

Abstract
A safer treatment for toxoplasmosis would be achieved by improving the selectivity and potency of dihydrofolate reductase (DHFR) inhibitors, such as pyrimethamine (1), for Toxoplasma gondii DHFR (TgDHFR) relative to human DHFR (hDHFR). We previously reported on the identification of meta-biphenyl analog 2, designed by in silico modeling of key differences in the binding pocket between TgDHFR and hDHFR. Compound 2 improves TgDHFR selectivity 6.6-fold and potency 16-fold relative to 1. Here, we report on the optimization and structure-activity relationships of this arylpiperazine series leading to the discovery of 5-(4-(3-(2-methoxypyrimidin-5-yl)phenyl)piperazin-1-yl)pyrimidine-2,4-diamine 3. 2-Methoxypyrimidine 3 has a TgDHFR IC50 of 1.57 ± 0.11 nM and a hDHFR to TgDHFR selectivity ratio of 196, making it 89-fold more potent and 16-fold more selective than 1. Compound 3 was highly effective in control of acute infection by highly virulent strains of T. gondii in the murine model and it possesses the best combination of selectivity, potency and prerequisite drug-like properties to advance into IND-enabling, pre-clinical development.
Graphical Abstract
INTRODUCTION
Toxoplasmosis is caused by the protozoan parasite Toxoplasma gondii (T. gondii), an obligate intracellular parasite capable of infecting a wide range of hosts and many different types of cells1. Approximately one third of the population worldwide is chronically infected with various strains of T. gondii2, with the seroprevalence being variable across the world. For example, T. gondii seroprevalence in the United States is estimated at about 10 to 15% of the population3, it is >60% in Brazil, the Philippines, and Madagascar; 40% to 60% in Egypt, Argentina, and several European countries, including France, Germany and Poland; and 20% to 40% in Australia, Chile, Saudi Arabia and Iran4–5. Infections occur through consuming contaminated food or water, mother-to-child (congenital/vertical) and iatrogenic (transplanted organs and blood transfusion) transmission6. The parasitic infection is kept in check by the immune system where it exists in a latent bradyzoite form contained within tissue cysts that are commonly found in skeletal muscle and the central nervous system (CNS)7. However, in cases where the immune system is compromised, such as in a developing fetus or in patients with HIV, undergoing cancer chemotherapy or immunosuppressive treatment for organ transplantation, the parasite can transition to an active, fast replicating, and tissue damaging tachyzoite form8. Depending upon localization of the tachyzoites, active infection can cause, among other conditions, myocarditis, blindness and encephalitis; and is associated with a high mortality rate for HIV patients, even for those on active anti-retroviral treatment8–11.
The recommended first-line treatment for toxoplasmosis is a combination therapy based on pyrimethamine (1) and sulfadiazine, supplemented with leucovorin (also known as folinic acid) to protect against bone marrow suyppression11–12. Pyrimethamine and sulfadiazine act synergistically on the folate metabolic pathway thereby inhibiting T. gondii proliferation and survival13–14. The drugs inhibit DHFR and dihydropteroate synthase (DHPS), respectively, and consequently block the synthesis of tetrahydrofolate, a key cofactor for thymidylate synthase. Thymidylate synthase is a methyl transferase that to produces deoxythymidine monophosphate from deoxyuridine monophosphate. In many organisms including T. gondii and humans, this pathway is the only means by which thymidine can be supplied, making it essential for DNA synthesis and cellular proliferation14–15. Importantly, although the combination of pyrimethamine and sulfadiazine can control actively proliferating forms such as tachyzoites, these agents have little effect on the semi-dormant bradyzoite stages within tissue cysts and hence do not cure infection16.
Adverse events associated with pyrimethamine-based therapy in toxoplasmosis often result in a need to reduce dosing or discontinue therapy17. The adverse events are mainly mechanism-based related to inhibition of folic acid metabolism in host tissues with high metabolic activity (e.g., epithelium and bone marrow)18. In mice, pyrimethamine has only about a 3-fold safety multiple between minimal effective dose and maximum tolerated dose19. Leucovorin, a form of tetrahydrofolate that is selectively taken up by human cells, is often co-administered to help alleviate the impact of mechanism-based toxicity to the host20–23. The use of pyrimethamine is also not suitable during the first trimester of pregnancy owing to its inhibition of human DHFR which can impact fetal organogenesis23. In addition, about 3% of the general population and 30% of patients with HIV/AIDS have a hypersensitivity reaction to the sulfonamide component of the treatment regimen24–26. Therefore, finding a standalone treatment would be of significant benefit to the HIV patient population most at risk of toxoplasmosis encephalitis and sulfa hypersensitivities. Finally, high doses of pyrimethamine can induce seizures, a likely consequence of off-target pharmacology27. The adverse events associated with pyrimethamine and sulfadiazine therapy could be reduced or eliminated by selectively inhibiting only the parasite DHFR. There have been numerous attempts to discover more selective inhibitors of TgDHFR, but with limited success28–32. Recently, the crystal structure of TgDHFR was solved33. This finding has enabled comparative modeling between the human and T. gondii enzymes, leading to the design and discovery of more selective TgDHFR small molecules such as TRC-19 (2)34.
Ideally, a next generation TgDHFR inhibitor would have sufficient selectivity so that maximal parasiticidal activity would occur without concomitant inhibition of human hDHFR. Therefore, we hypothesized it would be important that the compound exhibit TgDHFR to hDHFR selectivity of at least 150-fold, which is about 10-fold greater than that of pyrimethamine and possess low clearance to minimize maximal plasma concentrations while achieving a long duration of maximally effective exposure. Given the parasites ability to invade the CNS and cause significant harm upon reactivation of semi-dormant tissue cysts, it is also necessary that the compound readily penetrate the blood-brain barrier. Finally, improving TgDHFR potency can reduce effective dose levels and decrease the likelihood of compound related off-target pharmacology and associated side effects and toxicity.
In this study, we report on the SAR and lead optimization strategy underlying the discovery of 5-(4-(3-(2-methoxypyrimidin-5-yl)phenyl)piperazin-1-yl)pyrimidine-2,4-diamine 3, a selective and potent TgDHFR inhibitor with properties consistent with those outlined above supporting a standalone treatment for toxoplasmosis.
RESULTS AND DISCUSSION
Pyrimethamine selectivity and potency.
Pyrimethamine 1 is reported to be 7.6-fold more selective for TgDHFR compared to hDHFR with an IC50 of 760 ± 130 nM for inhibiting conversion of dihydrofolate to tetrahydrofolate35. In our hands, 1 has an IC50 for inhibiting TgDHFR of 139 ± 49 nM (Figure 1), which makes it 12-fold more potent for inhibiting TgDHFR over hDHFR. Although this level of selectivity is sufficient to make 1 a preferred DHFR inhibitor for the treatment of toxoplasmosis, as can be seen in Figure 1, it is not sufficient to preclude inhibition of hDHFR and associated dose limiting mechanism-based toxicity36. The concentration of 1 that gives significant inhibition of TgDHFR, i.e., > IC80, begins to also inhibit hDHFR by 5 to 35%. Based on these data, it seemed reasonable that at least an order of magnitude and preferably 20-fold increase in selectivity in a new compound would be sufficient to overcome the mechanism-based toxicity issues observed with 1 and possibly enable a standalone therapy.
Figure 1.

aDose response curve of 1 for inhibition of T. gondii DHFR and human DHFR
aThe shaded blue area represents the concentration of 1 with >80% inhibition of TgDHFR and 5 to 35% hDHFR.
Early profiling data for lead 2 and comparison to DHFR inhibitor reference standard drugs.
The structure of lead 234 and DHFR inhibitor reference compounds pyrimethamine (1), trimethoprim (4), methotrexate (5) and trimetrexate (6) are shown in Figure 2. The DHFR inhibitory activity and selectivity, kinetic solubility, human liver microsomal intrinsic clearance and permeability and efflux across an MDR1-MDCK cell monolayer for lead 2 and reference compounds 1, 4, 5 and 6 are shown in Table 1. Lead 2 has a TgDHFR IC50 of 8.76 ± 1.0 nM with 79-fold selectivity relative to the human enzyme. This is 6.6-fold more selective than 1. Although this improvement in selectivity was encouraging, in our view is insufficient to achieve a stand-alone treatment. Trimethoprim 4, sometimes used off label in combination with sulfamethoxazole for the treatment of toxoplasmosis, was about 240-fold less potent against TgDHFR than 1 with an IC50 of 33,100 nM and selectivity index of 15. Methotrexate 5 was far more potent than 1, 4 or 2 for inhibiting hDHFR with an IC50 of 4.74 nM. Interestingly, 5 has the opposite selectivity profile as compared to 1, where it is 17-fold less potent for inhibiting TgDHFR (IC50 of 78.3 nM), giving a selectivity ratio of 0.061. Trimetrexate 6, which structurally is a combination of the diaminopteridine head of 5 and trimethoxybenzyl tail of 4, is nearly equally active at TgDHFR and hDHFR with respective IC50’s of 1.35 and 4.07 nM, giving a selectivity index of 3.0.
Figure 2.

Reference compounds Pyrimethamine (1), Trimethoprim (4), Methotrexate (5), Trimetrexate (6) and initial lead TRC-19 (2).
Table 1.
Evaluation of Comparator Compounds on DHFR Inhibition, Kinetic Solubility, Clearance in Human Liver Microsomes and MDR1-MDCK Permeability
| Compound | TgDHFRa IC50 (nM) | hDHFRa IC50 (nM) | Selectivity Indexb | Solubilityc (μM) | HLM (mL/min/kg) | MDR1-MDCKd |
|---|---|---|---|---|---|---|
| 1 | 139 ± 14 | 1680 ± 230 | 12 | 184 | <8.6 | 36 (0.83) |
| 2 | 8.76 ± 1.0 | 689 ± 39 | 79 | 3.9 | 28.4 | 2.7 (0.62) |
| 4 | 33,100 ± 3400 | 503,000 ± 51,000 | 15 | -- | <8.6 | 7.5 (3.5) |
| 5 | 78.3 ± 6.7 | 4.74 ± 0.36 | 0.061 | -- | -- | -- |
| 6 | 1.35 ± 0.07 | 4.07 ± 0.11 | 3.0 | -- | -- | -- |
Average of at least 3 independent replicates ± SEM.
Selectivity index (SI) is the hDHFR IC50/TgDHFR IC50, determined within the same experiment.
Kinetic solubility.
A:B (papp 10−6 cm/sec) (ER), where ER is efflux ratio defined as permeability in A:B/B:A directions.
Further characterization of 1 and 2 was performed to benchmark and guide a lead optimization strategy (Table 1). Lead 2 has low kinetic and thermodynamic solubility (< 4 μM), high clearance in human liver microsomes (HLM) (i.e. > liver blood flow), and low to moderate MDCK-MDR1 permeability, all areas for improvement during lead optimization. Interestingly, 1, which was discovered and developed in the 1940s, has near ideal CNS drug properties in terms of solubility, HLM stability and MDR1-MDCK permeability. Properties for 1 and 2 are summarized in Table 2, with the main differences being an increase in both MW and log P for 2, which likely contributes to its undesirable solubility, metabolic stability and permeability profile. Based on these data, lead optimization was initially focused on understanding the SAR underlying selectivity and potency, while simultaneously designing analogs with lower cLog P and higher calculated solubility in an effort to improve physical chemical properties.
Table 2.
aProperties of 1 and 2
| Compound | MW | cLog P | Log Pb | TPSA | cSol (μM) |
|---|---|---|---|---|---|
| 1 | 248.7 | 3.00 | 2.71 | 76.8 | 135 |
| 2 | 346.4 | 3.74 | 3.38 | 83.2 | 3.09 |
Calculated properties were performed using ChemDraw® Professional software version 17.0.0.206 (121).
Measured LogP was performed at pH 11.0 in octanol/buffer.
Structure-activity relationships for analogs of 2.
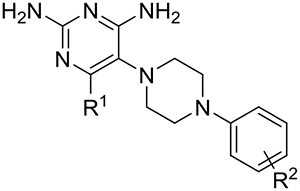
Structure activity relationship (SAR) data based on 2 for inhibition of TgDHFR and selectivity relative to hDHFR are shown in Tables 3–5. Initial optimization of 2 focused on a systematic approach to gain a better understanding of its SAR with emphasis on improving DHFR selectivity (Table 3). As a basis for comparison, unsubstituted analog 7 was prepared and found to lose potency (IC50 209 nM) but maintained a similar selectivity index (59-fold) relative to initial lead 2. Ortho-phenyl 8 and para-phenyl 9 were both about 4-fold less potent (IC50 ~ 30 nM) than 2 but differed in their selectivity profiles. Ortho-phenyl 8 maintained similar selectivity as 2 at 58-fold, whereas para-phenyl 9 had reduced selectivity at 11-fold. These data are consistent with modeling predictions of the binding cavity34. Replacing the distal meta-phenyl of 2 with an ortho-methyl 10 or ortho-chloro 13 resulted in complete loss of activity at the highest doses tested. This result is surprising considering ortho-phenyl analog 8 had a TgDHFR IC50 of 31 nM. Meta-methyl and meta-chloro analogs 11 and 14, respectively, were more potent and selective than corresponding para-substituted compounds 12 and 15, supporting the hypothesis based on modeling that meta-substituents would be preferred. Encouragingly, meta-methyl 11 while 8-fold less potent than 2, maintained equivalent selectivity of 79-fold. Based on these findings, design focused on meta-substituents to further probe this apparently important pocket for both selectivity and potency. Meta-methoxy 16, meta-trifluoromethyl 17, meta-trifluoromethoxy 18 and meta-cyclopropyl 19 all had reduced potency and selectivity as compared to meta-phenyl lead 2. Adding a methyl or ethyl to the 6-position of the diaminopyrimidine group, giving 20 and 21 respectively, maintained or slightly improved potency over 2, but with a slight loss in selectivity.
Table 3.
Structure-Activity Relationships of phenylpiperazine analogs on inhibition of TgDHFR and hDHFR
 |
|||||
|---|---|---|---|---|---|
| Compound | R2 | R1 |
TgDHFRa IC50 (nM) |
hDHFRa IC50 (nM) |
Selectivity Indexb |
| 2 | 3-phenyl | H | 8.76 ± 1.0 | 689 ± 39 | 79 |
| 7 | H | H | 209 ± 28 | 12,500 ± 1200 | 59 |
| 8 | 2-phenyl | H | 30.9 ± 2.2 | 1790 ± 190 | 58 |
| 9 | 4-phenyl | H | 31.5 ± 2.7 | 347 ± 23 | 11 |
| 10 | 2-Me | H | >10,000 | >30,000 | Not available |
| 11 | 3-Me | H | 75.3 ± 5.5 | 5980 ± 320 | 79 |
| 12 | 4-Me | H | 181 ± 14 | 3260 ± 110 | 18 |
| 13 | 2-Cl | H | >10,000 | >30,000 | Not available |
| 14 | 3-Cl | H | 39.8 ± 5.3 | 1540 ± 170 | 39 |
| 15 | 4-Cl | H | 45.9 ± 7.3 | 1090 ± 140 | 24 |
| 16 | 3-MeO | H | 57.8 ± 3.4 | 2710 ± 150 | 47 |
| 17 | 3-CF3 | H | 62.1 ± 5.2 | 541 ± 43 | 8.7 |
| 18 | 3-CF3O | H | 12.9 ± 1.1 | 819 ± 36 | 63 |
| 19 | 3-cyPropyl | H | 29.0 ± 1.7 | 1630 ± 45 | 56 |
| 20 | 3-phenyl | Me | 9.19 ± 0.94 | 237 ± 21 | 26 |
| 21 | 3-phenyl | Et | 3.91 ± 0.61 | 200 ± 16 | 51 |
Average of at least 3 independent replicates ± SEM.
Selectivity index (SI) is the hDHFR IC50/TgDHFR IC50.
Table 5.
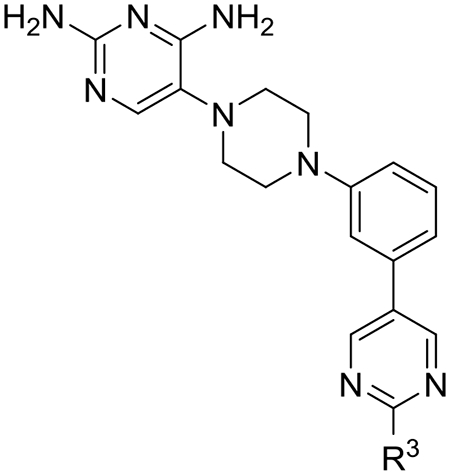
Evaluation of substitutions on the distal pyrimidine ring of 26 on DHFR inhibition, selectivity, solubility, human liver microsomal clearance and MDCK-MDR-1 permeability
 |
|||||||
|---|---|---|---|---|---|---|---|
| Compound | R3 |
TgDHFRa IC50 (nM) |
hDHFRa IC50 (nM) |
Selectivity indexb | Solubility (μM)c |
HLMd (CLint) |
MDR1-MDCKe |
| 29 | Me | 3.95 ± 0.26 | 935 ± 46 | 237 | 150 | <8.6 | 19.3 (1.2) |
| 3 | MeO | 1.57 ± 0.42 | 308 ± 71 | 196 | 186 | <8.6 | 20.1 (1.1) |
| 30 | CF3 | 32.7 ± 3.3 | 2800 ± 470 | 85.6 | 17 | 10.5 | -- |
| 31 | cypropyl | 9.92 ± 0.83 | 973 ± 120 | 98 | 12 | 17.4 | -- |
Average of at least 3 independent replicates ± SEM.
Selectivity index (SI) is the hDHFR IC50/TgDHFR IC50, determined within the same experiment.
Kinetic solubility.
HLM human liver microsomes, CLint intrinsic clearance (mL/min/kg),
A:B permeability (papp 10−6 cm/sec), where ER is efflux ratio defined as permeability in A:B/B:A directions.
Structure-activity relationships for heteroaryl analogs of meta-biphenyl 2.
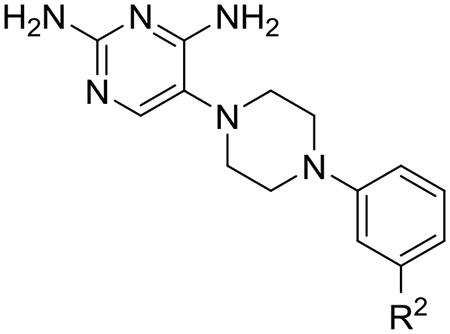
We then turned our focus toward improving the solubility and metabolic stability issues observed with early lead 2 by preparing a variety of heteroaryl analogs (Table 4). We hypothesized that the distal phenyl ring of 2 was the likely site of metabolism and that introducing nitrogen molecules into this ring would block metabolism and increase solubility. The 2-, 3- and 4-pyridine analogs 22, 23 and 24 maintained or slightly improved potency and selectivity, but without much improvement in solubility. The potency of 3-pyridine analog 23 is noteworthy at 2.7 nM, about 3.3-fold more potent than 2, and with a selectivity index of 112-fold. Addition of a second nitrogen into this ring to give pyrimidines, pyridazines and pyrazines provided analogs with variable improvements in potency, selectivity, solubility and HLM metabolic stability. 5-Pyrimidine analog 26 stood out with a 307-fold TgDHFR selectivity index and a TgDHFR IC50 of 5.2 nM, as well as markedly improved solubility (>200 uM) and metabolic stability (Clint < 8.6 mL/min/kg). The improvement in selectivity observed for 26 is nearly 4-times that of early lead 2 and 25-fold better than 1. 2-Pyrazine 27 and 4-pyridazine 28 had promising potency with IC50s of 10 and 6.0 nM respectively, and selectivity indexes of 129 and 185-fold respectively, but both were hampered with low kinetic solubility, precluding additional investigation with these chemotypes. In general, heteroaryl substitutions increase TgDHFR selectivity and maintain or slightly improve potency relative to 2. The two-fold improvement in selectivity observed for 5-pyrimidine analog 26 compared to the 3-pyridyl analog 23 may result from one of the nitrogen atoms on the distal pyrimidine ring being forced to make an unfavorable interaction in hDHFR. The 3-pyridyl analog 23, which can rotate the nitrogen away from such an unfavorable interaction is about 5-fold more potent at hDHFR than pyrimidine 26 (hDHFR IC50 1430 nM).
Table 4.
Evaluation of heteroaryl replacements of the distal phenyl of 2 on DHFR inhibition, selectivity, solubility, human liver microsomal clearance and MDCK-MDR1 permeability.
 |
|||||||
|---|---|---|---|---|---|---|---|
| Compound | R2 |
TgDHFRa IC50 (nM) |
hDHFRa IC50 (nM) |
Selectivityb | Solubilityc (μM) |
HLMd (CLint) |
MDR1-MDCKe |
| 22 | 2-pyridine | 10.3 ± 1.6 | 1360 ± 210 | 132 | 12 | 17.7 | -- |
| 23 | 3-pyridine | 2.68 ± 0.12 | 300 ± 55 | 112 | -- | -- | -- |
| 24 | 4-pyridine | 4.34 ± 0.43 | 468 ± 32 | 108 | 2.2 | 18.7 | 17.4 (0.86) |
| 25 | 2-pyrimidine | 27.3 ± 2.3 | 1300 ± 51 | 47.6 | 180 | <8.6 | 31.7 (0.80) |
| 26 | 5-pyrimidine | 4.75 ± 0.34 | 1430 ± 100 | 301 | >200 | <8.6 | 20.2 (1.2) |
| 27 | 2-pyrazine | 10.3 ± 0.39 | 1330 ± 50 | 129 | 1.6 | <8.6 | 19.3 (0.77) |
| 28 | 4-pyridazine | 6.00 ± 0.70 | 1100 ± 130 | 185 | 5.7 | <8.6 | 6.40 (3.8) |
Average of at least 3 independent replicates ± SEM.
Selectivity index (SI) is the hDHFR IC50/TgDHFR IC50, determined within the same experiment.
Kinetic solubility.
HLM human liver microsomes, CLint intrinsic clearance (mL/min/kg),
A:B permeability (papp 10−6 cm/sec), where ER is efflux ratio defined as permeability in A:B/B:A directions.
SAR for the distal 5-pyrimidine ring of 26.
Next, we evaluated if selectivity could be further improved by adding substitutions onto the 2-position of the distal 5-pyrimidine ring of 26 (Table 5). Based on the crystal structure and modeling (see Figures 3 and 4), it appeared that substitutions at this position were not likely to have steric effects as they pointed into solvent space. However, if the pyrimidine nitrogen atoms were making a favorable hydrogen bond interaction with TgDHFR His-27 and an unfavorable interaction with hDHFR Pro-26, then electron donating substituents would be predicted to enhance potency and selectivity. Methyl analog 29 maintained TgDHFR potency (IC50 3.95 nM) with only a slight reduction in selectivity (237-fold) relative to unsubstituted 5-pyrimidine 26. Methyl analog 29 had good solubility, HLM stability and MDCK-MDR1 permeability. An electron donating 2-methoxy substituent gave 3, with excellent TgDHFR potency with an IC50 of 1.57 nM, but a slightly reduced selectivity index of 196. Trifluoromethyl 30 and cyclopropyl 31 were significantly less potent against TgDHFR, less selective and had low kinetic solubility. The loss in selectivity with these analogs is principally driven more by loss of activity against TgDHFR rather than by gaining potency at hDHFR.
Figure 3.

Crystal Structure of TgDHFR with pyrimidines 29 and 3 bound.
Crystal structure data was visualized and modeled using MOE software from Chemical Consulting Group.
Figure 4.

Crystal Structure of TgDHFR with pyrimidines 29 and 3 bound.
Crystal structure data was visualized and modeled using MOE software from Chemical Consulting Group.
Further characterization of 3 demonstrated good kinetic solubility of 186 μM at pH 7.4, low human microsomal clearance (<8.6 mL/min/kg) and high permeability in MDR1-MDCK cells (20.1 × 10−6 cm/s) with a low efflux ratio of 1.1 (Table 5). In the CEREP 44 safety screen at a 10 μM dose level, 3 demonstrated good selectivity against all 44 targets tested, except for hERG where [3H] Dofetilide binding was inhibited by 60%.
Chemistry.
The compounds presented here were synthesized as shown in Schemes 1 and 2. In Scheme 1, substituted piperazines (36a-n) were condensed with 5-bromouracil 33 in the presence of KF and heating in DMSO to give the corresponding uracil derivatives 37a-n. Chlorination using POCl3 and DIPEA provided intermediates 38a-n, followed by amination reaction using NH3/EtOH at 145 °C to give final products 2, 7–18, 20, 21 and 32. 6-Methyl and 6-ethyl diaminopyrimidine targets 20 and 21 were synthesized as described above, but starting with the corresponding 6-alkyl-5-bromouracils 34 and 35 and condensing with biphenyl-piperazine 36a. Piperazine intermediates 36a-n are generally commercially available or can be prepared by various literature methods37–38.
Scheme 1.

aSynthesis of Compounds 2, 7-18, 20, 21 and 32
R2 = (a) 3-phenyl, (b) hydrogen, (c) 2-phenyl, (d) 4-phenyl, (e) 2-methyl, (f) 3-methyl, (g) 4-methyl, (h) 2-chloro, (i) 3-chloro, (j) 4-chloro, (k) 3-methoxy, (l) 3-trifluoromethyl, (m) 3-trifluoromethoxy, (n) 3-bromo
a Reagents and conditions: (a) KF, DMSO, 110 °C, 12 h; (b) POCl3, DIPEA, 110 °C, 5 h; (c) NH3, EtOH, 145 °C, 24 h
Scheme 2.

aSynthesis of Compounds 3, 19, 22–31
R2 = (o) 5-(2-methoxypyrimidine), (p) cyclopropyl, (q) 3-pyridine, (r) 4-pyridine, (s) 5-pyrimidine, (t) 4-pyridazine, (u) 5-(2-methylpyrimidine), (v) 5-(2-trifluoromethylpyrimidine), (w) 5-(2-cyclopropylpyrimidine), (x) 2-pyridine, (y) 2-pyrimidine and (z) 2-pyrazine.
a Reagents and conditions: (a) Pd(PPh3)4, R2-B(OH)2 (39o-w), Cs2CO3, Dioxane/H2O, 110 °C; or (b) Pd2(dba)3, Sn(nBu)3-R2 (40x-z), Xphos, dioxane, 100 °C
Analogs in Scheme 2 were prepared by either Suzuki coupling of 3-bromophenyl 32 with the corresponding boronic acids 39o-w to give targets 3, 19, 23, 24, 26 and 28–31, or Stille coupling of 3-bromophenyl 32 with Sn(Bu)3R2 40x-z to generate compounds 22, 25 and 27.
Crystal structure of TgDHFR.
To aide in the understanding of the observed selectivity and guide future lead optimization efforts, several X-ray crystal structures with lead compounds bound to TgDHFR were solved, including the complex of 5-pyrimidines 3 and 29 with TgDHFR (Figures 3 and 4). Although at a high-level these data are consistent with modeling predictions, the crystal structure provides more subtle clues into why the distal pyrimidine moiety enhances selectivity. The phenyl groups of 3 and 29 occupy the TgDHFR cavity created near F91 and G22 (Figure 4), while the residues present in hDHFR at these positions appear to be less accommodating. These TgDHFR phenylalanine residues (F32 and F91) create a more enclosed hydrophobic area around the phenyl ring connected to the piperazine. This configuration also positions the distal heteroaromatic ring into the glycine site, possibly enabling a hydrogen bonding interaction with His 27 in the T. gondii enzyme (Figure 3). The presence of a proline residue (Pro26) in hDHFR, at the analogous position of His 27 in TgDHFR, provides a hypothesis for the observed subtle improvement in selectivity with the 5-pyrimidines. The TgDHFR His27 may enable a favorable H-bond with a pyrimidine N, whereas this would not be feasible in the human enzyme. In addition, there could be an unfavorable interaction in the hDHFR between the pyrimidine nitrogen atoms of 3 and Pro 26. This interaction might help explain why 3-pyridine 23 is less selective than pyrimidine 3, as the pyridine N could rotate away from the proposed unfavorable interaction with Pro26 in the human enzyme. We hypothesize these are the likely reasons for the observed TgDHFR selectivity. However, it is also noted that, N64 in hDHFR blocks π-π interactions. Interactions at these residues are well supported by the literature and may also contribute to the observed TgDHFR selectivity39. A search of UniProt shows that there are no reported human polymorphisms related to these amino acids that would alter selectivity40.
Parasiticidal and anti-proliferative effects of 5-methoxypyrimidine 3 in cell-based models.
Optimized pyrimidine 3 and comparator drug 1 were evaluated in cell-based models to investigate translation of the respective DHFR inhibitory activities into parasiticidal and antiproliferative host cell effects. Lead 3 had a parasiticidal EC50 of 13 nM and 1 had an EC50 of 680 nM against the type I RH strain of T. gondii. The 52-fold improvement in potency of 3 over 1 in this assay is consistent with its 89-fold potency advantage for inhibiting TgDHFR. Antiproliferative effects of these compounds in human MCF-7 cells, measured using the MTT assay, were consistent with the human DHFR data, wherein 1 and 3 had EC50s of 11,600 nM and 7,300 nM, respectively. The selectivity of both 1 (17-fold) and 3 (560-fold) in the cell-based assays (Table 6) is in line with their DHFR selectivity profiles of 12 and 200-fold, respectively. These data are important as they demonstrate that 3 has anti-parasiticidal activity with similar efficacy as 1, but with substantially better potency and selectivity.
Table 6.
Parasiticidal activity for 1 and 3 in T. gondii infected human foreskin fibroblast cells as compared to the antiproliferative effects of 1 and 3 in MCF-7 cells
| Compound |
T. gondii Cell-Based EC50 (nM)a |
MCF-7 EC50 (nM)b |
Selectivity Indexc |
|---|---|---|---|
| 3 | 13 ± 3.8 | 7300 | 560 |
| 1 | 680 ± 210 | 11,700 | 17 |
EC50=half maximal response concentration, average of at least 3 experiments (± SD).
MCF-7 cell viability determined using the MTT assay in triplicate (n = 1).
Selectivity index calculated as the MCF-7 EC50/Tg cell-based EC50.
Comparison of potency against additional strains of Toxoplasma.
Given the enhanced potency of 3, we wanted to test it against different lineages of T. gondii. Consequently, we chose a series of low passage isolates that are representative of the major genotypes of T. gondii seen in human infection from different regions of the world. As we have previously described, the population structure of T. gondii is comprised of 6 major clades, each of which contains several related lineages41. The majority of human cases of toxoplasmosis in Europe and North America are due to type 2 strains42–44, and we typically use a laboratory strain called ME49 as representative for such strains. The ME49 strain was originally isolated from a sheep in the United States45, and it reflects the genotype of many strains found in food animals and which cause human toxoplasmosis. Type 1 strains are also reasonably abundant in North America, and they are of interest as they are more pathogenic in many hosts including immunocompromised humans44, 46 Although most lab studies use the type 1 RH strain, use of the GT1 strain is preferable as it preserves the entire lifecycle47. The GT1 strain was originally isolated from a goat in North America48, although it is genetically similar to a number of human isolates including the RH strain. In contrast to North America and Europe, strains in South America are dominated by highly pathogenic lines such as types 5 (reference strain RUB) and 10 (reference strain VAND)41. The VAND and RUB strains were isolated from severe human cases of toxoplasmosis that are characteristic of the Amazon region49–50. These strains have also been used as reference strains for the Toxoplasma comparative genomes paper51.
To monitor the effects of new antiparasitic compounds on different strains of T. gondii we generated fire fly luciferase (FLUC) expressing lines so that we could use a common luciferase-based growth assay to monitor growth inhibition. We compared the potency of 3 for its ability to inhibit the growth of T. gondii strains in vitro. As shown in Table 7, the potency of 3 was highly similar across these different lineages, with only 2 to 3-fold differences being observed in potency. The FLUC lines used here also carry a pyrimethamine resistance allele of DHFR that was used to generate the transgenic lines. This resistant DHFR allele confers highly level resistance to pyrimethamine as described previously52, but interestingly this does not lead to cross-resistance to 3.
Table 7.
aGrowth effects of 3 on different strains of T. gondii
| Strain | Host of origin | Geographic Distribution | Type | EC50 (nM) |
|---|---|---|---|---|
| GT1 | goat | North America | 1 | 18.0 ± 9.3 |
| ME49 | sheep | North America Europe | 2 | 8.3 ± 2.9 |
| RUB | human | South America | 5 | 7.6 ± 3.3 |
| VAND | human | South America | 10 | 11.2 ± 0.5 |
Average of at least 3 independent replicates ± SEM
Mouse pharmacokinetic and brain to plasma exposure studies.
2-Methoxypyrimidine 3 was selected over 2-methylpyrimidine 29 and unsubstituted pyrimidine 26 for further in vivo characterization owing to its substantially improved stability in mouse liver microsomes (MLM). Preclinical models of toxoplasmosis are primarily performed in mice and therefore a compound with sufficient oral bioavailability, and plasma and CNS exposure to support murine efficacy testing was desired. Fortunately, 3 had moderate mouse liver microsome (MLM) intrinsic clearance (Clint) of 56.3 mL/min/Kg, which is 63% of mouse liver blood flow (LBF), whereas 29 and 26 both had high predicted clearance with MLM Clint values of 431 and 2,700 mL/min/Kg, respectively, multiples above mouse LBF. The oral and IV PK parameters in mouse are summarized in Table 8. Compound 3 has low to moderate clearance of 10.6 mL/min/kg, a volume of distribution of 1.14 L/kg and a half-life of 3.9 h after a 1.0 mg/kg, iv dose. Oral bioavailability after a 0.83 mg/kg, po dose was 47.3% with a Cmax of 178 ng/mL 30 minutes after dosing. The unbound fraction (%) in mouse plasma is 8.7 ± 0.2 and in brain homogenate 2.4 ± 0.3, determined using an equilibrium dialysis method. Compound 3 was freely permeable into the mouse CNS at 0.5 hours after a 10 mg/kg oral dose, the concentration in brain was 2,560 ± 240 ng/g and in blood 1,610 ± 580 ng/mL giving a brain to blood ratio of 1.7 ± 0.6.
Table 8.
aPharmacokinetics of compound 3 in mouse
| Route | Dose (mg/kg) |
%F | Cmax, po (ng/mL) |
Tmax, po (h) |
AUC0-last, po (ng•h/mL) |
CL, iv (mL/min/kg) |
Vd, iv (L/kg) |
t½, iv (h) |
|---|---|---|---|---|---|---|---|---|
| IV | 1.0 | -- | -- | -- | -- | 10.6 | 1.14 | 3.9 |
| PO | 0.83 | 47.3 | 178 | 0.05 | 750 | -- | -- | -- |
Oral bioavailability (%F), area under the concentration-time curve from time zero to the last measurable concentration (AUC0-last), clearance (CL), maximum observed plasma concentration (Cmax), intravenous (IV), per os (PO), elimination half-life (t½), time to Cmax (Tmax), volume of distribution (Vd)
In vivo efficacy of 1 and 3 in a murine model of acute Toxoplasmosis.
Compounds 1 and 3 were tested independently in a murine model of acute toxoplasmosis and observed to improve survival in a dose-dependent manner (Figures 6a and 6b). Female CD-1 mice were inoculated intraperitoneally (IP) with 3,000 tachyzoites of the highly virulent RH strain of T. gondii on Day 0. In parallel, 3 negative control mice were inoculated with vehicle, lacking the parasite. The parasite strain used in this study was genetically modified to constitutively express the enzyme luciferase and enable infection monitoring via an in vivo imaging system (IVIS®)53. Mice were monitored for survival for 30 days with intermittent IVIS monitoring (data not shown). On day 4 of dosing, compound exposure was assessed by micro-sampling from tail vein from 3 mice per group at 1, 4 and 12 h post-dose and 3 mice per group at 2, 8 and 24 h post-dose (data shown in Figures 7a and 7b).
Figure 6a.

aKaplan-Meier survival curves of compound 1 in CD-1 female mice (n = 12 for drug treated groups, n = 9 for vehicle group) infected with 3,000 RH type 1 T. gondii tachyzoites on day 0 followed by oral administration of 1 beginning on day 1 through day 7.
a Mice were carefully monitored for signs of infection, including by intermittently imaging (data not shown).
Figure 6b.

aKaplan-Meier survival curves of compound 3 in CD-1 female mice (n = 12 for drug treated groups, n = 9 for vehicle group) infected with 3,000 RH type 1 T. gondii tachyzoites on day 0 followed by oral administration of 3 beginning on day 1 through day 7.
a Mice were carefully monitored for signs of infection, including by intermittently imaging (data not shown).
Figure 7a.

Day 4 [plasma]free exposure of 1 from the acute murine model of toxoplasmosis plotted as free plasma (nM) levels versus time.
Figure 7b.

Day 4 [plasma]free exposure of 3 from the acute murine model of toxoplasmosis plotted as free plasma (nM) levels versus time.
Compound 3 at doses of 10 mg/kg, QD or BID for 7 days, yielded 100% survival for 30 days (end of study), while doses of 1 and 3 mg/kg produced similar results on survival as comparator compound 1 did at 18 mg/kg BID and 54 mg/kg QD, respectively. Survival data for 1 in this study were consistent with published literature54–56. All mice treated with vehicle died within 6 to 7 days. It should be noted that the single animals that were lost in the 3 mg/kg compound 3 group and 54 mg/kg compound 1 group were believed to be due to handling errors during imaging rather than active T. gondii infection as no signs of infection or bioluminescence (data not shown) were observed for these animals during the study. Consistent with the improved in vitro potency of 3 relative to 1, it was also more potent in vivo producing similar efficacy at doses about 18-fold lower.
Cure rates were assessed by subinfection of naive mice with combined homogenized brain and lung tissue from mice that survived 30 days. The tissues from two surviving mice of the same dosing group were injected into one donor mouse. Infection with a single T. gondii parasite of the type I RH strain is believed to be 100% lethal in naive mice47; therefore, sub-inoculated naive mice that survived for 15 days and were without signs of infection, was indicative of a cure (i.e., the donor mice were devoid of parasite). It should be noted that the RH strain of T. gondii does not typically form tissue cysts. Therefore, a cure in this model may not translate to a cure with clinically relevant strains that form tissue cysts. Nonetheless, it is clear both 1 and 3 are strongly efficacious in eradicating the tissue damaging and life threating tachyzoite form of the parasite.
Compound 3 had a cure rate of 50% in the 1 and 3 mg/kg groups and 100% in the 10 mg/kg groups. Pyrimethamine 1 was 100% curative at the 54 mg/kg dose and 33% to 67% curative at the 18 mg/kg BID dose. These data suggest that inhibition of TgDHFR is sufficient for maximal efficacy as a stand-alone therapy (e.g., without coadministration of a DHPS inhibitor). Regarding the results for pyrimethamine 1, the human equivalent dose of 54 mg/kg in mouse (approximately 300 mg/day in a 70-kg individual) cannot be administered in patients owing to dose-limiting, mechanism-based safety issues.
For both compounds, the onset of effect on survival time occurred at exposures near or slightly above the T. gondii cell-based EC50 values (1.0 mg/kg for 3 and 18 mg/kg for 1), with maximal effects occurring at dose levels of 3 mg/kg and 10 mg/kg for 3 and 54 mg/kg for 1 (Figures 7a and 7b), which exceed the cell-based EC50s.
CONCLUSIONS
Rational drug design led to the identification of several highly selective and potent arylpiperazine-based TgDHFR inhibitors. The structure-activity relationship for these compounds provides proof-of-concept for the feasibility of discovering a next generation toxoplasmosis treatment with improved potency, selectivity, and safety. 2-Methoxypyrimidine 3, with 16-fold better selectivity for TgDHFR and 89-fold better potency as compared to pyrimethamine 1, and desirable DMPK and safety properties, was selected for exploration in preclinical models of toxoplasmosis. Compound 3 was effective in radical cure of acute infection with the highly virulent type I RH strain in the murine model and showed similar potency across multiple parasite lineages in cell-based parasite growth assays. Based on the efficacy observed, we hypothesize it will be feasible to achieve parasiticidal levels of 3 in patients that are below concentrations that would inhibit hDHFR, thus providing a safer and more efficacious therapy than is currently available and possibly obviating the need for sulfonamide combination therapy.
EXPERIMENTAL SECTION
Materials and Methods.
General.
All synthetic chemistry, DMPK, T. gondii DHFR crystal structures with 3 and 29, and DHFR in vitro pharmacology was performed at WuXi App Tec at their China facilities in Tianjin and Shanghai. Work with Toxoplasma gondii was performed at Evotec in Manchester, UK and in Prof. Sibley’s lab at Washington University, St. Louis. Proton NMR spectra were recorded on a Varian 400 MHz NMR. LCMS were taken on a quadrupole Mass Spectrometer on Shimadzu LCMS 2010 (Column: sepax ODS 50×2.0 mm, 5 μm) or Agilent 1200 HPLC, 1956 MSD (Column: Shim-pack XR-ODS 30×3.0 mm, 2.2 μm) operating in ES (+) ionization mode. All final compounds were greater than 95% pure based on HPLC UV% AUC. Structures were determined by 1H NMR and LC/MS. Final compounds in this manuscript are not known to interfere with the assays here in (i.e., they are not PAINS).
All animal studies were conducted at WuXi App Tec in China and Evotec in Manchester, UK and were performed in accordance with the institutional guidelines for these Countries and Laboratories.
LC/MS Methods
Method A:
Run on a Shimadzu LC-20AB with a MS 2010 detector using a Luna-C18(1) column (2.0*30mm, 3um) at 40 °C. Mobile phase A was 0.037% (v/v) aqueous TFA and mobile phase B was 0.018% (v/v) TFA in acetonitrile. The flow rate was 0.8 mL/min from 0.01 to 1.51 min, then 1.2 mL/min from 1.52 to 2.00 min. The gradient ran from 90% mobile phase A to 10% mobile phase A over 1.15 min then remained at 10% mobile phase A through 1.65 min then back to 90% mobile phase A at 1.66 min and was maintained at 90% mobile phase A through 2.0 min. The UV detection was 220 nm and the MS was measured in positive ion mode.
Method B:
Run on an Agilent 1200 with a MS 6120 detector using an Xbridge Shield RP18 column (2.1*50mm, 5um) at 40 °C. Mobile phase A was 10 mM aqueous NH4HCO3 and mobile phase B was acetonitrile. The flow rate was 1.0 mL/min from 0.01 to 2.48 min, then 1.2 mL/min from 2.50 to 3.00 min. The gradient ran from 90% mobile phase A to 20% mobile phase A over 2.00 min then remained at 20% mobile phase A through 2.48 min then back to 90% mobile phase A at 2.50 min and maintained at 90% mobile phase A through 3.0 min. The UV detection was 220 nm and the MS was measured in positive ion mode.
Method C:
Run on an Agilent 1200 with a MS 6120 detector using an Xbridge Shield RP18 column (2.1*50mm, 5um) at 40 °C. Mobile phase A was 10 mM aqueous NH4HCO3 and mobile phase B was acetonitrile. The flow rate was 1.0 mL/min from 0.01 to 2.50 min, then 1.2 mL/min from 2.51 to 3.00 min. The gradient ran from 70% mobile phase A to 10% mobile phase A over 1.50 min then remained at 10% mobile phase A through 2.50 min then back to 70% mobile phase A at 2.51 min and maintained at 70% mobile phase A through 3.0 min. The UV detection was 220 nm and the MS was measured in positive ion mode.
Method D:
Run on an Agilent 1200 with a MS 6120 detector using a Venusil XBP-C18 column (2.1*50mm, 5um) at 40 °C. Mobile phase A was 0.0375% aqueous TFA and mobile phase B was 0.018% TFA in acetonitrile. The flow rate was 0.8 mL/min from 0.01 to 4.5 min. The gradient was maintained at 99% mobile phase A from 0.00 min to 0.40 min, then the gradient ran from 99% mobile phase A to 10% mobile phase A over 3.00 min then to 0% mobile phase A over 0.45 min; then back to 99% mobile phase A over 0.01 min and maintained here for 0.55 min The UV detection was 220 nm and the MS was measured in positive ion mode.
Chemistry
The compounds in Figure 2, were sourced as follows. Compound 2 was prepared as described previously34. Pyrimethamine (1) was sourced from the drug manufacturer and 1H NMR and LC/MS data confirmed structure and purity of 99.9%. Trimethoprim (4) was purchased from a commercial supplier as the lactate salt and 1H NMR and LC/MS data confirmed structure with a purity of 98.7%. Methotrexate (5) was purchased from a commercial supplier and 1H NMR and LC/MS data confirmed structure with a purity near 100%. Trimetrexate (6) was purchased from a commercial supplier as the tri-hydrochloride salt and 1H NMR and LC/MS data confirmed structure with a purity of 95.2%.
The compounds in Tables 3, 4 and 5 were prepared according to Schemes 1 and 2.
Compounds 7–18 and 20 - 21 were prepared in a similar fashion as described below for the synthesis of 32.
5-(4-(3-Bromophenyl)piperazin-1-yl)pyrimidine-2,4-diamine (32) was prepared in 3 steps and 12.8% overall yield as follows.
Step 1. 5-(4-([3-Bromophenyl])piperazin-1-yl)pyrimidine-2,4(1H,3H)-dione (37n). To a mixture of 5-bromo-1H-pyrimidine-2,4-dione 33 (7.92 g, 42 mmol, 1.0 eq) and 1-(3-bromophenyl)piperazine 36n (10.0 g, 42 mmol, 1.0 eq) in DMSO (200 mL) was added potassium fluoride (5.0 g, 94 mmol, 2.0 eq). The resulting mixture was stirred at 130°C for 12 h, cooled to room temperature and poured into 300 mL of water. The precipitate was collected by suction filtration, washed with 200 mL of EtOH and dried in vacuo leaving 5-(4-([3-bromophenyl])piperazin-1-yl)pyrimidine-2,4(1H,3H)-dione 37n as a brown solid (10.6 g, 30.2 mmol, 73.5% yield) used as such in the next step. LCMS Method B (ESI+): Expected m/z 351 (M+1)+; found m/z 351 (M+1)+, RT: 1.07 Min.
Step 2. 5-(4-(3-Bromophenyl)piperazin-1-yl)-2,4-dichloropyrimidine (38n). A mixture of 5-(4-(3-bromophenyl)piperazin-1-yl)pyrimidine-2,4(1H,3H)-dione 37n (10 g, 28.5 mmol, 1.0 eq) and DIPEA (14.4 g, 111 mmol, 3.9 eq) in 70 mL of toluene was cooled to 10 °C and POCl3 (15.8 g, 103 mmol) was added over 1 h, keeping the reaction temperature 20°C. The mixture was stirred at 20°C for 1 h, heated to 95°C, and held at 95°C for 12 h. LCMS was consistent with the desired product MS. The reaction mixture was concentrated under reduced pressure to give a black residue, which was diluted with cold water (200 mL) and extracted with DCM (3 × 100 mL). The combined organic layers were washed with brine (2 × 100 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to give 5-(4-(3-bromophenyl)piperazin-1-yl)-2,4-dichloropyrimidine (38n) (6.4 g, 58% yield, 90.8% HPLC purity) as a yellow solid used as such in the next step. LC/MS Method C (ESI+): Expected m/z 389.1 (M+1)+; found m/z 389.0 (M+1)+, RT: 2.18 Min.
Step 3. 5-(4-(3-Bromophenyl)piperazin-1-yl)pyrimidine-2,4-diamine (32). A mixture of 5-(4-(bromophenyl)piperazin-1-yl)-2,4-dichloropyrimidine 38n (100.0 mg, 0.26 mmol, 1.0 eq) in NH3/EtOH (10 mL) was added to a steel bomb. The mixture was stirred at 145°C for 12 h. The suspension was cooled to room temperature and concentrated under reduced pressure to give a brown residue. The residue was purified by Prep-HPLC (TFA condition) to give 5-(4-(3-bromophenyl)piperazin-1-yl)pyrimidine-2,4-diamine 32 (86.5 mg, 249.6 μmol, 30.0% yield) as a white solid. LCMS Method D (ESI+): Expected m/z (349.0 and 351.1 M+1)+; found m/z 349.0 and 351.1 (M+1)+, Rt: 2.18 Min. 1H NMR (DMSO-d6 400MHz) ) δ = 7.57 (s, 1H), 7.19 – 7.12 (m, 1H), 7.09 (br s, 1H), 6.94 (br dd, J = 7.9, 18.4 Hz, 2H), 5.65 (s, 2H), 3.29 (br s, 4H), 2.85 (br s, 4H).
5-(4-Phenylpiperazin-1-yl)pyrimidine-2,4-diamine (7) was prepared in an analogous 3 step manner as described for 32 starting with 1-phenylpiperazine (36b) to give a light yellow solid (26.6% yield). 1H NMR (DMSO-d6) δ = 7.59 (s, 1H), 7.22 (br t, J=7.8 Hz, 2H), 6.96 (br d, J = 8.2 Hz, 2H), 6.78 (t, J = 7.0 Hz, 1H), 6.07 (br s, 2H), 5.61 (br s, 2H), 3.25 (br s, 4H), 2.88 (br t, J = 4.5 Hz, 4H). LC/MS (M + 1) Expected MW 271.1, Observed MW 271.1, % UV purity 99.9%.
5-(4-([1,1’-Biphenyl]-2-yl)piperazin-1-yl)pyrimidine-2,4-diamine (8) was prepared in an analogous 3 step manner as described for 32 above starting with 1-([1,1’-biphenyl]-2-yl)piperazine (36c) to give 8 in 7.0% yield as a white solid. 1H NMR (DMSO-d6) δ = 7.65 (br d, J = 7.6 Hz, 2H), 7.48 (s, 1H), 7.44 (br t, J = 7.2 Hz, 2H), 7.31 (br t, J = 7.2 Hz, 2H), 7.21 (br d, J = 7.2 Hz, 1H), 7.12 – 7.05 (m, 2H), 5.99 (br s, 1H), 5.60 (s, 2H), 2.88 (br s, 4H), 2.64 (br s, 4H). LC/MS (M + 1) Expected MW 347.2, Observed MW 347.2, UV purity 97.8%.
5-(4-([1,1’-Biphenyl]-4-yl)piperazin-1-yl)pyrimidine-2,4-diamine (9) was prepared in an analogous 3 step manner as described for 32 starting with 1-([1,1’-biphenyl]-4-yl)piperazine (36d) to give 9 in 10.6% yield as a white solid. 1H NMR (METHANOL-d4) δ = 7.60 – 7.50 (m, 5H), 7.39 (br t, J = 7.4 Hz, 2H), 7.30 – 7.23 (m, 1H), 7.10 (br d, J = 8.6 Hz, 2H), 3.48 – 3.36 (m, 4H), 3.03 (br d, J = 4.3 Hz, 4H). LC/MS (M + 1) Expected MW 347.2, Observed MW 347.2, UV Purity 96.7%.
5-(4-(o-Tolyl)piperazin-1-yl)pyrimidine-2,4-diamine (10) was prepared in an analogous 3 step manner as described for 32 starting with 1-(o-tolyl)piperazine (36e) to give 10 in 32.8% yield as a white solid. 1H NMR (METHANOL-d4) δ = 7.53 (s, 1H), 7.21 – 7.11 (m, 3H), 7.03 – 6.97 (m, 1H), 3.10 (br s, 4H), 3.03 (br d, J = 4.0 Hz, 4H), 2.34 (s, 3H). LC/MS (M + 1) Expected MW 285.2, Observed MW 285.1, UV Purity 99.4%.
5-(4-(m-Tolyl)piperazin-1-yl)pyrimidine-2,4-diamine (11) was prepared in an analogous 3 step manner as described for 32 starting with 1-(m-tolyl)piperazine (36f) to give 11 in 7.2% yield as a yellow solid. 1H NMR (DMSO-d6) δ = 7.59 (s, 1H), 7.10 (t, J = 7.8 Hz, 1H), 6.83 – 6.70 (m, 2H), 6.60 (br d, J = 7.4 Hz, 1H), 6.08 (br s, 2H), 5.62 (s, 2H), 3.23 (br s, 4H), 2.86 (br t, J = 4.3 Hz, 4H), 2.25 (s, 3H). LC/MS (M + 1) Expected MW 285.1, Observed MW 285.1, UV Purity 99.5%.
5-(4-(p-Tolyl)piperazin-1-yl)pyrimidine-2,4-diamine (12) was prepared in an analogous 3 step manner as described for 32 starting with 1-(p-tolyl)piperazine (36g) to give 12 in 2.3% yield as a white solid. 1H NMR (DMSO-d6) δ = 7.58 (s, 1H), 7.03 (d, J = 8.4 Hz, 2H), 6.86 (d, J = 8.4 Hz, 2H), 6.06 (br. s., 1H), 5.60 (s, 2H), 3.19 (br. s., 4H), 2.87 (d, J = 4.0 Hz, 4H), 2.20 (s, 3H). LC/MS (M + 1) Expected MW 285.1, Observed MW 285.1, UV Purity 98.1%.
5-(4-(2-Chlorophenyl)piperazin-1-yl)pyrimidine-2,4-diamine (13) was prepared in an analogous 3 step manner as described for 32 starting with 1-(2-chlorophenyl)piperazine (36h) to give 13 in 19.4% yield as a light yellow solid. 1H NMR (DMSO-d6) δ = 7.62 (s, 1H), 7.42 (br d, J = 7.8 Hz, 1H), 7.35 – 7.28 (m, 1H), 7.18 (br d, J = 7.8 Hz, 1H), 7.05 (br t, J= 7.4 Hz, 1H), 6.09 (br s, 2H), 5.63 (s, 2H), 3.11 (br s, 4H), 2.91 (br d, J = 3.9 Hz, 4H). LC/MS (M + 1) Expected MW 305.1, Observed MW 305.1, UV Purity 99.1%.
5-(4-(3-Chlorophenyl)piperazin-1-yl)pyrimidine-2,4-diamine (14) was prepared in an analogous 3 step manner as described for 32 starting with 1-(3-chlorophenyl)piperazine (36i) to give 14 in 6.6% yield as a solid. 1H NMR (DMSO-d6) δ = 11.76 (br, 1H), 8.43 (br s, 1H), 7.65 (br s, 1H), 7.56 (br s, 1H), 7.43 (br s, 2H), 7.23 (br t, J = 8.4 Hz, 1H), 6.98 (br s, 1H), 6.93 (br d, J = 8.4 Hz, 1H), 6.80 (br d, J = 7.6 Hz, 1H), 3.36 (br s, 4H), 2.86 (br s, 4H). LC/MS (M + 1) Expected MW 305.1, Observed MW 305.1, UV Purity 98.1%.
5-(4-(4-Chlorophenyl)piperazin-1-yl)pyrimidine-2,4-diamine (15) was prepared in an analogous 3 step manner as described for 32 starting with 1-(4-chlorophenyl)piperazine (36j) to give 15 in 71.9% yield as a white solid. 1H NMR (DMSO-d6) δ = 7.58 (s, 1H), 7.24 (br d, J = 9.0 Hz, 2H), 6.97 (br d, J = 9.0 Hz, 2H), 6.08 (br s, 1H), 5.61 (s, 2H), 3.26 (br s, 4H), 2.86 (br t, J = 4.3 Hz, 4H). LC/MS (M + 1) Expected MW 305.1, Observed MW 305.1, UV Purity 96.3%.
5-(4-(3-Methoxyphenyl)piperazin-1-yl)pyrimidine-2,4-diamine (16) was prepared in an analogous 3 step manner as described for 32 starting with 1-(3-methoxyphenyl)piperazine (36k) to give 16 in 24.2% yield as a white solid. 1H NMR (DMSO-d6) δ = 7.58 (s, 1H), 7.11 (t, J = 8.4 Hz, 1H), 6.54 (br d, J = 8.0 Hz, 1H), 6.47 (s, 1H), 6.37 (br d, J = 8.0 Hz, 1H), 6.08 (br s, 1H), 5.61 (s, 2H), 3.72 (s, 3H), 3.25 (br s, 4H), 2.86 (br d, J = 4.0 Hz, 4H). LC/MS (M + 1) Expected MW 301.1, Observed MW 301.1, UV Purity 98.7%.
5-(4-(3-(Trifluoromethyl)phenyl)piperazin-1-yl)pyrimidine-2,4-diamine (17) was prepared in an analogous 3 step manner as described for 32 starting with 1-(3-trifluoromethylphenyl)piperazine (36l) to give 17 in 18.8% yield as a yellow solid. 1H NMR (METHANOL-d4) δ = 7.50 (s, 1H), 7.44 – 7.39 (m, 1H), 7.23 (br d, J = 8.8 Hz, 1H), 7.20 (br s, 1H), 7.10 (br d, J = 7.5 Hz, 1H), 3.40 (br s, 4H), 3.01 (br d, J = 4.4 Hz, 4H). LC/MS (M + 1) Expected MW 339.1, Observed MW 339.1, UV Purity 99.5%.
5-(4-(3-(Trifluoromethoxy)phenyl)piperazin-1-yl)pyrimidine-2,4-diamine (18) was prepared in an analogous 3 step manner as described for 32 starting with 1-(3-trifluoromethoxyphenyl)piperazine (36m) to give 18 in 22.7% yield as a yellow solid. 1H NMR (METHANOL-d4) δ = 7.49 (s, 1H), 7.31 (t, J = 8.2 Hz, 1H), 6.98 (d, J = 7.9 Hz, 1H), 6.84 (s, 1H), 6.72 (d, J = 7.9 Hz, 1H), 3.38 (s, 4H), 2.99 (s, 4H). LC/MS (M + 1) Expected MW 355.1, Observed MW 355.1, UV Purity 100%.
5-(4-([1,1’-Biphenyl]-3-yl)piperazin-1-yl)-6-methylpyrimidine-2,4-diamine (20) was prepared in an analogous 3 step manner as described for 32 starting with 1-([1,1’-biphenyl]-3-yl)piperazine (36a) and 5-bromo-6-methyluracil (34) to give 20 in 31.8% yield as a yellow solid. 1H NMR (METHANOL-d4) d = 7.64 – 7.57 (m, 2H), 7.51 – 7.28 (m, 5H), 7.20 (br d, J = 7.5 Hz, 1H), 7.09 (br d, J = 7.9 Hz, 1H), 3.66 (br d, J = 11.5 Hz, 2H), 3.45 – 3.35 (m, 2H), 3.26 – 3.16 (m, 2H), 3.03 (br d, J = 11.5 Hz, 2H), 2.38 (s, 3H). LC/MS (M + 1) Expected MW 361.2, Observed MW 361.1, UV Purity 95.9%.
5-(4-([1,1’-Biphenyl]-3-yl)piperazin-1-yl)-6-ethylpyrimidine-2,4-diamine (21) was prepared in an analogous 3 step manner as described for 32 starting with 1-([1,1’-biphenyl]-3-yl)piperazine (36a) and 5-bromo-6-ethyluracil (35) to give 21 in 71.4% yield as a yellow solid. 1H NMR (METHANOL-d4) δ = 7.61 (d, J = 7.2 Hz, 2H), 7.43 (d, J = 8.0 Hz, 4H), 7.36 (d, J = 6.4 Hz, 1H), 7.30 (d, J = 7.2 Hz, 1H), 7.18 (d, J = 7.2 Hz, 1H), 3.65 (d, J = 9.2 Hz, 2H), 3.44–3.31 (m, 4H), 3.12 (d, J = 8.8 Hz, 2H), 2.73 (d, J = 7.6 Hz, 2H), 1.33 (t, J = 6.8 Hz, 3H). LC/MS (M + 1) Expected MW 375.2, Observed MW 375.1, UV Purity 98.7%.
Compounds 3, 19, 23, 24, 26, 28 and 30–31 were prepared in a similar fashion as described below for the synthesis of 29.
5-(4-(3-(2-methylpyrimidin-5-yl)phenyl)piperazin-1-yl)pyrimidine-2,4-diamine (29)
A mixture of 5-(4-(3-bromophenyl)piperazin-1-yl)pyrimidine-2,4-diamine 32 (1.0 g, 2.8 mmol, 1.0 eq), (2-methylpyrimidin-5-yl)boronic acid 39u (395 mg, 2.8 mmol, 1.0 eq), Cs2CO3 (1.4 g, 4.3 mmol, 1.5 eq), Pd(PPh3)4 (165 mg, 143 μmol, 0.05 eq) in dioxane (32 mL) and H2O (8.0 mL) was degassed and purged with N2 3 times, and then stirred at 100°C for 12 h under N2 atmosphere. Then it was stirred with silica S thiol Met at 20 °C, filtered and concentrated to give a residue. The residue was purified by prep-HPLC (TFA condition) to give 5-(4-(3-(2-methylpyrimidin-5-yl)phenyl)piperazin-1-yl)pyrimidine-2,4-diamine 29 (1.2 g, 2.5 mmol, 88.0% yield) as a white solid. 1H NMR, (METHANOL-d4) δ = 8.93 (s, 2H), 7.51 (s, 1H), 7.41 (t, J = 7.8 Hz, 1H), 7.26 – 7.24 (m, 1H), 7.16 – 7.09 (m, 2H), 3.44 (br s, 4H), 3.02 (br t, J = 4.8 Hz, 4H), 2.73 (s, 3H). LCMS expected m/z 363.2 (M+1)+; found m/z 363.1 (M+1)+. UV purity 99.0%.
5-(4-(3-(2-Methoxypyrimidin-5-yl)phenyl)piperazin-1-yl)pyrimidine-2,4-diamine (3) was prepared in an analogous manner as described for 29 starting with 32 and 5-(2-methoxypyrimidinyl)boronic acid (39o) to give 3 in 92.8% yield as a light yellow solid. 1H NMR (METHANOL-d4) δ = 8.81 (s, 2H), 7.51 (s, 1H), 7.39 (br t, J = 7.6 Hz, 1H), 7.22 (br s, 1H), 7.14 – 7.07 (m, 2H), 4.06 (s, 3H), 3.44 (br s, 4H), 3.03 (br s, 4H). Elemental analysis for C19H22N8O + 0.15 H2O: Theoretical C 59.83%, H 5.90%, N 29.39%. Found C 59.56%, H 5.83%, N 29.05%. LC/MS (M + 1) Expected MW 379.2, Observed MW 379.1, UV Purity 96.4%.
5-(4-(3-Cyclopropylphenyl)piperazin-1-yl)pyrimidine-2,4-diamine (19) was prepared in an analogous manner as described for 29 starting with 32 and cyclopropyl boronic acid (39p) to give 19 in 59.2% yield as solid. 1H NMR (METHANOL-d4) δ = 7.52 (br s, 1H), 7.20 (br d, J = 7.2 Hz, 1H), 6.97 – 6.83 (m, 2H), 6.73 (br s, 1H), 3.43 (br s, 4H), 3.06 (br s, 4H), 1.91 (br s, 1H), 0.96 (br d, J = 6.0 Hz, 2H), 0.68 (br s, 2H). LC/MS (M + 1) Expected MW 311.2, Observed MW 311.1, UV Purity 98.9%.
5-(4-(3-(Pyridin-3-yl)phenyl)piperazin-1-yl)pyrimidine-2,4-diamine (23) was prepared in an analogous manner as described for 29 starting with 32 and 3-pyridyl boronic acid (39q) to give 23 in 28.2% yield as a light yellow oil. 1H NMR (METHANOL-d4) δ = 9.08 (br s, 1H), 8.79 – 8.69 (m, 2H), 8.01 (br s, 1H), 7.52 (s, 1H), 7.49 – 7.44 (m, 1H), 7.36 (s, 1H), 7.24 (d, J = 7.6 Hz, 1H), 7.19 (dd, J = 2.0, 8.4 Hz, 1H), 3.48 (br s, 4H), 3.04 (br s, 4H). LC/MS (M + 1) Expected MW 348.2, Observed MW 348.2, UV Purity 98.2%.
5-(4-(3-(Pyridin-4-yl)phenyl)piperazin-1-yl)pyrimidine-2,4-diamine (24) was prepared in an analogous manner as described for 29 starting with 32 and 4-pyridyl boronic acid (39r) to give 24 in 11.5% yield. 1H NMR (METHANOL-d4) δ = 8.56 (br s, 2H), 7.73 – 7.62 (m, 3H), 7.39 (br d, J = 8.8 Hz, 1H), 7.33 (br s, 1H), 7.22 (br d, J = 7.2 Hz, 1H), 7.13 (br d, J = 7.6 Hz, 1H), 3.50 – 3.35 (m, 4H), 3.03 (br s, 4H). LC/MS (M + 1) Expected MW 348.2, Observed MW 348.2, UV Purity 95.3%.
5-(4-(3-(Pyrimidin-5-yl)phenyl)piperazin-1-yl)pyrimidine-2,4-diamine (26) was prepared in an analogous manner as described for 29 starting with 32 and 5-pyrimidinyl boronic acid (39s) to give 26 in 34.9% yield as a light yellow oil. 1H NMR (METHANOL-d4) δ = 9.14 (br s, 1H), 9.07 (br s, 2H), 7.52 (s, 1H), 7.49 – 7.42 (m, 1H), 7.34 (br s, 1H), 7.21 (br dd, J = 7.7, 18.7 Hz, 2H), 3.48 (br s, 4H), 3.05 (br s, 4H). LC/MS (M + 1) Expected MW 349.1, Observed MW 349.1, UV Purity 95.8%.
5-(4-(3-(Pyridazin-4-yl)phenyl)piperazin-1-yl)pyrimidine-2,4-diamine (28) was prepared in an analogous manner as described for 29 starting with 32 and 4-pyridazine-boronic acid pinacol ester (39t) to give 28 in 9.2% yield as a yellow solid. 1H NMR (METHANOL-d4) δ = 9.59 (s, 1H), 9.25 (d, J = 5.3 Hz, 1H), 8.10 (dd, J = 2.4, 5.5 Hz, 1H), 7.55 – 7.41 (m, 3H), 7.34 (br d, J = 7.9 Hz, 1H), 7.22 (br d, J = 8.4 Hz, 1H), 3.48 (br s, 4H), 3.04 (br d, J = 4.4 Hz, 4H). LC/MS (M + 1) Expected MW 349.2, Observed MW 349.1, UV Purity 100%.
5-(4-(3-(2-(Trifluoromethyl)pyrimidin-5-yl)phenyl)piperazin-1-yl)pyrimidine-2,4-diamine (30) was prepared in an analogous manner as described for 29 starting with 32 and 5-(2-trifluoromethylpyrimidine)-boronic acid (39v) to give 30 in 27.3% yield as a yellow solid. 1H NMR (METHANOL-d4) δ = 9.23 (s, 2H), 7.51 (s, 1H), 7.46 – 7.44 (m, 1H), 7.36 (s, 1H), 7.25 (d, J = 8.0 Hz, 1H), 7.18 (d, J = 6.4 Hz, 1H), 3.47 (br s, 4H), 3.03 (br s, 4H). LC/MS (M + 1) Expected MW 417.2, Observed MW 417.1, UV Purity 98.2%.
5-(4-(3-(2-Cyclopropylpyrimidin-5-yl)phenyl)piperazin-1-yl)pyrimidine-2,4-diamine (31) was prepared in an analogous manner as described for 29 starting with 32 and 5-(2-cyclopropylpyrimidine)-boronic acid (39w) to give 31 in 42.9% yield as a light yellow solid. 1H NMR (METHANOL-d4) δ = 8.89 (s, 2H), 7.52 (s, 1H), 7.43 – 7.41 (m, 1H), 7.28 (s, 1H), 7.19–7.13 (m, 2H), 3.46 (br s, 4H), 3.04 (br s, 4H), 2.30 – 2.25 (m, 1H), 1.17 – 1.15 (m, 4H). Expected MW 389.2, Observed MW 389.1, UV Purity 100%.
Compounds 25 and 27 were prepared in a fashion as described below for the synthesis of 22.
5-(4-(3-(pyridin-2-yl)phenyl)piperazin-1-yl)pyrimidine-2,4-diamine (22)
A mixture of 5-(4-(3-bromophenyl)piperazin-1-yl)pyrimidine-2,4-diamine 32 (100.0 mg, 286.3 μmol, 1.0 eq), tributyl(pyridin-2-yl)stannane 40x (106 mg, 286 μmol, 1 eq), Pd2(dba)3 (7.8 mg, 8.6 μmol, 0.03 eq), XPhos (23 mg, 49 μmol, 0.17 eq) in dioxane (8.0 mL) was degassed and purged with N2 3 times. The mixture was stirred at 100 °C for 12 h under N2 atmosphere, concentrated under reduced pressure and the residue was purified by prep-HPLC (TFA condition) to give 5-(4-(3-(pyridin-2-yl)phenyl)piperazin-1-yl)pyrimidine-2,4-diamine 22 (4.5 mg, 4.5% yield) as a yellow solid. LCMS Method D (ESI+): Expected m/z (348.2 M+1)+; found m/z 348.1 (M+1)+. 1H NMR (CD3OD-d6) δ = 8.60 (br d, J = 4.9 Hz, 1H), 7.94 – 7.88 (m, 1H), 7.87 – 7.82 (m, 1H), 7.63 (s, 1H), 7.58 (s, 1H), 7.42 – 7.34 (m, 3H), 7.12 (br s, 1H), 3.39 (br d, J = 11.0 Hz, 4H), 3.03 (br t, J = 4.6 Hz, 4H).
5-(4-(3-(Pyrimidin-2-yl)phenyl)piperazin-1-yl)pyrimidine-2,4-diamine (25) was prepared in an analogous manner as described for 22 starting with 32 and tributyl(pyrimidin-2-yl)stannane (40y) to give 25 in 23.5% yield as a white solid. 1H NMR ((METHANOL-d4) δ = 8.85 (br s, 2H), 8.07 (br s, 1H), 7.92 (br d, J = 6.6 Hz, 1H), 7.52 (br s, 1H), 7.40 (br d, J = 13.7 Hz, 2H), 7.21 (br s, 1H), 3.46 (br s, 4H), 3.05 (br s, 4H). Expected MW 349.2, Observed MW 349.2, UV Purity 97.1%.
5-(4-(3-(Pyrazin-2-yl)phenyl)piperazin-1-yl)pyrimidine-2,4-diamine (27) was prepared in an analogous manner as described for 22 starting with 32 and tributyl(pyrazin-2-yl)stannane (40z) to give 27 in 50.1% yield as a light yellow solid. 1H NMR (METHANOL-d4) δ = 9.19 (s, 1H), 8.73 (s, 1H), 8.60 (br d, J = 2.0 Hz, 1H), 8.30 (s, 1H), 8.11 (br d, J = 7.6 Hz, 1H), 7.73 – 7.65 (m, 2H), 7.62 (s, 1H), 3.85 (br s, 4H), 3.30 – 3.26 (m, 4H). Expected MW 349.2, Observed MW 349.2, UV Purity 99.1%.
DHFR assay for human and T. gondii.
All compounds were tested in duplicate and replicated in at least 3 independent experiments. Human and T. gondii DHFR assays were run in parallel using the same stocks to optimize selectivity data. Key comparator compounds 1 (n =12 for human and T. gondii) and 3 (n = 16 for human and T. gondii) were tested in free base form on the same days for optimal comparison. The protein expression and purification protocol was adapted from a previously published procedure33. T. gondii TS-DHFR was sub-cloned into a PET15b plasmid and transformed into Escherichia coli BL21 competent cells. Overnight cultured bacteria were inoculated into a 1 L LB culture media at a ratio of 1:100 at 37 °C. Upon reaching an OD 600 nm of 0.7, protein expression was induced with 0.5 mM isopropyl β-D-thiogalactoside at 16 °C overnight. Cells were then pelleted (~4.6 g) and re-suspended in buffer A (25 mM Tris-HCl, pH 7.3, 100 mM NaCl, 1 mM EDTA), before lysis by sonication. MTX agarose beads (~1 mL) were added to the lysate, and the beads were subsequently washed 2X with buffer A (~10 mL) and 1X with buffer B (~10 mL – 25 mM Tris-HCl, pH 7.3, 1 M KCl, 1 mM EDTA) prior to elution with buffer C (~6 mL, 25 mM Tris-HCl, 10 mM DTT, 10% glycerol, 2 mM H2F). The eluent containing the purified enzyme was then concentrated and the protein was transferred, using a PD-MiniTrap G-25 column, to the final storage buffer (25 mM TrisHCl, 10mM DTT, 10% glycerol). Purified hDHFR was obtained commercially from Sigma Aldrich; Dihydrofolate Reductase human (Sigma D6566). The protein was confirmed by sequencing.
The diaphorase-coupled assay for DHFR activity was adapted from a previously published procedure57. Compounds were added as solutions of DMSO at 100X the desired concentration to purified enzyme (1 μg/mL) suspended in assay buffer (150 mM KCl, 8.9 mM ß-mercaptoethanol in 40.0 mM sodium phosphate at pH 7.4) in 384-well format (corning 3573). Following a 15 min incubation at 25 °C, solutions of NADPH (1.6 μM, Sigma N7505) and DHF (10 μM, Sigma D7006) in assay buffer were added sequentially. The plate was then incubated at 25 °C for an additional 60 min prior to the addition of diaphorase (10 U/mL) and resazurin (5 μM, Sigma R7017). After a final 10 min incubation at 25 °C, fluorescence was measured using an EnVision plate reader (531 nmEx/590 nmEm). IC50 values were determined from the raw fluorometric data by non-linear regression using Graphpad Prism.
Parasite strains and cell lines
Tachyzoites of T. gondii strains were grown in monolayers of human foreskin fibroblasts (HFF) (ATCC SCRC 1041) maintained in complete medium (DMEM containing 10% FBS, 2 mM L-glutamine, 10 mM HEPES and 10 μg/ml gentamicin in 35 mM NaHCO3 solution), 37°C in 5% CO2. Following natural egress, tachyzoites were purified in HBSS containing 10 mM HEPES, 0.1 mM EGTA and separated from host debris using 3.0 micro polycarbonate membrane filters, followed by centrifugation at 400g.
Cell-based parasite growth assays using β-galactosidase
This work was performed at Evotec in Manchester, UK. Parasite growth inhibition assays were conducted using the type I RH strain, 2F clone that expresses bacterial β-galactosidase (β-gal), as described previously58. Compounds dissolved in DMSO at 10 mM stocks were diluted in medium to two times final concentrations and added to an equal volume of medium containing 5 × 102 parasites and incubated for 20 min. Mixtures of compounds (ranging from 10 μM to 0.01 nM) containing 0.1% (v/v) DMSO, or DMSO alone were added to monolayers of HFF cells grown in 96 well plates, centrifuged at 300 g for 5 min, and returned to culture at 37°C, 5% CO2. The cells were preincubated for 2 h in the presence of parasites. Varying concentrations of drugs were added to each well (100 μL volumes containing 0.2% DMSO in D10 media) to give a final concentration range of 0.02 to 10 μM in 0.1% DMSO. The plates were centrifuged at 300G for 5 min at room temperature and then incubated at 37°C in 5% CO2 for 72 h. At the end of the incubation period, the monolayer was lysed in 1% Triton X-100 and β-gal activity monitored using 1 mM chlorophenol red-β-D-galactopyranoside by absorption at 570 nm, as described previously58. Individual EC50 values were determined from three or more independent biological replicates and are reported as mean values.
Generation of firefly luciferase (FLUC) tagged strains
This work was performed at Washington University of St. Louis. Experiments were conducted with the ME49Δhx::FLUC strain of T. gondii, a transgenic line that expresses firefly luciferase (FLUC)59. To tag additional strains with FLUC, a reporter plasmid was created using a two-step Gibson Assembly approach. Fragment one was generated from PCR linearized pUPRT::Floxed DHFR-TS* plasmid (Addgene #100606) that includes both 5’ and 3’ uracil phosphoribosyltransferase (UPRT) flanking regions to drive locus specific integration at the UPRT gene and a floxed T. gondii DHFR selectable marker. Fragment two was synthesized using a gBlock (IDT DNA) containing the constitutive T. gondii alpha-tubulin promoter (608-bp) driving expression of the firefly luciferase reporter (1641-bp). The expression construct was targeted to the UPRT gene in T. gondii by co-transfection with the CRISPR plasmid pSAG1:CAS9, U6:sgUPRT. Parasites were sequentially selected in pyrimethamine (1.0 μM) followed by 5-fluorodeoxyracil (FUDR, 10 μM) and independent clones isolated by limiting dilution. All clones contain the floxed T.gondii DHFR selectable marker that can be excised using a plasmid expressing CRE recombinase.
In vitro growth assays using luciferase
To assess potency of compounds on different strains of T. gondii, values were determined form a 10-point dose-response curve. Briefly, 5×103 of luciferase expressing parasites (100 μL/well) were added to a 96-well plate that contained 100 μL of 2X compound concentration (to achieve 1X final compound concentration in 200 μL total well volume containing 0.1% DMSO) and allowed to replicate for 72 h prior to preparation for luciferase assay. All experimental steps, growth conditions and luciferase assay protocols were completed as described above. Compounds were tested using a 3-fold dilution series from 10 μM to 0.001 μM with all wells containing a final concentration of 0.1% DMSO. The EC50 data are presented as the average of three or more biological replicates (i.e. separate EC50 titrations) each with two technical replicates (i.e. separate wells).
Antiproliferative assay in MCF7 cells
This work was performed at Cyprotex in Watertown, MA. MCF-7 cells were plated on 96-well tissue culture treated polystyrene plates at 0.75×104 cells in 100 μL of MEM/EBSS (supplemented with 10% fetal bovine serum, sodium pyruvate, and antibiotics) per well. After an overnight incubation at 37°C, the cells were dosed with test compounds and controls at a range of concentrations and incubated for 72 h at 37°C. Pyrimethamine 1 was tested from 0.156 to 20 μM and Compound 3 was evaluated from 0.313 to 40 μM with an 8-point dose curve in triplicate on three separate plates. Cell viability was measured using the Promega CellTiter 96 Non-Radioactive Cell Proliferation Assay (MTT) kit by adding 15 μL of the Dye Solution to each well and incubating for 3 h at 37°C. After incubation, 100 μL of the Solubilization Solution/Stop Mix was added to each well. Plates were incubated at 37°C for 1 h, mixed on a plate shaker for 10 min and then absorbance was read at 570 nm. The EC50 was then calculated.
Pharmacokinetics (mouse)
Fasted female CD-1 mice (n = 3), 6–8 weeks old (supplied from Beijing Vital River Laboratory Animal Technology Co., Ltd) were administered a 0.5 mg/kg intravenous dose of compound 3, formulated as a clear 0.1 mg/mL solution in saline at pH 5. A second group (n=3) was administered a 0.83 mg/kg oral dose of compound 3 (targeted dose was 1.0 mg/kg), formulated as a 0.1 mg/mL homogenous opaque suspension with fine particles in 0.5% CMC. Plasma (EDTA-K2) from the iv dose was collected at 0.083, 0.50, 1.0, 2.0, 4.0, 8.0 and 24 h. Plasma (EDTA-K2) from the po dose was collected at 0.50, 1.0, 2.0, 4.0, 8.0, 12 and 24 h. Plasma preparation: An aliquot of 10 μL plasma was protein precipitated with 100 μL IS, the mixture was vortexed and centrifuged at 13000 rpm for 10 min, 4°C. The supernatant (25 μL) was then mixed with 25 μL water/ACN (v:v, 95:5) containing 0.1% formic acid, vortexed and centrifuged at 4 °C, 3 μL of supernatant was injected for LC-MS/MS analysis. Concentration of compound 3 was determined using an LC-MS/MS-AG (API 4000) instrument in ESI positive mode with SRM detection monitoring for [M+H]+ m/z transition 379.3/152.2 and using an internal standard 100 ng/mL tolbutamide [M+H]+ m/z transition 271.1/155.1. The UPLC method Mobile Phase A: 0.025% formic acid, 1 mM NH4OAc in water/ACN (v:v, 95:5) and Mobile Phase B: 0.025% formic acid, 1 mM NH4OAc in ACN/water (v:v, 95:5) used a gradient starting with 15% Mobile Phase B and going to 90% over 1.40 min, on a Acquity UPLC BEH C18 1.7 μm 2.1 × 50 mm column at 60 °C at a flow rate of 0.7 mL/min. The retention time of compound 3 was 0.55 min and tolbutamide 0.88 min. A calibration curve from 1.00 to 3000 ng/mL of compound 3 in male CD-1 mouse plasma (EDTA-K2) was made and the data analyzed using Phoenix WinNonlin 6.3 with a linear/log trapezoidal calculation method.
Brain to blood ratio (mouse)
This work was performed at Evotec, Manchester, UK. Fed, female CD1 mice (n = 3) were orally administered 10 mg/kg of the TFA-salt form of compound 3, formulated as a clear solution in 0.25% carboxymethyl cellulose (CMC). Blood (EDTA-K2) 20 μL was collected by tail prick of the lateral tail vein 0.5 h post-dose and diluted 2x with water for LC-MS/MS injection. Animals were then euthanized, and brain was collected and homogenized. The brain homogenates (40 μl) were run within the blood calibration curve and utilized the homogenization dilution factors for final quantitation but also incorporated a 2-fold adjustment for sample volume versus blood.
In vivo murine model of Toxoplasmosis
This work was performed at Evotec in Manchester, UK. Fed female CD-1 mice (60 mice per study; 12 mice per drug treatment group, 9 mice for vehicle and 3 mice as negative controls) were inoculated intraperitoneally with 3,000 tachyzoites of the highly virulent RH strain of T. gondii on Day 0. The T. gondii strain used in this study had been genetically modified to constitutively express the enzyme fire fly luciferase (FLUC) and enable infection monitoring via an in vivo bioluminescence imaging system53. Beginning 24 h after inoculation, mice were dosed with either compound 1 formulated at 0.6, 1.8 and 5.4 mg/mL in 0.25% CMC or the TFA salt form of compound 3 formulated in 0.25% CMC at 0.1 mg/mL, 0.3 mg/mL or 1.0 mg/mL as a clear solution and dosed at a volume of 10 mL/kg. On day 4 micro-sampling of blood from the tail vein was collected into K2EDTA tubes and evaluated by LC/MS-MS as described above for the mouse brain to blood experiment.
Supplementary Material
Figure 5.

Parasiticidal dose response curves of 1 and 3 in T. gondii infected human foreskin fibroblast cells.
Acknowledgement:
The authors are grateful to Rubin Ben-Harari and Nick Pelliccione for their contributions to the writing of this manuscript and support. The authors are also grateful to the chemists, in vitro pharmacologists and ADME/DMPK personal at WuXi for generating much of lead optimization data summarized in this paper.
Funding source: This study was funded by Vyera Pharmaceuticals. Contributions on this project from Dr. Sibley’s laboratory were supported by a research Agreement between Vyera Pharmaceuticals and Washington University.
Abbreviations (nonstandard) used:
- FLUC
fire fly luciferase
- CMC
carboxymethyl cellulose
- UPRT
uracil phosphoribosyltransferase
- DHFR
Dihydrofolate reductase
- CNS
central nervous system
- MTX
methotrexate
- HFF
human foreskin fibroblasts
Footnotes
Supporting Information: Methods for TgDHFR-TS protein purification and crystallization with 3 and 29 with diffraction and refinement statistics data. Methods for kinetic solubility, Log P, human and mouse liver microsome clearance, and MDCK-MDR1 permeability determinations. CEREP 44 cross-reactivity safety assessment data.
PDB ID Codes: Crystal structures of the TgDHFR-TS protein with bound compounds 3 and 29 have PDB ID codes 6n1t and 6n1s, respectively. Authors will release the atomic coordinates and experimental data upon article publication.
Disclosure Statement: AH, ST and AB are former employees of Vyera Pharmaceuticals.
REFERENCES:
- 1.Dubey JP; Lindsay DS; Speer CA, Structures of Toxoplasma gondii tachyzoites, bradyzoites, and sporozoites and biology and development of tissue cysts. Clin Microbiol Rev 1998, 11 (2), 267–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tenter AM; Heckeroth AR; Weiss LM, Toxoplasma gondii: from animals to humans. Int J Parasitol 2000, 30 (12–13), 1217–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones JL; Kruszon-Moran D; Rivera HN; Price C; Wilkins PP, Toxoplasma gondii seroprevalence in the United States 2009–2010 and comparison with the past two decades. Am J Trop Med Hyg 2014, 90 (6), 1135–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maenz M; Schluter D; Liesenfeld O; Schares G; Gross U; Pleyer U, Ocular toxoplasmosis past, present and new aspects of an old disease. Progress in retinal and eye research 2014, 39, 77–106. [DOI] [PubMed] [Google Scholar]
- 5.Pappas G; Roussos N; Falagas ME, Toxoplasmosis snapshots: global status of Toxoplasma gondii seroprevalence and implications for pregnancy and congenital toxoplasmosis. Int J Parasitol 2009, 39 (12), 1385–1394. [DOI] [PubMed] [Google Scholar]
- 6.Prusa A-R; Kasper DC; Sawers L; Walter E; Hayde M; Stillwaggon E, Congenital toxoplasmosis in Austria: Prenatal screening for prevention is cost-saving. PLoS Negl Trop Dis 2017, 11 (7), e0005648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dalimi A; Abdoli A, Latent toxoplasmosis and human. Iran J Parasitol 2012, 7 (1), 1–17. [PMC free article] [PubMed] [Google Scholar]
- 8.Montoya A; Boothroyd JC; Kovacs J, Toxoplasma gondii In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 8th ed.; Elsevier Saunders: Philadelphia, PA, 2015; pp 3122–3153. [Google Scholar]
- 9.Halonen SK; Weiss LM, Chapter 8 - Toxoplasmosis In Handbook of clinical neurology, Hector H. Garcia HBT; Oscar HDB, Eds. Elsevier: 2013; Vol. Volume 114, pp 125–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rabaud C; May T; Amiel C; Katlama C; Leport C; Ambroise-Thomas P; Canton P, Extracerebral toxoplasmosis in patients infected with HIV. A French National Survey. Medicine (Baltimore) 1994, 73 (6), 306–14. [DOI] [PubMed] [Google Scholar]
- 11.Akira S, The role of IL-18 in innate immunity. Current Opinion in Immunology 2000, 12, 59–63. [DOI] [PubMed] [Google Scholar]
- 12.Cuervo G; Simonetti AF; Alegre O; Sanchez-Salado JC; Podzamczer D, Toxoplasma myocarditis: a rare but serious complication in an HIV-infected late presenter. Aids 2016, 30 (14), 2253–4. [DOI] [PubMed] [Google Scholar]
- 13.Cuervo G; Simonetti AF; Alegre O; Sanchez-Salado JC; Podzamczer D, Toxoplasma myocarditis: a rare but serious complication in an HIV-infected late presenter. AIDS 2016, 30 (14), 2253–2254. [DOI] [PubMed] [Google Scholar]
- 14.Montoya JG; Liesenfeld O, Toxoplasmosis. Lancet 2004, 363 (9425), 1965–1976. [DOI] [PubMed] [Google Scholar]
- 15.Olliaro P, Drug resistance hampers our capacity to roll back malaria. Clin Infect Dis 2005, 41 Suppl 4, S247–57. [DOI] [PubMed] [Google Scholar]
- 16.McCabe RE, Antitoxoplasma chemotherapy In Toxoplasmosis: a comprehensive clinical guide, Joynson DHM; Wreghitt TG, Eds. Cambridge Univ. Press: Cambridge, 2001; pp 319–359. [Google Scholar]
- 17.Ben-Harari RR; Goodwin E; Casoy J, Adverse Event Profile of Pyrimethamine-Based Therapy in Toxoplasmosis: A Systematic Review. Drugs R D 2017, 17 (4), 523–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khan Assir MZ; Ahmad HI; Akram J; Yusuf NW; Kamran U, An Outbreak of Pyrimethamine Toxicity in Patients with Ischaemic Heart Disease in Pakistan. Basic & Clinical Pharmacology & Toxicology 2014, 115 (3), 291–296. [DOI] [PubMed] [Google Scholar]
- 19.Eyles DE; Coleman N, Tests of 2,4-diaminopyrimidines on toxoplasmosis. Public Health Rep. 1952, 67, 249–52. [PMC free article] [PubMed] [Google Scholar]
- 20.Felix JPF; Lira RPC; Zacchia RS; Toribio JM; Nascimento MA; Arieta CEL, Trimethoprim-Sulfamethoxazole Versus Placebo to Reduce the Risk of Recurrences of Toxoplasma Gondii Retinochoroiditis: Randomized Controlled Clinical Trial. American Journal of Ophthalmology 2014, 157 (4), 762–766.e1. [DOI] [PubMed] [Google Scholar]
- 21.Stanford MR; See SE; Jones LV; Gilbert RE, Antibiotics for toxoplasmic retinochoroiditis: an evidence-based systematic review. Ophthalmology 2003, 110 (5), 926–31; quiz 931–2. [DOI] [PubMed] [Google Scholar]
- 22.Taques IIGG; Barbosa TR; Martini A. d. C.; Pitchenin LC; Braga ÍA; de Melo ALT; Nakazato L; Dutra V; de Aguiar DM, Molecular assessment of the transplacental transmission of Toxoplasma gondii, Neospora caninum, Brucella canis and Ehrlichia canis in dogs. Comparative immunology, microbiology and infectious diseases 2016, 49, 47–50. [DOI] [PubMed] [Google Scholar]
- 23.Peters PJ; Thigpen MC; Parise ME; Newman RD, Safety and toxicity of sulfadoxine/pyrimethamine implications for malaria prevention in pregnancy using intermittent preventive treatment. Drug Saf. 2007, 30 (6), 481–501. [DOI] [PubMed] [Google Scholar]
- 24.Carr A; Cooper DA; Penny R, Allergic manifestations of human immunodeficiency virus (HIV) infection. J Clin Immunol 1991, 11 (2), 55–64. [DOI] [PubMed] [Google Scholar]
- 25.Phillips E; Mallal S, Drug hypersensitivity in HIV. Curr Opin Allergy Clin Immunol 2007, 7 (4), 324–30. [DOI] [PubMed] [Google Scholar]
- 26.Slatore CG; Tilles SA, Sulfonamide hypersensitivity. Immunol Allergy Clin North Am 2004, 24 (3), 477–90, vii. [DOI] [PubMed] [Google Scholar]
- 27.Amabeoku G; Chikuni O, GABAergic and dopaminergic systems may be involved in seizures induced by pyrimethamine in mice. Gen. Pharmacol 1994, 25 (6), 1269–77. [DOI] [PubMed] [Google Scholar]
- 28.Bolstad DB; Bolstad ESD; Frey KM; Wright DL; Anderson AC, Structure-Based Approach to the Development of Potent and Selective Inhibitors of Dihydrofolate Reductase from Cryptosporidium. J. Med. Chem 2008, 51 (21), 6839–6852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan DCM; Fu H; Forsch RA; Queener SF; Rosowsky A, Design, synthesis, and antifolate activity of new analogues of piritrexim and other diaminopyrimidine dihydrofolate reductase inhibitors with ω-carboxyalkoxy or ω-carboxy-1-alkynyl substitution in the side chain. J. Med. Chem 2005, 48 (13), 4420–4431. [DOI] [PubMed] [Google Scholar]
- 30.Jackson HC; Biggadike K; McKilligin E; Kinsman OS; Queener SF; Lane A; Smith J, 6,7-Disubstituted 2,4-diaminopteridines: novel inhibitors of Pneumocystis carinii and Toxoplasma gondii dihydrofolate reductase. Antimicrob. Agents Chemother 1996, 40 (6), 1371–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pelphrey PM; Popov VM; Joska TM; Beierlein JM; Bolstad ESD; Fillingham YA; Wright DL; Anderson AC, Highly efficient ligands for dihydrofolate reductase from cryptosporidium hominis and toxoplasma gondii inspired by structural analysis. J. Med. Chem 2007, 50 (5), 940–950. [DOI] [PubMed] [Google Scholar]
- 32.Rosowsky A; Hynes JB; Queener SF, Structure-activity and structure-selectivity studies on diaminoquinazolines and other inhibitors of Pneumocystis carinii and Toxoplasma gondii dihydrofolate reductase. Antimicrob. Agents Chemother. 1995, 39 (1), 79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharma H; Landau MJ; Vargo MA; Spasov KA; Anderson KS, First Three-Dimensional Structure of Toxoplasma gondii Thymidylate Synthase-Dihydrofolate Reductase: Insights for Catalysis, Interdomain Interactions, and Substrate Channeling. Biochemistry 2013, 52 (41), 7305–7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Welsch ME; Zhou J; Gao Y; Yan Y; Porter G; Agnihotri G; Li Y; Lu H; Chen Z; Thomas SB, Discovery of Potent and Selective Leads against Toxoplasma gondii Dihydrofolate Reductase via Structure-Based Design. ACS Med. Chem. Lett 2016, 7 (12), 1124–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Allegra CJ; Kovacs JA; Drake JC; Swan JC; Chabner BA; Masur H, Potent in vitro and in vivo antitoxoplasma activity of the lipid-soluble antifolate trimetrexate. J. Clin. Invest 1987, 79 (2), 478–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gomes TC; de AJHF; Lescano SAZ; Amato-Neto V, In vitro action of antiparasitic drugs, especially artesunate, against Toxoplasma gondii. Rev Soc Bras Med Trop 2012, 45 (4), 485–90. [DOI] [PubMed] [Google Scholar]
- 37.Gao R; Canney DJ, A Versatile and Practical Microwave-Assisted Synthesis of Sterically Hindered N-Arylpiperazines. J. Org. Chem 2010, 75 (21), 7451–7453. [DOI] [PubMed] [Google Scholar]
- 38.Ravilla L; Venkata Subba Naidu N; Nagarajan K, An efficient scale up process for synthesis of N-arylpiperazines. Tetrahedron Lett. 2015, 56 (30), 4541–4544. [Google Scholar]
- 39.Lamb KM; G-Dayanandan N; Wright DL; Anderson AC, Elucidating Features That Drive the Design of Selective Antifolates Using Crystal Structures of Human Dihydrofolate Reductase. Biochemistry 2013, 52 (41), 7318–7326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.UniProtKB, U. UniProtKB-P00374 (DYR_HUMAN) [database entry for human DHFR]. (accessed November 2, 2017).
- 41.Su CL; Khan A; Zhou P; Majumdar D; Ajzenberg D; Dardé ML; Zhu XQ; Ajioka JW; Rosenthal B; Dubey JP; Sibley LD, Globally diverse Toxoplasma gondii isolates comprise six major clades originating from a small number of distinct ancestral lineages. Proc. Natl. Acad. Sci. (USA) 2012, 109, 5844–5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ajzenberg D; Cogné N; Paris L; Bessieres MH; Thulliez P; Fillisetti D; Pelloux H; Marty P; Dardé ML, Genotype of 86 Toxoplasma gondii isolates associated with human congenital toxoplasmosis and correlation with clinical findings. J. Infect. Dis 2002, 186, 684–689. [DOI] [PubMed] [Google Scholar]
- 43.Ajzenberg D; Yera H; Marty P; Paris L; Dalle F; Menotti J; Aubert D; Franck J; Bessieres MH; Quinio D; Pelloux H; Delhaes L; Desbois N; Thulliez P; Robert-Gangneux F; Kauffmann-Lacroix C; Pujol S; Rabodonirina M; Bougnoux ME; Cuisenier B; Duhamel C; Duong TH; Filisetti D; Flori P; Gay-Andrieu F; Pratlong F; Nevez G; Totet A; Carme B; Bonnabau H; Darde ML; Villena I, Genotype of 88 Toxoplasma gondii isolates associated with toxoplasmosis in immunocompromised patients and correlation with clinical findings. J Infect Dis 2009, 199 (8), 1155–67. [DOI] [PubMed] [Google Scholar]
- 44.Howe DK; Sibley LD, Toxoplasma gondii comprises three clonal lineages: correlation of parasite genotype with human disease. J. Infect. Dis 1995, 172, 1561–1566. [DOI] [PubMed] [Google Scholar]
- 45.Lunde MN; Jacobs L, Antigenic differences between endozoites and cystozoites of Toxoplasma gondii. Journal of Parasitology 1983, 65, 806–808. [PubMed] [Google Scholar]
- 46.Khan A; Su C; German M; Storch GA; Clifford D; Sibley LD, Genotyping of Toxoplasma gondii strains from immunocompromised patients reveals high prevalence of type I strains. J. Clin. Micro 2005, 43, 5881–5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Su C; Howe DK; Dubey JP; Ajioka JW; Sibley LD, Identification of quantitative trait loci controlling acute virulence in Toxoplasma gondii. Proc. Natl. Acad. Sci. (USA) 2002, 99, 10753–10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dubey J, Mouse pathogenicity of Toxoplasma gondii isolated from a goat. American Journal of Veterinary Research 1980, 41, 427–429. [PubMed] [Google Scholar]
- 49.Carme B; Bissuel F; Ajzenberg D; Bouyne R; Aznar C; Demar M; Bichat S; Louvel D; Bourbigot AM; Peneau C; Neron P; Dardé ML, Severe acquired toxoplasmsis in immunocompetent adult patients in French Guiana. J . Clin. Microbiol 2002, 40, 4037–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Demar M; Hommel D; Djossou F; Peneau C; Boukhari R; Louvel D; Bourbigot AM; Nasser V; Ajzenberg D; Darde ML; Carme B, Acute toxoplasmoses in immunocompetent patients hospitalized in an intensive care unit in French Guiana. Clin Microbiol Infect 2012, 18 (7), E221–31. [DOI] [PubMed] [Google Scholar]
- 51.Lorenzi H; Khan A; Behnke MS; Namasivayam S; Swapna LS; Hadjithomas M; Karamycheva S; Pinney D; Brunk BP; Ajioka JW; Ajzenberg D; Boothroyd JC; Boyle JP; Darde ML; Diaz-Miranda MA; Dubey JP; Fritz HM; Gennari SM; Gregory BD; Kim K; Saeij JP; Su C; White MW; Zhu XQ; Howe DK; Rosenthal BM; Grigg ME; Parkinson J; Liu L; Kissinger JC; Roos DS; Sibley LD, Local admixture of amplified and diversified secreted pathogenesis determinants shapes mosaic Toxoplasma gondii genomes. Nat Commun 2016, 7, 10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reynolds MG; Roos DS, A biochemical and genetic model for parasite resistance to antifolates. Toxoplasma gondii provides insights into pyrimethamine and cycloguanil resistance in Plasmodium falciparum. J Biol Chem 1998, 273 (6), 3461–9. [DOI] [PubMed] [Google Scholar]
- 53.Behnke MS; Fentress SJ; Mashayekhi M; Li LL; Taylor GA; L.D. S, The polymorphic pseudokinase ROP5 controls virulence in Toxoplasma gondii by regulating the active kinase ROP18. PLoS Path. 2012, 8, e1002992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arribas JR; de Diego JA; Gamallo C; Vazquez JJ, A new murine model of severe acute toxoplasma encephalitis. J. Antimicrob. Chemother 1995, 36 (3), 503. [DOI] [PubMed] [Google Scholar]
- 55.Martins-Duarte ES; de Souza W; Vommaro RC, Toxoplasma gondii: The effect of fluconazole combined with sulfadiazine and pyrimethamine against acute toxoplasmosis in murine model. Exp. Parasitol 2013, 133 (3), 294–299. [DOI] [PubMed] [Google Scholar]
- 56.Piketty C; Derouin F; Rouveix B; Pocidalo JJ, In vivo assessment of antimicrobial agents against toxoplasma gondii by quantification of parasites in the blood, lungs, and brain of infected mice. Antimicrob. Agents Chemother. 1990, 34 (8), 1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kumar A; Zhang M; Zhu L; Liao RP; Mutai C; Hafsat S; Sherman DR; Wang M-W, High-throughput screening and sensitized bacteria identify an M. tuberculosis dihydrofolate reductase inhibitor with whole cell activity. PLoS One 2012, 7 (6), e39961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lourido S; Zhang C; Lopez MS; Tang K; Barks J; Wang Q; Wildman SA; Shokat KM; Sibley LD, Optimizing small molecule inhibitors of calcium-dependent protein kinase 1 to prevent infection by Toxoplasma gondii. J. Med. Chem 2013, 56, 3068–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tobin CM; Knoll LJ, A patatin-like protein protects Toxoplasma gondii from degradation in a nitric oxide-dependent manner. Infect. Immun 2012, 80 (1), 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.