

Abstract
Metformin is an antidiabetic drug. However, the pleiotropic beneficial effects of metformin in nondiabetic models still need to be defined. The objective of this study is to investigate the effect of metformin on angiotensin II (Ang II)-induced hypertension and cardiovascular diseases. Mice were infused with Ang II (400 ng/kg per min) with or without metformin for 2 weeks. Mice infused with angiotensin II displayed an increase in blood pressure associated with enhanced vascular endoplasmic reticulum (ER) stress markers, which were blunted after metformin treatment. Moreover, hypertension-induced reduction in phosphorylated AMPK, endothelial nitric oxide synthase (eNOs) phosphorylation, and endothelium-dependent relaxation (EDR) in mesenteric resistance arteries (MRA) were rescued after metformin treatment. Infusion of ER stress inducer (tunicamycin, Tun) in control mice induced ER stress in MRA and reduced phosphorylation of AMPK, eNOS synthase phosphorylation, and EDR in MRA without affecting systolic blood pressure (SBP). All these factors were reversed subsequently with metformin treatment. ER stress inhibition by metformin improves vascular function in hypertension. Therefore, metformin could be a potential therapy for cardiovascular diseases in hypertension independent of its effects on diabetes.
Keywords: Metformin, Angiotensin II, Hypertension, AMP-activated protein kinase, endoplasmic reticulum stress
Introduction
Metformin, an oral antidiabetic agent, is usually prescribed as a first-line drug to patients with type-2 diabetes mellitus (T2DM) by international guidelines [1]. Clinical studies on the effects of metformin on blood pressure (BP) have shown various findings from decreased to unaltered BP [2, 3]. Metformin has also been reported to lower BP in spontaneously hypertensive rats (SHR) [4]. The protective effect on BP in nondiabetic hypertensives is unclear. Thus, the molecular mechanisms that determine the effect of metformin on blood vessel responsiveness in relation to lowering blood pressure remain uncharacterized.
The renin–angiotensin–aldosterone system (RAAS) has a central role in vascular adaptive processes [5]. The stimulation of this system has been demonstrated in a range of cardiovascular disorders, especially in hypertension and hypertension with diabetes mellitus [6]. Angiotensin II (Ang II) is a potent vasoconstrictor that plays a key role in BP regulation [7]. Furthermore, Ang II is an important component of the RAAS system that alternatively controls BP [8]. ER stress has been involved in vascular endothelial dysfunction and cardiac damage in an Ang II-induced hypertension model [9]. However, several studies have suggested the importance of other mechanisms through which Ang II may regulate the progression of hypertension [10].
AMP-activated protein kinase (AMPK) is an evolutionarily conserved serine/threonine protein kinase that consists of α, β, and γ subunits. AMPK is reported to exert a direct vasorelaxant effect in isolated aortic rings [11]. Some studies have demonstrated that AMPK is essential in maintaining ER function in vascular smooth muscle cells (VSMCs), and that aberrant ER stress might play a causative role in the development and progression of hypertension [12]. In endothelial cells (ECs), AMPKα2 deficiency causes aberrant ER stress resulting in vascular dysfunction and atherosclerosis in vivo [13]. Published data indicate that metformin is a potent activator of AMPK [14]. The aim of the present study is to determine whether AMPK-suppressed ER stress by metformin is required to preserve endothelial function in an Ang II-induced hypertension model.
Materials and methods
General protocol in mice
All experiments were performed according to the guidelines of Animal Care Committee of Shanghai Jiao Tong University School of Medicine. Fifty mice (C57BL/6J, 8-week-old males) were purchased from the Shanghai Laboratory Animal Company (Shanghai, China), housed in groups of five mice, and maintained at a temperature of 23 °C with a 12 h light/dark cycle. Mice were fed on a solid standard diet (Na+ content 0.4 %) and water. Mice were divided in four groups: (1) sham mice (control mice infused with saline, n = 5), (2) sham mice treated with metformin (300 mg.kg−1 body weight per day) in drinking water for 2 weeks, n = 5, (3) control mice infused with Ang II (400 ng/kg/min) using subcutaneous mini-osmotic pumps for 2 weeks (HT, n = 5), and (4) control mice infused with Ang II and metformin (300 mg.kg−1 body weight per day) in drinking water for 2 weeks, n = 5. Body weight was recorded weekly during the experimental period. The dose of Ang II was selected based on our previous studies [15, 16], and the dose of metformin used was based on the literature [17].
Systolic blood pressure (SBP) was measured using the CODA tail-cuff blood pressure system (Kent Scientific Torrington, USA). Arterial blood pressure measurements were performed at the same time of day (between 9 and 11 a.m.) to avoid the influence of the circadian cycle. SBP was obtained by calculating the average of 10 measurements.
At the end of the treatment period, mice were anesthetized with isoflurane. Mesenteric resistance arteries (MRA) were then harvested immediately and placed in PSS solution (composition in mM: NaCl 118; KCl 4.7; CaCl2 2.5; KH2PO4 1.2; MgSO4×7H2O 1.2; NaHCO3 25 and glucose 11, pH = 7.4) to be processed appropriately for further studies.
In another set of experiments, we used 8-week-old C57BL/6J male mice divided into four groups: (1) sham group (control received saline, n = 5), (2) sham mice treated with metformin (300mg.kg−1 body weight per day) in drinking water for 2 weeks, n = 5, (3) control group that received intraperitoneal injection of tunicamycin (Tun, 1 mg/kg, two injections/week for 2 weeks, Sham + Tun, n = 5), and (4) control group that received tunicamycin and metformin (300 mg.kg−1 body weight per day) in drinking water for 2 weeks, n = 5. Systolic blood pressure was measured weekly during the treatment period. At the end of treatment, mice were anesthetized with isoflurane, and then mesenteric resistance arteries were immediately harvested and placed in PSS solution for reactivity and biochemistry assays.
Vascular reactivity
MRA from all groups of mice were carefully cleaned by removing adipose and connective tissue and were further cut into rings (2 mm in length). MRA were mounted in a small vessel dual-chamber myograph for measurement of isometric tension. After a 30-min equilibration period in PSS solution bubbled with carbogen at 37 °C and pH = 7.4, arteries were stretched to their optimal lumen diameter for active tension development. After a second 30-min equilibration period, arteries were stimulated with phenylephrine (PE, 10−5M) followed by acetylcholine (ACh, 10−6M) to assess the function of endothelial cells. After a 1-h incubation, cumulative concentration responses to phenylephrine (PE, 3.10−8 – 10−4M) and thromboxane analog (U46619,10−9–10−5M) were obtained. In another series of experiments, rings were preconstricted with PE (10−4mol/L) and steady maximal contraction. Cumulative concentration-response curves were obtained for ACh (1 × 10−8–3 × 10−5mol/L) and sodium nitroprosside SNP (1 × 10−8 –3 × 10−5mol/L).
Western blot analysis
Western blot analysis for total and phospho eNOS, phospho PERK, total and phospho eIF2-α, CHOP, GRP78, ATF6, and total and phospho AMPKα (1:1000 dilution, Cell Signaling Technology, Inc, USA) was performed in lysates of mesenteric arteries as previously described.
Immunofluorescence
MRA segments were frozen in Tissue Tek OCT embedding medium (Sakura Finetek Europe, The Netherlands). Transverse sections were cut 5-μm thick. After blockade in PBS containing 5% fetal bovine serum and 0.3% Triton X-100, sections were incubated with primary antibodies against 8-hydroxydeoxyguanosine (8-OHDG) (Abcam, Cambridge, MA), and von Willebrand factor antibody (Abcam, Cambridge, MA) used at a 1:50–1:200 dilution followed by a biotinylated secondary antibody for immunofluorescence. Immunofluorescent signals were viewed using an Eclipse 55i fluorescence microscope (x20), Nikon.
Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity assay
Superoxide anion levels generated by NADPH oxidase activity were measured in lysates of aorta and MRA using lucigenin chemiluminescence. Briefly, lysates were prepared in a sucrose buffer containing 50 mM KH2PO4, 1 mM EGTA, and 150 mM sucrose at a pH of 7.0 and the “Complete-C mini” protease inhibitor cocktail (Roche Diagnostics, IN) in a Tissue Dounce homogenizer on ice, and aliquots of the homogenates were immediately used. To start the assay, a volume of 100 μL of each lysate was used in a total volume of 1 mL of PBS buffer preheated at 37 °C that contained lucigenin (5 μM) and NADPH (100 μM). Blank samples were prepared using 100 μL of sucrose buffer. Lucigenin activity was measured every 30 s for 10 min in a luminometer (Turner biosystem 20/20, single tube luminometer) or until enzymatic activity reached a plateau. Data are expressed as % of the area under the curve of relative light units (RLU) normalized to protein content (μg protein) compared to sham.
Drugs
Phenylephrine hydrochloride, acetylcholine, NADPH, angiotensin II, and metformin were obtained from Sigma–Aldrich (USA). U46619 and tunicamycin were obtained from Tocris Bioscience. Stock solutions of drugs were prepared in ultrapure water and stored at −20 °C, and appropriate dilutions were made on the day of the experiment.
Statistical analysis
Data are expressed as the mean ± SEM. Concentration-response curves were analyzed using GraphPad Prism 4.0 software (GraphPad, USA) and adjusted to a logistic equation. Statistical calculations for significant differences were performed using one-way Student’s t-tests followed by post-hoc tests or two-way ANOVAs as appropriate. Comparisons are considered significant when p < 0.05.
Results
Metformin ameliorated angiotensin II-induced systolic blood pressure
We aimed to study the effect of metformin on SBP. Our data demonstrated that mice infused with Ang II for 2 weeks had significantly increased SBP compared to the sham group infused with saline (Fig. 1a). Interestingly, metformin treatment significantly reduced SBP in Ang II-infused mice (Fig. 1a). Body weight was not affected by treatment and was similar among groups (Fig. 1b).
Fig. 1.
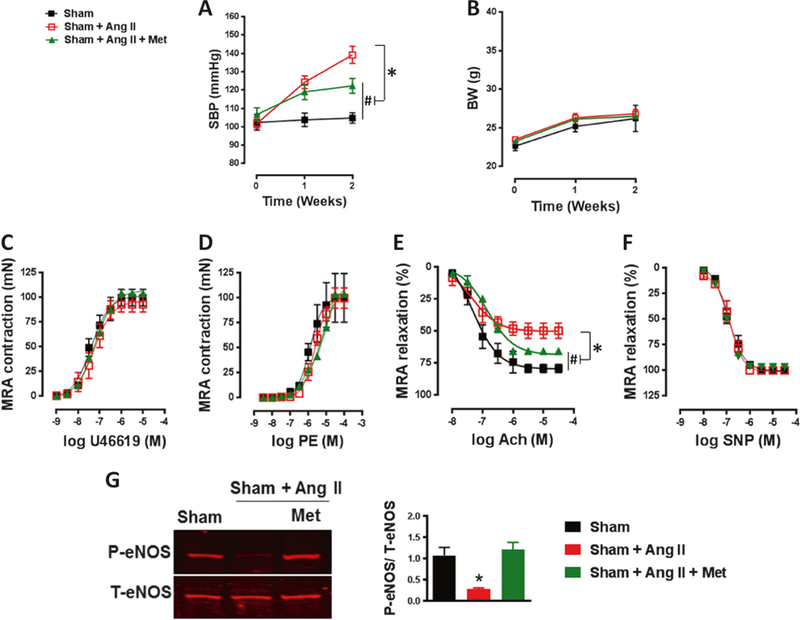
Metformin reduces systolic blood pressure and improves vascular function in hypertension. a The increased systolic blood pressure (SBP) in C57/Bl6 mice after Ang II infusion (400 ng/kg/min) was significantly reduced with metformin treatment (300mg.kg−1 body weight per day). #p < 0.05 vs Sham; *p < 0.05 vs Sham + Ang II. b Body weight (BW) was similar among groups. c, d Contractile response to thromboxane analogue (U46619) and phenylephrine (PE) in mesenteric resistance arteries (MRA) was similar among groups. e Endothelial-dependent relaxation to acetylcholine (Ach) was damaged in the MRA of C57/Bl6 mice after Ang II infusion (400 ng/kg/min) and was significantly improved with metformin treatment (300 mg.kg−1 body weight per day). #p < 0.05 vs Sham; *p < 0.05 vs Sham + Ang II. f Endothelial-independent relaxation to sodium nitroprusside (SNP) was similar among groups. g Immunoblots for P-eNOS and T-eNOS and quantification of the ratio of P-eNOS/T-eNOS showed decreased eNOS activity in the MRA of C57/Bl6 mice after Ang II infusion (400 ng/kg/min), which was restored with metformin treatment (300 mg.kg−1 body weight per day). *p < 0.05 vs Sham + Ang II
Metformin improved vascular function
To study the effects of metformin treatment on MRA reactivity, we subjected vessels to cumulative doses of thromboxane analogue (U46619) and PE. Our data indicate that there were no differences in the contractile response between the sham group and the groups infused with Ang II in the presence or absence of metformin (Fig. 1c, d). To determine the role of metformin in vascular endothelial dysfunction in hypertension, we examined EDR by using acetylcholine (Ach). Ang II-induced hypertension attenuated EDR in MRA compared with the sham group (Fig. 1e). Interestingly, metformin treatment significantly improved EDR (Fig. 1e). Endothelium-independent relaxation to SNP was similar among groups (Fig. 1f).
Since ACh produces NO through activation of eNOS, we analyzed the protein levels of total and phosphorylated eNOS. Our data indicate that Ang II-induced hypertension reduced eNOS phosphorylation in MRA, which was restored after metformin treatment (Fig. 1g). The expression of total eNOS protein level was similar among all groups of mice (Fig. 1g).
Metformin inhibited ER stress and restored p-AMPK expression
The aim of this experiment was to study the effect of metformin on AMPK kinase. Our data indicate that ER stress marker (BIP, p-eIF2α/T-eIF2α, p-PERK, CHOP, and ATF6) expression levels in MRA were significantly higher in the Ang II group compared to the sham group (Fig. 2a). Metformin treatment significantly reduced ER stress marker expression levels (Fig. 2a).
Fig. 2.
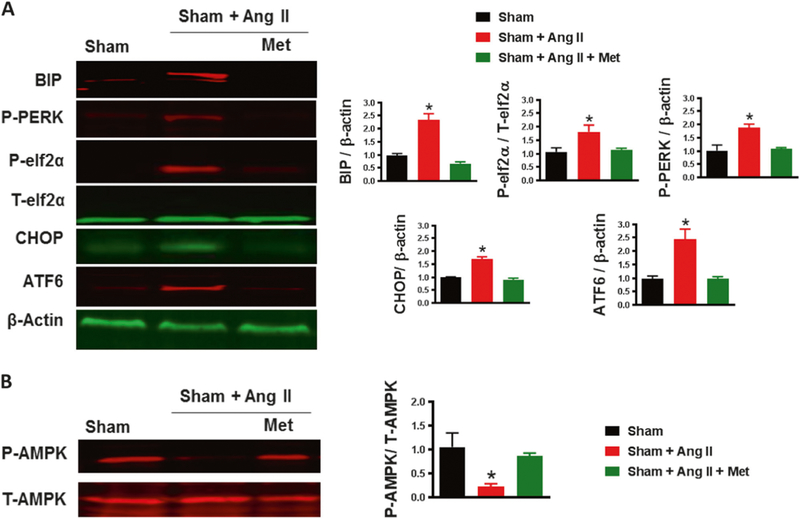
Metformin alleviates hypertension-induced ER stress by activating AMPK. a Immunoblots for ER stress markers (BIP, p-eIF2α/T-eIF2α, p-PERK, CHOP, and ATF6) and quantification showed increased ER stress markers in the MRA of C57/Bl6 mice after Ang II infusion (400 ng/kg/min) that was significantly reduced with metformin treatment (300mg.kg−1 body weight per day). *p < 0.05 vs Sham + Ang II. b Immunoblots for P-AMPK and T-AMPK and quantification of the ratio of P-AMPK/T-AMPK showed decreased P-AMPK in the MRA of C57/Bl6 mice after Ang II infusion (400 ng/kg/min), which was further restored with metformin treatment (300 mg.kg−1 body weight per day). *p < 0.05 vs Sham + Ang II
T-AMPK expression in MRA was similar among groups (Fig. 2b). However, the expression of p-AMPK was significantly reduced after exposure to Ang II compared to the sham group. p-AMPK expression was restored by treatment with metformin (Fig. 2b).
Metformin reduced NADPH activity
This experiment was performed to assess the effect of metformin on oxidative stress. Our results showed that NADPH oxidase activity was higher in the Ang II group compared to the sham group (Fig. 3a). Interestingly, metformin significantly reduced NADPH oxidase activity (Fig. 3a). To confirm our data, we proceeded with an immunostaining experiment using von Willebrand factor (vWF) as a marker for blood vessel endothelial cells and 8-OHDG as a marker for oxidative stress. Our data showed that 8-OHDG was significantly increased in the Ang II-infused mice compared to sham mice. Treatment with metformin significantly reduced expression of 8-OHDG (Fig. 3b).
Fig. 3.
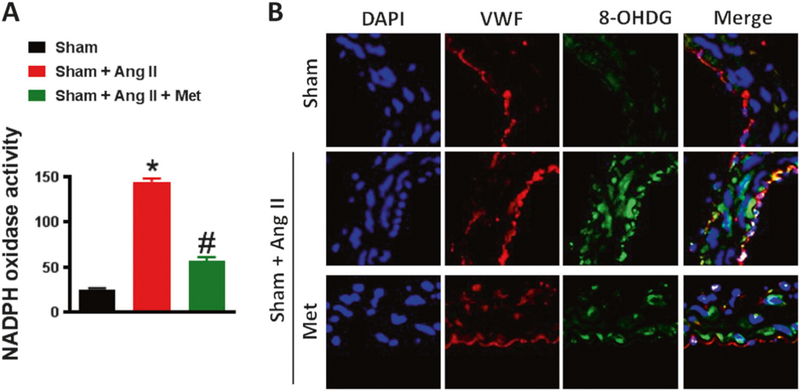
Metformin inhibits oxidative stress. a NADPH oxidase activity and b immunofluorescence for 8-Oxo-2′-deoxyguanosine (8-OHDG) show an increase in oxidative stress in the MRA of C57/Bl6 mice after Ang II infusion (400 ng/kg/min), which was significantly reduced with metformin treatment (300 mg.kg−1 body weight per day).*p < 0.05 vs Sham + Ang II. #p < 0.05 vs Sham
Metformin protects against tunicamycin-induced vascular dysfunction
To further determine the relationship between metformin, ER stress, AMPK, and vascular dysfunction independent of hypertension, we performed in vivo studies by treating mice with an ER stress inducer (tunicamycin) for 2 weeks. The results showed that treatment with tunicamycin had no effect on systolic blood pressure (Fig. 4a).
Fig. 4.
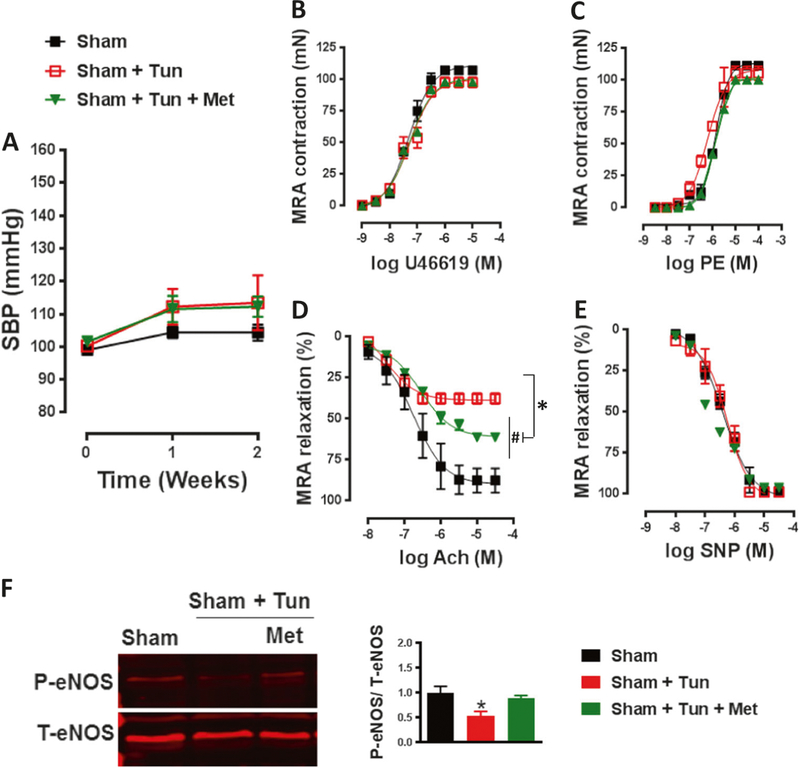
Metformin improves vascular function independent of hypertension. a Systolic blood pressure (SBP) was similar among groups and in the range of normotensive values. b, c Contractile response to thromboxane analogue (U46619) and phenylephrine (PE) in mesenteric resistance arteries (MRA) was similar among groups. d Endothelial-dependent relaxation to acetylcholine (Ach) was damaged in the MRA of C57/Bl6 mice after intraperitoneal injection of tunicamycin (Tun, 1 mg/kg, two injections/week for 2 weeks) and was significantly improved with metformin treatment (300mg.kg−1 body weight per day). #p < 0.05 vs Sham; *p < 0.05 vs Sham + Tun. e Endothelial-independent relaxation to sodium nitroprusside (SNP) was similar among groups. f Immunoblots for P-eNOS and T-eNOS and quantification of the ratio of P-eNOS/T-eNOS showed decreased eNOS activity in the MRA of C57/Bl6 mice after intraperitoneal injection of tunicamycin (Tun, 1 mg/kg, two injections/week for 2 weeks), which was restored with metformin treatment (300mg.kg−1 body weight per day).*p < 0.05 vs Sham + Tun
We did not detect any differences in contractile response to U46619 or PE among groups (Fig. 4b, c). EDR that was damaged after tunicamycin treatment was significantly improved with metformin treatment (Fig. 4d). Endothelium-independent relaxation to SNP was similar among groups (Fig. 4e). We then detected the expression of eNOS, and there was no difference in the expression of T-eNOS among groups (Fig. 4f). Phosphorylated eNOS was reduced in the tunicamycin mice compared to the sham mice. Interestingly, treatment with metformin completely restored the expression of p-eNOS (Fig. 4f).
Metformin inhibited tunicamycin-induced ER stress and restored p-AMPK expression
The rationale of this experiment is to see the effect of metformin on vasculature independent of hypertension. Our data showed that ER stress marker (BIP, p-PERK, and CHOP) expression levels in MRA were higher in the tunicamycin group compared to the sham group (Fig. 5a). Treatment with metformin significantly reduced ER stress marker expression levels (Fig. 5a).
Fig. 5.
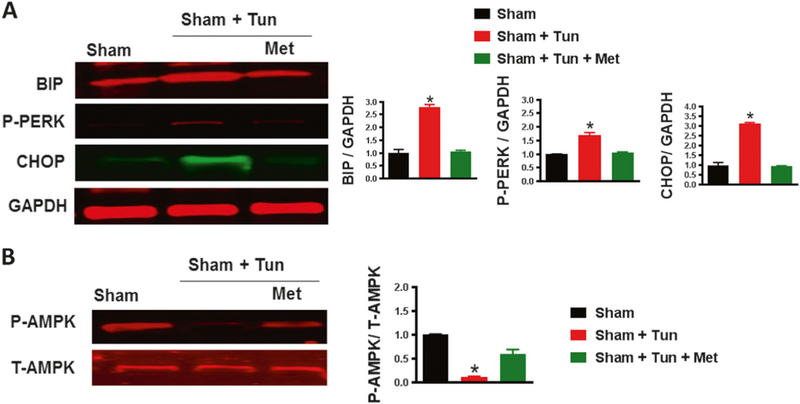
Metformin alleviates tunicamycin-induced ER stress by activating AMPK. a Immunoblots for ER stress markers (BIP, p-PERK, and CHOP) and quantification showing increases in ER stress markers in the MRA of C57/Bl6 mice after intraperitoneal injection of tunicamycin (Tun, 1 mg/kg, two injections/week for 2 weeks) were significantly reduced with metformin treatment (300mg.kg−1 body weight per day). *p < 0.05 vs Sham + Tun. b Immunoblots for P-AMPK and T-AMPK and quantification of the ratio of P-AMPK/T-AMPK showed decreased P-AMPK in the MRA of C57/Bl6 mice after intraperitoneal injection of tunicamycin (Tun, 1 mg/kg, two injections/week for 2 weeks), which was then restored with metformin treatment (300 mg.kg−1 body weight per day). *p < 0.05 vs Sham + Tun
T-AMPK expression in MRA was similar among groups (Fig. 5b). The expression of p-AMPK was largely inhibited by tunicamycin but was restored with metformin (Fig. 5b).
Metformin reduced NADPH activity induced by tunicamycin
This experiment was performed to elucidate the role of metformin in NADPH oxidase activity independent of increased SBP. Our data demonstrated that NADPH oxidase activity was significantly increased in mice treated with tunicamycin compared to the sham group. Moreover, metformin reduced NADPH oxidase activity (Fig. 6a). The data were further confirmed by immunostaining (Fig. 6b).
Fig. 6.
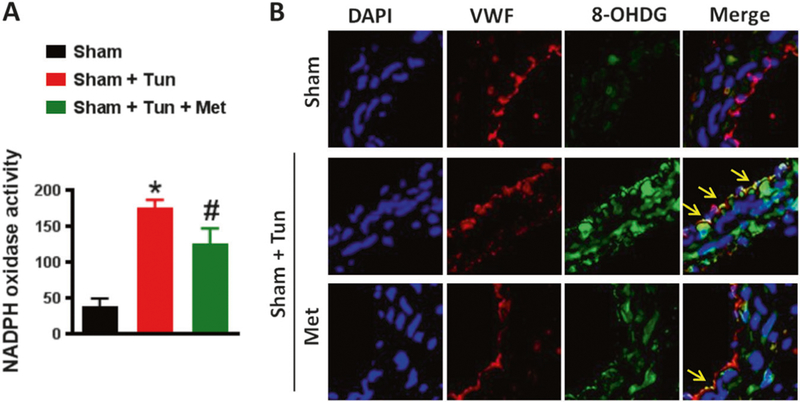
Metformin inhibits oxidative stress. a NADPH oxidase activity and b immunofluorescence for 8-Oxo-2′-deoxyguanosine (8-OHDG) show an increase in oxidative stress in the MRA of C57/Bl6 mice after intraperitoneal injection of tunicamycin (Tun, 1 mg/kg, two injections/week for 2 weeks). Oxidative stress induced by tunicamycin was significantly reduced with metformin treatment (300mg.kg−1 body weight per day). *p < 0.05 vs Sham + Tun. #p < 0.05 vs Sham
It is important to mention that the sham mice group treated with metformin showed no differences in any parameters compared to the sham group alone (data not shown). Thus, to examine the overall data, we did not include the sham group treated with metformin in the results for easier interpretation.
Discussion
In the present study, we demonstrated that metformin inhibited Ang II-induced aberrant ER stress and vascular dysfunction through activation of AMPK. Such evidence came from our in vivo data in which hypertensive mice treated with metformin had reduced blood pressure and ameliorated vascular reactivity function. These results suggest that metformin is an important drug that plays a crucial role in reversing vascular complications during hypertension.
It is well established that hypertension is associated with vascular endothelial dysfunction [18]. We and others have successfully demonstrated that Ang II-induced hypertension reduces EDR, AMPK, and eNOS phosphorylation and enhances ER stress in MRA [19, 20]. Interestingly, treatment with metformin significantly reduced SBP. Our data agree with two studies with SHR rats treated with metformin that show a significant reduction in SBP [21, 22]. Another meta-analysis study suggested that metformin could effectively lower SBP in nondiabetic patients [23]. Lowered blood pressure has also been reported in some clinical studies, such as in nondiabetic, obese, and hypertensive women who received metformin [2]. The results demonstrated the feasibility and benefits of metformin beyond its glycemic control properties in nondiabetic mouse models.
Recently, we and others have documented a link between ER stress, hypertension, and cardiovascular diseases [19]. Additionally, emerging evidence using human and animal models has shown that elevated ER stress is an important contributor to the development of hypertension and cardiovascular diseases [24]. Thus, inhibition of ER stress offers a promising therapeutic tool for treating ER stress-mediated endothelial dysfunction. In the present study, we demonstrated that the activation of ER-induced endothelial dysfunction by a nonchemical inducer (Ang II) and by a chemical inducer (tunicamycin) could be suppressed after treatment with metformin. The relationship between ER stress and metformin is well documented in diabetes [25]. However, little is known about this relationship in nondiabetic hypertensive models, specifically in endothelial cells. A study by Duan et al. demonstrated that metformin treatment alleviated Ang II-induced ER stress in vascular smooth muscle cells (SMC) [20]. One limitation in our study was not differentiating between ECs and SMCs since our western blot was performed using the entire vessel. We assume that treatment with metformin has beneficial effects on ECs and not on SMCs since we did not see any differences among groups in response to contractile agents or NO donor agents, which act directly on SMCs. However, after treatment with metformin, our findings indicate an improvement in the damaged relaxation to ACh, which primarily depends on NO release from the ECs. Additional studies are needed to confirm this point.
AMPK has recently emerged as a key regulator of ER stress in different cell types. [26, 27] It has been reported that AMPK protects cardiomyocytes against hypoxia via attenuation of ER stress and suppresses palmitate-induced ER stress in endothelial cells and liver cells [28–30]. In the vasculature, Duan et al. previously reported that metformin attenuates Ang II-triggered hypertension in mice by activating AMPKα2, and metformin was also shown to inhibit Ang II-induced ER stress in vascular smooth muscle cells [20]. Our data agree with these studies since our results show that hypertensive mice treated with metformin restored the phosphorylated AMPK expression in vessels. Since dysfunction in AMPK signaling contributes to the development of hypertension and cardiovascular diseases [12], increasing AMPK activity during metformin treatment in hypertension may be of therapeutic value.
Ang-II-induced hypertension is a well-studied model for endothelial dysfunction [31]. Metformin treatment showed improvement in EDR associated with an increase in p-eNOS. The relationship between metformin and NO bioavailability has been documented in diabetic models, but little information is known about the relationship between NO and metformin in hypertensive models [32]. Although studies have shown that treating SHR rats with metformin increases NO in plasma and kidneys, NO bioavailability in the vasculature has not yet been evaluated [33]. Similarly, Pitocco et al. showed that treatment of streptozotocin (STZ)-SHR rats with metformin upregulated NO and thereby improved EDR [34]. Increased eNOS activation after metformin treatment is also in agreement with the results of a previous study by Davis et al., which showed that metformin improves endothelial vascular function by increasing AMPK-dependent, hsp90-mediated eNOS activation in diabetes mellitus [32]. Furthermore, a study performed by Katakam et al. showed that metformin improves vascular function through a direct mechanism rather than by improving metabolic abnormalities [35]. Interestingly, while p-eNOS was restored, the EDR was partially recovered in our treated mice. A possible explanation is that superoxide anions, known to be induced during hypertension [36], neutralize endothelium-derived NO, which further results in diminished endothelium-dependent relaxation [37].
To evaluate this hypothesis, we measured the level of NADPH oxidase among groups. NADPH oxidases are the major sources of ROS in the arterial walls in pathological conditions, such as hypertension, diabetes, and aging. Therefore, NADPH oxidases are important contributors to oxidative stress and endothelial dysfunction [38]. Our data indicate that metformin reduced NADPH oxidase activity but not to the same level as the control. Therefore, the excessive O2 reduced the NO bioavailability, which could explain why a full relaxation did not occur in the hypertensive group treated with metformin. A study conducted by Anh et al. showed that endothelial cells treated with glucose in the presence of metformin was mediated by NADPH oxidase inhibition [39]. Additionally, studies have shown that metformin suppresses NADPH oxidase in podocytes [40]. Such data confirm the inhibitory role of metformin on NADPH oxidase in a nondiabetic model.
Treatment with metformin improved vascular function associated with a reduction in SBP. However, it was difficult to conclude whether the effect of metformin on AMPK, ER stress, NADPH oxidase, and eNOS was a direct or a consequence of the reduction in SBP. To confirm the direct effect of metformin on ER stress and to rule out that the beneficial effects of metformin on the vasculature were due to SBP reduction, we used a well-established model to induce ER stress in vivo without producing an increase in SBP. We treated mice with tunicamycin, which is a well-known ER stress inducer [18]. Our data indicate that mice treated with tunicamycin did not have increased SBP. SBP was in the same range as normotensive mice. These data agree with our previous study [12]. Our data indicate that metformin increases AMPK, inhibits tunicamycin-induced ER stress and NADPH oxidase, restores eNOS activity, and improves EDR. All these effects caused by metformin were independent of changes in SBP. Thus, metformin appears to improve vascular function independent of SBP. This study showed some of the beneficial pleiotropic effects of metformin independent of its effects on diabetes.
In summary, we demonstrated that metformin, independent of changes in SBP, improves vascular endothelium-dependent relaxation, increases eNOS activation, and suppresses ER stress and NADPH oxidase activity by activating AMPK. These results suggest that activation of metformin could be a useful therapeutic strategy for reversing vascular complications and cardiac damage from hypertension.
Footnotes
Compliance with ethical standards
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Conflict of interest The authors declare that they have no conflict of interest.
References
- 1.Pernicova I, Korbonits M. Metformin mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10:143–56. [DOI] [PubMed] [Google Scholar]
- 2.Giugliano D, De Rosa N, Di Maro G, Marfella R, Acapora R, Buoninconti R, et al. Metformin improves glucose, lipid metabolism, and reduces blood pressure in hypertensive, obese women. Diabetes Care. 1993;16:1387–90. [DOI] [PubMed] [Google Scholar]
- 3.He H, Zhao Z, Chen J, Ni y, Zhong J, Yan Z, et al. Metformin-based treatment for obesity-related hypertension: a randomized, double-blind, placebo-controlled trial. J Hypertens. 2012;30:1430–9. [DOI] [PubMed] [Google Scholar]
- 4.Muntzel MS, Nyeduala B, Barrett S. High dietary salt enhances acute depressor responses to metformin. Am J Hypertens. 1999;12:1256–9. [DOI] [PubMed] [Google Scholar]
- 5.Cody RJ. The sympathetic nervous system and the renin-angiotensin-aldosterone system in cardiovascular disease. Am J Cardiol. 1997;80:9J–14J. [DOI] [PubMed] [Google Scholar]
- 6.Ohishi M Hypertension with diabetes mellitus: physiology and pathology. Hypertens Res. 2018;41:389–93. [DOI] [PubMed] [Google Scholar]
- 7.Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, et al. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci Usa. 2006;103:17985–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campbell DJ. Do intravenous and subcutaneous angiotensin II increase blood pressure by different mechanisms? Clin Exp Pharmacol Physiol. 2013;40:560–70. [DOI] [PubMed] [Google Scholar]
- 9.Hasty AH, Harrison DG. Endoplasmic reticulum stress and hypertension—anew paradigm? J Clin Invest. 2012;122:3859–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deji N, Kume S, Araki S, Isshiki K, Araki H, Chin-Kanasaki M, et al. Role of angiotensin II-mediated AMPK inactivation on obesity-related salt-sensitive hypertension. Biochem Biophys Res Commun. 2012;418:559–64. [DOI] [PubMed] [Google Scholar]
- 11.Ford RJ, Rush JW. Endothelium-dependent vasorelaxation to the AMPK activator AICAR is enhanced in aorta from hypertensive rats and is NO and EDCF dependent. Am J Physiol Heart Circ Physiol. 2011;300:H64–H75. [DOI] [PubMed] [Google Scholar]
- 12.Liang B, Wang S, Wang Q, Zhang W, Viollet B, Zhu Y, et al. Aberrant endoplasmic reticulum stress in vascular smooth muscle increases vascular contractility and blood pressure in mice deficient of AMP-activated protein kinase-α2 in vivo. Arterioscler Thromb Vasc Biol. 2013;33:595–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dong Y, Zhang M, Liang B, Xie Z, Zhao Z, Asfa S, et al. Reduction of AMP-activated protein kinase alpha2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation. 2010;121:792–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kassan M, Ait-Aissa K, Radwan E, Mali V, Haddox S, Gabani M, et al. Essential Role of Smooth Muscle STIM1 in Hypertension and Cardiovascular Dysfunction. Arterioscler Thromb Vasc Biol. 2016;36:1900–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kassan M, Galan M, Partyka M, Trebak M, Matrougui K. Interleukin-10 released by CD4(+)CD25(+) natural regulatory T cells improves microvascular endothelial function through inhibition of NADPH oxidase activity in hypertensive mice. Arterioscler Thromb Vasc Biol. 2011;31:2534–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Linden MA, Lopez KT, Fletcher JA, Morris EM, Meers GM, Siddique S, et al. Combining metformin therapy with caloric restriction for the management of type 2 diabetes and nonalcoholic fatty liver disease in obese rats. Appl Physiol Nutr Metab. 2015;40:1038–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–4. [DOI] [PubMed] [Google Scholar]
- 19.Kassan M, Galán M, Partyka M, Saifudeen Z, Henrion D, Trebak M, et al. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arterioscler Thromb Vasc Biol. 2012;32:1652–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duan Q, Song P, Ding Y, Zou MH. Activation of AMP-activated protein kinase by metformin ablates angiotensin II-induced endoplasmic reticulum stress and hypertension in mice in vivo. Br J Pharmacol. 2017;174:2140–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamidi Shishavan M, Henning RH, van Buiten A, Goris M, Deelman LE, Buikema H. Metformin improves endothelial function and reduces blood pressure in diabetic spontaneously hypertensive rats independent from glycemia control: comparison to vildagliptin. Sci Rep. 2017;7:10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verma S, Bhanot S, McNeill JH. Metformin decreases plasma insulin levels and systolic blood pressure in spontaneously hypertensive rats. Am J Physiol. 1994;267:H1250–H1253. [DOI] [PubMed] [Google Scholar]
- 23.Zhou L, Liu H, Wen X, Peng Y, Tian Y, Zhao L. Effects of metformin on blood pressure in nondiabetic patients: a meta-analysis of randomized controlled trials. J Hypertens. 2017;35:18–26. [DOI] [PubMed] [Google Scholar]
- 24.Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004;110:705–12. [DOI] [PubMed] [Google Scholar]
- 25.Cheang WS, Tian XY, Wong WT, Lau CW, Lee SS, Chen ZY, et al. Metformin protects endothelial function in diet-induced obese mice by inhibition of endoplasmic reticulum stress through 5’ adenosine monophosphate-activated protein kinase-peroxisome proliferator-activated receptor δ pathway. Arterioscler Thromb Vasc Biol. 2014;34:830–6. [DOI] [PubMed] [Google Scholar]
- 26.Hwang SL, Jeong YT, Li X, Kim YD, Lu Y, Chang YC, et al. Inhibitory cross-talk between the AMPK and ERK pathways mediates endoplasmic reticulum stress-induced insulin resistance in skeletal muscle. Br J Pharmacol. 2013;169:69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim A, Im M, Ma JY. Ethanol extract of Remotiflori radix induces endoplasmic reticulum stress-mediated cell death through AMPK/mTOR signaling in human prostate cancer cells. Sci Rep. 2015;5:8394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu L, Zhou L, Wu X, Liu C, Fan Y, Li Q. Hypoxic pre-conditioning protects cardiomyocytes against hypoxia/reoxygenation injury through AMPK/eNOS/PGC-1α signaling pathway. Int J Clin Exp Pathol. 2014;7:7378–88. [PMC free article] [PubMed] [Google Scholar]
- 29.Li J, Wang Y, Wang Y, Wen X, Ma XN, Chen W, et al. Pharmacological activation of AMPK prevents Drp1-mediated mitochondrial fission and alleviates endoplasmic reticulum stress-associated endothelial dysfunction. J Mol Cell Cardiol. 2015;86:62–74. [DOI] [PubMed] [Google Scholar]
- 30.Jung TW, Lee SY, Hong HC, Choi HY, Yoo HJ, Baik SH, et al. AMPK activator-mediated inhibition of endoplasmic reticulum stress ameliorates carrageenan-induced insulin resistance through the suppression of selenoprotein P in HepG2 hepatocytes. Mol Cell Endocrinol. 2014;382:66–73. [DOI] [PubMed] [Google Scholar]
- 31.Yamamoto E, Kataoka K, Shintaku H, Yamashita T, Tokutomi Y, Dong YF, et al. Novel mechanism and role of angiotensin II induced vascular endothelial injury in hypertensive diastolic heart failure. Arterioscler Thromb Vasc Biol. 2007;27:2569–75. [DOI] [PubMed] [Google Scholar]
- 32.Davis BJ, Xie Z, Viollet B, Zou MH. Activation of the AMP-activated kinase by antidiabetes drug metformin stimulates nitric oxide synthesis in vivo by promoting the association of heat shock protein 90 and endothelial nitric oxide synthase. Diabetes. 2006;55:496–505. [DOI] [PubMed] [Google Scholar]
- 33.Tsai CM, Kuo HC, Hsu CN, Huang LT, Tain YL. Metformin reduces asymmetric dimethylarginine and prevents hypertension in spontaneously hypertensive rats. Transl Res. 2014;6:452–9. [DOI] [PubMed] [Google Scholar]
- 34.Pitocco D, Zaccardi F, Tarzia P, Milo M, Scavone G, Rizzo P, et al. Metformin improves endothelial function in type 1 diabetic subjects: a pilot, placebo-controlled randomized study. Diabetes Obes Metab. 2013;15:427–31. [DOI] [PubMed] [Google Scholar]
- 35.Katakam PV, Ujhelyi MR, Hoenig M, Miller AW. Metformin improves vascular function in insulin-resistant rats. Hypertension. 2000;35:108–12. [DOI] [PubMed] [Google Scholar]
- 36.Laplante MA, Wu R, Moreau P, de Champlain J. Endothelin mediates superoxide production in angiotensin II-induced hypertension in rats. Free Radic Biol Med. 2005;38:589–96. [DOI] [PubMed] [Google Scholar]
- 37.Gumusel B, Tel BC, Demirdamar R, Sahin-Erdemli I. Reactive oxygen species-induced impairment of endothelium-dependent relaxation in rat aortic rings: protection by L-arginine. Eur J Pharmacol. 1996;306:107–12. [DOI] [PubMed] [Google Scholar]
- 38.Konior A, Schramm A, Czesnikiewicz-Guzik M, Guzik TJ. NADPH oxidases in vascular pathology. Antioxid Redox Signal. 2014;20:2794–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.An H, Wei R, Ke J, Yang J, Liu Y, Wang X. Metformin attenuates fluctuating glucose-induced endothelial dysfunction through enhancing GTPCH1-mediated eNOS recoupling and inhibiting NADPH oxidase. J Diabetes Complicat. 2016;30:1017–24. [DOI] [PubMed] [Google Scholar]
- 40.Piwkowska A, Rogacka D, Jankowski M, Dominiczak MH, Stepiński JK, Angielski S. Metformin induces suppression of NAD(P)H oxidase activity in podocytes. Biochem Biophys Res Commun. 2010;393:268–73. [DOI] [PubMed] [Google Scholar]