

Abstract
Peptide cleanup is essential for the removal of contaminating substances that may be introduced during sample preparation steps in bottom-up proteomic workflows. Recent studies have described benefits of carboxylate-modified paramagnetic particles over traditional reversed-phase methods for detergent and polymer removal, but challenges with reproducibility have limited the widespread implementation of this approach among laboratories. To overcome these challenges, the current study systematically evaluated key experimental parameters regarding the use of carboxylate-modified paramagnetic particles and determined those that are critical for maximum performance and peptide recovery and those for which the protocol is tolerant to deviation. These results supported the development of a detailed, easy-to-use standard operating protocol, termed SP2, which can be applied to remove detergents and polymers from peptide samples while concentrating the sample in solvent that is directly compatible with typical LC-MS workflows. We demonstrate that SP2 can be applied to phosphopeptides and glycopeptides, and that the approach is compatible with robotic liquid handling for automated sample processing. Altogether, the results of this study and accompanying detailed operating protocols for both manual and automated processing are expected to facilitate reproducible implementation of SP2 for various proteomics applications and will especially benefit core or shared resource facilities where unknown or unexpected contaminants may be particularly problematic.
Keywords: peptide cleanup, contaminant removal, LC-MS/MS, bottom-up proteomics, glycopeptides, phosphopeptides, TMT
Graphical Abstract
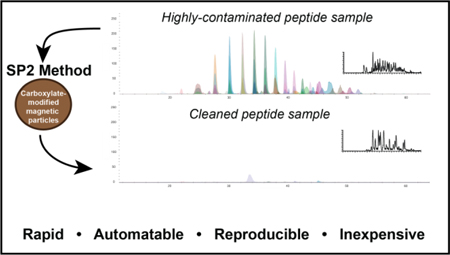
Introduction
In bottom-up proteomic workflows, peptide cleanup is essential for the removal of expected and unexpected contaminating substances that are introduced during sample preparation steps. Common contaminants include protein solubilizing agents (e.g. detergents and chaotropes) and polymeric contaminants from sample processing workflows (e.g. molecular weight cutoff filters and sample enrichment particles). If they remain in the sample during liquid chromatography mass spectrometry (LC-MS) analysis, these compounds unfavorably affect data quality, HPLC column integrity, and lead to deposition of contaminants on instrument components that adversely affect performance for subsequently analyzed samples.
While on-line cleanup methods have been described (1, 2), off-line cleanup strategies can permit more flexibility in sample preparation and do not require specialized instrument configurations. Off-line methods that can be performed at the protein level include protein precipitation to eliminate some contaminants from the protein sample prior to digestion, but the resulting protein pellet can be challenging to re-dissolve and digest, which can contribute to sample loss. The use of reversed-phase liquid chromatography C18 resin (C18) is a popular choice for peptide cleanup as this method is effective for removing salts and concentrating peptides and is available in a wide variety of easy-to-use formats (e.g. Stage-Tips, Sep-Pak Cartridges, Micro SpinColumns) (3). However, C18 will concentrate, rather than remove, polymeric species such as polyethyleneglycol (PEG) and common detergents (e.g. NP-40, SDS, Triton X). Moreover, because peptides are eluted from the C18 stationary phase in a high percentage of organic mobile phase (e.g. 60–80% acetonitrile), the eluent is commonly dried under vacuum to remove the organic content prior to injection into an LC-MS instrument, a step that adds time and can contribute to sample loss.
Alternative approaches to reversed-phase chromatography include normal-phase, where peptides are retained on the stationary phase in organic and eluted in aqueous mobile phase, and ion exchange, where peptides can be separated based on their charge. Popular normal-phase methods - which can be used in place of, or orthogonally to, reversed-phase methods -include strong cationic exchange, hydrophilic interaction chromatography (HILIC), and electrostatic repulsion-hydrophilic interaction chromatography (ERLIC) (4, 5, 6). In addition to these, the use of mixed mode chromatography for peptide cleanup in proteomic workflows has recently gained popularity. A report by Hughes et al., described Single-Pot Solid-Phase-enhanced Sample Preparation (SP3), an approach that utilizes HILIC- and ERLIC-like interactions with carboxylate-modified paramagnetic particles to perform protein capture, tryptic digestion, and peptide cleanup within a single sample tube (7). Advantages of the SP3 protocol include the suitability for processing small sample sizes, the ability to remove MS-incompatible detergents, and the capacity to elute in an aqueous mobile phase that is directly compatible with LC-MS analysis.
Despite its potential, the broad implementation of SP3 has been limited, and this may be due, in part, to challenges in reproducing the published method in other laboratories (personal communications and (8)). Acknowledging these challenges, the authors of the SP3 protocol sought to further clarify the operating parameters around the protein capture aspect of SP3 (9). However, as a C18 method was used for peptide cleanup in this most recent SP3 study, it does not address the challenges in implementing the use of carboxylate-modified particles for routine peptide cleanup. While other studies have made use of carboxylate-modified particles for peptide cleanup, notably Larkin et al. for the removal of unidentified impurities observed after C18 (10, 11), experimental details are not provided for this aspect. Moreover, while the carboxylate-modified particles used in SP3 are paramagnetic, and thus theoretically amenable to automation, a detailed evaluation of their suitability in this context has not been described. Altogether, although the use of carboxylate-modified paramagnetic particles for peptide cleanup has significant potential advantages over C18 methods, a comprehensive evaluation of key experimental details critical for successful implementation of manual and automated processing has not yet been described.
To address this gap, the current study aimed to systematically evaluate experimental parameters regarding the use of carboxylate-modified paramagnetic particles for peptide cleanup and to develop a detailed, easy-to-use operating protocol to promote reproducible implementation of this method among laboratories. This study focused only on peptide cleanup, as for many laboratories and experimental designs, it may not be practical or appropriate to implement the full SP3 protocol, which includes a step to capture protein on the carboxylate-modified particles. By evaluating more than 15 experimental variables, this study determined which parameters are most critical for maximizing peptide recovery and those for which the protocol is tolerant to deviation. To our knowledge, our study is the first to show that carboxylate-modified paramagnetic particles can be applied to phosphopeptides and glycopeptides and is the first to demonstrate the utility of this approach in an automated processing workflow using a liquid handling workstation. Based on these observations, we developed a detailed standard operating protocol for the routine application of carboxylate-modified paramagnetic particles for peptide cleanup. We have termed this protocol SP2. While SP2 is conceptually similar to SP3 in that the stationary phase is the same, there is no requirement for protein capture on the particles for SP2 and there are important specifications related to peptide binding and elution. The results of this study and accompanying detailed operating protocols for both manual and automated processing are expected to facilitate implementation of SP2 for various proteomics applications and will especially benefit core or shared resource facilities where unknown or unexpected contaminants may be particularly problematic.
Methods
Cell culture
HEK-293T cells were cultured at 37 °C with 5% CO2 in DMEM with 10% FBS (v/v), 1% GlutaMAX (v/v), and 1x Penn/Strep. Cells were passaged at 80% confluency using 2 min incubation with TrypLE to dissociate cells and a 1:8 split ratio.
Peptide Sample Preparation from Cell Lysate and Recombinant Sources
All experimental details are provided in Supporting Information. In brief, one 10 cm plate of HEK-293T cells was collected and subsequently digested in 50 mM Ammonium Bicarbonate (AmBic) containing 2x Invitrosol (40% v/v; Thermo Fisher Scientific, Waltham, MA). For glycopeptide samples, trypsin digested human thrombospondin-1, alpha-1-acid glycoprotein and transferrin (Sigma Aldrich, St. Louis, MO) were combined in an equimolar ratio and cleaned by either SP2 or C18. For phosphopeptide samples, HEK-293T cells were lysed in 100 mM AmBic containing 20% MeCN, 2x Invitrosol (40% v/v; Thermo Fisher Scientific, Waltham, MA), and 1% Phosphatase Inhibitor Cocktail 1 (Sigma Aldrich). Following tryptic digestion, peptides were cleaned by either SP2 or C18 and subsequently phosphopeptides were enriched sequentially using High-Select TiO2 and High-Select Fe-NTA phosphopeptide enrichment kits, according to the manufacturer’s instruction (Thermo Fisher Scientific).
Peptide Quantitation
Peptides, pre- and post-cleanup, were quantified using Pierce Quantitative Fluorometric Peptide Assay (Thermo Fisher Scientific) according to the manufacturer’s instructions on a Varioskan LUX Multimode Microplate Reader using SkanIt 5.0 software (Thermo Fisher Scientific).
Peptide Cleanup
All conditions were tested in technical triplicate and performed in a 1.7 mL low-binding snap-cap tube (Thermo Fisher Scientific) unless otherwise specified. Carboxylate-modified paramagnetic particle stock was prepared by mixing 200 μL each of Sera-Mag Hydrophylic and Sera-Mag Hydrophobic Magnetic Particles (GE Life Sciences 24152105050250 and 44152105050250), washing the mixture in 1 mL of HPLC-grade water four times, and storing in 400 μL LC-MS grade water (a stock concentration of 50 μg/μL). A magnetic tube rack (Permagen Labware, Peabody, MA) was used for sample processing. An overview of the method and variables tested is shown in Figure 1A. The following standard procedure was performed for all experiments and a single variable, as indicated in each experiment or figure, was evaluated per experiment: Carboxylate-modified paramagnetic particle stock was vortexed to ensure particles were well-suspended in solution. 6 μL of particle suspension was added to 5 μL of 3 mg/mL peptide sample and subsequently triturated to thoroughly mix sample and particles. 100% HPLC grade acetonitrile (MeCN) was added to the sample such that the final MeCN concentration was exactly 95%. The samples were triturated by pipette until the particles were well-suspended in solution (typically 3 – 5 times). The mixture was then allowed to settle for 8 min before being placed on the magnetic rack for an additional 2 min. The supernatant was aspirated to waste, taking care not to disturb the particles. The tube was removed from the magnetic rack and 180 μL of 100% HPLC-grade MeCN was added to cover the particles completely. The tube was returned to the magnetic rack, the mixture was allowed to settle for 1 min, and the supernatant was aspirated to waste. The tube was removed from the magnetic rack and the particles were reconstituted in 60 μL of 2% LC-MS MeCN in LC-MS water. The tube was vortexed in a Thermomixer at 1000 rpm for 5 min at 25 °C. Samples were centrifuged at 13000 rpm for three seconds. The tube was placed on the magnetic rack and the particles were allowed to collect for 4 min. The supernatant was transferred to a clean tube. 20 μL of each sample (10 μL per technical duplicate) were used to determine the peptide concentration.
Figure 1: Overview of SP2 workflow, variables tested, and assessment of peptide recovery with varying particle:peptide ratios and digestion buffers.
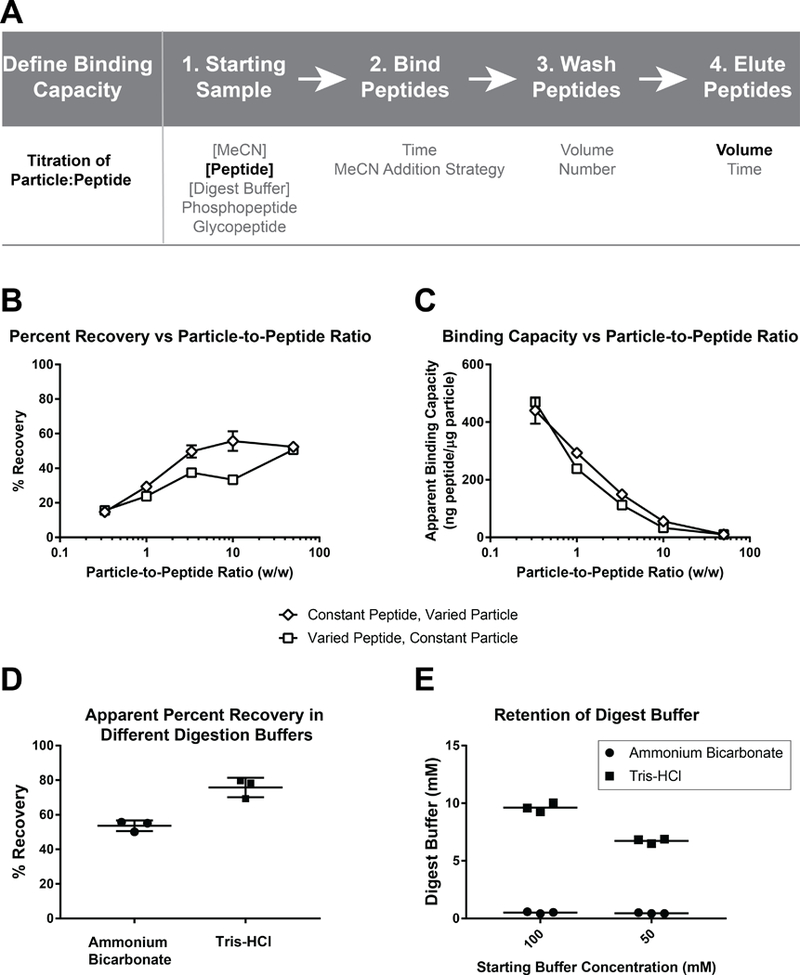
(A) Overview of the SP2 strategy and experimental variables tested at each stage of the protocol. The variables indicated by emboldened font were determined to be most critical for SP2 performance. (B) Percent recovery is plotted against the particle-to-peptide ratio demonstrating a positive, saturable relationship. (C) The apparent binding capacity is plotted against the particle-to-peptide ratio demonstrating a negative correlation. (D) The calculated percent recovery when peptides are digested in either AmBic or Tris buffer. (E) The calculated concentration of digestion buffer retained when either AmBic or Tris buffer is subjected to SP2. All conditions were tested in technical triplicate. The mean is shown as a symbol, in (B-C), or as line, in (D-E), ± the standard deviation.
Determination of Binding Capacity
The binding capacity of the particles was investigated using a pair of experiments in which the peptide-to-particle ratio was manipulated. These experiments were performed using the peptide standards from the Pierce Quantitative Fluorometric Peptide Assay to represent a simple mixture and using digested HEK-293T cell lysate in 50 mM AmBic with 2X (40% v/v) Invitrosol to represent a complex mixture. First, a constant amount of particles (10 μg) was used with a varying amount of peptide added (0.2 μg, 1 μg, 3 μg, 10 μg, 30 μg). Peptides were eluted with 20 μL, 40 μL, 60 μL, 80 μL, or 100uL of 2% MeCN in water, respectively. Next, a constant amount of digested peptide (15 μg) was used with a varying amount of particles added (15 μg, 50 μg, 150 μg, 450 μg, 750 μg). Peptides were eluted with 60 μL of 2% MeCN in water.
Determination of Buffer Removal Capacity
The percent recovery was measured for HEK-293T lysate which was digested in either 100mM Tris- or 100mM AmBic with 20% MeCN and 2X Invitrosol (40% v/v). The ability to remove AmBic or Tris from a non-peptide containing solution was also evaluated as a means to determine whether the particles can remove these commonly used digestion buffers. As a crude measure of buffer concentration, the Peptide Fluorometric Quantitation assay was used to generate a calibration curve from 0.2–100 mM each buffer for which quantitation values obtained for pre-cleaned vs. post-cleaned samples were compared.
Variations to the Manual Peptide Cleanup Protocol
The following list describes experimental details of the permutations that were applied at each step of the protocol (Figure 1A). For each experiment, digested HEK-293T cell lysate in 50 mM AmBic with 2X (40% v/v) Invitrosol was used.
1. Starting Sample
1.1. Sample Concentration:
15 μg of peptides was diluted with different volumes of water such that the starting sample concentration was varied (0.23, 0.5, 1.5, 7.7 μg/μL).
1.2. Concentration of Digest Buffer:
The peptide sample was diluted to 30 μL in different concentrations of digest buffer (50 mM AmBic with 40% v/v Invitrosol) diluted in water – (1x, 0.5x, 0.25x, 0x).
1.3. Organic concentration:
The starting peptide sample was diluted to 30 μL in different concentrations of MeCN in water (0, 5, 20, and 70%) before adding particles.
2. Binding Peptides
2.1. Addition of MeCN:
The peptide sample was diluted to 0.25 μg/μL in water so that >1 mL of MeCN would be required to bring the sample to 95% MeCN - necessitating more than 1 pipetting step. MeCN was added by four different strategies and mixing by trituration was only performed where specified: added 712.5 μL then 712.5 μL and mixed; added 1000 μL then 425 μL and mixed; added 425 μL then 1000 μL and mixed; added 425 and mixed then 1000 μL and mixed again.
2.2. Incubation time:
Various lengths of time for the capture step were compared (2, 4, 8, and 16 min).
3. Peptide Wash
3.1. Volume:
The wash step was performed with various volumes of MeCN (180, 360, 720, and 1000 μL).
3.2. Number:
Various numbers of wash steps were performed (1, 2, 3, and 4).
4. Peptide Elution
4.1. Time:
Various lengths of time in the Thermomixer (1, 2, 5, and 10 min) were compared for elution. In the elution step, particles were allowed to settle for 1 min before taking an aliquot for peptide quantitation.
4.2. Elution Volume:
4.2.1. 15 μg peptide and 6 μL particles:
The elution step was performed with various volumes of MeCN (6, 12, 18, 36, 54, and 72 μL). For all samples, eluent was diluted to 72 μL before peptide quantification.
4.2.2. 10 μg peptide and 15 μL particles:
The elution step was performed with various volumes of MeCN (15, 30, 45, 90, 135, and 180 μL). For all samples, eluent was diluted to 180 μL before peptide quantification.
4.2.3. 4 μg peptide and 6 μL particles:
The elution step was performed with various volumes of MeCN (6, 12, 18, 36, 54, and 72 μL). For all samples, eluent was diluted to 72 μL before peptide quantification.
5. Assessing the Performance of Cumulative Changes:
The baseline protocol, described above, was compared to a modified protocol which included a 2 min binding time, two 500 μL wash steps, a 1 min elution time, and 1 min collection times on the magnetic rack using six technical replicates each.
Automated SP2 using a Liquid Handling Workstation
All experiments were performed using an epMotion® 5073m liquid handling workstation (Eppendorf) and applications were developed using epBlue™ 40.6.2.6 control software (Eppendorf). Performance of the automated protocol was evaluated by observing whether the particles were disturbed during liquid dispensing and aspiration and determining percent recovery of peptides after cleanup. The automated processing proceeded similarly to the manual protocol, using a low-binding 96 deep well plate (96-DWP) in the place of low-binding Eppendorf tubes with one additional wash step (compared to the baseline protocol) included due to the 16 μL bottom-volume limit of the epMotion when using a 96 deep well plate (96-DWP). Parameters that were assessed on the epMotion included the length of time the 96-DWP was allowed to sit on the magnetic rack to ensure the particles were completely settled and different pipette tip sizes (i.e. 1000 μL tips were used for transferring large volumes but 50 μL pipette tips were used when aspirating liquid from the particles to ensure they were not disturbed). Finally, to account for the various hardware required throughout the protocol, the automated SP2 protocol was written as three separate applications which were implemented in succession. Application A: Peptide dispensing; Application B: Peptide cleanup; Application C: Peptide elution. Between Applications B and C, any liquid remaining on the particles was removed manually to ensure the appropriate concentration of MeCN for elution. To facilitate implementation in other laboratories, a detailed description of the methods can be found in the Supporting Information and all instrument methods files and a diagram of the stage setup are freely available for download at http://www.cellsurfer.net/sp2/.
Evaluating SP2 for removal of MS-incompatible Detergents
Four different samples were cleaned using the automated SP2 method: digested HEK cell lysate containing Invitrosol (40% v/v) and MeCN (20% v/v), sodium lauryl sarcosinate (SLS, 9 mM; Sigma Aldrich), sodium deoxycholate (SDC, 12 mM; Fisher Scientific), or SLS (9 mM) and SDC (12 mM). Peptide recoveries were evaluated by peptide quantitation and peptides were analyzed by LC-MS/MS to evaluate proteome coverage.
Evaluating Peptide Selectivity between SP2 and C18 methods
Digested HEK cell lysate (600 μg) was cleaned manually by SP2 by splitting the sample into fifteen 40 μg aliquots. Clean peptides were eluted in 40 μL of 50 mM triethyl ammonium bicarbonate (TEAB), the eluents were pooled, and the peptide concentration was measured by Pierce Quantitative Fluorometric Peptide Assay. Six aliquots (75 μg each) were prepared from the SP2-cleaned peptide solution, diluted to 100 μL with 50 mM TEAB, and labeled with TMTsixplex reagents (one channel/aliquot) per manufacturer’s instructions (Thermo Fisher Scientific). Three samples (channels 127, 129, 131) were cleaned by SP2 using the epMotion for automation and three samples (channels 126, 128, 130) were cleaned manually using C18 Micro SpinColumns (Harvard Apparatus). A detailed protocol for the C18 cleanup is provided in Supporting Information.
Evaluation of Optimized SP2 protocol for Manual and Automated Peptide Cleanup
Using the modified protocol (2 min binding time, two 500 μL wash steps, and a 1 min elution time) the percent recovery and coefficient of variation (CV) for recovery was compared between manual and automated processing of eight technical replicates of digested HEK lysate.
Mass Spectrometry
Samples were analyzed by LC-MS/MS using a Dionex UltiMate 3000 RSLCnano system (Thermo Fisher Scientific) in line with an Orbitrap Fusion Lumos MS (Thermo Fisher Scientific). All technical details are provided in Supporting Information. All MS data files are publicly available at MassIVE (MSV000083117; massive.ucsd.edu).
Data Analysis.
MS data were analyzed using Proteome Discoverer 2.2 (Thermo Fisher Scientific) for peptide and phosphopeptide database searching and Byonic for glycopeptide database searching. Using the contaminant molecular library in Skyline (12) rapid extraction of precursor signals using MS1 filtering enabled convenient viewing of data for >100 common contaminants. Statistical analyses were performed using either an unpaired t test, a one-way ANOVA with Tukey’s post hoc analysis, or a two-way ANOVA with Bonferroni’s multiple comparisons test. Hierarchical clustering of peptide data was performed in KNIME (v 3.6.0). All details for analysis of mass spectrometry data and statistical significance are provided in Supporting Information. Plots were generated in GraphPad Prism.
Results and Discussion
Investigating the Peptide Binding Capacity of the Carboxylate-Modified Particles
The peptide-binding capacity of the particles was investigated using a pair of experiments that manipulated the particle-to-peptide ratio (w/w) by either changing the amount of particles added to a fixed amount of peptide or by changing the amount of peptide added to a fixed amount of particles. The observed percent peptide recovery demonstrates a positive, saturable relationship with the particle-to-peptide ratio for the digested cell lysate (Figure 1B). The percent recovery for the complex mixture was saturated between a 3.33:1 – 10:1 particle-to-peptide (w/w) ratio. The apparent binding capacity (the total peptide (in ng) retained per μg of particle) had a negative relationship with the particle-to-peptide (w/w) ratio (Figure 1C). Similar results were observed for a simple peptide mixture, though a higher particle-to-peptide (w/w) ratio, between 15:1 – 30:1, was required to achieve saturation (Figure S1A–B). Although a greater total amount of peptide could be recovered by using more particles, the capture was less efficient at a higher particle-to-peptide ratio. Therefore, although the binding capacity at the lower end of the curve was ~450 ng/μg for the digested lysate and ~120 ng/μg for the simple peptide mixture, operation within this particle-to-peptide scheme results in a precipitous loss of sample. A more conservative apparent binding capacity can be calculated at the percent recovery curve inflection points, ~5:1 particle-to-peptide ratio for digested cell lysate and ~20:1 for the simple peptide mixture, where a more acceptable percentage (≥60%) of sample is recovered. These operational binding capacities, 50 and 200 ng/μg for simple and complex peptide mixtures, respectively, are markedly lower (500x – 2000x) than the reported binding capacity of the particles for protein (100 μg protein/μg particle) (7).
Evaluating the Capacity to Clean Peptides from Different Digestion Buffers
After determining a suitable particle-to-peptide ratio that is required to achieve a desirable percent recovery, we next evaluated the capacity of the particles to desalt samples containing two commonly used digestion buffers, AmBic or Tris. In these experiments, the apparent peptide recovery from the Tris buffer was significantly higher than that from AmBic (Figure 1D). However, we questioned whether the capacity of salts to be retained by HILIC- and ERLIC-like interactions led to interference with the peptide quantitation assay, which is sensitive to primary amines such as Tris. We then designed an experiment to test whether the digestion buffers were being retained during the SP2 protocol. First, exploiting the Peptide Fluorometric Quantitation Assay, which uses an amine-reactive reagent, calibration curves could be obtained for >500-fold concentration range for Tris and AmBic (Figure S1C–D). Two concentrations, 50mM and 100mM, of Tris and AmBic only (i.e. not containing peptides) were subjected to the SP2 cleanup and the relative amounts of buffer that remained in the sample after cleanup were determined with reference to the respective buffer-only calibration curves. Overall, 20% of the original concentration of Tris and <1% of AmBic remained in the sample after cleanup (Figure 1E). Therefore, the retained Tris could account for the difference in the apparent percent recovery of Tris and AmBic buffered peptide solutions as measured by the Peptide Fluorometric Assay. As these results suggest that additional cleanup of Tris would be required (e.g. another round of SP2 or C18), and that peptide quantitation measurements would be more prone to interference from the Tris buffered samples, all subsequent experiments were performed using only the AmBic buffered samples. These results are in contrast to the guidelines provided in the SP3 protocol, wherein Tris-HCl (up to a concentration of 1M) is listed as a contaminant that could be removed (7). This discrepancy may be due to our utilization of the cleanup method solely at the peptide level (i.e. perhaps the conditions to bind proteins are less favorable to the retention of salt than the conditions to bind peptides). While it is not clear which method was used to assess the removal of Tris in the previous report (7), the LC-MS/MS system included a trap column. Therefore, we suspect that if any Tris remains in the peptide sample following SP3, it would be removed during online desalting achieved by the trap. Altogether, based on our observations, we recommend that Tris-containing samples cleaned by SP2 undergo subsequent desalting, either offline or online using a trap column.
Evaluating the Effect of SP2 Procedural Variants on Percent Recovery
Several procedural variants were evaluated because they either reflect common differences in composition expected for proteomic samples or are important for establishing the amenability to automation. The first set of parameters investigated were related to peptide sample composition and included an analysis of how differences in sample composition could affect recovery. While the total amount of peptide is one important factor to determine prior to SP2 because of the measured impact of particle-to-peptide ratio on recovery, we also considered that peptide concentration may vary depending on the sample preparation strategy used. To test whether this variable affects recovery, a range of 0.23 – 7.7 μg/μL was tested and it was determined that the percent recovery was higher for more concentrated peptide samples (Figure 2A). This observation suggests either a peptide concentration-dependent effect on the efficiency of peptide capture, or a confounding effect driven by the relative concentration of buffer in the sample. Therefore, the relative level of buffer concentration-to-peptide was subsequently investigated by diluting the same amount of peptide in various dilutions of digest buffer-to-water. The percent recovery was not found to be affected by this relationship (Figure 2B). These results support a salt-independent relationship between peptide concentration and capture efficiency. As peptide concentration cannot always be controlled, especially in sample-limited studies, this is a limitation of SP2 that, to some extent, can be accounted for during the experimental design by minimizing lysis and digestion volumes.
Figure 2. Peptide recovery with varied peptide binding and elution conditions.

(A) Digested lysate (at 7.7 μg/μL), or dilutions of the digested lysate in water at 1.5 μg/μL, 0.5 μg/μL and 0.23 μg/μL were added such that the total peptide amount was 15 μg. (B) 15 μg of digested lysate was diluted to 0.5 μg/μL with various concentrations (1x, 0.58x, 0.38x, 0.17x) of digest buffer (50mM AmBic w/40% Invitrosol (v/v) in water). (C) 15 μg of digested lysate was diluted to 0.5 μg/μL in 0%, 5%, 20%, or 70% MeCN in water. (D) After mixing by trituration, peptide was allowed to bind for the indicated time. (E) Samples were washed with 180, 360, 720, or 1000 μL of LC-MS MeCN. (F) Samples were washed 1, 2, 3, or 4 times with 180 μL of LC-MS MeCN. (G) After vortexing, peptides were allowed to elute for 1, 2, 5, and 10 min. (H) Different ratios of eluent volume-to-particle amount (0.02, 0.04, 0.06, 0.15, 0.18, 0.24 μL/ μg) were tested. All conditions were tested in technical triplicate. The mean is shown as a line, in (A-F), or a symbol, in (G-H), ± the standard deviation.
MeCN may be added to tryptic digestions to increase enzymatic specificity and activity (13, 14). Moreover, in some cases SP2 could be implemented to remove contaminants that remain after C18 cleanup (10, 11). In these cases, the starting sample for SP2 would contain MeCN. For these reasons, the effect of the starting concentration of MeCN on percent recovery was investigated. Over the range of sample concentrations tested here (0 – 70%) no significant differences in recovery were observed (Figure 2C). Finally, as several variations of carboxylate-modified particles are commercially available, we explored whether the particle format or composition would affect recovery. Overall, percent recovery was comparable when using either the Sera-Mag or the Sera-Mag SpeedBeads (data not shown). Although the SpeedBeads were observed to aggregate faster once placed near the magnet, both formats were suitable for manual and automated processing.
The second set of parameters investigated were related to peptide capture and included an analysis of how differences in MeCN addition and binding time could affect the percent recovery. First, the manner in which MeCN is added had no significant effect on recovery, as long as the 95% MeCN solution containing the particles is eventually well-mixed (Figure S2A). Second, variations in the incubation time did not result in any significant effect within the range tested, 2 to 16 min (Figure 2D). Both of these variables are important factors to consider for automation as it may be difficult to precisely control the time that peptides are incubated with particles, and pipette volume restrictions may render it impossible to bring the peptide to a suitable concentration of MeCN in a single pipetting step (e.g. using a 1000 μL pipette would require multiple steps to deliver more than 1 mL).
The third set of parameters investigated were related to peptide washing and elution. Overall, once the peptides are bound to the particles, they appear to be retained quite well. No significant loss in sample was observed by increasing the number of washes from 1 to 4 or the volume of the wash from 180μL to 1000 μL (Figure 2E). The ability to wash the particles iteratively with excess volume is of benefit, particularly for automation, as the bottom-volume limits on liquid handlers typically lead to a small amount of supernatant left behind after aspiration. In this context, additional washes can be beneficial to ensure complete removal of contaminants. Peptides elute from the particles quickly and stay in solution, as no significant changes were observed in comparison to the 5 min baseline when the elution time was decreased to 1 min or increased to 10 min (Figure 2G). Rather, the parameter which had the most significant effect on percent recovery was the elution volume. Specifically, higher recovery is obtained with larger elution volumes (Figure 2H). Percent recovery was positively correlated with increasing elution volume and reached saturation when elution volume-to-particle ratio (μL/μg) ranged 0.18:1 to 0.24:1 (Figure 2H). Accordingly, if the particles are stored at 50 μg/μL stock, as described, a 9:1 – 12:1 ratio (v/v) of elution-to-particle solution volume is optimal for maximum peptide recovery. The same trend was observed using a different particle-to-peptide ratio and a different amount of beads (Figure S2B). HILIC, which is similar in principle to SP2, is understood to work by partitioning analytes into surface-phase interactions by hydrophilic interactions. Therefore, it stands to reason that the reverse reaction (i.e. dissociation of an analyte from the particle surface) could be driven by an increase in the volume of the hydrophilic phase.
Finally, a modified version of the protocol which included several individual changes that demonstrated no significant effect on recovery (2 min binding time, two 500 μL wash steps, and 1 min elution time) was compared to our baseline protocol (8 min binding time, one 180 μL wash step, and 5 min elution time). Percent recovery using the modified protocol was statistically indistinguishable from the baseline protocol (Figure S2C), and could be completed in under 5 min, (i.e. one-third of the total preparation time of baseline protocol). Altogether, these results suggest that SP2 is tolerant to many procedural variables with only a few variables that must be adjusted to maximize percent recovery (Figure 1A). In addition, the flexibility of the procedure renders it suitable for automation.
Evaluating SP2 Utility for Glycopeptides, Phosphopeptides, and a “Real World” Sample
Post-translational modifications alter the physiochemical properties of peptides. For this reason, we investigated the capacity of the SP2 protocol to capture and elute glycosylated and phosphorylated peptides. For glycopeptide analyses, tryptic digests of three different human glycoproteins with known glycosites were cleaned with the manual SP2 protocol. Peptide backbone and glycan fragmentation was achieved with targeted mass triggered EThCD and both non-glycosylated and intact glycopeptides were observed in this approach. For each protein, ≥ 88% of all possible tryptic peptides were observed (Figure 3A), and all canonical N-glycosites were confirmed through the detection of intact N-glycopeptides (Figure 3B; Supporting Information). Compared to an equivalent sample cleaned by C18 (n = 3 each with 3 technical injections) there was no significant difference in the number of glycopeptide spectral matches (Figure 3C). SP2 cleanup is also compatible with downstream phosphopeptide enrichment, as evidenced by the identification of 2320 phosphoproteins on average (n = 3, each with 3 technical injections) from 75 μg SP2-cleaned starting peptide material (Figure S3). The number of identified proteins and the observed specificity (phosphoproteins/total proteins) were statistically indistinguishable from three matched replicates of C18-cleaned samples. Though not utilized as a feature of the workflow in this study, the starting buffer of the phosphoenrichment kit is capable of eluting material from the SP2 particles; an advantage in time and number of sample transfer steps compared to C18. Altogether, these data suggest that glycopeptides and phosphopeptides bind and elute from the carboxylate-modified particles under the same conditions as used for non-modified peptide cleanup and that SP2 represents a viable alternative for workflows that utilize C18.
Figure 3. Application of SP2 for glycopeptide cleanup.
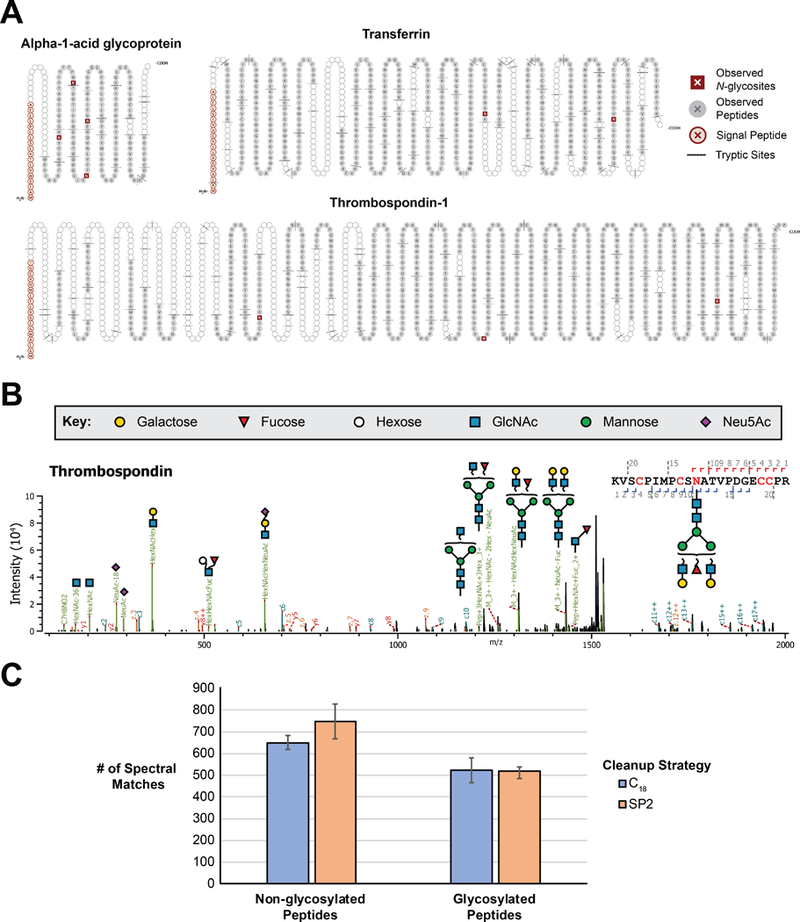
(A) Images depicting the sequence coverage and N-glycosites observed for tryptic digestion of three glycoproteins cleaned by SP2. Images were generated using Protter (15). (B) Annotated EThcD MS/MS spectra of intact N-glycopeptides identified in Thrombospondin with high confidence in glycan localization and composition assignment. (C) Number of spectral matches for non-glycosylated and glycosylated peptides are shown for cleaning equivalent amounts of digested glycoprotein by either C18 or SP2. The mean is plotted as a column ± standard deviation.
Demonstrating the utility of SP2 for removal of unexpected contaminants in a “real-world’ scenario, Figure 4 shows extracted ion chromatograms for contaminants within a TMT-labeled immunoprecipitation sample submitted to our MS Center for analysis (i.e. represents a common scenario where a non-expert submits samples to a core facility). Prior to receiving the sample, proteins were enriched following manufacturer’s instructions for Pierce Biotinylated Protein Interaction Pull-Down Kit, with the exception that following the recommended protocol, the samples were washed four times with acetate-NaCl buffer, pH 5.0, prior to elution in an effort to remove any remaining detergent. Following standard tryptic digestion, C18 cleanup, TMT-labeling, and another C18 cleanup, a portion of the sample was analyzed by LC-MS/MS, but significant contaminants were observed throughout the chromatogram (Figure 4A). To determine whether the sample could be salvaged, the remaining sample was cleaned by SP2, which yielded a sample with dramatically improved chromatography and reduced level of detectable contaminants (Figure 4A–B). All contaminants detected in the C18-only cleaned sample were reduced by SP2 cleanup (Figure 4B), with 50% of the contaminants experiencing >100-fold reduction (Figure 4C, Supporting Information). Although SP2 cleaning did not result in a significant increase in the number of identified proteins, there are time and financial savings afforded when the level of contaminants loaded onto the system are drastically reduced. Overall, this results in less downtime due to system maintenance, improved instrument performance, and higher sample throughput. Altogether, these data demonstrate the utility of SP2 when unanticipated contaminants are present in peptide samples, even those that have been cleaned by C18 methods.
Figure 4. SP2 cleanup of a “real world” sample.

(A). Extracted ion chromatograms for contaminants within a TMT-labeled immunoprecipitation sample following standard tryptic digestion, C18 cleanup, TMT-labeling, and another C18 cleanup (top). A portion of the remaining C18-cleaned sample subsequently cleaned by SP2 (bottom). (B) Peak area intensities for contaminants remaining after C18 or SP2 cleaning. (C) A histogram of the observed fold reduction in contaminants after SP2 cleanup.
Operational Considerations for Performing SP2 using an Automated Liquid Handling Workstation
Having found SP2 applicable for the cleanup of complex mixtures, glycopeptides, and phosphopeptides, we investigated the ability to automate the protocol using an epMotion® 5073m liquid handling platform. The development of an automated workflow often requires attention to details specific to the platform, including monitoring the behavior of all materials and reagents and considering physical constraints that are not present in manual handling. When using magnetic particles, for example, conditions and materials which favor rapid and tight pelleting of the particles are important to minimize carryover during supernatant aspiration steps. For SP2, experimental details found to be important for automation include the speed at which liquids are aspirated and dispensed and the length of time the plate rests on the magnet to allow the particles to form a tight pellet. The choice of pipette tip was also found to be important, and in general, a 50 μL pipette tip was chosen for aspirating liquids away from the particles, as larger pipette tips such as 1000 μL caused the pellet of magnetic particles to be disturbed. Other important changes introduced to the protocol to adapt it to the workstation were related to physical constraints of the system. For example, as the epMotion® is unable to triturate small volumes (< 16 μL), the 96-DWP was shaken at 500 rpm for 1 min on the built-in Thermomixer.
Evaluating SP2 for Removal of MS-Incompatible Detergents
Sample preparation strategies commonly include detergents that are incompatible with MS and SP3 has been previously applied to remove SDS, Triton-X100, NP-40, and SDC (7) from protein samples. Here we evaluated SP2 for the removal of SDC and SLS, either alone or in combination, in comparison to an MS-compatible surfactant, Invitrosol, from peptide samples to determine if the presence of detergent affected the reproducibility of recovery or the features of the recovered peptides. Using the automated SP2 method, similar recoveries, peptides, and proteins were identified among all four conditions tested (Figure S4). Specifically, when using 20 μL of particles to clean 20 μg of digested peptide, percent recovery averaged 60% across replicates, with CVs ranging 2 – 9% (Figure S4A). An average of 3652 proteins were identified by 2 or more unique peptides per experiment (Figure S4B), and the majority of proteins identified (2993, 73%) are found in all experiments, (Figure S4C). Finally, an average of 27761 peptides were identified in each condition, and the differences in peptides identified among surfactants was no more significant than the differences among replicates indicated by the failure of replicates to cluster regardless of the distance measurement applied (Figure S4D). Altogether, these data suggest SP2 is similarly applicable for the removal of harsh detergents like SDC, SLS and that the detergent background has no apparent effect on the recovered peptides.
Evaluating Peptide Selectivity Preferences of SP2 vs C18
To further assess reproducibility of the automated SP2 method and determine if peptide recovery with this format is comparable to the more popular C18 method, a quantitative multiplex experiment using TMTsixplex labeled HEK-293T lysate was used. Three replicate samples were cleaned by the automated SP2 protocol and three were cleaned manually by C18 (experimental design is shown in Figure S4A). The CV for the percent recoveries, assessed by the total peptide signal, for the different cleanup strategies was 7.9% and 7.7% for C18 and SP2, respectively. Histograms representing the individual peptide abundance CVs when comparing among SP2 and C18 channels individually are highly similar, with 94 and 91% of peptides, respectively, having <20% CV (Figure 5A). However, when the CVs of individual peptide abundances were compared across all six channels, the peptide abundances were found to vary to a greater extent (Figure 5A), though most peptides (87%) were still below a 20% CV. Viewing these data in terms of abundance ratios between samples demonstrates a similar trend while further demonstrating that there are no clear sample outliers causing the CV values to be skewed (Figure S4B). Visualizing the data as a volcano plot reveals that only a small percentage of peptides were observed to have significantly different abundances between SP2 and C18 (Figure S4C). Only 90 peptides (1.3%) crossed the thresholds of 2 and 0.05 for fold change and p-value, respectively.
Figure 5. Evaluating the peptide selectivity of SP2 and C18 methods.

(A) A histogram for the coefficients of variation (CVs) for individual peptide abundances in samples subjected to SP2, C18, or to either method. (B) Histograms for peptide abundance CVs subjected to each sample preparation strategy (i.e. C18 or SP2) broken down by grand average of hydropathy (GRAVY), net charge, or length.
To investigate whether there exists a discrepancy in the reproducibility of SP2 and C18 for peptides with different physiochemical properties, the distribution of CVs was calculated for peptides grouped by grand average of hydropathy (GRAVY) score, net charge, and number of residues (Figure 5B). Reproducibility for peptides cleaned by C18 was affected by each of these characteristics such that hydrophilic, highly negative, and lengthy peptides were more variable. (Figure 5B). Though the number of residues was a factor in the reproducibility of peptides cleaned by either method (where longer peptides have higher CVs), SP2 reproducibility appeared less dependent on net charge and was largely independent of GRAVY score. These results demonstrate that while these methods are highly comparable for most peptides, each cleanup strategy offers improved reproducibility for peptides with particular properties. These results are largely consistent with those reported in Hughes et al., where no difference was demonstrated in the frequency of peptides with particular properties identified using either SP3 or FASP (followed by C18). However, here we show by careful analysis of the individual peptide abundance CVs that SP2, as compared to C18, provides better reproducibility for peptides with specific features (long, hydrophilic, and highly negative).
Reproducibility and Timing of SP2 Use
Overall, the SP2 protocol developed here is highly reproducible for peptide cleanup. Across eight technical replicates, the CV of peptide recovery was 7.4% when performed manually and 4.4% when performed using the automated protocol on the epMotion®, although the average percent recovery was not significantly different between the manual and automated modes of operation (Figure 6). Using the manual SP2 protocol, a single MS-ready peptide sample can be prepared from digested lysate in less than 10 min and a set of 8 samples can be prepared in less than 20 min. The automated protocol requires ~40 min to prepare 8 samples, and while this protocol is longer than the manual alternative, it is time that the operator does not have to spend at the bench. Altogether, based on the results from these studies, it is clear that there are distinct advantages of the SP2 method for peptide cleanup prior to LC-MS/MS analysis compared to alternative strategies. Finally, to facilitate the implementation of SP2 in other laboratories, a comprehensive SOP based on the results of the current study is provided in Supporting Information and can also be downloaded at http://www.cellsurfer.net/sp2/. The SOP contains the detailed experimental protocols for both manual and automated processing along with a substantial number of observations, suggestions, and considerations for customization to assist new users in its implementation.
Figure 6. Comparing percent recovery and reproducibility between manual and automated SP2.

The percent recovery is plotted for eight technical replicates of digested lysate cleaned by SP2 using both the manual and automated modes of operation. The mean is shown as a horizontal bar ± standard deviation.
Considerations for Selecting SP2 as a Peptide Cleanup Strategy
Contaminant removal is critically important for the acquisition of high-quality data and long-term instrument maintenance. There are several factors to consider when selecting a peptide cleanup strategy, including sample composition, availability/starting amount of sample, reproducibility required, and cost. Whereas SP2 and strong cation exchange can efficiently remove detergents and polymers, these molecular classes can be concentrated, rather than removed, by C18. However, as we found in this study, SP2 does not efficiently remove Tris, which poses potential issues for accurate peptide quantitation and when using instrument configurations that do not include trap columns. Furthermore, SP2 does not appear well-suited for very dilute samples, though this variable can be controlled to some extent by experimental design. All peptide-based cleanup strategies avoid the difficulties and losses associated with protein precipitation/resuspension and demonstrate good reproducibility, but as noted in this manuscript, there may be subtle differences in reproducibility in particular classes of peptides. SP2 offers the advantage of eluting peptides in aqueous solutions that are immediately amenable to LC-MS/MS or other enrichment strategies (e.g. phosphoenrichment) without requiring organic solvent removal. As SP2 uses magnetic particles in suspension, particle carryover into the peptide samples is a concern for contaminating the LC-MS/MS platform. However, this risk can be mitigated by additional magnetic separations or centrifugation steps. Finally, cost can be a significant variable to consider, especially for large study designs, and SP2 is more cost-effective compared to alternatives. For example, SP2, C18, and Phoenix (an in-Stage Tip based strong cationic exchange) conservatively cost an estimated $0.01, $0.06, and $0.19, respectively to clean 1 μg of sample. Altogether, the selection of a cleanup strategy must fit the constraints of the study design, and here we present SP2 as a viable alternative to previously described methods.
Conclusion
The SP2 protocol developed here is a fast, straightforward, and scalable strategy for the removal of contaminants and concentration of peptide samples prior to LC-MS/MS analyses. Although conceptually similar to the SP3 protocol, SP2 has been developed specifically for the cleanup of peptide samples and key findings from these studies which are in contrast to the SP3 publication include: 1) the binding capacity was markedly lower for peptides than for proteins; 2) Tris was not efficiently removed from peptide samples; and 3) peptides characterized as long, hydrophobic, or highly negative are more reproducibly processed with SP2 than with C18. Altogether, the results of this study and accompanying detailed operating protocols for both manual and automated processing are expected to facilitate reproducible implementation of SP2 for various proteomics applications and will especially benefit core or shared resource facilities where unknown or unexpected contaminants may be particularly problematic. Of course, we advocate that laboratories validate this protocol independently for their samples. However, as the current study demonstrates the utility of SP2 among a broad range of sample conditions, the SOP is expected to benefit both established laboratories and those new to this field.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health [R01-HL126785 and R01-HL134010 to RLG; F31-HL140914 to MW] and the Center for Biomedical Mass Spectrometry Research at the Medical College of Wisconsin. Funding sources had no involvement in study design, data collection, interpretation, analysis or publication. Mass spectrometry analyses were performed using instrumentation in the Center for Biomedical Mass Spectrometry Research at the Medical College of Wisconsin. We are grateful to Dr. Deepali Rathore for assistance with phosphopeptide experimental design, Dr. Amanda Rae Buchberger for analysis with KNIME, and Chase Castro for assistance with preliminary experiments in SP2 method optimization.
Footnotes
Supporting Information
The following supporting information is available free of charge at ACS website: https://pubs.acs.org/
References
- 1.Vissers JP; Chervet JP; Salzmann JP, Sodium dodecyl sulphate removal from tryptic digest samples for on-line capillary liquid chromatography/electrospray mass spectrometry. J Mass Spectrom 1996, 31, (9), 1021–7. [DOI] [PubMed] [Google Scholar]
- 2.Serra A; Gallart-Palau X; Dutta B; Sze SK, Online Removal of Sodium Dodecyl Sulfate via Weak Cation Exchange in Liquid Chromatography-Mass Spectrometry Based Proteomics. J Proteome Res 2018, 17, (7), 2390–2400. [DOI] [PubMed] [Google Scholar]
- 3.Wiśniewski JR; Zougman A; Nagaraj N; Mann M, Universal sample preparation method for proteome analysis. Nature Methods 2009, 6, 359. [DOI] [PubMed] [Google Scholar]
- 4.Kulak NA; Pichler G; Paron I; Nagaraj N; Mann M, Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nature Methods 2014, 11, 319. [DOI] [PubMed] [Google Scholar]
- 5.Alpert AJ, Hydrophilic-interaction chromatography for the separation of peptides, nucleic acids and other polar compounds. Journal of Chromatography A 1990, 499, 177–196. [DOI] [PubMed] [Google Scholar]
- 6.Alpert AJ, Electrostatic repulsion hydrophilic interaction chromatography for isocratic separation of charged solutes and selective isolation of phosphopeptides. Anal Chem 2008, 80, (1), 62–76. [DOI] [PubMed] [Google Scholar]
- 7.Hughes CS; Foehr S; Garfield DA; Furlong EE; Steinmetz LM; Krijgsveld J, Ultrasensitive proteome analysis using paramagnetic bead technology. Molecular Systems Biology 2014, 10, (10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sielaff M; Kuharev J; Bohn T; Hahlbrock J; Bopp T; Tenzer S; Distler U, Evaluation of FASP, SP3, and iST Protocols for Proteomic Sample Preparation in the Low Microgram Range. Journal of Proteome Research 2017, 16, (11), 4060–4072. [DOI] [PubMed] [Google Scholar]
- 9.Moggridge S; Sorensen PH; Morin GB; Hughes CS, Extending the Compatibility of the SP3 Paramagnetic Bead Processing Approach for Proteomics. Journal of Proteome Research 2018, 17, (4), 1730–1740. [DOI] [PubMed] [Google Scholar]
- 10.Larsbrink J; Tuveng TR; Pope PB; Bulone V; Eijsink VGH; Brumer H; McKee LS, Proteomic data on enzyme secretion and activity in the bacterium Chitinophaga pinensis. Data in Brief 2017, 11, 484–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larsbrink J; Tuveng TR; Pope PB; Bulone V; Eijsink VGH; Brumer H; McKee LS, Proteomic insights into mannan degradation and protein secretion by the forest floor bacterium Chitinophaga pinensis. Journal of Proteomics 2017, 156, 63–74. [DOI] [PubMed] [Google Scholar]
- 12.Rardin MJ, Rapid Assessment of Contaminants and Interferences in Mass Spectrometry Data Using Skyline. J Am Soc Mass Spectrom 2018, 29, (6), 1327–1330. [DOI] [PubMed] [Google Scholar]
- 13.Waas M; Bhattacharya S; Chuppa S; Wu X; Jensen DR; Omasits U; Wollscheid B; Volkman BF; Noon KR; Gundry RL, Combine and conquer: surfactants, solvents, and chaotropes for robust mass spectrometry based analyses of membrane proteins. Anal Chem 2014, 86, (3), 1551–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strader MB; Tabb DL; Hervey WJ; Pan C; Hurst GB, Efficient and specific trypsin digestion of microgram to nanogram quantities of proteins in organic-aqueous solvent systems. Anal Chem 2006, 78, (1), 125–34. [DOI] [PubMed] [Google Scholar]
- 15. Omasits U; Ahrens CH; Muller S; Wollscheid B, Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics 2014, 30, (6), 884–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.