

Abstract
Background
Human microRNAs (miRNAs) have diverse functions in biology, and play a role in nearly every biological process. Here we report that miR-520d-5p (520d-5p) causes undifferentiated cancer cells to adopt benign or normal status in vivo in immunodeficient mice via demethylation and P53 upregulation. Further we found that 520-5p causes normal cells to elongate cellular lifetime and mesenchymal stem cell-like status with CD105 positivity. We hypothesized that ectopic 520d-5p expression reduced mutations in undifferentiated type of hepatoma (HLF) cells through synergistic modulation of methylation-related enzymatic expression.
Methods
To examine whether there were any changes in mutation status in cells treated with 520d-5p, we performed next generation sequencing (NGS) in HLF cells and human iPSC-derivative cells in pre-mesenchymal stem cell status. We analyzed the data using both genome-wide and individual gene function approaches.
Results
520d-5p induced a shift towards a wild type or non-malignant phenotype, which was regulated by nucleotide mutations in both HLF cells and iPSCs. Further, 520d-5p reduced mutation levels in both the whole genome and genomic fragment assemblies.
Conclusions
Cancer cell genomic mutations cannot be repaired in most contexts. However, these findings suggest that applied development of 520d-5p would allow new approaches to cancer research and improve the quality of iPSCs used in regenerative medicine.
Electronic supplementary material
The online version of this article (10.1186/s12885-019-5786-y) contains supplementary material, which is available to authorized users.
Keywords: Mutation, iPSC, Progenitor MSC, Hsa-miR-520d-5p, Genomic conversion
Background
miRNAs are small, non-coding RNAs of only 19–25 bases in length, which control gene expression by targeting mRNA for degradation or inhibiting translation. Aberrant epigenetics and coding gene mutations are known to play a role in the development and progression of cancer [1–4]. DNA methylation, histone acetylation, histone methylation and miRNA play important roles in epigenetics [5–7]. Some miRNAs are known to be dysregulated in multiple cancers, and are classified as onco-miRs, suppressor-miRs, or metasta-miRs [8–10]. Target gene expression is negatively correlated with miRNA levels. miRNA and other epigenetic mechanisms underlie network formation, and are involved with the prevention or progression of neoplasia. Thus, examining miRNA alterations in cancers is important to predict patient outcomes and develop novel therapies. However, no previous reports have identified that miRNAs can alter differentiation in cancers. This notion is not generally accepted, but in the present report we provide supporting evidence of this phenomenon.
Next-generation sequencing (NGS) technology has produced a substantial volume of biological data, and has shed light on the path towards personalized medicine. Although the acquisition cost of high-throughput genome sequencing has decreased, the analysis and interpretation of these large-scale sequencing data continues to be prohibitive [11–13]. Many aligner and variant programs have been developed to identify variants in GS data, which have been developed and incorporated into diverse pipelines. A typical pipeline contains an aligner and a variant program, wherein the aligner program maps the sequencing reads to a reference genome, and the variant program identifies variant sites and assigns a genotype to the subject(s). The performances of different aligners have been studied extensively [11, 14, 15]. NGS is a powerful tool for identifying rare and de novo variants, disease mapping, and quantification of expression levels. For analysis, NGS reads are first aligned to a reference genome, and subsequently subjected to variant analysis after necessary quality control procedures. Whole exome sequencing of thousands of human cancers has led to the unexpected discovery of many inactivating mutations of genes that control the epigenome [16]. These mutations have the potential to disrupt DNA methylation patterns, histone modifications, and nucleosome positioning and therefore, gene expression. Genetic alteration of the epigenome therefore contributes to cancer, just as epigenetic processes can cause point mutations and impairment of DNA repair functions.
We previously reported that 520d-5p can convert undifferentiated status of cancer to benign or normal status via demethylation and P53 upregulation, hence reprogramming cancer cells to human mesenchymal stem cells (hMSCs) in conditions that can maintain differentiation [17, 18]. Furthermore, 520d-5p reprograms fibroblasts into CD105-positive cells, extending cellular lifespan [19] and preventing fatal DNA damage via a non-canonical nuclear stress and demethylation process [20]. Therefore, we postulated that this small molecule may alter epigenetic status, and in the present study evaluated 520d-5p influence on mutations in cancer cells using NGS, including exome analysis.
Methods
Cell preparation
For DNA extraction, we used three cell lines and lentiviral vectors. hiPSCs (HPS0002: 253G1), mesenchymal stem cells (hMSC), and undifferentiated type of hepatoma cells (HLF) were provided by the RIKEN BioResource Center Cell Bank [21] and the Cell Resource Center for Biomedical Research, Institute of Development, Takara Bio (Kusatsu, Shiga, Japan), and Aging and Cancer Tohoku University, respectively. HLF, MSC, and iPSC (253G1) were cultured in RPMI1640 medium supplemented with 10% FBS and 1% penicillin/streptomycin, in Mesenchymal Stem Cell Growth Medium 2 (Takara Bio, Kusatsu, Shiga, Japan), and in ReproStem medium (ReproCell, Tokyo, Japan) with 10 ng/ml of bFGF-2, respectively. Additionally, for the transfection to HLF cells or hMSCs, the human mesangial cell line 293FT (Invitrogen Japan K.K., Tokyo, Japan) was used for producing 520d-5p expressing-lentiviral particles. 293FT cells were cultured in DMEM supplemented with 10% FBS, 0.1 mM MEM nonessential amino acid solution, 2 mM L-glutamine and 1% penicillin/streptomycin. Also, we induced hMSCs from iPSCs using STEMdiff Mesenchymal Progenitor kit (STEMCELL technologies, Seattle, WA, USA) and defined them as 520d/MSC after they were transfected by miR-520d-5p.
Lentiviral vector construct
To examine the effects of miR-520d-5p overexpression, we transfected pMIRNA1-miR-520d-5p/GFP (20 μg; System Biosciences, Mountain View, CA, USA) or the mock vector pCDH (20 μg) into 293FT and HLF cells (5 × 106 cells/10 cm culture dish). To investigate the HLF cells and hMSCs with miR-520d-5p expression, we harvested viral particles produced in the medium cultured 293FT cells, the cells were centrifuged at 170,000 x g (120 min, 4 °C). The viral pellets were collected, and viral copy numbers were measured with a Lenti-X™ qRT-PCR Titration kit (Clontech, Mountain View, CA, USA). For 293FT or HLF cell infection, one million lentiviral copies were used per 10-cm culture dish. We transfected 50 nM synthesized oligonucleotides into 293FT cells with FuGENE HD Transfection Reagent (Roche Diagnostics, Basel, Switzerland). pCDH/lenti/GFP-treated cells were used as controls. 520d-5p-transfected hMSCs were generated by the lentiviral transfection of 520d-5p to hMSCs after the differentiation induction from iPSCs to hMSCs using hPSC-Derived MSC differentiation system and reagents (Veritas Corporation, Tokyo, Japan).
Next-generation sequencing (NGS) analysis
The following cell lines were used in NGS analysis: HLF, mock/HLF, 520d/HLF [3 days after transfection (3D) and 5 days after transfection (5D)], 520d/HLF [7 days after transfection (7D)], 520d/HLF (R1 and R2), sorted with both GFP and alkaline phosphatase (ALP), iPSC (253G1), 520d-transfected and 253G1-derived progenitor hMSC (520d/hMSC), DNA extraction was performed using Qiagen DNeasy kit according to manufactures instruction (QIAGEN, Tokyo, Japan). Exome Sequencing with a Next-Generation Sequencer. Three replicates per group were analyzed (n = 3). Genomic DNA was processed using the SureSelectXT Human All Exon v5 + UTRs (Agilent Technologies, Inc.), and sequenced on the Illumina HiSeq 2500 platform with 101 bp paired-end reads (HSS, Sapporo, Japan). The paired-end reads in FastQ format were mapped to reference genome HG19 using BWA-0.7.10. The mapping files in SAM (Sequence Alignment/Map) format were converted to BAM (binary version of SAM) format and sorted by SAMtools-1.2. Local realignment around known indels was performed by GATK-Lite-2.3.0 on the sorted BAM files. Picardtools-1.133 [22, 23] was used to remove PCR duplicates. Finally, base quality score recalibration was performed using GATK again. RNA Sequencing with a Next-Generation Sequencer. Total RNA was processed using the TruSeq Stranded mRNA Library Prep Kit (Illumina), and sequenced on the Illumina HiSeq 2500 platform with 101 bp paired-end reads. The paired-end reads in FastQ format were mapped to reference genome HG19 using TopHat.
Mutation analysis
Samtools mpileup [options: -d 10000 -L 10000 -B -t DP, DV, SP, DP4, DPR] was piped with bcftools call [options: -A -v -m -f GQ] to produce variant call format (VCF) files. VCF files were further filtered to select variants that had at least two reads supporting the variants and that showed a minimum allele frequency of 0.8 and a maximum read depth of 35 at called sites. For high-throughput, deep sequencing of selected cDNA such as Tp53 were subjected to Sanger sequencing to screen for nonsynonymous mutations [24–26].
RT2 PCR array analysis
To examine epigenetics-related genes that 520d-5p may regulate, PCR array analysis was performed using RT2 profiler PCR array (real-time PCR technology), according to manufacturers’ instruction (Qiagen Japan, Tokyo, Japan).
Results
Mutation analysis in HLF, mock/HLF, 3 days after transfection (3D), 5 days after transfection (5D), 7 days after transfection (7D), R1 (see ref. [17]), R2 (see ref. [17]), hMSC, 520d/hMSC or hiPSC showed that 520d-5p gradually decreased the total number of mutations, including point mutations, two-nucleotide mutations and three-nucleotide mutations (Figs. 1, 2). Mutations in genomic fragment assemblies were reduced more prominently than mutations in chromosomes assemblies (Figs. 1, 2 and Table 1). Four sites in chromosomes are shown in Fig. 1, and two sites in fragmented genomes are shown in Figs. 2 and 3. Furthermore, when we examined the frequency of genomic alterations in each chromosome, the reduction in mutation frequency was most prominently observed in chromosome X (Table 2). Interestingly, the increased number of frequencies seemed to be maintained with the reduction in number, except for in chromosome X.
Fig. 1.

A frequency of allele changes per chromosome is shown. Mutation analysis in HLF, mock/HLF, 3D, 5D, 7D, R1, R2, hMSC, hiPSC by NGS. This area graph showed that the total number of mutations were getting reduced in HLF derivatives regarding a frequency of allele changes per chromosome. ★ indicates the representative sites in Fig. 3
Fig. 2.

A frequency of allele changes of fragmented genomic sequence in which information was not decided on chromosome. Mutation analysis in HLF, mock/HLF, 3D, 5D, 7D, R1, R2, hMSC, hiPSC regarding a frequency of allele changes of fragmented genomic sequence by NGS. This area graph showed that the total number of mutations in genomic fragment assembly was reduced prominently. ★ indicates the representative sites in Fig. 3
Table 1.
Details for genomic fragments that information was not decided on chromosome and shown at the bottom of the area graph in Fig. 2
| MT (1–65) | |
| GL00026 (66–90) | |
| GL000229 (91–108) | |
| GL000231 (109–126) | |
| GL000239 (127–129) | |
| GL000201 (130–131) | |
| GL000246 (132) | |
| GL000249 (133) | |
| GL000238 (134–138) | |
| GL000234 (139–160) | |
| GL000232 (161–163) | |
| GL000240 (164–165) | |
| GL000241 (166–180) | |
| GL000243 (181–183) | |
| GL000230 (184–250) | |
| GL000237 (251–385) | |
| GL000204 (386) | |
| GL000198 (387–394) | |
| GL000208 (395–402) | |
| GL000191 (403–414) | |
| GL000228 (415–420) | |
| GL000214 (421–566) | |
| GL000221 (567–573) | |
| GL000209 (574–643) | |
| GL000218 (644–722) | |
| GL000220 (723–1345) | |
| GL000213 (1346–1347) | |
| GL000211 (1348) | |
| GL000199 (1349–1356) | |
| GL000217 (1357–1365) | |
| GL000216 (1366–1395) | |
| GL000205 (1396–1547) | |
| GL000219 (1548–1614) | |
| GL000224 (1615–1729) | |
| GL000223 (1730–1746) | |
| GL000195 (1747–1966) | |
| GL000212 (1967–2107) | |
| GL000222 (2108–2124) | |
| GL000193 (2125–2146) | |
| GL000194 (2147–2186) | |
| GL000225 (2187–2267) | |
| GL000192 (2268) |
Fig. 3.
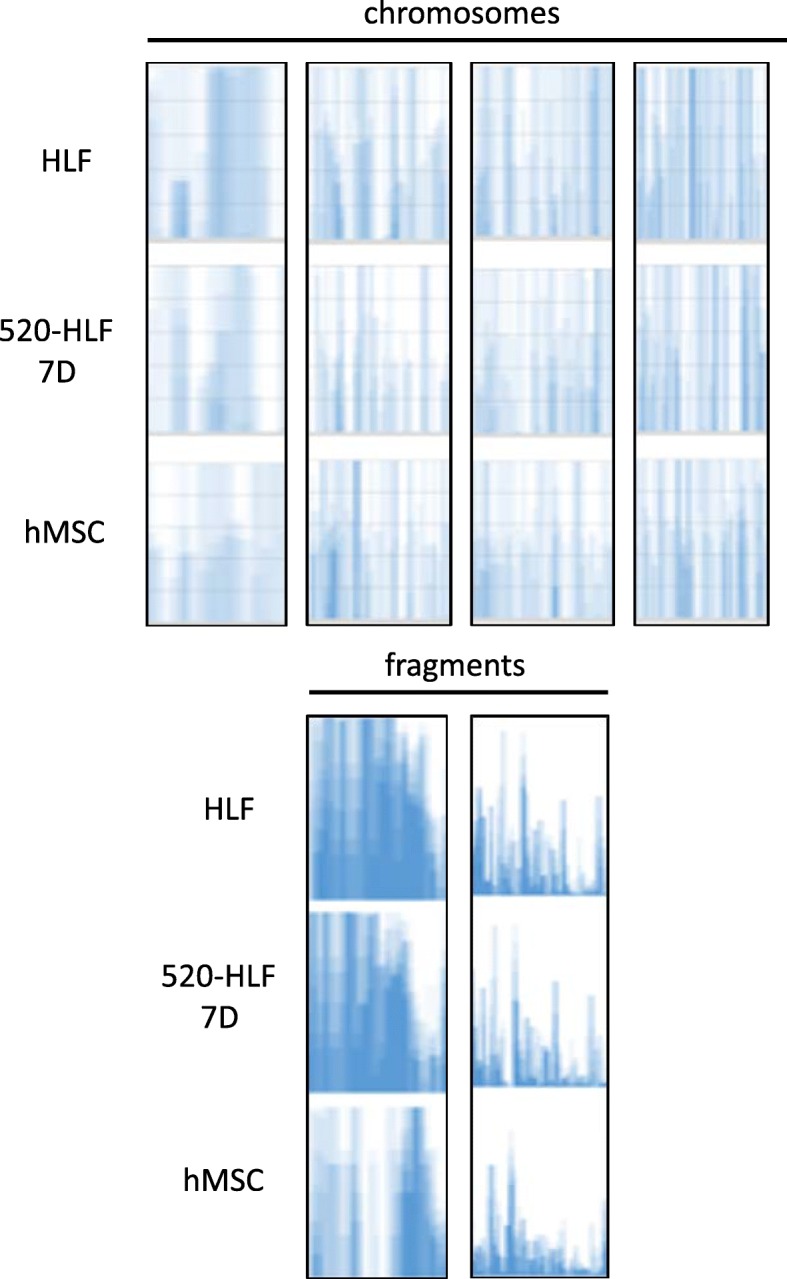
To show the genomic changes, 4 sites (upper) in Figs. 1 and 2 sites (bottom) in Fig. 2 were chosen and zoomed up in representative cells (HLF, 520d-5p 7D, and hMSC). The changes in genomic fragments were less than those of chromosomes. The chosen sites are shown as ★ in Figs. 1 and 2
Table 2.
The change (%) of frequency number in genomic alterations on each chromosome by 520d-5p between mock/HLF and 7D-transfectants
| Status of frequency | chr. 1 | chr. 2 | chr. 3 | chr. 4 | chr. 5 | chr. 6 | chr. 7 | chr. 8 |
| reduction | 8.5 | 9.4 | 10.8 | 7.2 | 5.4 | 14.5 | 3.7 | 10.4 |
| no changes | 83.2 | 81.3 | 78.8 | 85.6 | 89.4 | 71.4 | 92.7 | 79.6 |
| gain | 8.3 | 9.3 | 10.4 | 7.2 | 5.2 | 14.1 | 3.6 | 10.0 |
| Status of frequency | chr. 9 | chr. 10 | chr. 11 | chr. 12 | chr. 13 | chr. 14 | chr. 15 | chr. 16 |
| reduction | 3.9 | 11.4 | 9.7 | 11.0 | 3.1 | 4.8 | 4.4 | 12.5 |
| no changes | 92.3 | 76.9 | 80.5 | 77.9 | 93.0 | 90.5 | 91.4 | 74.7 |
| gain | 3.8 | 11.7 | 9.8 | 11.1 | 3.9 | 4.7 | 4.2 | 12.8 |
| Status of frequency | chr. 17 | chr. 18 | chr. 19 | chr. 20 | chr. 21 | chr. 22 | chr. 23 | chr. X |
| reduction | 9.3 | 11.0 | 4.4 | 11.6 | 20.3 | 12.5 | 3.8 | 30.7 |
| no changes | 82.1 | 79.0 | 91.7 | 77.0 | 59.7 | 75.2 | 92.6 | 43.8 |
| gain | 8.6 | 10.0 | 3.9 | 11.4 | 20.0 | 12.3 | 3.6 | 25.5 |
Conversions to wild-type nucleotides were observed in many genes. For example, NGS identified that the Rb1 gene had four exon mutations. In two of the mutations, 520d-5p transfection caused base switching by 5D, and a base in the original site was wild type at 7D. The left and right bases were converted from T to C and C to T, respectively. The base conversion effect on three sites from 49,002,200 bp to 49,002,400 bp by lentiviral induction was reversible, and the mutations resolved after 7D (Fig. 4). Also, lysine acetyltransferase 1 (Kat8) had two mutations in HLF, but nucleotide mutations at two locations in Kat8 (location: chr16:31,126,985-31,44,714) were reverted to wild type nucleotides by 520d-5p (Fig. 5). The nucleotide at location (31,188,324 in reference) in Kat8 converted thymine in HLF and mock/HLF to adenine in 3D, 5D, 7D, R1, R2, and hiPSC. Also, Tp53 had eleven mutations in HLF, and five of eleven point mutations were converted to wild type (Fig. 6a), while six mutation sites were not reverted to wild type (no conversions: N.C.) (Fig. 6b). The cell cycle regulator Rb1 had four mutations in HLF and two of four mutations were converted to wild type (Fig. 4). Representative conversions of mutations in Kat8/Myst1 (91,134,500 bp-91,135,000 bp on chr16 p11.2) are shown in Additional file 1: Figure S1. Allelic changes in representative genes related to DNA methylation, histone modification, DNA repair, oncogenesis, tumor suppression, stemness, and chromatin remodeling, and known target genes of 520d-5p, are shown in Additional file 2: Tables S1-S7.
Fig. 4.

Representative conversion of RB1 after 520d-5p transfection. HLF originally had possible mutated regions at two sites, and alterations in 5 sites could be seen after transfection by 520d-5p. However, transfectants in 7D did not have any of them there. Conversion could be observed and maintained in R1 and R2 (sorted transfectants)
Fig. 5.

Representative conversion in Kat8 after 520d-5p transfection. Kat8 had two point mutations in HLF in RNA-seq, but this region had a conversion to wild type nucleotide since 3D after transfection by 520d-5p (left rectangle). All samples had the same and unchangeable nucleotide status in HLF to hiPSC (right rectangle). This figure was drawn based on the alignment data of RNA-Seq analysis
Fig. 6.

Analysis regarding the effect of 520d-5p on Tp53 in mutated sites. Eleven mutated regions were found. a Five converted mutations to wild type status although the definite rule of conversion was not confirmed. b Remaining sites had no effects on mutation status in HLF and transfectants (N.C.). This figure was drawn based on the alignment data of RNA-Seq analysis. *means a mutated region
We were unable to detect a common mechanistic rule for conversion of mutations to wild type nucleotides. Mutations in 520d/hMSC derived from hiPSCs were clearly less than those of hMSCs (Fig. 7a), independent of individual differences. Approximately 60 % (60.3%; 301,733 out of 500,182) of nucleotide changes (allele frequency) in hiPSCs were reduced in hMSCs (520d/hMSC) both induced from hiPSCs and transfected by 520d-5p. 11.7% of mutations (58,519 out of 301,733) increased in 520d/hMSC (Fig. 7b). However, 38.6% of mutations (22,561/58519) of the change of allelic frequency in 520d/hMSC appeared to be nucleotide changes secondary to differentiation to hMSCs, as the 0.5 to 1.5 group indicated similar to allele frequency in hMSCs in volatility (Fig. 7c). The ratio of allele frequency was calculated by (520d/hMSC induced from hiPSCs/hMSC). Allelic changes in representative genes related to stemness are shown in Additional file 2: Table S7. Kras, Cmyc, or Bcl2 in iPSC-derivatives had no changes in individual genes. P21, Tp53, and Braf had diminished resolution of mutated regions (Additional file 1: Figure S2-a, c, d). Tp53 and Stat3 (a target gene of 520d-5p) had alterations accompanied with differentiation (Additional file 1: Figure S2b). Additionally, mutations in two genes involved in DNA repair (Abl2 and Atr) were decreased by 520d-5p transfection (Additional file 1: Figure S3).
Fig. 7.

a ALT1 allele frequency in hiPSC, 520d/progenitor hMSC, or hMSC. hiPSC had non-negligible number of mutated sites, but the total number of mutations were reduced and some of them were converted to wild type during the differentiation process after 520d-5p transfection. b Circle graph showing allelic status induced by 520d-5p transfection. The decrease of ALT1 allele frequency was observed in 60.3% (301,733/500,189) of mutated sites. 11.1% (58,519/500,189) gained the new mutated regions. c. Scatter plot showing alterations of nucleotides during the differentiation towards hMSC status in the increased mutations (585,199 sites in 2)). Ratio of allele frequency 520d-5p-transfected hMSC progenitor cells / normal hMSC) was estimated. A large parts of alteration of increased sites in progenitor cells seemed to be an alteration accompanied by the differentiation process towards hMSC status according to the standardization to nucleotides status observed in hMSC
PCR array analysis of epigenetics-related genes in 520d/HLFs (5D, 7D) and hiPSCs revealed that only two genes (Hat1/Kat1 and Myst1/Kat8) were commonly downregulated by 520d-5p transfection (Additional file 1: Figure S4).
Discussion
Previously, we reported that 520d-5p induces transformation of undifferentiated cancer cells into non-malignant status [17]. We have subsequently sought to determine the mechanism for this transformation. On the other hand, earlier studies concluded that cancer cell mutations do not revert to wild type status, and that cancer cell mutations are unaltered by anti-cancer agents or other factors, although one prior study suggested that BRCA mutations in triple-negative breast cancer correlate with miRNA expression [27, 28]. This study is the first to comprehensively demonstrate that genomic alterations or mutations reversibly changed to wild type. Here we demonstrated 520d-5p modulated cancer cell transformation from a malignant to benign phenotype in vivo using immunodeficient mice. This transformation was induced in the presence of genomic level mutations including exons, introns, and non-coding lesions.
Prior reports demonstrated that 520d-5p directly targets the 3’UTRs of eleven genes, including Elavl2, Tead1, Gatad2b, Atm, Casp3, Tada3, Stat3, Twist1, Sirpa, Sp1 and Cthrc1, and more than 9000 genes are predicted targets of 520d-5p [17, 19, 20, 29–31].
Together, current and previous finding demonstrate that 520d-5p has comprehensive effects on DNA and RNA demethylation, indicating that synergistic epigenetic alterations may be implicated in the cellular conversion phenomena via this reprogramming process. In the present study, we found that mutations greater than two-base alterations were not observed in undifferentiated hepatoma cells or iPSC derivatives expressing 520d-5p, suggesting that the effect of this miRNA is limited to one-base conversions, and appears to most prominently affect chromosome X (Fig. 8). However, we were unable to detect any specific rule for base conversion, for example T in reference to C in the samples, or C to T. Mutations and conversion status in genes related to DNA methylation, histone modification, DNA repair, oncogenesis, stemness, as well as chromatin remodeling and 520d-5p target genes and tumor suppressor genes, are summarized in Additional file 2: Tables S1-S7. However, the mechanism for these conversions remains unclear. We presume that the in-depth mechanism includes a synergistic process by RNA demethylation and methyltransferase accompanied with mutation reductions, independent of nucleotide type, as we suggested previously (17–20).
Fig. 8.

The improvement to wild type of ALT1 in HLF by the transfection of miR-520d-5p A bar graph showed that 520d-5p induced some of the ALT1 in transfectants, to wild type status
In comparison between hiPSCs and 520d-5p-transfected hMSCs induced from hiPSCs, we are interested in the effect of 520d-5p on hiPSCs that are not completely initialized. We attempted to differentiate hiPSCs towards hMSCs with four transfections of 520d-5p. As the result, we found that mutations in hiPSCs derived from original cells could be altered (Fig. 7a). These mutations had a dominant reduction (Fig. 7a, b), although new allelic alterations involved in the differentiation from hiPSC to hMSC increased (Fig. 7c). Because we identified that 520d-5p induced demethylation in HLF in previous studies [17, 19], we comprehensively examined epigenetics-related genes using a PCR array and identified indirect downregulation of Hat1 and Kat8, which are lysine acetyltransferases [32–35]. However, the downstream functions of 520d-5p remain incompletely understood. To gain further insights and validate that our results are reliable in future studies, we will use additional approaches such as capillary sequencing, as there are fewer reading sites in the mutated alignments of other genes.
Conclusions
These findings suggest that in cancer cells, malignant properties can be reprogrammed and reversed by induction of nucleotide conversion. These findings also suggest that 520d-5p has potential in regenerative medicine, as transfection improved the quality of hiPSCs.
Additional files
Figure S1. Representative conversion in Kat8 after 520d-5p transfection. Figure S2. Comparative NGS analysis between hiPSC, 520d/hMSC progenitor cells, or hMSC. Figure S3. Comparative NGS analysis between hiPSC, 520d/hMSC progenitor cells, or hMSC regarding representative DNA repair genes. Figure S4. PCR array using RT2 Profiler PCR array system regarding epigenetics-related genes in 520d/HLF (5D, 7D) and hiPSC. (PDF 835 kb)
Summary of nucleotides alterations in respective genes regarding of our interest (Tables S1–S7). (DOCX 26 kb)
Acknowledgements
We all deeply thank Noriko Itaba, Ph.D. in Tottori University who supported for hiPSC differentiation procedure. The cell lines used in this study were provided by the Cell Resource Center for Biomedical Research, Institute of Development, Aging and Cancer, Tohoku University, Japan, and the American Type Culture Collection (ATCC).
Data availability
The authors declare that all other data supporting the findings of this study are available within the article and its Supplementary Information Files.
Additional files
Abbreviations
- 520d-5p
Hsa-miR-520d-5p
- hiPSC
Human induced pluripotent stem cell
- hMSC
Human mesenchymal stem cell
- miRNA
MicroRNA
- NGS
Next generation sequencing
Authors’ contributions
The author(s) have made the following declarations regarding their contributions: NM and KM. conceived and designed the experiments. YI and KM prepared the samples for NGS. MK, YI, KM, and YM performed the experiments and analyzed the data. MK, NM and KM approved the final manuscript. All authors read and approved the final manuscript.
Funding
This work (cell culture and NGS analysis) was supported by a Grant-in-Aid of Research for Promoting Technological Seeds B (development type) and a Visionary Research of the Takeda Science Foundation, respectively.
Availability of data and materials
The datasets generated for this study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
All authors are aware of the consent and agree with the submission. The authors declare no conflict of interest or competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 2.Daugaard I, Hansen TB. Biogenesis and function of ago-associated RNAs. Trends Genet. 2017;33:208–219. doi: 10.1016/j.tig.2017.01.003. [DOI] [PubMed] [Google Scholar]
- 3.Vishnoi A, Rani S. MiRNA Biogenesis and Regulation of Diseases: An Overview. Methods Mol Biol. 2017;1509:1–10. doi: 10.1007/978-1-4939-6524-3_1. [DOI] [PubMed] [Google Scholar]
- 4.Ciardiello C, Cavallini L, Spinelli C, Yang J, Reis-Sobreiro M, de Candia P, Minciacchi VR, Di Vizio D. Focus on Extracellular Vesicles: New Frontiers of Cell-to-Cell Communication in Cancer. Int J Mol Sci. 2016;17:175. doi: 10.3390/ijms17020175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biswas S, Rao CM. Epigenetics in cancer: fundamentals and beyond. Pharmacol Ther. 2017;173:118–134. doi: 10.1016/j.pharmthera.2017.02.011. [DOI] [PubMed] [Google Scholar]
- 6.Wang S, Wu W, Claret FX. Mutual regulation of microRNAs and DNA methylation in human cancers. Epigenetics. 2017;12:187–197. doi: 10.1080/15592294.2016.1273308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saito Y, Jones PA. Epigenetic activation of tumor suppressor microRNAs in human cancer cells. Cell Cycle. 2006;5:2220–2222. doi: 10.4161/cc.5.19.3340. [DOI] [PubMed] [Google Scholar]
- 8.Sachdeva M, Zhu S, Wu F, Wu H, Walia V, Kumar S, Elble R, Watabe K, Mo YY. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc Natl Acad Sci U S A. 2009;106:3207–3212. doi: 10.1073/pnas.0808042106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dykxhoorn DM. MicroRNAs and metastasis: little RNAs go a long way. Cancer Res. 2010;70:6401–6406. doi: 10.1158/0008-5472.CAN-10-1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takei Y, Takigahira M, Mihara K, Tarumi Y, Yanagihara K. The metastasis-associated microRNA miR-516a-3p is a novel therapeutic target for inhibiting peritoneal dissemination of human scirrhous gastric cancer. Cancer Res. 2011;71:1442–1453. doi: 10.1158/0008-5472.CAN-10-2530. [DOI] [PubMed] [Google Scholar]
- 11.Nielsen R, Paul JS, Albrechtsen A, Song YS. Genotype and SNP calling from next-generation sequencing data. Nat Rev Genet. 2011;12:443–451. doi: 10.1038/nrg2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marziali A, Akeson M. New DNA sequencing methods. Annu Rev Biomed Eng. 2001;3:195–223. doi: 10.1146/annurev.bioeng.3.1.195. [DOI] [PubMed] [Google Scholar]
- 13.Ståhlberg A, Krzyzanowski PM, Egyud M, Filges S, Stein L, Godfrey TE. Simple multiplexed PCR-based barcoding of DNA for ultrasensitive mutation detection by next-generation sequencing. Nat Protoc. 2017;12:664–682. doi: 10.1038/nprot.2017.006. [DOI] [PubMed] [Google Scholar]
- 14.Ruffalo M, LaFramboise T, Koyutürk M. Comparative analysis of algorithms for next-generation sequencing read alignment. Bioinformatics. 2011;27:2790–2796. doi: 10.1093/bioinformatics/btr477. [DOI] [PubMed] [Google Scholar]
- 15.Pattnaik S, Vaidyanathan S, Pooja DG, Deepak S, Panda B. Customisation of the exome data analysis pipeline using a combinatorial approach. PLoS One. 2012;7:e30080. doi: 10.1371/journal.pone.0030080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bao S, Jiang R, Kwan W, Wang B, Ma X, Song YQ. Evaluation of next-generation sequencing software in mapping and assembly. J Hum Genet. 2011;56:687. doi: 10.1038/jhg.2011.91. [DOI] [PubMed] [Google Scholar]
- 17.Tsuno S, Wang X, Shomori K, Hasegawa J, Miura N. Hsa-miR-520d induces hepatoma cells to form normal liver tissues via a stemness-mediated process. Sci Rep. 2014;24:3852. doi: 10.1038/srep03852. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Ishihara Y, Tsuno S, Kuwamoto S, Yamashita T, Endo Y, Miura K, Miura Y, Sato T, Hasegawa J, Miura N. Tumor-suppressive effects of atelocollagen-conjugated hsa-miR-520d-5p on un-differentiated cancer cells in a mouse xenograft model. BMC Cancer. 2016;16:415. doi: 10.1186/s12885-016-2467-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishihara Y, Tsuno S, Kuwamoto S, Yamashita T, Endo Y, Hasegawa J, Miura N. Hsa-miR-520d converts fibroblasts into CD105+ populations. Drugs RD. 2014;14:253–264. doi: 10.1007/s40268-014-0064-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishihara Y, Tsuno S, Ping B, Ashizaki T, Nakashima M, Miura K, Miura Y, Yamashita T, Hasegawa J, Miura N. Hsa-miR-520d-5p promotes survival in human dermal fibroblasts exposed to a lethal dose of UV irradiation. npj Aging Mech Dis. 2016;2:16029. doi: 10.1038/npjamd.2016.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoi T, Okita K, Mochiduki Y, Takizawa N, Yamanaka S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 2008;26:101–106. doi: 10.1038/nbt1374. [DOI] [PubMed] [Google Scholar]
- 22.Nielsen R, Paul JS, Albrechtsen A, Song YS. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 2011;12:443–451. doi: 10.1038/nrg2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wysoker A, Tibbetts K, Fennell T. picardTools 1.5. 2011. [Google Scholar]
- 24.Falk MJ, Pierce EA, Consugar M, Xie MH, Guadalupe M, Hardy O, Rappaport EF, Wallace DC, LeProust E, Gai X. Mitochondrial disease genetic diagnostics: optimized whole-exome analysis for all MitoCarta nuclear genes and the mitochondrial genome. Discov Med. 2012;14:389–399. [PMC free article] [PubMed] [Google Scholar]
- 25.Kawazu M, Ueno T, Kontani K, Ogita Y, Ando M, Fukumura K, Yamato A, Soda M, Takeuchi K, Miki Y, et al. Transforming mutations of RAC guanosine triphosphatases in human cancers. Proc Natl Acad Sci U S A. 2013;110:3029–3034. doi: 10.1073/pnas.1216141110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takahashi Y, Mori J, Kami M. BRAF mutations in hairy-cell leukemia. N Engl J Med. 2011;365:960–961. doi: 10.1056/NEJMc1108130. [DOI] [PubMed] [Google Scholar]
- 27.Erturk E, Cecener G, Tezcan G, Egeli U, Tunca B, Gokgoz S, Tolunay S, Tasdelen I. BRCA mutations cause reduction in miR-200c expression in triple negative breast cancer. Gene. 2015;556:163–169. doi: 10.1016/j.gene.2014.11.047. [DOI] [PubMed] [Google Scholar]
- 28.Afghahi A, Timms KM, Vinayak S, Jensen KC, Kurian AW, Carlson RW, Chang PJ, Schackmann E, Hartman AR, Ford JM, et al. Tumor BRCA1 Reversion Mutation Arising During Neoadjuvant Platinum-Based Chemotherapy in Triple-Negative Breast Cancer Is Associated with Therapy Resistance. Clin Cancer Res. 2017;23:3365–3370. doi: 10.1158/1078-0432.CCR-16-2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li T, Guo H, Zhao X, Jin J, Zhang L, Li H, Lu Y, Nie Y, Wu K, Shi Y, et al. Gastric Cancer Cell Proliferation and Survival Is Enabled by a Cyclophilin B/STAT3/miR-520d-5p Signaling Feedback Loop. Cancer Res. 2017;77:1227–1240. doi: 10.1158/0008-5472.CAN-16-0357. [DOI] [PubMed] [Google Scholar]
- 30.Tsukerman P, Yamin R, Seidel E, Khawaled S, Schmiedel D, Bar-Mag T, Mandelboim O. MiR-520d-5p directly targets TWIST1 and downregulates the metastamiR miR-10b. Oncotarget. 2014;5:12141–12150. doi: 10.18632/oncotarget.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan L, Yu J, Tan F, Ye GT, Shen ZY, Liu H, Zhang Y, Wang JF, Zhu XJ, Li GX. SP1-mediated microRNA-520d-5p suppresses tumor growth and metastasis in colorectal cancer by targeting CTHRC1. Am J Cancer Res. 2015;5:1447–1459. [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang S, Liu X, Zhang Y, Cheng Y, Li Y. RNAi screening identifies KAT8 as a key molecule important for cancer cell survival. Int J Clin Exp Pathol. 2013;6:870–877. [PMC free article] [PubMed] [Google Scholar]
- 33.Xue L, Hou J, Wang Q, Yao L, Xu S, Ge D. RNAi screening identifies HAT1 as a potential drug target in esophageal squamous cell carcinoma. Int J Clin Exp Pathol. 2014;7:3898–3907. [PMC free article] [PubMed] [Google Scholar]
- 34.Loidl P. Histone acetylation: facts and questions. Chromosoma. 1994;103:441–449. doi: 10.1007/BF00337382. [DOI] [PubMed] [Google Scholar]
- 35.Franciosi F, Goudet G, Tessaro I, Papillier P, Dalbies-Tran R, Reigner F, Deleuze S, Douet C, Miclea I, Lodde V, et al. In vitro maturation affects chromosome segregation, spindle morphology and acetylation of lysine 16 on histone H4 in horse oocytes. Reprod Fertil Dev. 2017;29:721–730. doi: 10.1071/RD15350. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Representative conversion in Kat8 after 520d-5p transfection. Figure S2. Comparative NGS analysis between hiPSC, 520d/hMSC progenitor cells, or hMSC. Figure S3. Comparative NGS analysis between hiPSC, 520d/hMSC progenitor cells, or hMSC regarding representative DNA repair genes. Figure S4. PCR array using RT2 Profiler PCR array system regarding epigenetics-related genes in 520d/HLF (5D, 7D) and hiPSC. (PDF 835 kb)
Summary of nucleotides alterations in respective genes regarding of our interest (Tables S1–S7). (DOCX 26 kb)
Data Availability Statement
The authors declare that all other data supporting the findings of this study are available within the article and its Supplementary Information Files.
Additional files
The datasets generated for this study are available from the corresponding author on reasonable request.