mutation of MTA1 catalytic residues leads to loss of methylation, which is also inconsistent with contaminating methyltransferase activity.
MTA1c preferentially methylates ApT dinucleotides in dsDNA
We next investigated the substrate preferences of MTA1c. First, in vitro transcription was performed to generate dsRNA and ssRNA from the input dsDNA substrate. We found that MTA1c methylates dsDNA but not dsRNA or ssRNA of the same sequence, indicating that it is selective for DNA over RNA (Figure S3H). We then generated a series of dsDNA substrates by annealing oligonucleotide pairs of different length and sequence. All of these substrates are bona fide Tetrahymena genomic DNA sequences. In each case, MTA1c can methylate the annealed dsDNA but not ssDNA (Figures 2G and S3I).
Since 6mA methylation mainly lies in ApT dinucleotides in vivo (Figures 1D and S2D), we asked whether MTA1c preferentially methylates this motif. To test this, we used a 27 bp dsDNA substrate with two ApT dinucleotides in its native sequence (Figure 2G). We disrupted one or both ApT motifs (Figure 2G) by mutually swapping the 5’ A with a neighboring base 5’-CAT-3’ → 5’-ACT-3’. Disrupting both ApT dinucleotides resulted in >10-fold reduced methylation, while disrupting only one motif led to a 2–4 fold loss (Figures 2G and S3K).
Given that 6mA occurs on both strands of genomic DNA in vivo (Figure 1E and S2E), we asked whether pre-existing methylation of one strand affects MTA1c activity. DNA oligonucleotides were nonspecifically methylated with 6mA using EcoGII (Murray et al., 2018), a bacterial 6mA methyltransferase. After rigorous purification, samples were annealed to an unmethylated, complementary strand to yield hemimethylated dsDNA (Figure S3F). MTA1c activity was 3–3.5 fold higher on hemimethylated substrates, relative to unmethylated dsDNA (Figure 2G). This effect was similar between dsDNA substrates pre-methylated on the sense or antisense strand, consistent with the lack of an overt strand bias in 6mA locations in vivo (Figure 1E and S2E). Importantly, the increase in MTA1c activity cannot be attributed to contaminating EcoGII in hemimethylated substrates, since no activity was observed in the absence of MTA1c (Figure S3J). Thus, pre-existing 6mA methylation stimulates MTA1c, indicative of a positive feedback loop.
We then asked whether MTA1c activity is modulated not only by the dinucleotide motif sequence per se, but also by flanking sequences. This may manifest as the wide variation in frequency of DNA 4-mers containing a methylated ApT dinucleotide 5’-NA*TN-3’ in vivo (Figure S3M). To test this, we used a dsDNA substrate containing two ApT dinucleotides, both within a 5’-CATT-3’. Swapping of the ApT motif with the adjacent downstream DNA residue produced substrates containing 5’-TATA-3’ (Figure S3L). Substrates with this change at both locations had 4-fold less MTA1c activity, and an intermediate effect when only one dinucleotide was altered (Figures S3L). These data indicate that 5’-CATT-3’ is the preferred methylation substrate, consistent with the higher frequency of methylated 5’-CA*TT-3’ versus 5’-TA*TA-3’ in both Tetrahymena and Oxytricha genomic DNA (Figure S3M). The difference in frequency of methylated sequences cannot simply be attributed to the higher frequency of the 4nt 5’-CATT-3’ motif vs. 5’-TATA-3’ in the genome, because the opposite trend is observed (Figure S3N). Thus, MTA1c is sensitive to variation in DNA sequences flanking the ApT dinucleotide motif.
MTA1 is necessary for 6mA methylation in vivo
Having established that MTA1c is a 6mA methyltransferase, we tested the role of MTA1c in mediating 6mA methylation in vivo in Oxytricha, for which we have ease of generating mutants. The genome-wide localization of 6mA is conserved between Oxytricha and Tetrahymena (Figures 1 and S2A–F), implying similar underlying enzymatic machinery. Indeed, all four component genes – MTA1, MTA9, p1, and p2 – are clearly conserved between both species (Figure S2G–J). The DPPW catalytic motif is also completely conserved in Tetrahymena and Oxytricha MTA1 but not MTA9, suggesting that MTA1 is the likely catalytic subunit of MTA1c in both ciliates (Figure S3E). To abrogate MTA1c function, we disrupted the Oxytricha MTA1 gene by inserting an ectopic DNA sequence 49 bp downstream of the start codon, resulting in a frameshift mutation and loss of the C-terminal MTase domain (Figure 3A). Oxytricha has two MTA1 paralogs, named MTA1 and MTA1-B (Figures 2A and S2G). We focused on MTA1 because MTA1-B is not expressed in vegetative Oxytricha cells (Swart et al., 2013), which we used to profile 6mA locations via SMRT-seq. Dot blot analysis confirmed a significant reduction in bulk 6mA levels in mutant lines (Figure 3B). We then examined 6mA positions at high resolution using SMRT-seq to understand how the DNA methylation landscape is altered in mta1 mutants. Notably, these mutants exhibit genome-wide loss of 6mA, with complete abolishment of the dimethylated ApT motif, and reduction in frequency of all other methylated dinucleotide motifs (Figures 3C–E). These findings are consistent across all biological replicates and are robust to wide variation in SMRT-seq parameters for calling 6mA modifications (Figure S4B–D). It cannot be attributed to variation in sequencing coverage between wild type and mutant lines. The loss of methylated ApT dinucleotides in mta1 mutants is consistent with our in vitro data suggesting that MTA1c primarily methylates ApT sites (Figures 2G and S3K). The Inter Pulse Duration ratio (degree of polymerase slowing during PacBio sequencing due to presence of a modified base) and estimated fractional methylation also decreased significantly at called 6mA sites in mta1 mutants (p < 2.2 × 10−16, Wilcoxon rank-sum test) (Figure S4A). MTA1 is therefore necessary for a significant proportion of in vivo 6mA methylation events in Oxytricha.
Figure 3. Genome-wide loss of 6mA in mta1 mutants.
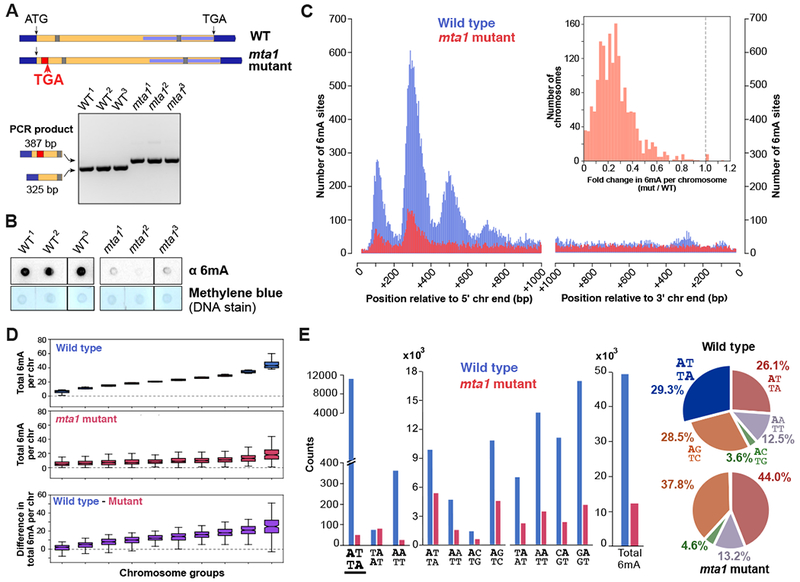
(A) Schematic depicting the disruption of Oxytricha MTA1 open reading frame. Flanking dark blue bars: 5’ and 3’ UTR; yellow = open reading frame; red = retention of 62 bp ectopic DNA segment; grey bar = intron; internal light blue bar = annotated MT-A70 domain; “ATG” = start codon; “TGA” = stop codon. Agarose gel analysis shows PCR confirmation of ectopic DNA retention.
(B) Dot blot analysis of RNase-treated genomic DNA.
(C) Histogram of 6mA counts near 5’ and 3’ Oxytricha chromosome ends. Inset depicts histogram of fold change in total 6mA in each chromosome, between mutant and wild type cell lines.
(D) Chromosomes are sorted into 10 groups according to total 6mA in wild type cells (blue boxplots). For each group, the total 6mA per chromosome in mutants, and the difference in total 6mA per chromosome is plotted below. Boxplot features are as described in Figure 1C.
(E) Motif distribution in wild type and mta1 mutants. Loss of ApT dimethylated motif is underlined.
What are the phenotypic consequences of 6mA loss in vivo? It has been proposed that DNA methylation – including 6mA and cytosine methylation – is involved in nucleosome organization (Fu et al., 2015; Huff and Zilberman, 2014). We thus asked whether nucleosome organization is altered in mta1 mutants. We quantified nucleosome “fuzziness”, defined as the standard deviation of MNase-seq read locations surrounding the called nucleosome peak (Lai and Pugh, 2017; Mavrich et al., 2008). A poorly positioned nucleosome consists of a shallow and wide peak of MNase-seq reads, manifested by a high fuzziness score. Nucleosomes were first grouped according to the change in flanking 6mA between wild type and mta1 mutant cells (Figure S5A–G). The nucleosomes that experience large changes in flanking 6mA exhibit significantly greater increase in fuzziness, compared to nucleosomes with little change in flanking 6mA (Figure S5A and S5D). Such nucleosomes also exhibit changes in occupancy that are consistent with an increase in fuzziness (Figures S5A and S5E). These results are robust to variation in MNase digestion (Figure S7C–D). On the other hand, nucleosome linkers do not change in length or occupancy, even though 6mA is lost from these regions (Figures S5B,C,F,G). We conclude that 6mA exerts subtle effects on nucleosome organization in vivo.
6mA disfavors nucleosome occupancy across the genome in vitro but not in vivo
Multiple factors, including 6mA, DNA sequence, and chromatin remodeling complexes, may collectively contribute to nucleosome organization in vivo. The effect of 6mA could therefore be masked by these elements. We next sought to determine whether 6mA directly impacts nucleosome organization. To this end, we assembled chromatin in vitro using Oxytricha gDNA, which contains cognate 6mA. To obtain a matched negative control lacking DNA methylation, 98 complete chromosomes were amplified using PCR (Figure 4A), purified and subsequently mixed together in stoichiometric ratios to obtain a “mini-genome” (Figure 4B). These chromosomes collectively reflect overall genome properties, including AT content, chromosome length, and transcriptional activity (Table S4). Native genomic DNA (containing 6mA) and amplified mini-genome DNA (lacking 6mA) were each assembled into chromatin in vitro using Xenopus or Oxytricha histone octamers (Figures S6A–F) and analyzed using MNase-seq. We computed nucleosome occupancy from the native genome and mini-genome samples across 199,795 overlapping DNA windows, spanning all base-pairs in the 98 chromosomes. This allowed the direct comparison of nucleosome occupancy in each window of identical DNA sequence, with and without 6mA (Figures 4C and 4D). Windows exhibit lower nucleosome occupancy with increasing 6mA, confirming the quantitative nature of this effect. Furthermore, similar trends were observed for both native Oxytricha and recombinant Xenopus histones, suggesting that the effects of 6mA on nucleosome organization arise mainly from intrinsic features of the histone octamer rather than from species-specific variants (Figure 4C and 4D). These results are also robust to the extent of MNase digestion of reconstituted chromatin (Figure S7A).
Figure 4. Effects of 6mA on nucleosome organization in vitro and in vivo.
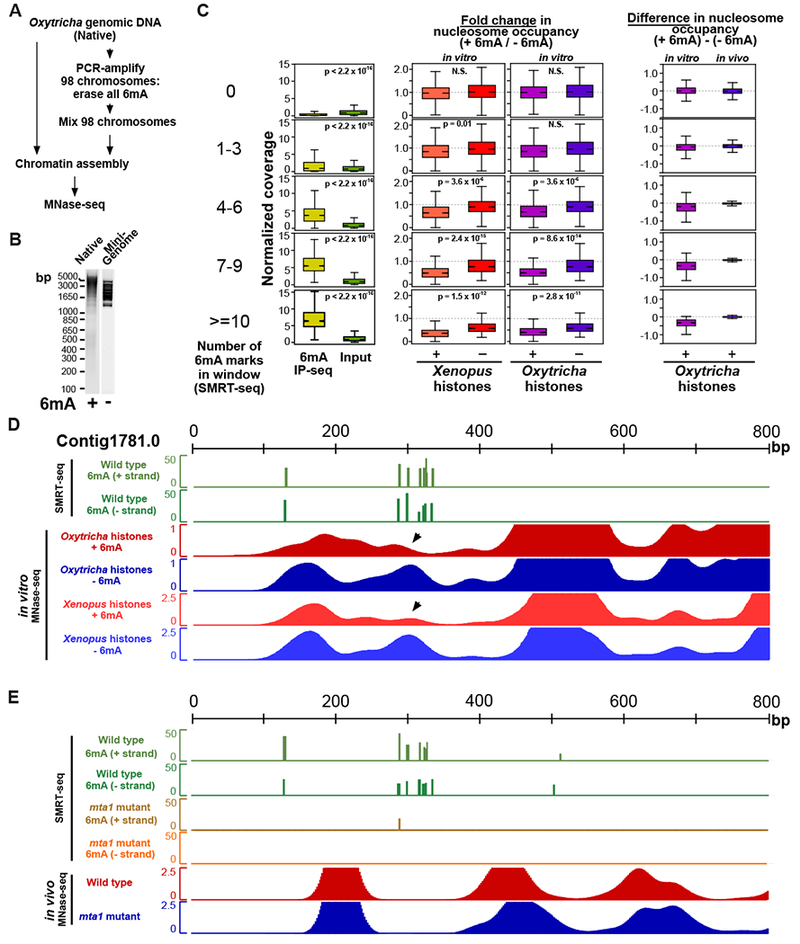
(A) Experimental workflow for the generation of mini-genome DNA.
(B) Agarose gel analysis of Oxytricha gDNA (‘Native’) and mini-genome DNA before chromatin assembly.
(C) Methylated regions exhibit lower nucleosome occupancy in vitro but not in vivo. Overlapping 51bp windows were analyzed across 98 chromosomes. For each window, the change in nucleosome occupancy in the absence versus presence of 6mA was calculated. Boxplot features are as described in Figure 1C. P-values were calculated using a two-sample unequal variance t-test. “N.S”: non-significant, with p > 0.05.
(D) Reduction in nucleosome occupancy at methylated loci in vitro (black arrowheads). For in vitro MNase-seq, “+ 6mA” refers to chromatin assembled on Oxytricha gDNA, while “−6mA” denotes chromatin assembled on mini-genome DNA. Vertical axis for SMRT-seq data denotes confidence score [−10 log(p-value)] of detection of 6mA, while that for in vitro MNase-seq data denotes nucleosome occupancy.
(E) No change in nucleosome occupancy in linker regions despite loss of 6mA in mta1 mutants. Vertical axes are the same as panel (D).
We then directly compared the impact of 6mA on nucleosome occupancy in vitro and in vivo. Loss of 6mA in vitro is achieved by mini-genome construction, while loss in vivo is achieved by the mta1 mutation. For each overlapping DNA window, we calculated the difference in nucleosome occupancy: 1) between native genome and mini-genome DNA in vitro, and 2) between wild type and mta1 mutants in vivo (Figure 4C). Nucleosome occupancy is indeed lower in the presence of 6mA methylation in vitro (Figures 4C and 4D). In contrast, no change in nucleosome occupancy is observed in vivo (Figures 4C and 4E). This result is consistent with our earlier analysis of linker occupancy in mta1 mutants (Figure S5C and S5G). We note that highly methylated DNA windows show greater change in 6mA relative to mta1 mutants (Figure 3D). Yet, these windows do not change in nucleosome occupancy in vivo. We conclude that 6mA methylation locally disfavors nucleosome occupancy in vitro, but that this intrinsic effect can be overcome by endogenous chromatin factors in vivo.
Modular synthesis of epigenetically defined chromosomes
The above experiments used kinetic signatures from SMRT-seq data to infer the presence of 6mA marks in genomic DNA. We next sought to confirm that 6mA is directly responsible for disfavoring nucleosomes in vitro, and to understand how this effect could be overcome by cellular factors. 6mA-containing oligonucleotides were annealed and subsequently ligated with DNA building blocks to form full length chromosomes. Importantly, these chromosomes contain 6mA at all locations identified by SMRT-seq in vivo. The representative chromosome, Contig1781.0, is 1.3kb, contains a clearly defined TSS, and encodes a single highly transcribed gene with a predicted RING finger domain. The length and gene structure are characteristic of typical Oxytricha chromosomes (Figure 5A). We independently validated the location of 6mA in vivo by sequencing chromosomal DNA immunoprecipitated with an anti-6mA antibody (Figure 5A).
Figure 5. Modular synthesis of full-length Oxytricha chromosomes.
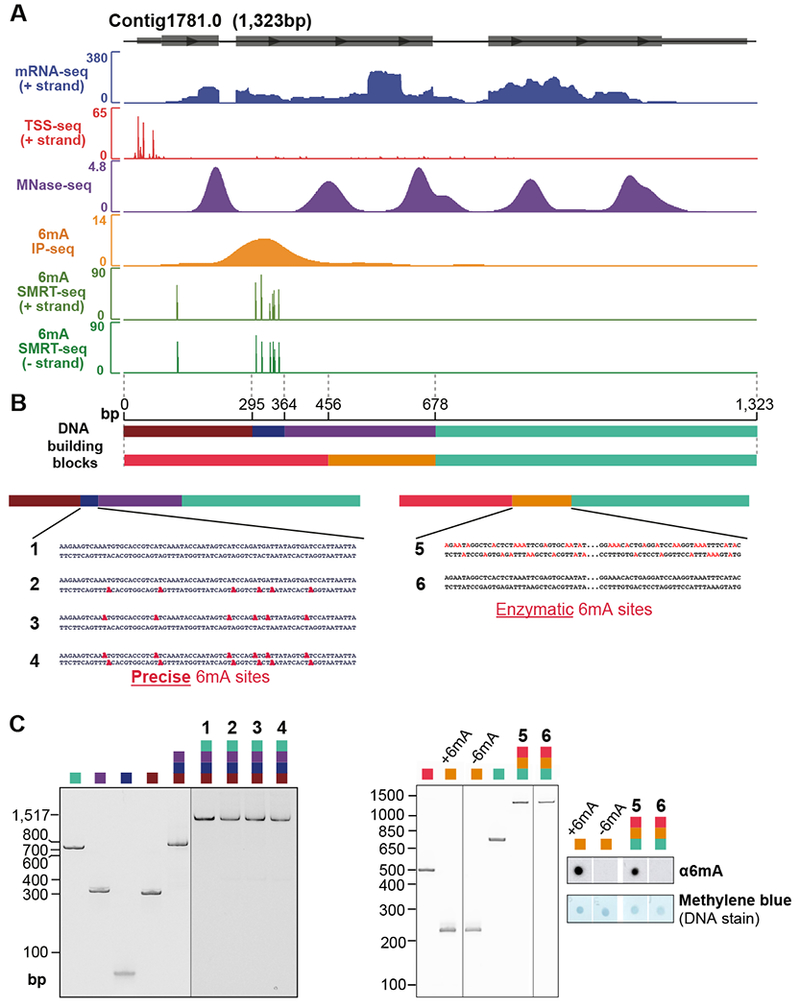
(A) Features of the chromosome selected for synthesis. Grey boxes represent exons. All data tracks represent normalized coverage except for SMRT-seq, which represents the confidence score [−10 log(p-value)] of detection of each methylated base.
(B) Schematic of chromosome construction. Different colors denote DNA building blocks ligated to form the full-length chromosome. Precise 6mA sites (bold red) represent cognate 6mA positions revealed by SMRT-seq in native genomic DNA. These are introduced via oligonucleotide synthesis. For chromosome 5, 6mA sites (non-bold red) represent possible locations ectopically installed by a bacterial 6mA methyltransferase, EcoGII. Intervening sequence within chromosomes 5 and 6 is represented as “…”
(C) Native polyacrylamide gel analysis and anti-6mA dot blot analysis of building blocks and purified synthetic chromosomes.
Four chromosome variants were synthesized, with cognate 6mA sites on neither, one, or both DNA strands (chromosomes 1–4 in Figures 5B–C). Chromatin was assembled by salt dialysis with either Oxytricha or Xenopus nucleosomes and subsequently digested with MNase to obtain mononucleosomal DNA (Figures 6A and S6G). Tiling qPCR was used to quantify nucleosome occupancy at ~50bp increments along the entire length of the synthetic chromosome (Figure 6B). The fully methylated locus exhibits a ~46% reduction in nucleosome occupancy relative to the unmethylated variant, while hemimethylated chromosomes containing half the number of 6mA marks showed intermediate nucleosome occupancy at the corresponding region (Figure 6B). The reduction in nucleosome occupancy was confined to the methylated region, and not observed across the rest of the chromosome. Similar trends were observed when chromatin was assembled using the NAP1 histone chaperone (Figure S7F, top panel), indicating that this effect is not an artifact of the salt dialysis method. Furthermore, moving 6mA to an ectopic location (chromosome 5 in Figures 5B–C) decreases nucleosome occupancy at that site (Figure 6C). We conclude that 6mA directly disfavors nucleosome occupancy in a local, quantitative manner in vitro.
Figure 6. Quantitative modulation of nucleosome occupancy by 6mA.
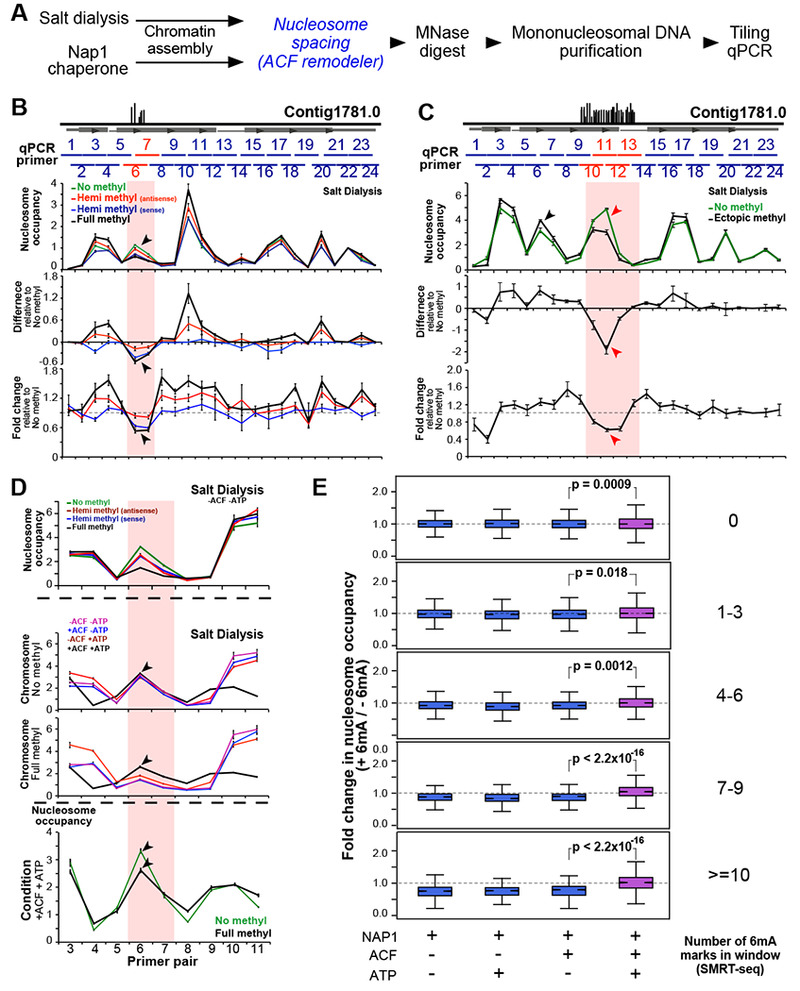
(A) Experimental workflow. Chromatin is assembled using either salt dialysis or the NAP1 histone chaperone. Italicized blue steps are selectively included.
(B) Tiling qPCR analysis of synthetic chromosome with cognate 6mA sites. Horizontal grey box represents annotated gene, and vertical black lines depict native 6mA positions. Horizontal blue bars span ~100bp regions amplified by qPCR. Red horizontal lines represent the region containing 6mA. ‘Hemi methyl’ chromosomes contain 6mA on the antisense and sense strands, respectively, while the ‘Full methyl’ chromosome has 6mA on both strands. Black arrowheads: decrease in nucleosome occupancy specifically at the 6mA cluster.
(C) Tiling qPCR analysis of ectopically methylated synthetic chromosome. Vertical black lines illustrate possible 6mA sites installed enzymatically. Red arrowheads: decrease in nucleosome occupancy in the ectopically methylated region. Black arrowheads: position of cognate 6mA sites (not in this construct).
(D) Tiling qPCR analysis of chromatin from panel B that is subsequently incubated with ACF and/or ATP. ACF equalizes nucleosome occupancy between the 6mA cluster and flanking regions in the presence of ATP (black line). Nucleosome occupancy at the methylated region is not restored to the same level as the unmethylated control (black arrowheads).
(E) MNase-seq analysis of chromatin is assembled on native gDNA (“+” 6mA) and mini-genome DNA (“−“ 6mA) using NAP1 +/− ACF and ATP. P-values were calculated using a two-sample unequal variance t-test.
Chromatin remodelers restore nucleosome occupancy over 6mA sites
Nucleosome occupancy in vivo is influenced not only by DNA sequences, but also by trans-acting factors such as ATP-dependent chromatin remodeling factors (Struhl and Segal, 2013). We used synthetic, methylated chromosomes to test how the well-studied chromatin remodeler ACF responds to 6mA in native DNA. ACF generates regularly spaced nucleosome arrays in vitro and in vivo (Clapier and Cairns, 2009; Ito et al., 1997). Its catalytic subunit ISWI is conserved across eukaryotes, including Oxytricha and Tetrahymena (Table S2). Synthetic chromosomes were assembled into chromatin by salt dialysis as before, then incubated with ACF in the presence of ATP (Figures S6H and 6D). We find that ACF partially – but not completely – restores nucleosome occupancy over the methylated locus in an ATP-dependent manner (Figure 6D). This effect is observed when ACF was added to chromatin assembled by salt dialysis or the NAP1 histone chaperone (Figure 6D and S7F). ACF also restores nucleosome occupancy over methylated loci in native genomic DNA (Figures 6E and S6I), indicating that the effect is not restricted to a single chromosome. This result is robust to the extent of MNase digestion (Figure S7B). Although the heterologous system used here may differ from endogenous chromatin assembly factors in Oxytricha, our experiment illustrates the principle that trans-acting factors can counteract or even overcome the effect of 6mA on nucleosome organization.
Disruption of MTA1 impacts gene expression and sexual development
Since mta1 mutants exhibit genome-wide loss of 6mA, we assayed these cells for transcriptional changes by poly(A)+ RNAseq. Only a small minority of genes show significant changes in gene expression (10% FDR, Figure 7A). To examine the methylation status of these differentially expressed genes, we grouped them according to “starting” methylation level, as defined by the total number of 6mA marks near the TSS in wild type cells. Genes exhibit two distinct transcriptional responses: those with low starting levels of 6mA exhibit a small change in 6mA between wild type and mutant cells (Figure 3D) and tend to be significantly upregulated in mutant lines (p = 2.8 × 10−9, Fisher’s exact test; and Figure 7B). Surprisingly, genes with high starting 6mA are not enriched in differentially expressed genes (p > 0.1, Fisher’s exact test), even though they exhibit greater loss of 6mA in mutants (Figure 3D). Steady state RNAseq levels are therefore largely robust to drastic changes in 6mA levels. Since most, but not all, 6mA is lost from mta1 mutants (Figure 3C), it is also possible that residual DNA methylation across the genome sufficiently buffers genes from changes in transcription.
Figure 7. Effects of 6mA on gene expression and cell viability in vivo.
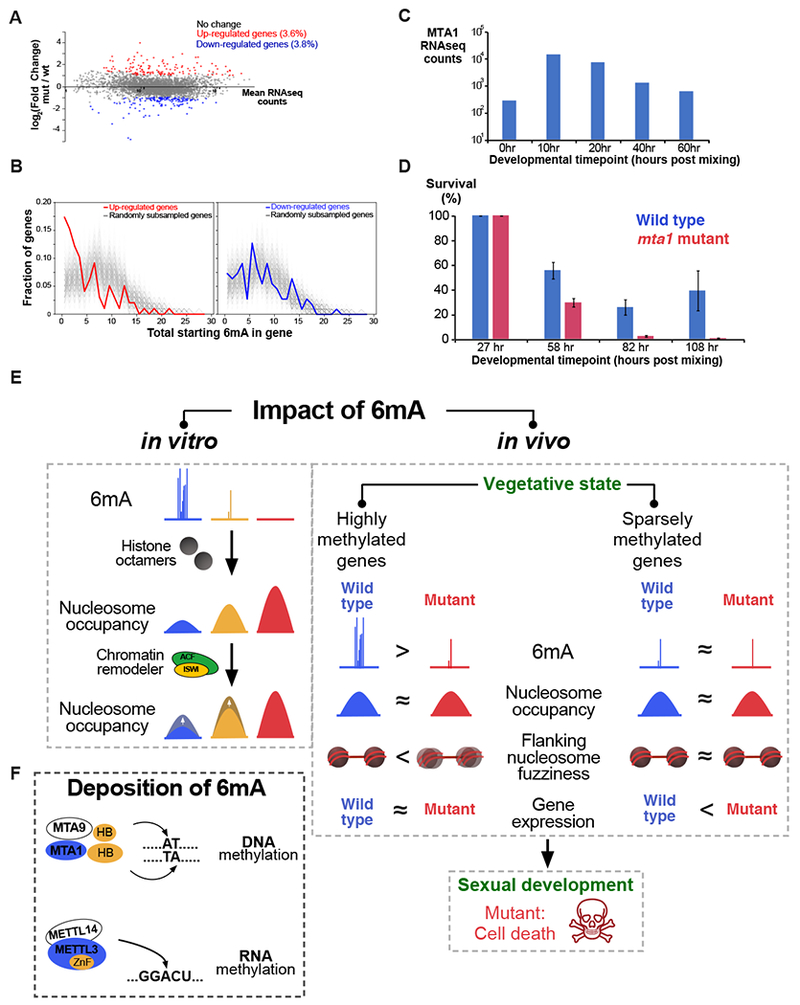
(A) Horizontal axis: the mean RNAseq counts across all biological replicates from wild type and mta1 mutant data for each gene. Vertical axis: log2(fold change) in gene expression (mutant / wild type).
(B) Upregulated genes tend to be sparsely methylated compared to randomly subsampled genes (grey lines). See Methods for analysis procedure.
(C) RNAseq analysis of MTA1 expression during the sexual cycle of Oxytricha. RNAseq timecourse data are from (Swart et al., 2013). The total duration of the sexual cycle is ~60 hr.
(D) Survival analysis of Oxytricha cells during the sexual cycle. The total cell number at each timepoint is normalized to 27 hr data to obtain the percentage survival. Error bars represent s.e.m. (n = 4).
(E) Model illustrating the impact of 6mA methylation by MTA1c on nucleosome organization and gene expression.
(F) Comparison of DNA and RNA N6-adenine methyltransferases. Blue denotes catalytic subunit, yellow denotes subunit with predicted DNA or RNA binding domain.
Because the aforementioned phenotypic changes were assayed in vegetative Oxytricha cells, we asked whether MTA1 may play roles outside of this developmental state. MTA1 transcript levels are markedly upregulated in the sexual cycle, as assayed by poly(A)+ RNAseq (Figure 7C). Strikingly, mta1 mutants fail to complete the sexual cycle when induced to mate, and display complete lethality (Figure 7D). Our data do not exclude the possibility that m6A RNA methylation, in addition to 6mA DNA methylation, is also impacted by MTA1 loss during development. Further studies would clarify the role of MTA1 in these pathways.
Discussion
Here, we identify MTA1c as a conserved, hitherto undescribed 6mA methyltransferase. It consists of two MT-A70 proteins (MTA1 / MTA9) and two homeobox-like proteins (p1 / p2). The composition of MTA1c provides immediate insights into how it specifically methylates DNA (Figure 7F). MTA1 likely mediates transfer of the methyl group from SAM to the acceptor adenine moiety, given that it contains conserved amino acid residues implicated in catalysis and SAM binding (Figure S3E). Indeed, we show that these residues are necessary for its activity (Figure 2E). While MTA1 constitutes the catalytic center, it lacks a CCCH-type zinc finger domain that is necessary for RNA binding in the canonical m6A methyltransferase METTL3. Instead, nucleic acid binding is likely assumed by the homeobox-like domains in p1 and p2, which are known to specifically engage dsDNA through helix-turn-helix motifs.
The observation that MTA1c is more active in the presence of pre-methylated DNA templates is reminiscent of the CpG methyltransferase DNMT1. Yet, MTA1c and DNMT1 exhibit distinct protein domain architectures. Further biochemical studies are required to elucidate the molecular basis of this property. A distinct MT-A70 protein, named TAMT-1, was recently reported to act as a 6mA methyltransferase in Tetrahymena, (Luo et al., 2018), suggesting that multiple enzymes mediate 6mA deposition. It remains to be determined how MTA1c and TAMT-1 collectively mediate DNA methylation at various developmental stages, and whether cross-talk occurs between these enzymes.
In addition to identifying the ciliate 6mA methyltransferase, we investigated the function of 6mA in vitro by building epigenetically defined chromosomes. We show that 6mA directly disfavors nucleosome occupancy in a local, quantitative manner, independent of DNA sequence (Figure 7E). Our experiments do not reveal exactly how 6mA disfavors nucleosome occupancy. Early studies suggest that 6mA destabilizes dA:dT base pairing, leading to a decrease in the melting temperature of DNA (Engel and von Hippel, 1978). Whether this or some other property of 6mA contributes to lowered nucleosome stability awaits further investigation.
Intriguingly, nucleosome organization exhibits only subtle changes after genome-wide loss of 6mA (Figure 7E). Only a small set of genes (<10%) is transcriptionally dysregulated. It is possible that residual 6mA in mta1 mutants could mask relevant phenotypes. Nonetheless, our results caution against interpreting 6mA function solely based on correlation with genomic elements. We also find that 6mA intrinsically disfavors nucleosomes in vitro, but – crucially – this effect can be overridden by distinct factors in vitro and in vivo. We propose that phased nucleosome arrays are first established in vivo, which then restrict MTAI-mediated methylation to linker regions due to steric hindrance. This in turn decreases the fuzziness of flanking nucleosomes, reinforcing chromatin organization. Therefore, 6mA tunes nucleosome organization in vivo. Our data do not support the hypothesis that nucleosome phasing is established by pre-deposited 6mA.
More broadly, our work showcases the utility of Oxytricha chromosomes for advancing chromatin biology. By extending current technologies (Müller et al., 2016), it should be feasible to introduce both modified nucleosomes and DNA methylation in a site-specific manner on full-length chromosomes. Such ‘designer’ chromosomes will serve as powerful tools for studying DNA-templated processes such as transcription within a fully native DNA environment.
STAR Methods
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to Laura Landweber (Laura.Landweber@columbia.edu).
Experimental model and subject details
Oxytricha trifallax
Vegetative Oxytricha trifallax strain JRB310 was cultured at a density of 1.5 × 107 cells/L to 2.5 × 107 cells/L in Pringsheim media (0.11mM Na2HPO4, 0.08mM MgSO4, 0.85mM Ca(NO3)2, 0.35mM KCl, pH 7.0) and fed daily with Chlamydomonas reinhardtii. Cells were filtered through cheesecloth to remove debris and collected on a 10μm Nitex mesh for subsequent experiments.
Tetrahymena thermophila
Stock cultures of vegetative Tetrahymena thermophila strain SB210 were maintained in Neff medium (0.25% w/v proteose peptone, 0.25% w/v yeast extract, 0.5% glucose, 33.3μM FeCl3). These cultures were inoculated into SSP medium (2% w/v proteose peptone, 0.1% w/v yeast extract, 0.2% w/v glucose, 33μM FeCl3) and grown to log-phase (~3.5 × 105 cells/mL) through constant shaking at 125 rpm / 30°C.
Method details
in vivo MNase-seq
3 × 105 vegetative Oxytricha cells were fixed in 1% w/v formaldehyde for 10 min at room temperature with gentle shaking, and then quenched with 125mM glycine. Cells were lysed by dounce homogenization in lysis buffer (20mM Tris pH 6.8, 3% w/v sucrose, 0.2% v/v Triton X-100, 0.01% w/v spermidine trihydrochloride) and centrifuged in a 10%-40% discontinuous sucrose gradient (Lauth et al., 1976) to purify macronuclei. The resulting macronuclear preparation was pelleted by centrifugation at 4000 × g, washed in 50ml TMS buffer (10mM Tris pH 7.5, 10mM MgCl2, 3mM CaCl2, 0.25M sucrose), resuspended in a final volume of 300μL, and equilibriated at 37°C for 5 min. Chromatin was then digested with MNase (New England Biolabs) at a final concentration of 15.7 Kunitz Units / μL at 37°C for 1 min 15 sec, 3 min, 5 min, 7 min 30sec, 10 min 30 sec, and 15 min respectively. Reactions were stopped by adding ½ volume of PK buffer (300mM NaCl, 30mM Tris pH 8, 75mM EDTA pH 8, 1.5% w/v SDS, 0.5mg/mL Proteinase K). Each sample was incubated at 65°C overnight to reverse crosslinks and deproteinate samples. Subsequently, nucleosomal DNA was purified through phenol:chloroform:isoamyl alcohol extraction and ethanol precipitation. Each sample was loaded on a 2% agarose-TAE gel to check the extent of MNase digestion. The sample exhibiting ~80% mononucleosomal species was selected for MNase-seq analysis, in accordance with previous guidelines (Zhang and Pugh, 2011). Mononucleosome-sized DNA was gel-purified using a QIAquick gel extraction kit (QIAGEN). Illumina libraries were prepared using an NEBNext Ultra II DNA Library Prep Kit (New England Biolabs) and subjected to paired-end sequencing on an Illumina HiSeq 2500 according to manufacturer’s instructions. All vegetative Tetrahymena MNase-seq data were obtained from (Beh et al., 2015).
poly(A)+ RNA-seq and TSS sequencing
Oxytricha cells were lysed in TRIzol reagent (Thermo Fisher Scientific) for total RNA isolation according to manufacturer’s instructions. Poly(A)+ RNA was then purified using the NEBNext Poly(A) mRNA Magnetic Isolation Module (New England Biolabs). Oxytricha poly(A)+ RNA was prepared for RNA-seq using the ScriptSeq v2 RNA-Seq Library Preparation Kit (Illumina). Tetrahymena poly(A)+ RNA-seq data was obtained from (Xiong et al., 2012). The 5’ ends of capped RNAs were enriched from vegetative Oxytricha total RNA using the RAMPAGE protocol (Batut et al., 2013), and used for library preparation, Illumina sequencing and subsequent transcription start site determination (ie. “TSS-seq”). These data were used to plot the distribution of Oxytricha TSS positions in Figure 1A. TSS positions used for analysis outside of Figure 1A were obtained from (Swart et al., 2013) and (Beh et al., 2015).
For RNAseq analysis of genes grouped according to “starting” methylation level level: total 6mA was counted between 100 bp upstream to 250 bp downstream of the TSS. Genes with high starting methylation have total 6mA in the 90th percentile and higher. Genes with low starting methylation have total 6mA at or below the 10th percentile.
Immunoprecipitation and Illumina sequencing of methylated DNA (6mA IP-seq)
Genomic DNA was isolated from vegetative Oxytricha cells using the Nucleospin Tissue Kit (Takara Bio USA, Inc.). DNA was sheared into 150bp fragments using a Covaris LE220 ultra-sonicator (Covaris). Samples were gel-purified on a 2% agarose-TAE gel, blunted with DNA polymerase I (New England Biolabs), and purified using MinElute spin columns (QIAGEN). The fragmented DNA was dA-tailed using Klenow Fragment (3′ -> 5′ exo-) (New England Biolabs) and ligated to Illumina adaptors following manufacturer’s instructions. Subsequently, 2.2μg of adaptor-ligated DNA containing 6mA was immunoprecipitated using an anti-N6-methyladenosine antibody (Cedarlane Labs) conjugated to Dynabeads Protein A (Invitrogen). The anti-6mA antibody is commonly used for RNA applications, but has also been demonstrated to recognize 6mA in DNA (Fioravanti et al., 2013; Xiao and Moore, 2011). The immunoprecipitated and input libraries were treated with proteinase K, extracted with phenol:chloroform, and ethanol precipitated. Finally, they were PCR-amplified using Phusion Hot Start polymerase (New England Biolabs) and used for Illumina sequencing.
Sample preparation for SMRT-seq
Vegetative Oxytricha macronuclei were isolated as described in the subheading “in vivo MNase-seq” of this study. Vegetative Tetrahymena macronuclei were isolated by differential centrifugation (Beh et al., 2015). Oxytricha and Tetrahymena cells were not fixed prior to nuclear isolation. Genomic DNA was isolated from Oxytricha and Tetrahymena macronuclei using the Nucleospin Tissue Kit (Macherey-Nagel). Alternatively, whole Oxytricha cells instead of macronuclei were used. SMRT-seq according to manufacturer’s instructions, using P5-C3 and P6-C4 chemistry, as in (Chen et al., 2014),. Oxytricha and Tetrahymena macronuclear DNA were used for SMRT-seq in Figures 1 and S2A–F, while Oxytricha whole cell DNA was used for all other Figures. Since almost all DNA in Oxytricha cells is derived from the macronucleus (Prescott, 1994), similar results are expected between the use of purified macronuclei or whole cells.
Illumina data processing
Reads from all biological replicates were merged before downstream processing. All Illumina sequencing data were quality trimmed (minimum quality score = 20) and length-filtered (minimum read length = 40nt) using Galaxy (Blankenberg et al., 2010; Giardine et al., 2005; Goecks et al., 2010). MNase-seq and 6mA IP-seq reads were mapped to complete chromosomes in the Oxytricha trifallax JRB310 (August 2013 build) or Tetrahymena thermophila SB210 macronuclear reference genomes (June 2014 build) using Bowtie2 (Langmead and Salzberg, 2012) with default settings, while poly(A)+ RNA-seq and TSS-seq reads were mapped using TopHat2 (Mortazavi et al., 2008) with August 2013 Oxytricha gene models or June 2014 Tetrahymena gene models, with default settings.
MNase-seq datasets were generated by paired-end sequencing. Within each MNase-seq dataset, the read pair length of highest frequency was identified. All read pairs with length +/− 25bp from this maximum were used for downstream analysis. On the other hand, 6mA IP-seq datasets were generated by single-read sequencing. 6mA IP-seq single-end reads were extended to the mean fragment size, computed using cross-correlation analysis (Kharchenko et al., 2008). The per-basepair coverage of Oxytricha MNase-seq read pair centers and extended 6mA IP-seq reads were respectively computed across the genome. Subsequently, the per-basepair coverage values were normalized by the average coverage within each chromosome to account for differences in DNA copy number (and hence, read depth) between Oxytricha chromosomes (Swart et al., 2013). The per-basepair coverage values were then smoothed using a Gaussian filter of standard deviation = 15. This smoothed data is denoted as “normalized coverage” or “nucleosome occupancy”. Tetrahymena MNase-seq data were processed similarly to Oxytricha, except that DNA copy number normalization was omitted as Tetrahymena chromosomes have uniform copy number (Eisen et al., 2006).
For the MNase-seq analysis in Figures 4C, 6E, S7A, and S7B, nucleosome occupancy and 6mA IP-seq coverage were calculated within overlapping 51bp windows across the 98 assayed chromosomes. Windows were binned according to the number of 6mA residues within. The in vitro MNase-seq coverage from chromatinized native gDNA (“+” 6mA) was divided by the corresponding coverage from chromatinized minigenome DNA (“−“ 6mA) to obtain the fold change in nucleosome occupancy in each window. Alternatively, a subtraction was performed on these datasets to obtain the difference in nucleosome occupancy in vitro. Identical DNA sequences were compared for each calculation. These data are labeled as (“+” histones) in Figures 4C and S7A. Naked native gDNA and mini-genome DNA were also MNase-digested, sequenced and analyzed in the same manner to control for MNase sequence preferences (“−“ histones). Nucleosome occupancy in vivo corresponds to normalized MNase-seq coverage from wild type and mta1 mutant cells.
Nucleosome positions were iteratively called as local maxima in normalized MNase-seq coverage, as previously described (Beh et al., 2015). “Consensus” +1, +2, +3 nucleosome positions downstream of the TSS were inferred from aggregate MNase-seq profiles across the genome (Figure 1A for Oxytricha and Figure S2A for Tetrahymena). Each gene was classified as having a +1, +2, +3 and/or +4 nucleosome if there is a called nucleosome dyad within 75 bp of the consensus nucleosome position.
RNA-seq and TSS-seq read coverage were calculated without normalization by DNA copy number since there is no correlation between Oxytricha DNA and transcript levels (Swart et al., 2013).
Oxytricha TSSs were called from TSS-seq data using CAGEr (Haberle et al., 2015); with clusterCTSS parameters (threshold = 1.6, thresholdIsTpm = TRUE, nrPassThreshold = 1, method = “paraclu”, removeSingletons = TRUE, keepSingletonsAbove = 5). Only TSSs with tags per million counts > 0.1 were used for downstream analysis. Tetrahymena TSSs were obtained from (Beh et al., 2015).
SMRT-seq data processing
We processed SMRT-seq data with SMRTPipe v1.87.139483 in the SMRT Analysis 2.3.0 environment using, in order, the P_Fetch, P_Filter (with minLength = 50, minSubreadLength = 50, readScore = 0.75, and artifact = −1000), P_FilterReports, P_Mapping (with gff2Bed = True, pulsemetrics = DeletionQV, IPD, InsertionQV, PulseWidth, QualityValue, MergeQV, SubstitutionQV, DeletionTag, and loadPulseOpts = byread), P_MappingReports, P_GenomicConsensus (with algorithm = quiver, outputConsensus = True, and enableMapQVFilter = True), P_ConsensusReports, and P_ModificationDetection (with identifyModifcations = True, enableMapQVFilter = False, and mapQvThreshold = 10) modules. All other parameters were set to the default. The Oxytricha August 2013 reference genome build was used for mapping Oxytricha SMRT-seq reads, with Contig10040.0.1, Contig1527.0.1, Contig4330.0.1, and Contig54.0.1 removed, as they are perfect duplicates of other Contigs in the assembly. Tetrahymena SMRT-seq reads were mapped to the June 2014 reference genome build. Only chromosomes with high SMRT-seq coverage (>= 80× for Oxytricha; >=100× for Tetrahymena) were used for all 6mA-related analyses.
Chromosome synthesis
Synthetic Contig1781.0 chromosomes were constructed from “building blocks” of native chromosome sequence (Figures 5B and 5C). The dark blue building block in Figure 5B was prepared by annealing synthetic oligonucleotides, while all other building blocks were generated by PCR-amplification from genomic DNA using Phusion DNA polymerase (New England Biolabs). All oligonucleotides used for annealing and PCR amplification are listed in Table S6. The PCR-amplified building blocks contain terminal restriction sites for BsaI (New England Biolabs), a type IIS restriction enzyme that cuts distal from these sites. BsaI cleaves within the native DNA sequence, generating custom 4nt 5’ overhangs and releasing the non-native BsaI restriction site as small fragments that are subsequently purified away. The BsaI-generated overhangs are complementary only between adjacent building blocks, conferring specificity in ligation and minimizing undesired by-products. After BsaI digestion, PCR building blocks were purified by phenol:chloroform extraction and ethanol precipitation. Building blocks were then sequentially ligated to each other using T4 DNA ligase (New England Biolabs) and purified by phenol:chloroform extraction and ethanol precipitation. Size selection after each ligation step was performed using polyethylene glycol (PEG) precipitation or Ampure XP beads (Beckman Coulter) to enrich for the large ligated product over its smaller constituents. The size of individual building blocks and their corresponding order of ligation were designed to maximize differences in size between ligated products and individual building blocks. This increases the efficiency in size selection of products over reactants. Chromosomes 1 and 6 in Figure 5B was generated by full length PCR from genomic DNA. To prepare chromosomes 2-4 in Figure 5B, the red, dark blue, and purple blocks were first ligated in a 3-piece reaction and purified from the individual components. This product was subsequently ligated with the turquoise building block to obtain the full length chromosome. To prepare chromosomes 5 in Figure 5B, the red, orange, and emerald building blocks were ligated in a 3-piece reaction and subsequently purified. All chromosomes were subjected to Sanger sequencing to verify ligation junctions. 6mA was installed in synthetic chromosomes using annealed oligonucleotides, or by incubation of DNA building blocks with EcoGII methyltransferase (New England Biolabs).
Verification of synthetic chromosome sequences
All chromosomes were dA-tailed using Klenow Fragment (3′ -> 5′ exo-) (New England Biolabs), cloned using a TOPO TA cloning kit (Thermo Fisher) or StrataClone PCR Cloning Kit (Agilent Technologies), transformed into One Shot TOP10 chemically competent E. coli, and sequenced using flanking T7, T3, M13F, or M13R primers.
Preparation of Oxytricha histones
Vegetative Oxytricha trifallax strain JRB310 was cultured as described in the subheading: “Experimental model and subject details” of this study. Cells were starved for 14 hr and subsequently harvested for macronuclear isolation as described in the subheading: “in vivo MNase-seq” of this study. However, formaldehyde fixation was omitted. Purified nuclei were pelleted by centrifugation at 4000 × g, resuspended in 0.421mL 0.4N H2SO4 per 106 input cells, and nutated for 3 hr at 4°C to extract histones. Subsequently, the acid-extracted mixture was centrifuged at 21,000 × g for 15 min to remove debris. Proteins were precipitated from the cleared supernatant using trichloroacetic acid (TCA), washed with cold acetone, then dried and resuspended in 2.5% v/v acetic acid. Individual core histone fractions were purified from crude acid-extracts using semi-preparative RP-HPLC (Vydac C18, 12 micron, 10 mM × 250 mm) with 40-65% HPLC solvent B over 50 min (Figure S6A). The identity of each purified histone fraction was verified by western analysis (Figure S6C) using antibodies: anti-H2A (Active Motif #39111), anti-H2B (Abcam #ab1790), anti-H3 (Abcam #ab1791), anti-H4 (Active Motif #39269).
Preparation of recombinant Xenopus histones
All RP-HPLC analyses were performed using 0.1% TFA in water (HPLC solvent A), and 90% acetonitrile, 0.1% TFA in water (HPLC solvent B) as the mobile phases. Wild-type Xenopus H4, H3 C110A, H2B and H2A proteins were expressed in BL21(DE3) pLysS E.coli and purified from inclusion bodies through ion exchange chromatography (Debelouchina et al., 2016). Purified histones were characterized by ESI-MS using a MicrOTOF-Q II ESI-Qq-TOF mass spectrometer (Bruker Daltonics). H4: calculated 11,236 Da, observed 11,236.1 Da; H3 C110A: calculated 15,239 Da, observed 15,238.7 Da; H2A: calculated 13,950 Da, observed 13,949.8 Da; H2B: calculated 13,817 Da, observed 13,816.8 Da.
Preparation of histone octamers
Oxytricha and Xenopus histone octamers were respectively refolded from core histones using established protocols (Beh et al., 2015; Debelouchina et al., 2016). Briefly, lyophilized histone proteins (Xenopus modified or wild-type; Oxytricha acid-extracted) were combined in equimolar amounts in 6 M guanidine hydrochloride, 20 Mm Tris pH 7.5 and the final concentration was adjusted to 1mg/mL. The solution was dialyzed against 2M NaCl, 10mM Tris, 1mM EDTA, and the octamers were purified from tetramer and dimer species using size-exclusion chromatography on a Superdex 200 10/300 column (GE Healthcare Life Sciences). The purity of each fraction was analyzed by SDS-PAGE. Pure fractions were combined, concentrated and stored in 50% v/v glycerol at −20°C.
Preparation of mini-genome DNA
98 full-length chromosomes were individually amplified from Oxytricha trifallax strain JRB310 genomic DNA using Phusion DNA polymerase (New England Biolabs). Primer pairs are listed in Table S6. Amplified chromosomes were separately purified using a MinElute PCR purification kit (QIAGEN), and then mixed in equimolar ratios to obtain “mini-genome” DNA. The sample was concentrated by ethanol precipitation and adjusted to a final concentration of ~1.6mg/mL.
Preparation of native genomic DNA for chromatin assemblystarry
Macronuclei were isolated from vegetative Oxytricha trifallax strain JRB310 as described in the subheading “in vivo MNase-seq” of this study. However, cells were not fixed prior to nuclear isolation. Genomic DNA was purified using the Nucleospin Tissue kit (Macherey-Nagel). Approximately 200μg of genomic DNA was loaded on a 15%-40% linear sucrose gradient and centrifuged in a SW 40 Ti rotor (Beckman Coulter) at 160,070 × g for 22.5hr at 20°C. Sucrose solutions were in 1M NaCl, 20mM Tris pH 7.5, 5mM EDTA. Individual fractions from the sucrose gradient were analyzed on 0.9% agarose-TAE gels. Fractions containing high molecular weight DNA that migrated at the mobility limit were discarded as such DNA species were found to interfere with downstream chromatin assembly. All other fractions were pooled, ethanol precipitated, and adjusted to 0.5mg/mL DNA.
Chromatin assembly and preparation of mononucleosomal DNA
Chromatin assemblies were prepared by salt gradient dialysis as previously described (Beh et al., 2015; Luger et al., 1999), or using mouse NAP1 histone chaperone and Drosophila ACF chromatin remodeler as previously described (An and Roeder, 2003; Fyodorov and Kadonaga, 2003). Details of each chromatin assembly procedure are listed below. To reduce sample requirements while maintaining adequate DNA concentrations for chromatin assembly, synthetic chromosomes were first mixed with a hundred-fold excess of “buffer” DNA (PCR-amplified Oxytricha Contig17535.0). We verified that nucleosome occupancy in the methylated region (qPCR primer pairs 6 and 7) of the synthetic chromosome is unaffected by the presence of buffer DNA (Figure S7E). Native and mini-genome DNA were not mixed with buffer DNA prior to chromatin assembly.
For chromatin assembly through salt dialysis: histone octamers and (synthetic chromosome + buffer) DNA were mixed in a 0.8:1 mass ratio, while histone octamers and (native or mini-genome) DNA were mixed in a 1.3:1 mass ratio, each in a 50μL total volume. Samples were first dialyzed into start buffer (10mM Tris pH 7.5, 1.4M KCl, 0.1mM EDTA pH 7.5, 1mM DTT) for 1 hr at 4°C. Then, 350mL end buffer (10mM Tris pH 7.5, 10mM KCl, 0.1mM EDTA, 1mM DTT) was added at a rate of 1mL/min with stirring. The assembled chromatin was dialyzed overnight at 4°C into 200mL end buffer, followed by a final round of dialysis in fresh 200mL end buffer for 1 hr at 4°C. The assembled chromatin was then adjusted to 50mM Tris pH 7.9, 5mM CaCl2 and digested with MNase (New England Biolabs) to mainly mononucleosomal DNA as previously described (Beh et al., 2015).
For chromatin assembly using mouse NAP1 and Drosophila ACF: NAP1 was recombinantly expressed and purified as described in (An and Roeder, 2003). ACF was purchased from Active Motif. 0.49μM NAP1 and 58nM histone octamer were first mixed in a 302μl reaction volume containing 62mM KCl, 1.2% w/v polyvinyl alcohol (Sigma Aldrich), 1.2% w/v polyethylene glycol 8000 (Sigma Aldrich), 25mM HEPES-KOH pH 7.5, 0.1mM EDTA-KOH, 10% v/v glycerol, and 0.01% v/v NP-40. The NAPI-histone mix was incubated on ice for 30 min. Meanwhile, “AM” mix was prepared, consisting of 20mM ATP (Sigma Aldrich), 200mM creatine phosphate (Sigma Aldrich), 33.3mM MgCl2, 33.3μg/μl creatine kinase (Sigma Aldrich) in a 56μl reaction volume. After the 30 min incubation, 5.29 μl of 1.7 μM ACF complex (Active Motif) and the “AM” mix were sequentially added to the NAP1-histone mix. Then, 10.63μl of native or mini-genome DNA (2.66μg) was added, resulting in a 374μl reaction volume. The final mixture was incubated at 27°C for 2.5 hr to allow for chromatin assembly. Subsequently, CaCl2 was added to a final concentration of 5mM, and the chromatin was digested with MNase (New England Biolabs) to mainly mononucleosomal DNA as previously described (Beh et al., 2015).
Mononucleosome-sized DNA from MNase-digested chromatin was gel-purified and used for tiling qPCR on a Viia 7 Real-Time PCR System with Power SYBR Green PCR master mix (Thermo Fisher), or in vitro MNase-seq on an Illumina HiSeq 2500, according to the manufacturer’s instructions. qPCR primer sequences are listed in Table S6.
Tiling qPCR analysis of nucleosome occupancy
qPCR data were analyzed using the ΔΔCt method (Livak and Schmittgen, 2001). At each locus along the synthetic chromosome, ΔCt = (Ct at locus of interest) – (Ct at qPCR primer pair 22, far from the methylated region). See Figure 6B for location of qPCR primer pair 22. Separate ΔCt values were calculated from mononucleosomal DNA and the corresponding naked, undigested synthetic chromosome. The ΔΔCt value was calculated from this pair of ΔCt values. This controls for potential variation in PCR amplification efficiency, especially over methylated regions. The fold change in mononucleosomal DNA relative to naked chromosomal DNA at a particular locus is calculated as 2−ΔΔCt, and denotes ‘nucleosome occupancy’ for all presented qPCR data.
ACF spacing assay
ATP-dependent nucleosome spacing was performed in accordance with a previous study (Lieleg et al., 2015). Chromatin was assembled by salt gradient dialysis as described above, and then adjusted to 20mM HEPES-KOH pH 7.5, 80mM KCl, 0.5mM EGTA, 12% v/v glycerol, 10mM (NH4)2SO4, 2.5mM DTT. Samples were then incubated for 2.5 hr at 27°C with 3mM ATP, 30mM creatine phosphate, 4mM MgCl2, 5 ng/μl creatine kinase, and 11 ng/μL ACF complex (Active Motif). Remodeled chromatin was then adjusted to 5mM CaCl2 and subjected to MNase digestion, mononucleosomal DNA purification, and qPCR analysis as described above.
Phylogenetic analysis
The MTA1 amino acid sequence (UniProt ID: J9IF92_9SPIT) was queried against the NCBI nr database using PSI-BLAST (Altschul et al., 1997; Schäffer et al., 2001) (maximum e-value = 1e−4; enable short queries and filtering of low complexity regions). Retrieved hits were collapsed using CD-HIT (Huang et al., 2010) with minimum sequence identity = 0.97 to remove redundant sequences. The resulting sequences were added to existing MT-A70 alignments from (Greer et al., 2015) using MAFFT (--add) (Katoh et al., 2017; Kuraku et al., 2013). Gaps and duplicate sequences were removed from the merged alignment. Only sequences corresponding to the taxa in Figure 2B were retained. The alignment was then used for phylogenetic tree construction using MrBayes in the CIPRES Science Gateway (Miller et al., 2010) with 5 × 106 generations. Protein sequences used for MrBayes analysis are given in Table S5.
The above procedure was also used for constructing phylogenetic trees from p1 (UniProt ID: Q22VV9_TETTS) and p2 (UniProt ID: I7M8B9_TETTS). However, protein sequences were aligned using MAFFT without adding to an existing alignment.
Preparation of nuclear extracts with DNA methyltransferase activity
Vegetative Tetrahymena cells were grown in SSP medium to log-phase (~3.5 × 105 cells/mL) and collected by centrifugation at 2,300 × g for 5 min in an SLA-3000 rotor. The supernatant was discarded, and cells were resuspended in medium B (10mM Tris pH 6.75, 2mM MgCl2, 0.1M sucrose, 0.05% w/v spermidine trihydrochloride, 4% w/v gum Arabic, 0.63% w/v 1-octanol, and 1mM PMSF). Gum arabic (Sigma Aldrich) is prepared as a 20% w/v stock and centrifuged at 7,000 × g for 30 min to remove undissolved clumps. For each volume of cell culture, one-third volume of medium B was added to the Tetrahymena cell pellet. Cells were resuspended and homogenized in a chilled Waring Blender (Waring PBB212) at high speed for 40 sec. The resulting lysate was subsequently centrifuged at 2,750 × g for 5 min in an SLA-3000 rotor to pellet macronuclei. The nuclear pellet was washed twice with medium B and then five times in MM medium (10mM Tris-HCl pH 7.8, 0.25M sucrose, 15mM MgCl2, 0.1% w/v spermidine trihydrochloride, 1mM DTT, 1mM PMSF). Macronuclei were pelleted between wash steps by centrifuging at 2,500 × g for 5 min in an SLA-3000 rotor. Finally, the total number of washed macronuclei was counted with a hemocytometer using a Zeiss ID03 microscope. Nuclear proteins were extracted by vigorously resuspending the pellet in MMsalt buffer (10mM Tris-HCl pH 7.8, 0.25M sucrose, 15mM MgCl2, 350mM NaCl, 0.1% w/v spermidine trihydrochloride, 1mM DTT, 1mM PMSF). 1mL MMsalt buffer was added per 2.33 × 108 macronuclei. The viscous mixture was nutated for 45 min at 4°C, and then cleared at 175,000 × g for 30 min at 4°C in a SW 41 Ti rotor. Following this, the supernatant was dialyzed in a Slide-A-Lyzer 3.5K MWCO cassette (Thermo Fisher) overnight at 4°C against two changes of MMminus medium (10mM Tris-HCl pH 7.8, 15mM MgCl2, 1mM DTT, 0.5mM PMSF). The dialysate was then centrifuged at 7,197 × g for 1 hr at 4°C to remove precipitates, and dialyzed overnight in a Slide-A-Lyzer 3.5K MWCO cassette (Thermo Fisher) at 4°C against two changes of MN3 buffer (30mM Tris-HCl pH 7.8, 1mM EDTA, 15mM NaCl, 20% v/v glycerol, 1mM DTT, 0.5mM PMSF). The final dialysate was cleared by centrifugation at 7,197g for 1.5 hr at 4°C, flash frozen, and stored at −80°C. This nuclear extract was used for all subsequent biochemical fractionation and 6mA methylation assays.
Partial purification of MTA1c from nuclear extracts
Tetrahymena nuclear extracts were passed through a HiTrap Q HP column (GE Healthcare) and eluted using a linear gradient of 15mM to 650mM NaCl in 30mM Tris-HCl pH 7.8, 1mM EDTA, 20% v/v glycerol, 1mM DTT, 0.5 mM PMSF, over 30 column volumes. Each fraction was assayed for DNA methyltransferase activity using radiolabeled SAM as described in the next section. The DNA methyltransferase activity eluted in two peaks, at ~60mM and ~365mM NaCl, termed the “low salt sample” and “high salt sample”. Fractions corresponding to each peak were pooled and passed through a HiTrap Heparin HP column (GE Healthcare). Bound proteins were eluted using a linear gradient of 60 mM to 1M NaCl (for the low salt sample) or 350mM to 1M NaCl (for the high salt sample) over 30 column volumes. Fractions with DNA methyltransferase activity were respectively pooled and dialyzed into 10mM sodium phosphate pH 6.8, 100mM NaCl, 10% v/v glycerol, 0.3mM CaCl2, 0.5mM DTT (for the low salt sample); or 30mM Tris-HCl pH 7.8, 1mM EDTA, 200mM NaCl, 10% v/v glycerol, 1mM DTT, 0.2mM PMSF (for the high salt sample). The dialyzed low salt sample was passed through a Nuvia cPrime column (Bio-Rad) and eluted using a linear gradient of 100 mM to 1M NaCl in 50 mM sodium phosphate pH 6.8, 10% v/v glycerol, 0.5 mM DTT. Separately, the dialyzed high salt sample was fractionated using a Superdex 200 10/300 GL column (GE Healthcare) in 30mM Tris-HCl pH 7.8, 1mM EDTA, 200mM NaCl, 10% v/v glycerol, 1mM DTT. Fractions from the Nuvia cPrime and Superdex 200 columns were dialyzed into 30mM Tris-HCl pH 7.8, 1mM EDTA, 15mM NaCl, 20% v/v glycerol, 1mM DTT, 0.5mM PMSF and assayed for DNA methyltransferase activity. Those with qualitatively low, medium, and high activity were subjected to mass spectrometry to identify candidate methyltransferase proteins (Figure 2D and Table S3). This experiment identified four proteins that co-purify with DNA methyltransferase activity – MTA1, MTA9, p1, and p2 – and are collectively termed as “MTA1c” in our study. All four proteins are necessary for 6mA methylation in vitro.
Recombinant expression of MTA1, MTA9, p1, and p2 proteins
Full length MTA1, MTA9, p1, and p2 open reading frames were codon-optimized for bacterial expression and cloned into a pET-His6-SUMO vector using ligation independent cloning. Protein sequences are listed in Table S7. The vector was a gift from Scott Gradia (Addgene plasmid #29659; http://n2t.net/addgene:29659; RRID: Addgene_29659). Mutations in the MTA1 open reading frame was introduced using the Q5® Site-Directed Mutagenesis Kit (New England Biolabs). For recombinant expression, pET-His6-SUMO-MTA1 (wild type and mutant) was transformed into SHuffle T7 competent E. coli (New England Biolabs); pET-His6-SUMO-MTA9 was transformed into Lemo (DE3) competent E. coli (New England Biolabs); pET-His6-SUMO-p1 and pET-His6-SUMO-p2 were transformed into BL21(DE3) competent E. coli (New England Biolabs). IPTG induction was performed at 16°C overnight. Induced cells were resuspended in 25ml of lysis buffer B (50mM Tris pH 7.8, 300mM NaCl, 5% v/v glycerol, 10mM imidazole, 5mM BME, 1mM PMSF, 0.5× ProBlock Gold Bacterial protease inhibitor cocktail [GoldBio]). The cells were sonicated at 35% amplitude for a total of 4 minutes, with a 10 sec off, 10 sec cycle using a Model 505 Sonic Dismembrator (Fisherbrand). Lysates were cleared by centrifugation at 30,000g for 30 min at 4°C, mixed with pre-washed Ni-NTA agarose (Invitrogen), and nutated for 45min at 4°C. The resin was subsequently washed with lysis buffer and eluted in 50mM Tris pH 7.8, 300mM NaCl, 5%v/v glycerol, 400mM glycerol, 5mM BME, 1× ProBlock Gold bacterial protease inhibitor cocktail [GoldBio]). Eluates were dialyzed into lysis buffer B and then digested with TEV protease (gift from S.H. Sternberg) at 4°C overnight. The resulting mixture was passed through a fresh batch of Ni-NTA agarose (Invitrogen) to remove cleaved affinity tags. The flow-through containing each recombinant protein was flash frozen and used for all downstream methyltransferase assays.
Methyltransferase assays
Generation of DNA and RNA substrates
A 954bp dsDNA PCR product was used in all assays involving Tetrahymena nuclear extract. This substrate was amplified by PCR from Tetrahymena thermophila strain SB210 macronuclear SB210 genomic DNA using PCR primers metGATC_F2 and metGATC_R2 (Table S6). The resulting product was purified using Ampure XP beads (Beckman Coulter). This 954bp region of the genome contains a high level of 6mA in vivo. Thus, the underlying DNA sequence may be intrinsically amenable to methylation by Tetrahymena MTA1. Note that the amplified 954bp product is devoid of DNA methylation as unmodified dNTPs were used for PCR. Separately, a 350bp dsDNA PCR product was used in all assays involving recombinant MTA1, MTA9, p1 and p2. This sequence lacks 5’-NATC-3’ motifs, and was used to reduce background DNA methylation from contaminating Dam methyltransferase in recombinant protein preparations. The 350bp dsDNA PCR product was amplified from Tetrahymena thermophila strain SB210 macronuclear SB210 genomic DNA using the PCR primers noGATC2_F and noGATC2_R (Table S6), and purified using Ampure XP beads (Beckman Coulter).
For short DNA substrates (< 50bp), oligonucleotides were purchased from Integrated DNA Technologies and either directly used as ssDNA, or annealed with its complementary sequence to obtain dsDNA. To prepare hemimethylated 27bp dsDNA in Figure 2G, either strand was methylated using EcoGII methyltransferase (New England BioLabs) before annealing with the complementary sequence.
To generate ~350nt ssRNA and ~350bp dsRNA, the aforementioned 350bp dsDNA was first PCR-amplified using primers containing T7 overhangs (primer pairs T7noGATC2_F2 / noGATC2_R and T7noGATC2_F2 / T7noGATC2_R2 respectively; see Table S6 for primer sequences). Each PCR product was used as a template for in vitro transcription using the HiScribe T7 High Yield RNA Synthesis Kit (New England Biolabs). The synthesized RNA was rigorously treated with DNase (ThermoFisher) purified using acid phenol:chloroform extraction, followed by two rounds of chloroform extraction. Each sample was subsequently ethanol precipitated and resuspended in water for use in methyltransferase assays.
Radioactive methyltransferase assay
For experiments involving nuclear extract, 2.18μg of 954bp dsDNA substrate was mixed with 4-8μl nuclear extract and 0.64μM 3H-labeled S-adenosyl-L-methionine ([3H]SAM) in 33mM Tris-HCl pH 7.5, 6mM EDTA, 4.3mM BME, in a 15μl reaction volume. For experiments involving recombinant MTA1c protein components (ie. MTA1, MTA9, p1, and/or p2), ~3μM oligonucleotide ssDNA / annealed dsDNA is used. Alternatively, 1.3μg of 350bp dsDNA substrate (or an equimolar amount ~350nt ssRNA, or ~350bp dsDNA) was used in place of DNA oligonucleotide substrates. ssRNA was heated at 90°C for 2 min and snap cooled to minimize secondary structures before mixing with other components of the methyltransferase assay. All samples were incubated overnight at 37°C, and subsequently spotted onto 1cm × 1cm squares of Hybond-XL membrane (GE Healthcare). Membranes were then washed thrice with 0.2M ammonium bicarbonate, once with distilled water, twice with 100% ethanol, and finally air-dried for 1 hr. Each membrane was immersed in 5mL Ultima Gold (PerkinElmer) and used for scintillation counting on a TriCarb 2910 TR (PerkinElmer).
Non-radioactive methyltransferase assay
For assays involving nuclear extract: 5.5μg of 954bp DNA substrate was mixed with 2μl nuclear extract and 0.2mM S-adenosyl-L-methionine (NEB) in 33mM Tris-HCl pH 7.5, 6mM EDTA, 4.3mM BME in a 15μl reaction volume. For assays involving recombinant MTA1c protein components (ie. MTA1, MTA9, p1, and/or p2), 2.6μg of 350bp DNA substrate was mixed with 540nM MTA1, 90nM MTA9, 1.5μM p1, 1.0μM p2 proteins. The band of expected size in each recombinant protein preparation was compared against a series of BSA standards to calculate protein concentration. All methylation reactions were incubated at 37°C overnight, then purified using a MinElute purification kit (QIAGEN), denatured at 95°C for 10 min, and snap cooled in an ice water bath. Samples were spotted on a Hybond N+ membrane (GE Healthcare), air dried for 5 min and UV-cross-linked with 120,000 μJ / cm2 exposure using an Ultra-Lum UVC-515 Ultraviolet Multilinker. The cross-linked membrane was blocked in 5% milk in TBST (containing 0.1% v/v Tween) and incubated with 1:1,000 anti-N6-methyladenosine antibody (Synaptic Systems) at 4°C overnight. The membrane was then washed three times with TBST, incubated with 1:3,000 Goat anti-rabbit HRP antibody (Bio-Rad) at room temperature for 1 hr, washed another three times with 1× TBST, and developed using Amersham ECL Western Blotting Detection Kit (GE Healthcare). This dot blot assay was used to measure 6mA levels in Figures 2F, 3B, 5C and S3C.
Quantitative mass spectrometry analysis of dA and 6mA
10.5μg Oxytricha or Tetrahymena macronuclear genomic DNA was first digested to nucleosides by mixing with 14μl DNA degradase plus enzyme (Zymo Research) in a 262.5μl reaction volume. Samples were incubated at 37°C overnight, then 70°C for 20 min to deactivate the enzyme.
The internal nucleoside standards 15N5-dA and D3-6mA were used to quantify endogenous dA and 6mA levels in ciliate DNA. 15N5-dA was purchased from Cambridge Isotope Laboratories, while D3-6mA was synthesized as described in the following section. Nucleoside samples were spiked with 1 ng/μl 15N5-dA and 200 pg/μl D3-6mA in an autosampler vial. Samples were loaded onto a 1mm × 100mm C18 column (Ace C18-AR, Mac-Mod) using a Shimadzu HPLC system and PAL auto-sampler (20μl / injection) at a flow rate of 70μl / min. The column was connected inline to an electrospray source couple to an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher). Caffeine (2 pmol/μl in 50% Acetonitrile with 0.1% FA) was injected as a lock mass through a tee at the column outlet using a syringe pump at 0.5μl/min (Harvard PHD 2000). Chromatographic separation was achieved with a linear gradient from 10% to 99% B (A: 0.1% Formic Acid, B: 0.1% Formic Acid in Acetonitrile) in 5 min, followed by 5 min wash at 100% B and equilibration for 10 min with 1% B (total 20 min program). Electrospray ionization was achieved using a spray voltage of 4.50 kV aided by sheath gas (Nitrogen) flow rate of 18 (arbitrary units) and auxiliary gas (Nitrogen) flow rate of 2 (arbitrary units). Full scan MS data were acquired in the Orbitrap at a resolution of 60,000 in profile mode from the m/z range of 190-290. A parent mass list was utilized to acquire MS/MS spectra at a resolution of 7500 in the Orbitrap. LC-MS data were manually interpreted in Xcalibur’s Qual browser (Thermo, Version 2.1) to visualize nucleoside mass spectra and to generate extracted ion chromatograms by using the theoretical [M+H] within a range of ±2ppm. Peak areas were extracted in Skyline (Ver. 3.5.0.9319).
Synthesis of D3-6mA nucleoside
2′-Deoxyadenosine and CD3I were purchased from Sigma Aldrich. Flash chromatography was performed on a Biotage Isolera using silica columns (Biotage SNAP Ultra, HP-Sphere 25μm). Semi-preparative RP-HPLC was performed on a Hewlett-Packard 1200 series instrument equipped with a Waters XBridge BEH C18 column (5μm, 10 × 250 mm) at a flow rate of 4mL/min, eluting using A (0.1% formic acid in H2O) and B (0.1% formic acid in 9:1 MeCN/H2O). 1H NMR spectra were recorded on a Bruker UltraShield Plus 500 MHz instrument. Data for 1H NMR are reported as follows: chemical shift (δ ppm), multiplicity (s = singlet, br = broad signal, d = doublet, dd = doublet of doublets) and coupling constant (Hz) where possible. 13C NMR spectra were recorded on a Bruker UltraShield Plus 500 MHz.
D3-6mA (2′Deoxy-6-[D3]-methyladenosine) were synthesized and purified according to (Schiffers et al., 2017). After an initial purification by flash column chromatography, the methylated compounds were further purified by semipreparative RP-HPLC (linear gradient of 0% to 20% B over 30 min) affording the desired compounds in 14% and 10% yields respectively after lyophilization.
2′Deoxy-6-[D3]-methyladenosine:
1H NMR (500 MHz, D2O) δ 7.98 (s, 1H), 7.77 (s, 1H), 6.17 (m, 1H), 4.54 (m, 1H), 4.10 (m, 1H), 3.79 (dd, J = 12.7, 3.2 Hz, 1H), 3.71 (dd, J = 12.7, 4.3 Hz, 1H), 2.60 (m, 1H), 2.44 (ddd, J = 14.0, 6.3, 3.3 Hz, 1H).
13C NMR (126 MHz, D2O) δ 154.0, 151.5, 146.1, 138.9, 118.4, 87.3, 84.3, 71.1, 61.6, 39.2, 26.4 ppm. (Peak at 26.4 ppm appears as a broad signal. C-D coupling is not resolved).
HR-MS (ESI+): m/z calculated for [C11H13D3N5O3]+ ([M+H]+): 269.1436, found 269.1421.
Mass spectrometry analysis of proteins in Tetrahymena nuclear extracts
Samples where topped up to 200μl with 50mM ammonium bicarbonate pH 8. TCEP was added to 5mM final concentration and left to incubate at 60°C for 10 min. 15mM chloroacetamide was then added and left to incubate in the dark at room temperature for 30 min. 1μg of Trypsin Gold (Promega) was added to each sample and incubated end-over-end at 37°C for 16 hr. An additional 0.25μg of Trypsin Gold was added and incubated end-over-end at 37°C for 3 hr. Samples were acidified by adding TFA to 0.2% final concentration, and desalted using SDB stage-tips (Rappsilber et al., 2007). Samples were dried completely in a speedvac and resuspended in 20μl of 0.1% formic acid pH 3. 5μl was injected per run using an Easy-nLC 1200 UPLC system. Samples were loaded directly onto a 45cm long 75μm inner diameter nano capillary column packed with 1.9um C18-AQ (Dr. Maisch, Germany) mated to metal emitter inline with an Orbitrap Fusion Lumos (Thermo Scientific, USA). The mass spectrometer was operated in data dependent mode with the 120,000 resolution MS1 scan (AGC 4e5, Max IT 50ms, 400-1500 m/z) in the Orbitrap followed by up to 20 MS/MS scans with CID fragmentation in the ion trap. Dynamic exclusion list was invoked to exclude previously sequenced peptides for 60 sec if sequenced within the last 30 sec, and maximum cycle time of 3 sec was used. Peptides were isolated for fragmentation using the quadrupole (1.6 Da window). ns was utilized. Ion-trap was operated in Rapid mode with AGC 2e3, maximum IT of 300 msec and minimum of 5000 ions.
Raw files were searched using Byonic (Bern et al., 2012) and Sequest HT algorithms (Eng et al., 1994) within the Proteome Discoverer 2.1 suite (Thermo Scientific, USA). 10ppm MS1 and 0.4Da MS2 mass tolerances were specified. Caramidomethylation of cysteine was used as fixed modification, while oxidation of methionine, pyro-Glu from Gln and deamidation of asparagine were specified as dynamic modifications. Trypsin digestion with maximum of 2 missed cleavages were allowed. Files were searched against the Tetrahymena themophila macronuclear reference proteome (June 2014 build), supplemented with common contaminants (27,099 total entries).
Scaffold (version Scaffold_4.8.7, Proteome Software Inc., Portland, OR) was used to validate MS/MS based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 93.0% probability. Peptide Probabilities from Sequest and Byonic were assigned by the Scaffold Local FDR algorithm. Protein identifications were accepted if they could be established at greater than 99.0% probability to achieve an FDR less than 1.0% and contained at least 3 identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm (Nesvizhskii et al., 2003). Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony.
Generation of mta1 mutant lines
A frameshift mutation in the MTA1 gene was created by inserting a small non-coding DNA segment immediately downstream of the MTA1 start codon (Figures 3A and S5H). This non-coding DNA segment belongs to a class of genetic elements that are normally eliminated during the sexual cycle (Chen et al., 2014). When ssRNA homologous to such DNA segments is injected into Oxytricha cells undergoing sexual development, the DNA is erroneously retained (Khurana et al., 2018). This results in disruption of the MTA1 open reading frame. The ectopic DNA segment is propagated through subsequent cell divisions after completion of the sexual cycle. RNAseq analysis confirmed the presence of the ectopic insertion in mta1 mutant transcripts but not wild type controls (Figure S5H).
ssRNA was generated by in vitro transcription using a Hi-Scribe T7 High Yield RNA Synthesis Kit (New England Biolabs). The DNA template for in vitro transcription consists of the ectopic DNA segment flanked by 100-200bp cognate MTA1 sequence. Following DNase treatment, ssRNA was acid-phenol:chloroform extracted and ethanol precipitated. After precipitation, ssRNA was resuspended in nuclease-free water (Ambion) to a final concentration of 1 to 3 mg/mL for injection.
ssRNA microinjections
Oxytricha cells were mated by mixing 3mL of each mating type, JRB310 and JRB510, along with 6mL of fresh Pringsheim media. At 10 to 12 hr post mixing, pairs were isolated and placed in Volvic water with 0.2% bovine serum albumin (Jackson ImmunoResearch Laboratories) (Fang et al., 2012). ssRNA constructs were injected into the macronuclei of paired cells under a light microscope as previously described with DNA constructs (Nowacki et al., 2008). After injection, cells were pooled in Volvic water. At 60 to 72 hr post mixing, the pooled cells were singled out to grow clonal injected cell lines. As clonal population size grew, lines were transferred to 10 cm petri dishes and grown in Pringsheim media. Only water from the “Volvic” brand has been empirically tested in our laboratory to support Oxytricha growth. Similar products from other vendors have not been tested.
Survival analysis of Oxytricha mta1 mutants
This experiment was performed in Figure 7D. Wild type or mutant Oxytricha cells were mixed at 0 hr to induce mating. Since not all cells enter the sexual cycle, mated cells are separated from unmated vegetative cells at 15 hr and transferred into a separate dish. The cells are allowed to rest for 12 hr to account for cell death during transfer. The number of surviving mated cells is counted from 27 hr onwards. The total cell number at each timepoint is normalized to 27 hr data to obtain the percentage survival. An increase in survival at 108 hr is observed in wild type samples because the cells have completed mating and reverted to the vegetative state, where they can proliferate and increase in number.
Quantification and statistical analysis
All statistical tests were performed in Python (v2.7.10) or R (v3.2.5), and described in the respective Figure and Table legends.
Data and Software Availability
Oxytricha SMRT-seq data are deposited in SRA under the accession numbers SRX2335608, SRX2335607, and GSE94421. Tetrahymena SMRT-seq and all Oxytricha Illumina data are deposited in NCBI GEO under accession number GSE94421.
Supplementary Material
Figure S1. Mass spectrometry analysis of 6mA in ciliate DNA, Related to Figure 1.
(A) Oxytricha and Tetrahymena genomic DNA were digested into nucleosides using degradase enzyme mix, followed by analysis using reverse-phase HPLC and mass spectrometry. Isotopically labeled dA and 6mA standards (15N5-dA and D3-6mA) were mixed with each sample to allow quantitative measurement of endogenous dA and 6mA concentrations. MS/MS analysis of labeled dA and 6mA standards confirmed the mass of the nucleobase. Eluted peaks with expected masses of dA and 6mA, and with highly similar retention times (RT) to internal standards are detected in Oxytricha and Tetrahymena nucleosides.
(B) Quantitation of dA and 6mA levels in Oxytricha and Tetrahymena gDNA using internal isotopically labeled nucleoside standards. The detected level of 6mA in Tetrahymena gDNA agrees with earlier reports (Gorovsky et al., 1973; Pratt and Hattman, 1981). The calculated abundance of 6mA relative to (dA + 6mA) in Oxytricha is ~0.71%, which is similar to the estimate from SMRT-seq base calls (0.78 – 1.04%). Note that the calculation from SMRT-seq data is expected to be an overestimate because 6mA is scored at being present or absent at each site in the genome for this purpose. In actual fact, 6mA sites may be partially methylated (Figure S4A). Neither 6mA nor dA was detected from LC-MS analysis of Oxytricha culture media, arguing against spurious signal arising from contamination or overall technical handling. Our PacBio and LC-MS measurements of % 6mA in Oxytricha are both similar to thin layer chromatography analysis of nucleotides (0.6 – 0.7%) from a distinct but closely related species, Oxytricha fallax (Rae and Spear, 1978).
Table S5. Protein sequences for phylogenetic tree construction, related to Figure 2.
FASTA-formatted list of amino acid sequences used to construct the MT-A70, p1, and p2 phylogenetic trees in Figures 2A, S2G, S2H, and S2I.
Table S7. Recombinant protein sequences, related to Figure 2.
Sequences were manually curated by mapping RNAseq reads to reference gene annotations and verifying the accuracy of predicted exon boundaries.
Figure S2. Analysis of 6mA and methyltransferase components in Tetrahymena, Related to Figures 1 and 2.
(A) Tetrahymena MNase-seq data from (Beh et al., 2015), while SMRT-seq data were generated in this study. Meta-chromosome plots overlaying in vivo MNase-seq (nucleosome occupancy) and SMRT-seq (6mA), relative to annotated transcription start sites. 6mA lies mainly within nucleosome linker regions, between the +1, +2, +3, and +4 nucleosomes.
(B) Histograms of the total number of 6mA marks within each linker in Tetrahymena genes. Calculations are performed as described in Figure 1B. Distinct linkers are highlighted with horizontal bold blue lines.
(C) Relationship between transcriptional activity and total number of 6mA marks in Tetrahymena genes. Analysis is performed as in Figure 1C. RNA-seq data was obtained from (Xiong et al., 2012).
(D) Composite analysis of 441,618 methylation sites reveals that 6mA occurs within an 5’-ApT-3’ dinucleotide motif in Tetrahymena, consistent with previous experiments (Bromberg et al., 1982; Wang et al., 2017) and similar to Oxytricha.
(E) Distribution of various 6mA dinucleotide motifs across the genome.
(F) Organization of transcription (mRNA-seq), nucleosome organization (MNase-seq), and 6mA (SMRT-seq) in a Tetrahymena gene.
(G) All sequences used for phylogeny construction are listed in Table S5. Abbreviations: Cel: Caenorhabditis elegans; Ath: Arabidopsis thaliana; Sra: Syncephalastrum racemosum; Hve: Hesseltinella vesiculosa; Are: Absidia repens ; Dre: Danio rerio; Has: Homo sapiens; Ssc: Sus scrofa; Mmu: Mus musculus; Xla: Xenopus laevis; Dme: Drosophila melanogastep Cre: Chlamydomonas reinhardtii; Ltr: Lobosporangium transversale; Lpe: Linderina pennispora; Bme: Basidiobolus meristosporus; Pfi: Piromyces finnis; Aro: Anaeromyces robustus; Tth: Tetrahymena thermophila; Otri: Oxytricha trifallax. This Bayesian phylogenetic tree of MT-A70 proteins is the same as in Figure 2A, except that all sequences are now included and labeled. TAMT-1 proteins are named according to (Luo et al., 2018).
(H) Bayesian phylogenetic tree of p1 proteins.
(I) Bayesian phylogenetic tree of p2 proteins. Dashed box depicts outgroup consisting of vertebrate SNAPC4 genes. These genes bear weak similarity to the homeobox-like domain of p2 proteins, but do not group phylogenetically with them and are therefore unlikely to be functionally homologs.
(J) Phylogenetic distribution of ApT 6mA motif and various proteins, as depicted in Figure 2B, but now also including TAMT-1, p1, and p2 proteins. Filled boxes denote the presence of a particular protein in a taxon. Open dashed boxes indicate the presence of SNAPC4 genes in vertebrates.
(K) Gene expression profiles of Tetrahymena MTA1, MTA9, p1 and p2. Microarray counts represent poly(A)+ expression levels, and are obtained from TetraFGD (Miao et al., 2009; Xiong et al., 2011). MTA1, MTA9, p1 and p2 were found in our study to co-elute with 6mA methylase activity. On the other hand, TAMT-1 is a putative DNA methyltransferase described by (Luo et al., 2018). The horizontal axis categories beginning with “S” and “C” represent the number of hours since the onset of starvation and conjugation (mating), respectively. “Low”, “Med”, and “High” denote relative cell densities during log-phase growth. Blue and orange traces represent data from two biological replicates. Green and red shaded regions show the peaks in poly(A)+ RNA expression in vegetative growth and conjugation, respectively, for MTA1, MTA9, p1 and p2. Note that their expression pattern differs from TAMT-1.
Figure S3. Further characterization of 6mA methyltransferase activity and MTA1c, Related to Figure 2.
(A) Fractionation of nuclear extracts on a Q sepharose column results in two distinct peaks of DNA methyltransferase activity, denoted as “Low Salt sample” and “High Salt sample” by black horizontal bars. FT denotes column flow-through. The DNA methyltransferase assay is performed as in Figure 2E. The salt concentration at which individual fractions elute from the column is plotted against DNA methyltransferase activity of each fraction (counts per minute). Inset shows DNA methyltransferase activity of the input nuclear extract, flowthrough from the Q sepharose column, and blank control (nuclear extract buffer). Orange and blue plots denote replicates derived from independent preparations of nuclear extract.
(B) DNA methyltransferase assay showing that the activity from nuclear extracts is heat-sensitive and requires addition of DNA and SAM. Error bars represent s.e.m. (n = 3).
(C) Dot blot showing that nuclear extracts mediate 6mA methylation. Note that the low salt sample has substantial DNase activity, resulting in a lower amount of DNA available for dot blot analysis. DNA substrate, nuclear extract, and SAM cofactor were mixed as in panels A and B. The DNA was subsequently purified and used for dot blot analysis.
(D) Domain organization of Tetrahymena MTA1, MTA9, p1, and p2. Protein domains are predicted using hmmscan on the EMBL-EBI webserver (Finn et al., 2015). “aa” denotes amino acids. Start and end coordinates of each domain are stated below each polypeptide.
(E) Sequence alignment of human (Hsa) METTL3 with Tetrahymena (Tth) and Oxytricha (Otri) MTA1 / MTA9, within the MT-A70 domain. Horizontal black bars underscore the DPPW catalytic motif, and the N549 / Q550 residues in human METTL3 that interact with the ribose moiety of the SAM cofactor. Note that the DPPW catalytic motif is conserved in MTA1 but not MTA9.
(F) Dot blot analysis of hemimethylated dsDNA substrates. Sense or antisense oligonucleotides were first individually methylated using the EcoGII bacterial 6mA methyltransferase. Each methylated ssDNA was subsequently purified and annealed with an unmethylated complementary strand to form hemimethylated constructs.
(G) SDS-PAGE analysis of recombinant proteins. Full length proteins were expressed and purified from E. coli. Bands of expected size are indicated with a black arrowhead.
(H) Methyltransferase assay using radiolabeled SAM on DNA and RNA substrates, coupled with gel analysis of nucleic acid integrity. ssRNA and dsRNA were produced by in vitro transcription from the 350bp dsDNA template using T7 RNA polymerase, and subsequently purified before use in this assay. Methyltransferase activity on equimolar amounts of each substrate was measured after incubation at 37°C for 6 hr, and depicted as either scintillation counts (Counts per minute), or normalized to the 350bp dsDNA sample (Relative activity). Only dsDNA, and not dsRNA or ssRNA, was methylated. Activity measurements are represented as scintillation counts (counts per minute). In addition, aliquots from each reaction containing DNA or RNA substrate and recombinant MTA1c (ie. MTA1, MTA7, p1 and p2 proteins) were withdrawn at 0, 1, 2, 3, or 6 hr during the 37°C incubation, purified using phenol:chloroform extraction and ethanol precipitation, and subsequently analyzed on a non-denaturing agarose gel. Both dsDNA and dsRNA substrates remained intact after 6 hrs. The ssRNA migrates more diffusely on a nondenaturing agarose gel, with some decrease in size over time, suggesting partial degradation and/or RNA folding; however, there is no detectable methylation of ssRNA despite a significant presence on the agarose gel after 6 hr at 37°C. It is unlikely that this species is too short to be methylated, since MTA1c can methylate significantly shorter substrates such as 27bp dsDNA (Figure 2G, S3I, S3J, S3K). Error bars represent s.e.m. (n = 3).
(I) DNA methyltransferase assay using radiolabeled SAM, on ssDNA oligonucleotides or annealed dsDNA substrates. All four recombinant MTA1c protein components – MTA1, MTA9, p1, and p2 – were included in each sample. Activity measurements are represented as scintillation counts (counts per minute). dsDNA substrates were prepared by annealing ssDNA oligonucleotides, as in Figure 2G. Sense ssDNA nucleotide sequences are depicted in the 5’→3’ direction, while antisense ssDNA is depicted as 3’→5’. Error bars represent s.e.m. (n = 3).
(J) Control [3H]SAM assay using hemimethylated dsDNA. Reactions depicted in red represent hemimethylated dsDNA incubated with [3H]SAM in the absence of recombinant MTA1c (MTA1, MTA9, p1, and p2 proteins). These reactions showed no methyltransferase activity, verifying that there is no contaminating EcoGII methyltransferase in hemimethylated dsDNA preparations. Activity measurements are shown as scintillation counts, or as “Relative Activity” (normalized against the sample containing unmethylated DNA substrate, [3H]SAM, and MTA1c protein). Hemimethylated dsDNA substrates in this panel are the same as those used in Figure 2G. The unmethylated dsDNA substrate used in this panel is the same as the top-most dsDNA substrate in Figure 2G, with two uninterrupted ApT dinucleotides. Error bars represent s.e.m. (n = 3).
(K) DNA methyltransferase assay using radiolabeled SAM, on dsDNA substrates with disrupted ApT dinucleotides. All four recombinant MTA1c protein components – MTA1, MTA9, p1, and p2 – were included in each sample. Activity measurements are normalized against the parent dsDNA construct with two uninterrupted ApT dinucleotides (top-most construct in this panel). ApT dinucleotide positions are labeled in bold red. Note that the parent dsDNA construct is identical to that in Figure S3L. Error bars represent s.e.m. (n = 3).
(L) DNA methyltransferase assay using radiolabeled SAM, on dsDNA substrates with shifted ApT dinucleotides. All four recombinant MTA1c protein components – MTA1, MTA9, p1, and p2 – were included in each sample. Activity measurements are normalized against the parent dsDNA construct with two uninterrupted ApT dinucleotides (top-most construct in this panel). The parent construct is identical to that in Figure S3K. ApT dinucleotides are labeled in bold red. The adjacent nucleotides are labelled in bold black to highlight the 4-mer sequence that contains each ApT dinucleotide. Error bars represent s.e.m. (n = 3).
(M) Motif frequencies of all 4-mer sequences containing methylated ApT dinucleotides in the Tetrahymena and Oxytricha genomes. A* denotes 6mA. The 4-mers TA*TA and CA*TT are colored in red and blue, respectively, to highlight their large difference in genomic frequencies.
(N) Motif frequencies of 4-mer sequences – regardless of methylation state – in Tetrahymena and Oxytricha. These were calculated from genomic sequence between the 5’ chromosome end and the +4 nucleosome peak (Oxytricha), or between the TSS and the +4 nucleosome peak (Tetrahymena). Analysis was restricted to these regions in order to serve as “background” frequencies for comparison to A*T methylated 4-mers, which are also mainly found downstream of TSSs. The 4-mers TATA and CATT are colored in red and blue, respectively, to facilitate comparison with methylated TA*TA and CA*TT in panel M.
Figure S4. Supplemental SMRT-seq data analyses, related to Figures 1 and 3.
(A) Top two panels depict PacBio coverage (horizontal axis) plotted against fractional methylation at each called 6mA site (vertical axis). Bottom left panel is a histogram of fractional methylation of all 6mA sites. Bottom right panel is a histogram of IPD ratios of all 6mA sites. Mutant datasets show significantly lower fractional methylation and IPD ratios at 6mA sites than wild type data.
(B) Wild type SMRT-seq data are randomly subsampled 15 times, such that the resulting coverage is lower than mta1 mutant data. The difference in PacBio coverage between mutant and subsampled wildtype data is calculated for each chromosome, and is collectively represented as an olive boxplot (top panel). This set of calculations is repeated 15 times for each subsampled dataset, resulting in a series of 15 boxplots. The difference in PacBio coverage between mutant and fully sampled wild type data is represented as a violet boxplot. Separately, the difference in total 6mA marks per chromosome is calculated for respective datasets, and boxplots are shown in the bottom panel. Mutant datasets consistently yield lower numbers of called 6mA marks than subsampled wild type, despite the former having higher coverage than the latter.
(C) Scatterplot of total number of 6mA marks per chromosome in wild type versus mutant data. PacBio cutoffs for calling 6mA marks are varied as shown. A greater number of 6mA marks per chromosome are consistently detected in wild type than mutant data.
(D) Boxplot of PacBio chromosome coverage in individual wild type and mutant biological replicates (left panel). Only chromosomes with 100-150× PacBio coverage are shown. The total number of 6mA marks in each of these chromosomes are plotted in the right panel. Wild type replicates show consistently higher numbers of 6mA marks per chromosome than mutant replicates.
Figure S5. Analysis of nucleosome organization and confirmation of ectopic DNA insertion in mta1 mutants, Related to Figures 3 and 4.
Description of analysis in panels A-G: Nucleosomes are grouped according to their “starting” 6mA level, defined as the total number of 6mA marks +/− 200 bp from the nucleosome dyad in wild type cells (WT). The dyad is assigned to be the peak position of MNase-seq reads. Similarly, linkers are grouped according to their “starting” methylation level, defined as the total number of 6mA marks between two flanking nucleosome dyads (or between the 5’ chromosome end and the terminal nucleosome) in wild type cells. Loci with high starting 6mA have methylation greater than or equal to the 90th percentile of starting 6mA levels, and show greater changes in methylation between mutant and wild type cells (Figure 3D). Those with low starting 6mA are in the lowest 10th percentile. If 6mA impacts nucleosome organization in vivo, then loci with high starting 6mA should show a greater change in nucleosome organization. Possible effects are illustrated in panels A – C. Vertical green lines depict 6mA marks, while blue and red peaks denote nucleosome occupancy. The plots shown in panels A – C illustrate the idealized result if 6mA disfavors nucleosomes in vivo. Actual effects are shown in panels D – G. “Wild type” is abbreviated as WT. Analyses are restricted to the 5’ chromosome end.
(A) 6mA loss may result in an increase in nucleosome fuzziness (highlighted with bold red double-sided arrow). The effect should be greater for nucleosomes with high starting 6mA due to greater change in 6mA between mutant and wild type cells (“Change in nucleosome fuzziness” Box). Nucleosomes should, in turn, exhibit lower occupancy near the peak position, and higher occupancy in flanking regions (“Change in Nucleosome occupancy” Box; highlighted with red arrowheads and plotted +/−73bp from the dyad). Nucleosome fuzziness is calculated as the standard deviation of MNase-seq read locations +/− 73bp from the dyad.
(B) 6mA loss from nucleosome linker regions may result in a decrease in linker length (highlighted with bold red bracket). If so, the magnitude of decrease in linker length should be greater for linkers with high starting 6mA (“Change in linker length” Box).
(C) 6mA loss may result in an increase in occupancy directly over the methylated linker region (highlighted with bold red bracket). If so, the magnitude of increase in linker occupancy should be greater for regions with high starting 6mA (“Change in linker occupancy” Box). Linker occupancy denotes the average MNase-seq coverage +/−25bp from the midpoint between flanking nucleosome dyads or chromosome end. As an example, for the +1/+2 nucleosome linker, occupancy is calculated +/−25bp from the midpoint of the +1 and +2 nucleosome dyad positions. Since nucleosome linker length in Oxytricha is ~200bp (Figure S5F, bottom panels), the genomic window used to calculate linker occupancy has minimal overlap with that for calculating nucleosome fuzziness and occupancy in panel A.
(D) Impact of 6mA loss on nucleosome fuzziness. For each nucleosome, the change in fuzziness between mutant and wild type cells is calculated. Boxplots represent the distribution of changes in fuzziness scores. “MNase-seq” denotes sequencing of nucleosomal DNA obtained from Oxytricha chromatin in vivo, while “Control gDNA-seq” represents sequencing of MNase-digested, naked genomic DNA in vitro. Boxplot features are as described in Figure 1C. Distributions are compared using a Wilcoxon rank-sum test. N.S denotes “non-significant”, with p > 0.01.
(E) Impact of 6mA loss on nucleosome occupancy. For each nucleosome, the difference in nucleosome occupancy between mutant and wild type cells is calculated at individual basepairs +/−73bp around the nucleosome dyad. Data are averaged and depicted as line plots. The change in occupancy at the dyad is compared between nucleosomes with high and low starting 6mA using a Wilcoxon rank-sum test.
(F) Impact of 6mA loss on linker length. Three types of linkers are analyzed: between the 5’ chromosome end and +1 nucleosome dyad, between the +1 and +2 nucleosome dyads, and between the +2 and +3 nucleosome dyads. For each linker, the difference in its length between mutant and wild type cells is calculated. The resulting distribution of linker length differences is plotted as a histogram (top-most row of this panel). Distributions of linker length differences are compared using two-sample unequal variance t-test. N.S. indicates “not significant”, with p > 0.01. Separately, the respective distributions of linker lengths in mutant and wild type cells are plotted in the bottom two rows of this panel. The median linker length from each group is included as an inset.
(G) Impact of 6mA loss on linker occupancy. Linkers are binned as in panel F. For each linker, the difference in occupancy between mutant and wild type cells is calculated. The resulting distribution of changes in linker occupancy is represented as a boxplot. Distributions are compared using two-sample unequal variance t-test. N.S. indicates “not significant”, with p > 0.01. Boxplot features are as described in Figure 1C.
(H) Poly(A)+ RNAseq analysis of wild type and mta1 mutants. “ATG” denotes start codon of MTA1 gene. A 62bp ectopic DNA insertion results in a frameshift mutation in the MTA1 coding region. Three wild type (WT1, WT2, WT3) and mutant (mta11, mta12, mta13) biological replicates are analyzed. Short horizontal bars represent RNAseq reads, which are ~75 nt in length and mapped to the reference sequence. For a read to be successfully mapped, it must have no more than 2 mismatches relative to the reference sequence. Unmapped reads are discarded. Blue and red bars denote RNAseq reads that map to native and ectopic regions, respectively. RNAseq reads overlapping the ectopic region are detected in mutant but not wild type replicates. These reads span junctions between the ectopic and flanking coding regions, confirming the site of ectopic insertion.
Figure S6. Gel analysis of histone octamers and assembled chromatin, Related to Figures 4 and 6.
Description for panels A-D: Xenopus unmodified core histones were recombinantly expressed. Oxytricha histones were acid-extracted from vegetative nuclei. Oxytricha and Xenopus histones were subsequently refolded into octamers and purified through size exclusion chromatography.
Description for panels E-I: Xenopus or Oxytricha histone octamers were assembled on DNA and subsequently digested with MNase to obtain ~150bp mononucleosome-sized fragments (labeled with red arrowheads). The resulting products were visualized by agarose gel electrophoresis. Mononucleosomal DNA was gel-excised and analyzed using Illumina sequencing or tiling qPCR analysis in Figures 4, 6, and S7.
(A) Reverse-phase HPLC purification of acid-extracted Oxytricha histones. Fractions 1-5 were individually collected and analyzed by Coomassie staining and western blotting.
(B) SDS-PAGE analysis of purified Oxytricha histone fractions.
(C) Western blot analysis of Oxytricha histone fractions 1-5. The fraction that is most enriched in each type of histone is colored in red. Arrowheads indicate likely histone bands.
(D) SDS-PAGE analysis of purified Oxytricha and Xenopus histone octamers.
(E) Chromatin was assembled on PCR-amplified Oxytricha mini-genome DNA, digested with MNase, and analyzed by agarose gel electrophoresis.
(F) Chromatin was assembled on native Oxytricha genomic DNA, digested with MNase, and analyzed by agarose gel electrophoresis.
(G) Chromatin was assembled with synthetic chromosome DNA, digested with MNase, and visualized by agarose gel electrophoresis. All assemblies with synthetic chromosomes were performed in the presence of an approximately 100-fold mass excess of buffer DNA relative to synthetic chromosome (see Methods). This applies to panels G, H, and I. Representative assemblies with the unmethylated chromosome are shown. Methylated chromosome assemblies were separately performed in place of the unmethylated variant.
(H) Chromatin was assembled on unmethylated synthetic chromosomes by salt dialysis and subsequently incubated with ACF and/or ATP. The resulting mixture was digested with MNase and visualized by agarose gel electrophoresis. Regularly spaced nucleosomes (labeled with red dots) are observed only when chromatin was incubated with both ACF and ATP.
(I) Chromatin assembled on unmethylated synthetic chromosomes using the NAP1 histone chaperone in the presence of ACF and/or ATP. The resulting mixture was digested with MNase and visualized by agarose gel electrophoresis. Nucleosomes are regularly spaced (labeled with red dots) in the presence of both ACF and ATP, although less apparent than in panel H.
Figure S7. Control MNase-seq and tiling qPCR analysis, Related to Figures 4 and 6.
(A) Same analysis as Figure 4C, showing that 6mA quantitatively disfavors nucleosome occupancy in vitro but not in vivo. Here, the extent of MNase digestion was 40% of that in Figure 4C. P-values were calculated using a two-sample unequal variance t-test. N.S denotes “non-significant”, with p > 0.05.
(B) Same analysis as Figure 6E, showing that the ACF complex restores nucleosome occupancy over methylated DNA in an ATP-dependent manner in vitro. Here, the extent of MNase digestion was 25% of that in Figure 6E. P-values were calculated using a two-sample unequal variance t-test. N.S denotes “non-significant”, with p > 0.05.
(C) Same analysis as Figure S5D, showing that nucleosomes with high starting 6mA show larger changes in fuzziness. Here, the extent of MNase digestion was 40% of that in Figure S5D. Distributions are compared using a Wilcoxon rank-sum test. N.S denotes “non-significant”, with p > 0.01.
(D) Same analysis as Figure S5E, showing that nucleosomes with high starting 6mA exhibit characteristic changes in nucleosome occupancy at and around the nucleosome dyad. Here, the extent of MNase digestion was 40% of that in Figure S5E. The change in dyad occupancy is compared between nucleosomes with high and low starting 6mA using a Wilcoxon rank-sum test. N.S denotes “non-significant”, with p > 0.01.
(E) Tiling qPCR analysis of nucleosome occupancy in spike-in and homogeneous synthetic chromosome preparations. The blunt, unmethylated synthetic chromosome (construct #1 in Figure 5B) was used for chromatin assembly with (“Spike-in”) or without (“Homogeneous”) a 100-fold excess of buffer DNA. In the latter case, an equivalent mass of synthetic chromosome was added in place of buffer DNA to maintain the same DNA concentration for chromatin assembly. The tiling qPCR assay was performed as in Figure 6B. Shaded red bars depict the regions where 6mA modulates nucleosome occupancy in separate methylated chromosomes analyzed in Figures 6B and 6C. Note that methylated chromosomes were not used to generate qPCR data for this Figure. Black arrowheads indicate no decrease in nucleosome occupancy in these regions when buffer DNA is used. Thus, the decrease in nucleosome occupancy in methylated chromosomes reported in Figure 6 cannot be attributed to spike-in versus homogeneous addition of DNA for chromatin assembly. Error bars in all panels represent s.e.m. (n = 3-4).
(F) Chromatin was assembled on synthetic chromosomes using the NAP1 histone chaperone in the presence of ACF and/or ATP, instead of salt dialysis. qPCR analysis was performed as in Figure 6B. Methylated chromosomes used in this experiment contain 6mA in native sites. The addition of ACF and ATP results in a partial restoration of nucleosome occupancy over the methylated region. These results are similar to Figure 6D, where chromatin was assembled by salt dialysis instead of NAP1.
Highlights.
The ciliate methyltransferase MTA1c mediates DNA N6-adenine methylation (6mA)
6mA directly disfavors nucleosome occupancy in vitro
Synthesis of complete, epigenetically defined Oxytricha chromosomes
Use of designer eukaryotic chromosomes reveals a role for adenine methylation in nucleosome positioning.
Acknowledgments
We thank T. Srikumar and S. Kyin for assistance with nucleoside mass spectrometry; I. Pelczer for assistance with NMR measurements of nucleoside standards; W. Wang, J. Wiggins, and J. Miller for assistance with Illumina sequencing; C.D. Allis, V. Zakian, T. Bestor, S.H. Sternberg, W. Jack, R. Neme, G. Dann, K. Jani, A. Mostafavi and G. Liszczak for helpful discussions; I. Fernandez and A. Sobolevsky for discussions and access to FPLC equipment, and J. Wang, B. Dul, and F. Song for laboratory support. This work was funded by NIH grants R01-GM59708, R35-GM122555, and R01-GM109459 to L.F.L and R01-GM107047 to T.W.M.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, and Lipman DJ (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammermann D, Steinbrück G, Baur R, and Wohlert H (1981). Methylated bases in the DNA of the ciliate Stylonychia mytilus. Eur. J. Cell Biol 24, 154–156. [PubMed] [Google Scholar]
- An W, and Roeder RG (2003). Reconstitution and Transcriptional Analysis of Chromatin In Vitro. Methods Enzymol 377, 460–474. [DOI] [PubMed] [Google Scholar]
- Batut P, Dobin A, Plessy C, Carninci P, and Gingeras TR (2013). High-fidelity promoter profiling reveals widespread alternative promoter usage and transposon-driven developmental gene expression. Genome Res 23, 169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beh LY, Müller MM, Muir TW, Kaplan N, and Landweber LF (2015). DNA-guided establishment of nucleosome patterns within coding regions of a eukaryotic genome. Genome Res 25, 1727–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bern M, Kil YJ, and Becker C (2012). Byonic: Advanced Peptide and Protein Identification Software. Curr. Protoc. Bioinformatics. 13, 13.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, and Taylor J (2010). Galaxy: a web-based genome analysis tool for experimentalists. Curr. Protoc. Mol. Biol 19, 19.10.1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracht JR, Fang W, Goldman AD, Dolzhenko E, Stein EM, and Landweber LF (2013). Genomes on the edge: programmed genome instability in ciliates. Cell 152, 406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg S, Pratt K, and Hattman S (1982). Sequence specificity of DNA adenine methylase in the protozoan Tetrahymena thermophila. J. Bacteriol 150, 993–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownell JE, Zhou J, Ranalli T, Kobayashi R, Edmondson DG, Roth SY, and Allis CD (1996). Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell 84, 843–851. [DOI] [PubMed] [Google Scholar]
- Cassidy-Hanley DM. (2012). Tetrahymena in the Laboratory: Strain Resources, Methods for Culture, Maintenance, and Storage. In Methods in Cell Biology, pp. 237–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Bracht JR, Goldman AD, Dolzhenko E, Clay DM, Swart EC, Perlman DH, Doak TG, Stuart A, Amemiya CT, et al. (2014). The Architecture of a Scrambled Genome Reveals Massive Levels of Genomic Rearrangement during Development. Cell 158, 1187–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapier CR, and Cairns BR (2009). The biology of chromatin remodeling complexes. Annu. Rev. Biochem 78, 273–304. [DOI] [PubMed] [Google Scholar]
- Cummings DJ, Tait A, and Goddard JM (1974). Methylated bases in DNA from Paramecium aurelia. Biochim. Biophys. Acta - Nucleic Acids Protein Synth 374, 1–11. [DOI] [PubMed] [Google Scholar]
- Debelouchina GT, Gerecht K, and Muir TW (2016). Ubiquitin utilizes an acidic surface patch to alter chromatin structure. Nat. Chem. Biol 13, 105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen JA, Coyne RS, Wu M, Wu D, Thiagarajan M, Wortman JR, Badger JH, Ren Q, Amedeo P, Jones KM, et al. (2006). Macronuclear genome sequence of the ciliate Tetrahymena thermophila, a model eukaryote. PLoS Biol 4, e286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, and Yates JR (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Engel JD, and von Hippel PH (1978). Effects of methylation on the stability of nucleic acid conformations. Studies at the polymer level. J. Biol. Chem 253, 927–934. [PubMed] [Google Scholar]
- Fang W, Wang X, Bracht JR, Nowacki M, and Landweber LF (2012). Piwi-interacting RNAs protect DNA against loss during Oxytricha genome rearrangement. Cell 151, 1243–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Clements J, Arndt W, Miller BL, Wheeler TJ, Schreiber F, Bateman A, and Eddy SR (2015). HMMER web server: 2015 update. Nucleic Acids Res 43, W30–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioravanti A, Fumeaux C, Mohapatra SS, Bompard C, Brilli M, Frandi A, Castric V, Villeret V, Viollier PHP, Biondi EEG, et al. (2013). DNA Binding of the Cell Cycle Transcriptional Regulator GcrA Depends on N6-Adenosine Methylation in Caulobacter crescentus and Other Alphaproteobacteria. PLoS Genet 9, e1003541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Luo G-Z, Chen K, Deng X, Yu M, Han D, Hao Z, Liu J, Lu X, Doré LC, et al. (2015). N6-methyldeoxyadenosine marks active transcription start sites in Chlamydomonas. Cell 161, 879–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyodorov DV, and Kadonaga JT (2003). Chromatin assembly in vitro with purified recombinant ACF and NAP-1. Methods Enzymol 371, 499–515. [DOI] [PubMed] [Google Scholar]
- Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J, et al. (2005). Galaxy: a platform for interactive large-scale genome analysis. Genome Res 15, 1451–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goecks J, Nekrutenko A, and Taylor J (2010). Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 11, R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorovsky MA, Hattman S, and Pleger GL (1973). (6 N)methyl adenine in the nuclear DNA of a eucaryote, Tetrahymena pyriformis. J. Cell Biol 56, 697–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschling DE, and Cech TR (1984). Chromatin structure of the molecular ends of Oxytricha macronuclear DNA: phased nucleosomes and a telomeric complex. Cell 38, 501–510. [DOI] [PubMed] [Google Scholar]
- Greer EL, Blanco MA, Gu L, Sendinc E, Liu J, Aristizábal-Corrales D, Hsu C-H, Aravind L, He C, and Shi Y (2015). DNA Methylation on N6-Adenine in C. elegans. Cell 161, 868–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberle V, Forrest ARR, Hayashizaki Y, Carninci P, and Lenhard B (2015). CAGEr: precise TSS data retrieval and high-resolution promoterome mining for integrative analyses. Nucleic Acids Res 43, e51–e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattman S, Kenny C, Berger L, and Pratt K (1978). Comparative study of DNA methylation in three unicellular eucaryotes. J. Bacteriol 135, 1156–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JR, Liebert K, Bekes M, Jeltsch A, and Cheng X (2006). Structure and Substrate Recognition of the Escherichia coli DNA Adenine Methyltransferase. J. Mol. Biol 358, 559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Niu B, Gao Y, Fu L, and Li W (2010). CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26, 680–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Dong X, Gong Z, Qin L-Y, Yang S, Zhu Y-L, Wang X, Zhang D, Zou T, Yin P, et al. (2018). Solution structure of the RNA recognition domain of METTL3-METTL14 N6-methyladenosine methyltransferase. Protein Cell. 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff JT, and Zilberman D (2014). Dnmt1-independent CG methylation contributes to nucleosome positioning in diverse eukaryotes. Cell 156, 1286–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Bulger M, Pazin MJ, Kobayashi R, and Kadonaga JT (1997). ACF, an ISWI-containing and ATP-utilizing chromatin assembly and remodeling factor. Cell 90, 145–155. [DOI] [PubMed] [Google Scholar]
- Iyer LM, Zhang D, and Aravind L (2016). Adenine methylation in eukaryotes: Apprehending the complex evolutionary history and functional potential of an epigenetic modification. Bioessays 38, 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karrer KM, and VanNuland TA (1999). Nucleosome positioning is independent of histone H1 in vivo. J. Biol. Chem 274, 33020–33024. [DOI] [PubMed] [Google Scholar]
- Katoh K, Rozewicki J, and Yamada KD (2017). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Tolstorukov MY, and Park PJ (2008). Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat. Biotechnol 26, 1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana JS, Wang X, Chen X, Perlman DH, and Landweber LF (2014). Transcription-Independent Functions of an RNA Polymerase II Subunit, Rpb2, During Genome Rearrangement in the Ciliate, Oxytricha trifallax. Genetics 197, 839–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana JS, Clay DM, Moreira S, Wang X, and Landweber LF (2018). Small RNA-mediated regulation of DNA dosage in the ciliate Oxytricha. RNA 24, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koziol MJ, Bradshaw CR, Allen GE, Costa ASH, Frezza C, and Gurdon JB (2015). Identification of methylated deoxyadenosines in vertebrates reveals diversity in DNA modifications. Nat. Struct. Mol. Biol 23, 24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuraku S, Zmasek CM, Nishimura O, and Katoh K (2013). aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res 41, W22–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai WKM, and Pugh BF (2017). Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nat. Rev. Mol. Cell Biol 18, 548–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughlin TJ, Henry JM, Phares EF, Long MV, and Olins DE (1983). Methods for the Large-Scale Cultivation of an Oxytricha (Ciliophora: Hypotrichida). J. Protozool 30, 63–64. [Google Scholar]
- Lauth MR, Spear BB, Heumann J, and Prescott DM (1976). DNA of ciliated protozoa: DNA sequence diminution during macronuclear development of Oxytricha. Cell 7, 67–74. [DOI] [PubMed] [Google Scholar]
- Lawn RM, Heumann JM, Herrick G, and Prescott DM (1978). The gene-size DNA molecules in Oxytricha. Cold Spring Harb. Symp. Quant. Biol 42 Pt 1, 483–492. [DOI] [PubMed] [Google Scholar]
- Liang Z, Shen L, Cui X, Bao S, Geng Y, Yu G, Liang F, Xie S, Lu T, Gu X, et al. (2018). DNA N 6 -Adenine Methylation in Arabidopsis thaliana. Dev. Cell 45, 406–416.e3. [DOI] [PubMed] [Google Scholar]
- Lieleg C, Ketterer P, Nuebler J, Ludwigsen J, Gerland U, Dietz H, Mueller-Planitz F, and Korber P (2015). Nucleosome spacing generated by ISWI and CHD1 remodelers is constant regardless of nucleosome density. Mol. Cell. Biol 35, 1588–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. (2014). A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol 10, 93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Zhu Y, Luo G-Z, Wang X, Yue Y, Wang X, Zong X, Chen K, Yin H, Fu Y, et al. (2016). Abundant DNA 6mA methylation during early embryogenesis of zebrafish and pig. Nat. Commun 7, 13052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Taverna SD, Muratore TL, Shabanowitz J, Hunt DF, and Allis CD (2007). RNAi-dependent H3K27 methylation is required for heterochromatin formation and DNA elimination in Tetrahymena. Genes Dev 21, 1530–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, and Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Luger K, Rechsteiner TJ, and Richmond TJ (1999). Preparation of nucleosome core particle from recombinant histones. Methods Enzymol 304, 3–19. [DOI] [PubMed] [Google Scholar]
- Luo G-Z, Blanco MA, Greer EL, He C, and Shi Y (2015). DNA N6-methyladenine: a new epigenetic mark in eukaryotes? Nat. Rev. Mol. Cell Biol 16, 705–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo G-Z, Hao Z, Luo L, Shen M, Sparvoli D, Zheng Y, Zhang Z, Weng X, Chen K, Cui Q, et al. (2018). N6-methyldeoxyadenosine directs nucleosome positioning in Tetrahymena DNA. Genome Biol 19, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrich TN, Ioshikhes IP, Venters BJ, Jiang C, Tomsho LP, Qi J, Schuster SC, Albert I, and Pugh BF (2008). A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res 18, 1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao W, Xiong J, Bowen J, Wang W, Liu Y, Braguinets O, Grigull J, Pearlman RE, Orias E, and Gorovsky MA (2009). Microarray analyses of gene expression during the Tetrahymena thermophila life cycle. PLoS One 4, e4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MA, Pfeiffer W, and Schwartz T (2010). Creating the CIPRES Science Gateway for Inference of Large Phylogenetic Trees. [Google Scholar]
- Mondo SJ, Dannebaum RO, Kuo RC, Louie KB, Bewick AJ, LaButti K, Haridas S, Kuo A, Salamov A, Ahrendt SR, et al. (2017). Widespread adenine N6-methylation of active genes in fungi. Nat. Genet 49, 964–968. [DOI] [PubMed] [Google Scholar]
- Mortazavi A, Williams BBA, McCue K, Schaeffer L, et al. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. [DOI] [PubMed] [Google Scholar]
- MQller MM, Fierz B, Bittova L, Liszczak G, and Muir TW (2016). A two-state activation mechanism controls the histone methyltransferase Suv39h1. Nat. Chem. Biol 12, 188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray IA, Morgan RD, Luyten Y, Fomenkov A, Corrêa IR, Dai N, Allaw MB, Zhang X, Cheng X, and Roberts RJ (2018). The non-specific adenine DNA methyltransferase M.EcoGII. Nucleic Acids Res 46, 840–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, and Aebersold R (2003). A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem 75, 4646–4658. [DOI] [PubMed] [Google Scholar]
- Nowacki M, Vijayan V, Zhou Y, Schotanus K, Doak TG, and Landweber LF (2008). RNA-mediated epigenetic programming of a genome-rearrangement pathway. Nature 451, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, and Conrad NK (2017). The U6 snRNA m6A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell 169, 824–835.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt K, and Hattman S (1981). Deoxyribonucleic acid methylation and chromatin organization in Tetrahymena thermophila. Mol. Cell. Biol 1, 600–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott DM (1994). The DNA of ciliated protozoa. Microbiol. Rev 58, 233–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, and Ishihama Y (2007). Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc 2, 1896–1906. [DOI] [PubMed] [Google Scholar]
- Rae PM, and Spear BB (1978). Macronuclear DNA of the hypotrichous ciliate Oxytricha fallax. Proc. Natl. Acad. Sci. U. S. A 75, 4992–4996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäffer AA, Aravind L, Madden TL, Shavirin S, Spouge JL, Wolf YI, Koonin EV, and Altschul SF (2001). Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res 29, 2994–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffers S, Ebert C, Rahimoff R, Kosmatchev O, Steinbacher J, Bohne A-V, Spada F, Michalakis S, Nickelsen J, Müller M, et al. (2017). Quantitative LC-MS Provides No Evidence for m6 dA or m4 dC in the Genome of Mouse Embryonic Stem Cells and Tissues. Angew. Chem. Int. Ed. Engl 56, 11268–11271. [DOI] [PubMed] [Google Scholar]
- Śledź P, and Jinek M (2016). Structural insights into the molecular mechanism of the m(6)A writer complex. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl BD, Ohba R, Cook RG, and Allis CD (1999). Methylation of histone H3 at lysine 4 is highly conserved and correlates with transcriptionally active nuclei in Tetrahymena. Proc. Natl. Acad. Sci. U. S. A 96, 14967–14972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl K, and Segal E (2013). Determinants of nucleosome positioning. Nat. Struct. Mol. Biol 20, 267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swart EC, Bracht JR, Magrini V, Minx P, Chen X, Zhou Y, Khurana JS, Goldman AD, Nowacki M, Schotanus K, et al. (2013). The Oxytricha trifallax Macronuclear Genome: A Complex Eukaryotic Genome with 16,000 Tiny Chromosomes. PLoS Biol 11, e1001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna SD, Coyne RS, and Allis CD (2002). Methylation of histone h3 at lysine 9 targets programmed DNA elimination in tetrahymena. Cell 110, 701–711. [DOI] [PubMed] [Google Scholar]
- Wada RK, and Spear BB (1980). Nucleosomal organization of macronuclear chromatin in Oxytricha fallax. Cell Differ 9, 261–268. [Google Scholar]
- Wang P, Doxtader KA, and Nam Y (2016a). Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol. Cell 63, 306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, Gong Z, Wang Q, Huang J, Tang C, et al. (2016b). Structural basis of N6-adenosine methylation by the METTL3–METTL14 complex. Nature 534, 575–578. [DOI] [PubMed] [Google Scholar]
- Wang Y, Chen X, Sheng Y, Liu Y, and Gao S (2017). N6-adenine DNA methylation is associated with the linker DNA of H2A.Z-containing well-positioned nucleosomes in Pol II-transcribed genes in Tetrahymena. Nucleic Acids Res 45, 11594–11606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warda AS, Kretschmer J, Hackert P, Lenz C, Urlaub H, Höbartner C, Sloan KE, and Bohnsack MT (2017). Human METTL16 is a N6-methyladenosine (m6A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep 18, 2004–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Mizzen CA, Cook RG, Gorovsky MA, and Allis CD (1998). Phosphorylation of histone H3 at serine 10 is correlated with chromosome condensation during mitosis and meiosis in Tetrahymena. Proc. Natl. Acad. Sci. U. S. A 95, 7480–7484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TP, Wang T, Seetin MG, Lai Y, Zhu S, Lin K, Liu Y, Byrum SD, Mackintosh SG, Zhong M, et al. (2016). DNA methylation on N6-adenine in mammalian embryonic stem cells. Nature 532, 329–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao R, and Moore DD (2011). DamIP: Using Mutant DNA Adenine Methyltransferase to Study DNA-Protein Interactions In Vivo In Current Protocols in Molecular Biology, (NJ, USA: John Wiley & Sons, Inc.), p. Unit21.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C-L, Zhu S, He M, Chen D, Zhang Q, Chen Y, Yu G, Liu J, Xie S-Q, Luo F, et al. (2018). N6-Methyladenine DNA Modification in the Human Genome. Mol. Cell 71, 306–318.e7. [DOI] [PubMed] [Google Scholar]
- Xiong J, Lu X, Lu Y, Zeng H, Yuan D, Feng L, Chang Y, Josephine B, Gorovsky M, Fu C, et al. (2011). Tetrahymena Gene Expression Database (TGED): A resource of microarray data and co-expression analyses for Tetrahymena. Sci. China Life Sci 54, 65–67. [DOI] [PubMed] [Google Scholar]
- Xiong J, Lu X, Zhou Z, Chang Y, Yuan D, Tian M, Zhou Z, Wang L, Fu C, Orias E, et al. (2012). Transcriptome analysis of the model protozoan, Tetrahymena thermophila, using Deep RNA sequencing. PLoS One 7, e30630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao B, Cheng Y, Wang Z, Li Y, Chen L, Huang L, Zhang W, Chen D, Wu H, Tang B, et al. (2017). DNA N6-methyladenine is dynamically regulated in the mouse brain following environmental stress. Nat. Commun 8, 1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao B, Li Y, Wang Z, Chen L, Poidevin M, Zhang C, Lin L, Wang F, Bao H, Jiao B, et al. (2018). Active N 6 -Methyladenine Demethylation by DMAD Regulates Gene Expression by Coordinating with Polycomb Protein in Neurons. Mol. Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yerlici VT, and Landweber LF (2014). Programmed Genome Rearrangements in the Ciliate Oxytricha. Microbiol. Spectr 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, and Pugh BF (2011). High-resolution genome-wide mapping of the primary structure of chromatin. Cell 144, 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Huang H, Liu D, Cheng Y, Liu X, Zhang W, Yin R, Zhang D, Zhang P, Liu J, et al. (2015). N6-Methyladenine DNA Modification in Drosophila. Cell 161, 893–906. [DOI] [PubMed] [Google Scholar]
- Zhou C, Wang C, Liu H, Zhou Q, Liu Q, Guo Y, Peng T, Song J, Zhang J, Chen L, et al. (2018). Identification and analysis of adenine N6-methylation sites in the rice genome. Nat. Plants 4, 554–563. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Mass spectrometry analysis of 6mA in ciliate DNA, Related to Figure 1.
(A) Oxytricha and Tetrahymena genomic DNA were digested into nucleosides using degradase enzyme mix, followed by analysis using reverse-phase HPLC and mass spectrometry. Isotopically labeled dA and 6mA standards (15N5-dA and D3-6mA) were mixed with each sample to allow quantitative measurement of endogenous dA and 6mA concentrations. MS/MS analysis of labeled dA and 6mA standards confirmed the mass of the nucleobase. Eluted peaks with expected masses of dA and 6mA, and with highly similar retention times (RT) to internal standards are detected in Oxytricha and Tetrahymena nucleosides.
(B) Quantitation of dA and 6mA levels in Oxytricha and Tetrahymena gDNA using internal isotopically labeled nucleoside standards. The detected level of 6mA in Tetrahymena gDNA agrees with earlier reports (Gorovsky et al., 1973; Pratt and Hattman, 1981). The calculated abundance of 6mA relative to (dA + 6mA) in Oxytricha is ~0.71%, which is similar to the estimate from SMRT-seq base calls (0.78 – 1.04%). Note that the calculation from SMRT-seq data is expected to be an overestimate because 6mA is scored at being present or absent at each site in the genome for this purpose. In actual fact, 6mA sites may be partially methylated (Figure S4A). Neither 6mA nor dA was detected from LC-MS analysis of Oxytricha culture media, arguing against spurious signal arising from contamination or overall technical handling. Our PacBio and LC-MS measurements of % 6mA in Oxytricha are both similar to thin layer chromatography analysis of nucleotides (0.6 – 0.7%) from a distinct but closely related species, Oxytricha fallax (Rae and Spear, 1978).
Table S5. Protein sequences for phylogenetic tree construction, related to Figure 2.
FASTA-formatted list of amino acid sequences used to construct the MT-A70, p1, and p2 phylogenetic trees in Figures 2A, S2G, S2H, and S2I.
Table S7. Recombinant protein sequences, related to Figure 2.
Sequences were manually curated by mapping RNAseq reads to reference gene annotations and verifying the accuracy of predicted exon boundaries.
Figure S2. Analysis of 6mA and methyltransferase components in Tetrahymena, Related to Figures 1 and 2.
(A) Tetrahymena MNase-seq data from (Beh et al., 2015), while SMRT-seq data were generated in this study. Meta-chromosome plots overlaying in vivo MNase-seq (nucleosome occupancy) and SMRT-seq (6mA), relative to annotated transcription start sites. 6mA lies mainly within nucleosome linker regions, between the +1, +2, +3, and +4 nucleosomes.
(B) Histograms of the total number of 6mA marks within each linker in Tetrahymena genes. Calculations are performed as described in Figure 1B. Distinct linkers are highlighted with horizontal bold blue lines.
(C) Relationship between transcriptional activity and total number of 6mA marks in Tetrahymena genes. Analysis is performed as in Figure 1C. RNA-seq data was obtained from (Xiong et al., 2012).
(D) Composite analysis of 441,618 methylation sites reveals that 6mA occurs within an 5’-ApT-3’ dinucleotide motif in Tetrahymena, consistent with previous experiments (Bromberg et al., 1982; Wang et al., 2017) and similar to Oxytricha.
(E) Distribution of various 6mA dinucleotide motifs across the genome.
(F) Organization of transcription (mRNA-seq), nucleosome organization (MNase-seq), and 6mA (SMRT-seq) in a Tetrahymena gene.
(G) All sequences used for phylogeny construction are listed in Table S5. Abbreviations: Cel: Caenorhabditis elegans; Ath: Arabidopsis thaliana; Sra: Syncephalastrum racemosum; Hve: Hesseltinella vesiculosa; Are: Absidia repens ; Dre: Danio rerio; Has: Homo sapiens; Ssc: Sus scrofa; Mmu: Mus musculus; Xla: Xenopus laevis; Dme: Drosophila melanogastep Cre: Chlamydomonas reinhardtii; Ltr: Lobosporangium transversale; Lpe: Linderina pennispora; Bme: Basidiobolus meristosporus; Pfi: Piromyces finnis; Aro: Anaeromyces robustus; Tth: Tetrahymena thermophila; Otri: Oxytricha trifallax. This Bayesian phylogenetic tree of MT-A70 proteins is the same as in Figure 2A, except that all sequences are now included and labeled. TAMT-1 proteins are named according to (Luo et al., 2018).
(H) Bayesian phylogenetic tree of p1 proteins.
(I) Bayesian phylogenetic tree of p2 proteins. Dashed box depicts outgroup consisting of vertebrate SNAPC4 genes. These genes bear weak similarity to the homeobox-like domain of p2 proteins, but do not group phylogenetically with them and are therefore unlikely to be functionally homologs.
(J) Phylogenetic distribution of ApT 6mA motif and various proteins, as depicted in Figure 2B, but now also including TAMT-1, p1, and p2 proteins. Filled boxes denote the presence of a particular protein in a taxon. Open dashed boxes indicate the presence of SNAPC4 genes in vertebrates.
(K) Gene expression profiles of Tetrahymena MTA1, MTA9, p1 and p2. Microarray counts represent poly(A)+ expression levels, and are obtained from TetraFGD (Miao et al., 2009; Xiong et al., 2011). MTA1, MTA9, p1 and p2 were found in our study to co-elute with 6mA methylase activity. On the other hand, TAMT-1 is a putative DNA methyltransferase described by (Luo et al., 2018). The horizontal axis categories beginning with “S” and “C” represent the number of hours since the onset of starvation and conjugation (mating), respectively. “Low”, “Med”, and “High” denote relative cell densities during log-phase growth. Blue and orange traces represent data from two biological replicates. Green and red shaded regions show the peaks in poly(A)+ RNA expression in vegetative growth and conjugation, respectively, for MTA1, MTA9, p1 and p2. Note that their expression pattern differs from TAMT-1.
Figure S3. Further characterization of 6mA methyltransferase activity and MTA1c, Related to Figure 2.
(A) Fractionation of nuclear extracts on a Q sepharose column results in two distinct peaks of DNA methyltransferase activity, denoted as “Low Salt sample” and “High Salt sample” by black horizontal bars. FT denotes column flow-through. The DNA methyltransferase assay is performed as in Figure 2E. The salt concentration at which individual fractions elute from the column is plotted against DNA methyltransferase activity of each fraction (counts per minute). Inset shows DNA methyltransferase activity of the input nuclear extract, flowthrough from the Q sepharose column, and blank control (nuclear extract buffer). Orange and blue plots denote replicates derived from independent preparations of nuclear extract.
(B) DNA methyltransferase assay showing that the activity from nuclear extracts is heat-sensitive and requires addition of DNA and SAM. Error bars represent s.e.m. (n = 3).
(C) Dot blot showing that nuclear extracts mediate 6mA methylation. Note that the low salt sample has substantial DNase activity, resulting in a lower amount of DNA available for dot blot analysis. DNA substrate, nuclear extract, and SAM cofactor were mixed as in panels A and B. The DNA was subsequently purified and used for dot blot analysis.
(D) Domain organization of Tetrahymena MTA1, MTA9, p1, and p2. Protein domains are predicted using hmmscan on the EMBL-EBI webserver (Finn et al., 2015). “aa” denotes amino acids. Start and end coordinates of each domain are stated below each polypeptide.
(E) Sequence alignment of human (Hsa) METTL3 with Tetrahymena (Tth) and Oxytricha (Otri) MTA1 / MTA9, within the MT-A70 domain. Horizontal black bars underscore the DPPW catalytic motif, and the N549 / Q550 residues in human METTL3 that interact with the ribose moiety of the SAM cofactor. Note that the DPPW catalytic motif is conserved in MTA1 but not MTA9.
(F) Dot blot analysis of hemimethylated dsDNA substrates. Sense or antisense oligonucleotides were first individually methylated using the EcoGII bacterial 6mA methyltransferase. Each methylated ssDNA was subsequently purified and annealed with an unmethylated complementary strand to form hemimethylated constructs.
(G) SDS-PAGE analysis of recombinant proteins. Full length proteins were expressed and purified from E. coli. Bands of expected size are indicated with a black arrowhead.
(H) Methyltransferase assay using radiolabeled SAM on DNA and RNA substrates, coupled with gel analysis of nucleic acid integrity. ssRNA and dsRNA were produced by in vitro transcription from the 350bp dsDNA template using T7 RNA polymerase, and subsequently purified before use in this assay. Methyltransferase activity on equimolar amounts of each substrate was measured after incubation at 37°C for 6 hr, and depicted as either scintillation counts (Counts per minute), or normalized to the 350bp dsDNA sample (Relative activity). Only dsDNA, and not dsRNA or ssRNA, was methylated. Activity measurements are represented as scintillation counts (counts per minute). In addition, aliquots from each reaction containing DNA or RNA substrate and recombinant MTA1c (ie. MTA1, MTA7, p1 and p2 proteins) were withdrawn at 0, 1, 2, 3, or 6 hr during the 37°C incubation, purified using phenol:chloroform extraction and ethanol precipitation, and subsequently analyzed on a non-denaturing agarose gel. Both dsDNA and dsRNA substrates remained intact after 6 hrs. The ssRNA migrates more diffusely on a nondenaturing agarose gel, with some decrease in size over time, suggesting partial degradation and/or RNA folding; however, there is no detectable methylation of ssRNA despite a significant presence on the agarose gel after 6 hr at 37°C. It is unlikely that this species is too short to be methylated, since MTA1c can methylate significantly shorter substrates such as 27bp dsDNA (Figure 2G, S3I, S3J, S3K). Error bars represent s.e.m. (n = 3).
(I) DNA methyltransferase assay using radiolabeled SAM, on ssDNA oligonucleotides or annealed dsDNA substrates. All four recombinant MTA1c protein components – MTA1, MTA9, p1, and p2 – were included in each sample. Activity measurements are represented as scintillation counts (counts per minute). dsDNA substrates were prepared by annealing ssDNA oligonucleotides, as in Figure 2G. Sense ssDNA nucleotide sequences are depicted in the 5’→3’ direction, while antisense ssDNA is depicted as 3’→5’. Error bars represent s.e.m. (n = 3).
(J) Control [3H]SAM assay using hemimethylated dsDNA. Reactions depicted in red represent hemimethylated dsDNA incubated with [3H]SAM in the absence of recombinant MTA1c (MTA1, MTA9, p1, and p2 proteins). These reactions showed no methyltransferase activity, verifying that there is no contaminating EcoGII methyltransferase in hemimethylated dsDNA preparations. Activity measurements are shown as scintillation counts, or as “Relative Activity” (normalized against the sample containing unmethylated DNA substrate, [3H]SAM, and MTA1c protein). Hemimethylated dsDNA substrates in this panel are the same as those used in Figure 2G. The unmethylated dsDNA substrate used in this panel is the same as the top-most dsDNA substrate in Figure 2G, with two uninterrupted ApT dinucleotides. Error bars represent s.e.m. (n = 3).
(K) DNA methyltransferase assay using radiolabeled SAM, on dsDNA substrates with disrupted ApT dinucleotides. All four recombinant MTA1c protein components – MTA1, MTA9, p1, and p2 – were included in each sample. Activity measurements are normalized against the parent dsDNA construct with two uninterrupted ApT dinucleotides (top-most construct in this panel). ApT dinucleotide positions are labeled in bold red. Note that the parent dsDNA construct is identical to that in Figure S3L. Error bars represent s.e.m. (n = 3).
(L) DNA methyltransferase assay using radiolabeled SAM, on dsDNA substrates with shifted ApT dinucleotides. All four recombinant MTA1c protein components – MTA1, MTA9, p1, and p2 – were included in each sample. Activity measurements are normalized against the parent dsDNA construct with two uninterrupted ApT dinucleotides (top-most construct in this panel). The parent construct is identical to that in Figure S3K. ApT dinucleotides are labeled in bold red. The adjacent nucleotides are labelled in bold black to highlight the 4-mer sequence that contains each ApT dinucleotide. Error bars represent s.e.m. (n = 3).
(M) Motif frequencies of all 4-mer sequences containing methylated ApT dinucleotides in the Tetrahymena and Oxytricha genomes. A* denotes 6mA. The 4-mers TA*TA and CA*TT are colored in red and blue, respectively, to highlight their large difference in genomic frequencies.
(N) Motif frequencies of 4-mer sequences – regardless of methylation state – in Tetrahymena and Oxytricha. These were calculated from genomic sequence between the 5’ chromosome end and the +4 nucleosome peak (Oxytricha), or between the TSS and the +4 nucleosome peak (Tetrahymena). Analysis was restricted to these regions in order to serve as “background” frequencies for comparison to A*T methylated 4-mers, which are also mainly found downstream of TSSs. The 4-mers TATA and CATT are colored in red and blue, respectively, to facilitate comparison with methylated TA*TA and CA*TT in panel M.
Figure S4. Supplemental SMRT-seq data analyses, related to Figures 1 and 3.
(A) Top two panels depict PacBio coverage (horizontal axis) plotted against fractional methylation at each called 6mA site (vertical axis). Bottom left panel is a histogram of fractional methylation of all 6mA sites. Bottom right panel is a histogram of IPD ratios of all 6mA sites. Mutant datasets show significantly lower fractional methylation and IPD ratios at 6mA sites than wild type data.
(B) Wild type SMRT-seq data are randomly subsampled 15 times, such that the resulting coverage is lower than mta1 mutant data. The difference in PacBio coverage between mutant and subsampled wildtype data is calculated for each chromosome, and is collectively represented as an olive boxplot (top panel). This set of calculations is repeated 15 times for each subsampled dataset, resulting in a series of 15 boxplots. The difference in PacBio coverage between mutant and fully sampled wild type data is represented as a violet boxplot. Separately, the difference in total 6mA marks per chromosome is calculated for respective datasets, and boxplots are shown in the bottom panel. Mutant datasets consistently yield lower numbers of called 6mA marks than subsampled wild type, despite the former having higher coverage than the latter.
(C) Scatterplot of total number of 6mA marks per chromosome in wild type versus mutant data. PacBio cutoffs for calling 6mA marks are varied as shown. A greater number of 6mA marks per chromosome are consistently detected in wild type than mutant data.
(D) Boxplot of PacBio chromosome coverage in individual wild type and mutant biological replicates (left panel). Only chromosomes with 100-150× PacBio coverage are shown. The total number of 6mA marks in each of these chromosomes are plotted in the right panel. Wild type replicates show consistently higher numbers of 6mA marks per chromosome than mutant replicates.
Figure S5. Analysis of nucleosome organization and confirmation of ectopic DNA insertion in mta1 mutants, Related to Figures 3 and 4.
Description of analysis in panels A-G: Nucleosomes are grouped according to their “starting” 6mA level, defined as the total number of 6mA marks +/− 200 bp from the nucleosome dyad in wild type cells (WT). The dyad is assigned to be the peak position of MNase-seq reads. Similarly, linkers are grouped according to their “starting” methylation level, defined as the total number of 6mA marks between two flanking nucleosome dyads (or between the 5’ chromosome end and the terminal nucleosome) in wild type cells. Loci with high starting 6mA have methylation greater than or equal to the 90th percentile of starting 6mA levels, and show greater changes in methylation between mutant and wild type cells (Figure 3D). Those with low starting 6mA are in the lowest 10th percentile. If 6mA impacts nucleosome organization in vivo, then loci with high starting 6mA should show a greater change in nucleosome organization. Possible effects are illustrated in panels A – C. Vertical green lines depict 6mA marks, while blue and red peaks denote nucleosome occupancy. The plots shown in panels A – C illustrate the idealized result if 6mA disfavors nucleosomes in vivo. Actual effects are shown in panels D – G. “Wild type” is abbreviated as WT. Analyses are restricted to the 5’ chromosome end.
(A) 6mA loss may result in an increase in nucleosome fuzziness (highlighted with bold red double-sided arrow). The effect should be greater for nucleosomes with high starting 6mA due to greater change in 6mA between mutant and wild type cells (“Change in nucleosome fuzziness” Box). Nucleosomes should, in turn, exhibit lower occupancy near the peak position, and higher occupancy in flanking regions (“Change in Nucleosome occupancy” Box; highlighted with red arrowheads and plotted +/−73bp from the dyad). Nucleosome fuzziness is calculated as the standard deviation of MNase-seq read locations +/− 73bp from the dyad.
(B) 6mA loss from nucleosome linker regions may result in a decrease in linker length (highlighted with bold red bracket). If so, the magnitude of decrease in linker length should be greater for linkers with high starting 6mA (“Change in linker length” Box).
(C) 6mA loss may result in an increase in occupancy directly over the methylated linker region (highlighted with bold red bracket). If so, the magnitude of increase in linker occupancy should be greater for regions with high starting 6mA (“Change in linker occupancy” Box). Linker occupancy denotes the average MNase-seq coverage +/−25bp from the midpoint between flanking nucleosome dyads or chromosome end. As an example, for the +1/+2 nucleosome linker, occupancy is calculated +/−25bp from the midpoint of the +1 and +2 nucleosome dyad positions. Since nucleosome linker length in Oxytricha is ~200bp (Figure S5F, bottom panels), the genomic window used to calculate linker occupancy has minimal overlap with that for calculating nucleosome fuzziness and occupancy in panel A.
(D) Impact of 6mA loss on nucleosome fuzziness. For each nucleosome, the change in fuzziness between mutant and wild type cells is calculated. Boxplots represent the distribution of changes in fuzziness scores. “MNase-seq” denotes sequencing of nucleosomal DNA obtained from Oxytricha chromatin in vivo, while “Control gDNA-seq” represents sequencing of MNase-digested, naked genomic DNA in vitro. Boxplot features are as described in Figure 1C. Distributions are compared using a Wilcoxon rank-sum test. N.S denotes “non-significant”, with p > 0.01.
(E) Impact of 6mA loss on nucleosome occupancy. For each nucleosome, the difference in nucleosome occupancy between mutant and wild type cells is calculated at individual basepairs +/−73bp around the nucleosome dyad. Data are averaged and depicted as line plots. The change in occupancy at the dyad is compared between nucleosomes with high and low starting 6mA using a Wilcoxon rank-sum test.
(F) Impact of 6mA loss on linker length. Three types of linkers are analyzed: between the 5’ chromosome end and +1 nucleosome dyad, between the +1 and +2 nucleosome dyads, and between the +2 and +3 nucleosome dyads. For each linker, the difference in its length between mutant and wild type cells is calculated. The resulting distribution of linker length differences is plotted as a histogram (top-most row of this panel). Distributions of linker length differences are compared using two-sample unequal variance t-test. N.S. indicates “not significant”, with p > 0.01. Separately, the respective distributions of linker lengths in mutant and wild type cells are plotted in the bottom two rows of this panel. The median linker length from each group is included as an inset.
(G) Impact of 6mA loss on linker occupancy. Linkers are binned as in panel F. For each linker, the difference in occupancy between mutant and wild type cells is calculated. The resulting distribution of changes in linker occupancy is represented as a boxplot. Distributions are compared using two-sample unequal variance t-test. N.S. indicates “not significant”, with p > 0.01. Boxplot features are as described in Figure 1C.
(H) Poly(A)+ RNAseq analysis of wild type and mta1 mutants. “ATG” denotes start codon of MTA1 gene. A 62bp ectopic DNA insertion results in a frameshift mutation in the MTA1 coding region. Three wild type (WT1, WT2, WT3) and mutant (mta11, mta12, mta13) biological replicates are analyzed. Short horizontal bars represent RNAseq reads, which are ~75 nt in length and mapped to the reference sequence. For a read to be successfully mapped, it must have no more than 2 mismatches relative to the reference sequence. Unmapped reads are discarded. Blue and red bars denote RNAseq reads that map to native and ectopic regions, respectively. RNAseq reads overlapping the ectopic region are detected in mutant but not wild type replicates. These reads span junctions between the ectopic and flanking coding regions, confirming the site of ectopic insertion.
Figure S6. Gel analysis of histone octamers and assembled chromatin, Related to Figures 4 and 6.
Description for panels A-D: Xenopus unmodified core histones were recombinantly expressed. Oxytricha histones were acid-extracted from vegetative nuclei. Oxytricha and Xenopus histones were subsequently refolded into octamers and purified through size exclusion chromatography.
Description for panels E-I: Xenopus or Oxytricha histone octamers were assembled on DNA and subsequently digested with MNase to obtain ~150bp mononucleosome-sized fragments (labeled with red arrowheads). The resulting products were visualized by agarose gel electrophoresis. Mononucleosomal DNA was gel-excised and analyzed using Illumina sequencing or tiling qPCR analysis in Figures 4, 6, and S7.
(A) Reverse-phase HPLC purification of acid-extracted Oxytricha histones. Fractions 1-5 were individually collected and analyzed by Coomassie staining and western blotting.
(B) SDS-PAGE analysis of purified Oxytricha histone fractions.
(C) Western blot analysis of Oxytricha histone fractions 1-5. The fraction that is most enriched in each type of histone is colored in red. Arrowheads indicate likely histone bands.
(D) SDS-PAGE analysis of purified Oxytricha and Xenopus histone octamers.
(E) Chromatin was assembled on PCR-amplified Oxytricha mini-genome DNA, digested with MNase, and analyzed by agarose gel electrophoresis.
(F) Chromatin was assembled on native Oxytricha genomic DNA, digested with MNase, and analyzed by agarose gel electrophoresis.
(G) Chromatin was assembled with synthetic chromosome DNA, digested with MNase, and visualized by agarose gel electrophoresis. All assemblies with synthetic chromosomes were performed in the presence of an approximately 100-fold mass excess of buffer DNA relative to synthetic chromosome (see Methods). This applies to panels G, H, and I. Representative assemblies with the unmethylated chromosome are shown. Methylated chromosome assemblies were separately performed in place of the unmethylated variant.
(H) Chromatin was assembled on unmethylated synthetic chromosomes by salt dialysis and subsequently incubated with ACF and/or ATP. The resulting mixture was digested with MNase and visualized by agarose gel electrophoresis. Regularly spaced nucleosomes (labeled with red dots) are observed only when chromatin was incubated with both ACF and ATP.
(I) Chromatin assembled on unmethylated synthetic chromosomes using the NAP1 histone chaperone in the presence of ACF and/or ATP. The resulting mixture was digested with MNase and visualized by agarose gel electrophoresis. Nucleosomes are regularly spaced (labeled with red dots) in the presence of both ACF and ATP, although less apparent than in panel H.
Figure S7. Control MNase-seq and tiling qPCR analysis, Related to Figures 4 and 6.
(A) Same analysis as Figure 4C, showing that 6mA quantitatively disfavors nucleosome occupancy in vitro but not in vivo. Here, the extent of MNase digestion was 40% of that in Figure 4C. P-values were calculated using a two-sample unequal variance t-test. N.S denotes “non-significant”, with p > 0.05.
(B) Same analysis as Figure 6E, showing that the ACF complex restores nucleosome occupancy over methylated DNA in an ATP-dependent manner in vitro. Here, the extent of MNase digestion was 25% of that in Figure 6E. P-values were calculated using a two-sample unequal variance t-test. N.S denotes “non-significant”, with p > 0.05.
(C) Same analysis as Figure S5D, showing that nucleosomes with high starting 6mA show larger changes in fuzziness. Here, the extent of MNase digestion was 40% of that in Figure S5D. Distributions are compared using a Wilcoxon rank-sum test. N.S denotes “non-significant”, with p > 0.01.
(D) Same analysis as Figure S5E, showing that nucleosomes with high starting 6mA exhibit characteristic changes in nucleosome occupancy at and around the nucleosome dyad. Here, the extent of MNase digestion was 40% of that in Figure S5E. The change in dyad occupancy is compared between nucleosomes with high and low starting 6mA using a Wilcoxon rank-sum test. N.S denotes “non-significant”, with p > 0.01.
(E) Tiling qPCR analysis of nucleosome occupancy in spike-in and homogeneous synthetic chromosome preparations. The blunt, unmethylated synthetic chromosome (construct #1 in Figure 5B) was used for chromatin assembly with (“Spike-in”) or without (“Homogeneous”) a 100-fold excess of buffer DNA. In the latter case, an equivalent mass of synthetic chromosome was added in place of buffer DNA to maintain the same DNA concentration for chromatin assembly. The tiling qPCR assay was performed as in Figure 6B. Shaded red bars depict the regions where 6mA modulates nucleosome occupancy in separate methylated chromosomes analyzed in Figures 6B and 6C. Note that methylated chromosomes were not used to generate qPCR data for this Figure. Black arrowheads indicate no decrease in nucleosome occupancy in these regions when buffer DNA is used. Thus, the decrease in nucleosome occupancy in methylated chromosomes reported in Figure 6 cannot be attributed to spike-in versus homogeneous addition of DNA for chromatin assembly. Error bars in all panels represent s.e.m. (n = 3-4).
(F) Chromatin was assembled on synthetic chromosomes using the NAP1 histone chaperone in the presence of ACF and/or ATP, instead of salt dialysis. qPCR analysis was performed as in Figure 6B. Methylated chromosomes used in this experiment contain 6mA in native sites. The addition of ACF and ATP results in a partial restoration of nucleosome occupancy over the methylated region. These results are similar to Figure 6D, where chromatin was assembled by salt dialysis instead of NAP1.
Data Availability Statement
Oxytricha SMRT-seq data are deposited in SRA under the accession numbers SRX2335608, SRX2335607, and GSE94421. Tetrahymena SMRT-seq and all Oxytricha Illumina data are deposited in NCBI GEO under accession number GSE94421.