

Abstract
A major mystery of many types of neurological and psychiatric disorders, such as Alzheimer’s disease (AD), remains the underlying, disease-specific neuronal damage. Because of the strong interconnectivity of neurons in the brain, neuronal dysfunction necessarily disrupts neuronal circuits. In this article, we review evidence for the disruption of large-scale networks from imaging studies of humans and relate it to studies of cellular dysfunction in mouse models of AD. The emerging picture is that some forms of early network dysfunctions can be explained by excessively increased levels of neuronal activity. The notion of such neuronal hyperactivity receives strong support from in vivo and in vitro cellular imaging and electrophysiological recordings in the mouse, which provide mechanistic insights underlying the change in neuronal excitability. Overall, some key aspects of AD-related neuronal dysfunctions in humans and mice are strikingly similar and support the continuation of such a translational strategy.
Keywords: default mode network, hippocampus, slow-wave oscillations, memory consolidation, hyperactivity, imaging
INTRODUCTION
In 1907, the psychiatrist Alois Alzheimer reported postmortem pathological changes in the brain of a patient with a peculiar dementia (Alzheimer 1907, Alzheimer et al. 1995). These brain alterations, which would later be known as Alzheimer’s disease (AD), included miliary foci and changes to the neurofibrils, which were later identified as amyloid-β (Aβ) plaques and neurofibrillary tangles, respectively (see Serrano-Pozo et al. 2011). Aβ peptides are derived from the neuronal amyloid precursor protein (APP) following enzymatic cleavage by β- and γ-secretases (Hardy & Selkoe 2002, Selkoe & Hardy 2016). In AD, Aβ peptides accumulate extracellularly and form soluble oligomers, fibrils, and finally amyloid plaques. Although the plaques were at first considered the primary cause of the disease, many recent studies have emphasized the potential toxicity of soluble oligomers (Barry et al. 2011, Borlikova et al. 2013, Freir et al. 2001, Jin et al. 2011, Klyubin et al. 2008, Li et al. 2009, Shankar et al. 2008, Yang et al. 2017). In addition to Aβ, other cleavage products of APP, such as the APP intracellular domain, the soluble APP, and eta-amyloid (Aη), might play a role in AD (Müller et al. 2008, 2017; Willem et al. 2015).
Because pathological brain changes and clinical symptoms are often not directly related, modern definitions make a conceptual distinction between AD, which refers to patients with neuropathological changes but without overt clinical symptoms, and full-blown Alzheimer’s dementia (Jack et al. 2011). The most striking clinically observed symptom is memory loss. Brain regions prominently involved in the processing of memory, such as the hippocampus and the neocortex, are affected very early by the disease pathology (Braak & Braak 1991, Buckner et al. 2005). Accumulating evidence indicates that the neuropathological changes are paralleled by distinct changes in brain function, such as changes in neuronal plasticity and excitability and the disruption of large-scale neuronal circuits.
IMPAIRED LARGE-SCALE CIRCUITS
Early efforts to detect alterations in functional brain networks in patients living with AD used EEG recordings (reviewed in Jeong 2004). However, EEG recordings could not provide unambiguous pathophysiological information. The situation had changed decisively with the implementation of brain-imaging methods, primarily MRI and positron emission tomography (PET). In the 1970s and early 1980s, PET studies demonstrated a decreased level of brain metabolism in patients with Alzheimer’s dementia by using radiolabeled fluorodeoxyglucose (FDG) (Ferris et al. 1980), among other techniques. The main conclusion of these studies was that neurons display a pronounced decrease in their activity in AD. Since then, other researchers have confirmed this observation repeatedly (see Figure 1a), and the reduced metabolism of specific brain areas, including the posterior cingulate cortex and precuneus, in FDG-PET is now used as a staging tool for the progression of AD (Dubois et al. 2014, McKhann et al. 2011). In parallel to the use of PET imaging, MRI became available for the analysis of brain changes in patients with AD and demonstrated marked brain atrophy, especially in the hippocampus, validating earlier pathological observations of postmortem brain sections (Seab et al. 1988).
Figure 1.
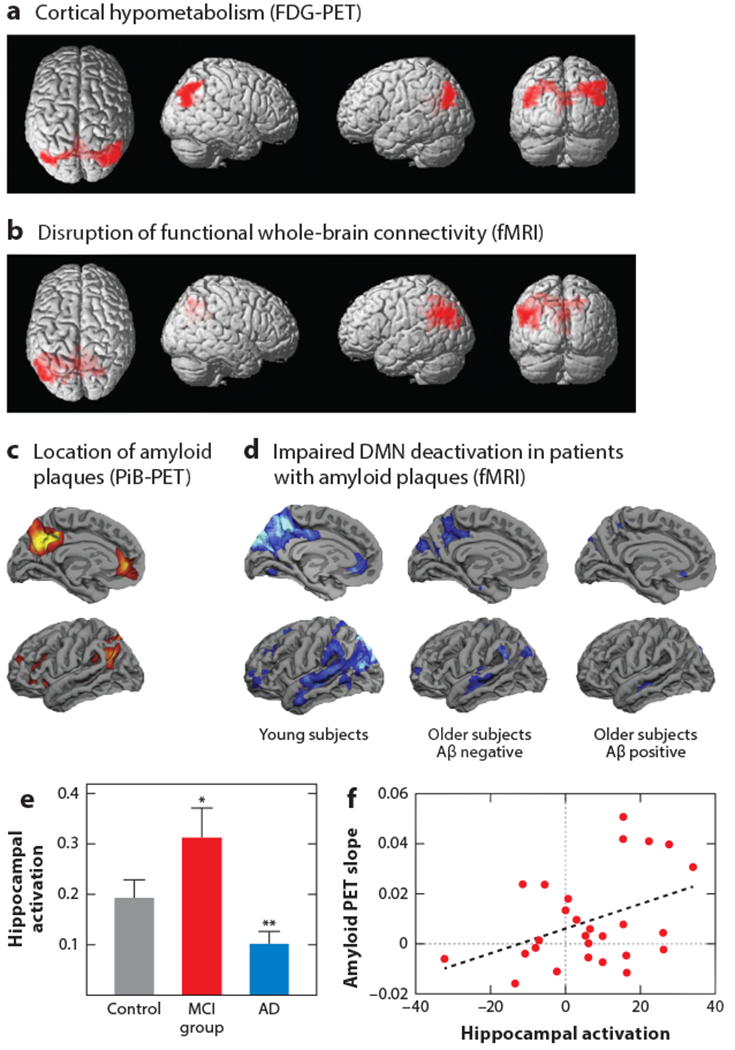
Circuit disruptions in AD—evidence from human studies. (a) FDG-PET imaging reveals cortical areas with decreased glucose metabolism (red). (b) Resting-state fMRI indicates disrupted cortical hubs. Voxel-based statistical group comparisons between MCI and PiB-negative healthy controls. Panels a and b adapted from Drzezga et al. (2011) with permission from Oxford University Press. (c) Cortical distribution of Aβ plaque load in PiB-positive subjects. Group-wise comparison with PiB-negative older subjects. (d) fMRI cortical maps of task-specific DMN deactivation (blue) in young (left), older Aβ-negative (middle), and older Aβ-positive subjects (right). Panels c and d adapted from Sperling et al. (2009) with permission from Elsevier. (e) Task-related hippocampal activation is increased in the MCI group compared with controls and patients with AD. Group mean fMRI activation data; activation is reported as the number of voxels within each region of interest that shows task-specific activation (*, P < 0.03; **, P < 0.005). Panel e adapted from Dickerson et al. (2005) with permission from Wolters Kluwer. (f) Graph of the PiB-PET imaging–derived slope of Aβ plaque accumulation versus fMRI-determined hippocampal activation at baseline. Panel f adapted from Leal et al. (2017). Abbreviations: Aβ, amyloid-β; AD, Alzheimer’s disease; DMN, default mode network; FDG, fluorodeoxyglucose; fMRI, functional MRI; MCI, mild cognitive impairment; PET, positron emission tomography; PiB, Pittsburgh compound B.
In addition to structural MRI, functional MRI (fMRI), which measures changes in the blood-oxygen-level-dependent (BOLD) signal, became increasingly popular for the analysis of changes in the function of neurons and circuits in patients with AD. Thus, Lustig et al. (2003) and Greicius et al. (2004) were able to relate AD pathology to changes in neuronal activity in certain cortical regions. In later studies, fMRI was frequently used to investigate impaired functional connectivity between different regions of the brain. The imaging palette was completed when a variety of PET tracers for Aβ, including the widely used PET tracer Pittsburgh compound B (PiB) (e.g., Klunk et al. 2004; for review see Morris et al. 2016) and, more recently, tau (Chien et al. 2014, Johnson et al. 2016), were developed.
A widely studied network considered to be impaired in AD is the default mode network (DMN). The DMN spans different cortical areas, including the precuneus, the posterior cingulate cortex, and the lateral and inferior parietal cortices, as well as regions of the temporal and medial prefrontal cortices (Raichle et al. 2001). These brain regions are functionally strongly connected and can be further divided into subnetworks (see Uddin et al. 2009). The DMN is usually active by default, especially during internal processes such as daydreaming, introspection, and mind wandering (Buckner et al. 2008). It becomes deactivated when the attention of the subject is shifted to outwardly directed tasks such as learning new information (Daselaar et al. 2004). Whether the hippocampus is part of the DMN (Buckner et al. 2008, Ward et al. 2014) remains controversial. Therefore, we review AD-related defects in the hippocampal network separately below. The DMN is a highly conserved circuit in different mammalian species, including human and nonhuman primates and even rodents (see Buckner et al. 2008, Lu et al. 2012). The DMN is of particular interest for the study of AD. First, it includes many areas affected by the hypometabolism detected through FDG-PET (Figure 1a). Second, it widely overlaps with areas of early Aβ plaque deposition (Figure 1c). Additional relevance of the DMN for AD results from its role in memory encoding and retrieval. Indeed, various alterations in the DMN directly correlate with the corresponding memory impairments. Finally, fMRI studies revealed striking functional impairments in wide parts of the DMN (Figure 1b,d), which are discussed in more detail in the following section.
A striking impairment of the DMN in AD is its decreased functional connectivity, which is revealed by resting-state fMRI. Accumulating evidence suggests that the functional disruption of DMN circuits occurs much earlier than the onset of clinical symptoms or obvious memory impairments. A decline in functional connectivity can be encountered in patients with normal cognition but distinct Aβ plaque deposition, as indicated by PiB-PET (Hedden et al. 2009, Sheline et al. 2010b). Similarly, there is evidence that the DMN connectivity is defective in carriers of familial early-onset Alzheimer’s disease (FAD) mutations at time points when cognition is still normal (Chhatwal et al. 2013). A disrupted connectivity in parts of the DMN can be observed in carriers of the apolipoprotein E (APOE) ε4 allele, the most important genetic risk factor for development of late-onset AD, even before detectable plaque deposition, cognitive impairment, or both (Sheline et al. 2010a). A recent study reported that the disrupted cortical connectivity can already be observed during the earliest accumulation of Aβ in the brain (Palmqvist et al. 2017).
AD is a neurodegenerative disease that develops slowly over several decades, and determining the relationship between the impaired connectivity and the cognitive decline is often challenging. Some studies suggest an AD-state-dependent correlation between impaired memory function and the increasingly impaired connectivity in parts of the DMN (Bai et al. 2011, Dillen et al. 2017, Liu et al. 2014). Other studies report a link between the extent of cognitive decline and the level of hyper- or hypoconnectivity in different parts of the DMN, implying that different subnetworks fail at different times during the disease (Damoiseaux et al. 2012). That parts of the DMN show pathologically increased connectivity at the beginning of the disease compared with that of healthy controls might indicate that these parts are in fact compensating for the failure of other parts of the circuit before they themselves eventually degenerate (Jones et al. 2016). Recent work suggests that poor DMN connectivity is a predictor of subsequent cognitive decline in the setting of elevated Aβ among clinically normal older individuals (Buckley et al. 2017).
Another major functional impairment of the DMN in AD, which parallels the disrupted functional connectivity, is the perturbed activity pattern of the DMN. More specifically, in contrast to the DMN activity in healthy young adults, which is high at rest and deactivates when the subjects successfully perform a task such as face-name matching (Sperling et al. 2009), the deactivation is reduced in healthy elderly people and largely absent in patients with AD (Lustig et al. 2003) (Figure 1d). Such DMN-specific deficits can be detected by fMRI well before symptom onset. Indeed, deactivation deficits are already present in cognitively normal APOE ε4 carriers (Persson et al. 2008, Pihlajamäki et al. 2010) as well as in subjects with Aβ plaques but without cognitive impairment (Sperling et al. 2009). During the progression of AD, the deactivation deficits of the DMN become gradually more pronounced and can switch occasionally into a paradoxical task-specific hyperactivation of the DMN (Sperling et al. 2009). The positive correlation between the connectivity and the activity of the DMN becomes visible in memory tests. In healthy subjects there is a high correlation between task-specific DMN deactivation and memory performance (Miller et al. 2008), whereas in patients with AD memory-related DMN deactivation is impaired (see Sperling et al. 2010).
Among the widely distributed cortical areas of the DMN, the posterior parietal cortex (PPC) and the precuneus, as well as parts of the prefrontal cortex, have a particularly high degree of susceptibility to early Aβ plaque deposition (Figure 1c) (Palmqvist et al. 2017). Intriguingly, these are also the cortical areas that are particularly strongly connected to many brain regions within the DMN (Buckner et al. 2008, Utevsky et al. 2014) and express strong levels of activity at rest (Buckner et al. 2005, Lustig et al. 2003). These findings might indicate that the combination of these factors predispose those areas to vulnerability for AD pathology. Independent evidence for these conclusions comes from PET imaging demonstrating decreased cortical glucose metabolism at sites with a strong decrease of connectivity, as illustrated in Figure 1a,b (Drzezga et al. 2011).
In conclusion, PET and fMRI results provide evidence for the disruption of neuronal circuits in AD, but we are still faced with the question of whether the circuit disruption is a cause or a consequence of the disease. A related question is whether Aβ plaques are directly responsible for the AD pathophysiology. Here, it is noteworthy that the DMN dysfunction can occur before Aβ plaque formation (Sheline et al. 2010a) or when plaques just start to form (Palmqvist et al. 2017). However, the absence of detectable Aβ plaques does not exclude increased concentrations of soluble Aβ oligomers or fibrils in those subjects. Another relevant observation may be that Aβ plaques are likely to cluster in cortical hubs with a particularly high functional connectivity (Myers et al. 2014). Finally, the increased task-specific DMN activity might directly determine an increased deposition of Aβ plaques. This hypothesis is supported by evidence from studies of mouse models suggesting a link between an increased level of neuronal activity and Aβ plaque deposition (reviewed in more detail below).
In summary, although changes in DMN connectivity and activity are now widely accepted as a useful diagnostic tool, a better understanding of the pathophysiology underlying circuit disruption in AD is needed.
HIPPOCAMPAL HYPERACTIVITY: CAUSE OR CONSEQUENCE?
The hippocampus is a brain region that has a key role in many forms of learning and memory (see Eichenbaum 2017) and, perhaps not surprisingly, is especially prone to various pathological changes in AD, including early formation of tau tangles and brain atrophy as well as, later, Aβ plaque deposition (Braak & Braak 1991, Devanand et al. 2007, Thal et al. 2002; see Serrano-Pozo et al. 2011). In line with the early onset of the structural changes, evidence exists for hippocampal dysfunction at the early stages of AD and a perturbation of the functional connections between the hippocampus and other brain regions.
There are indications that early stages of AD are characterized by increased or decreased functional connectivity between the hippocampus and other brain areas (Wang et al. 2006). In patients with AD and mild cognitive impairment (MCI), the most robust finding is decreased connectivity between the hippocampus and the PPC and precuneus regions (Allen et al. 2007, Tahmasian et al. 2015, Zhou et al. 2008). Also, studies using MRI and the diffusion tensor imaging method revealed that the structural connectivity between the hippocampus and other brain regions is significantly decreased in patients with AD (Fletcher et al. 2013, Nir et al. 2013). Together, these data support the notion of increasing functional and structural decoupling of the hippocampus that disrupts memory circuits.
What is the activity status of the hippocampus in AD? In healthy subjects, hippocampal activity increases during memory encoding of associative memory tasks such as face-name matching (Sperling et al. 2003, Zeineh et al. 2003), and memory deficits correlate especially with increased activation of the dentate gyrus and the CA3 area (Yassa et al. 2011). During the progression of AD, there seems to be a bimodal distribution of the hippocampal activity levels in memory tasks (Figure 1e). Thus, early in the disease or even before symptom onset, there is pronounced hippocampal hyperactivity. For example, subjects at risk for developing AD such as APOE ε4 carriers (Bookheimer et al. 2000) as well as patients carrying mutations associated with FAD (Quiroz et al. 2010) showed increased task-specific activity of the hippocampus before symptom onset. In addition, hippocampal hyperactivity was observed in subjects with Aβ plaque deposition but without memory impairment (Mormino et al. 2012). During disease progression, which is associated with an early decline of cognitive performance, patients suffering from relatively mild memory loss usually still exhibit increased task-related hippocampal activity (Dickerson et al. 2005, Huijbers et al. 2015). However, as the cognitive decline worsens, in the stage of AD, there is evidence for a strong reduction in task-related hippocampal activity (Pariente et al. 2005) (Figure 1e). The time course of the changes in the levels of hippocampal activity with the progression of the disease was confirmed in a prospective study. Patients with MCI that initially had increased hippocampal activity at baseline developed a decrease in hippocampal activation over time. Notably, the speed of the decrease in hippocampal activity correlated with the rate of worsening memory performance (O’Brien et al. 2010). In summary, the hypothesis of the inverse U-shaped trend (Sperling et al. 2010) (Figure 1e), in which initial hyperactivity is followed by a gradual switch to hypoactivity, is receiving increasing support.
The molecular and cellular mechanisms for these changes in AD were recently studied in mouse models of AD. In these mice, neuronal hyperactivity in the hippocampus occurs before the formation of amyloid plaques (Busche et al. 2012) (see section titled Analysis of Synaptic and Cellular Impairments). Moreover, other studies of mice provided evidence that higher firing rates can promote the production of Aβ (Cirrito et al. 2005, Dolev et al. 2013, Kamenetz et al. 2003, Yuan & Grutzendler 2016). Brain areas that are more active seem to be more prone to Aβ plaque deposition (Bero et al. 2011). Hyperactive neurons in the vicinity of newly formed amyloid plaques (Busche et al. 2008) could then close the vicious circle of hyperactivity and amyloid accumulation. Such a possible link between hyperactivity and Aβ plaque deposition finds support in human studies. Recent reports of patients with MCI indicated that hippocampal activity is associated with subsequent amyloid plaque deposition as well as hippocampal atrophy and memory decline (Huijbers et al. 2015, Leal et al. 2017). Most remarkably, the level of hippocampal hyperactivity predicted the slope of further amyloid plaque accumulation (Figure 1f). Furthermore, the slope of Aβ plaque accumulation indicates the magnitude of cognitive decline during the next three to four years (Leal et al. 2017). In line with these observations, the pharmacological treatment of excessive hippocampal activity can ameliorate memory deficits. Recently, two studies indicated that the treatment of hippocampal hyperactivity, as monitored in the dentate gyrus and CA3 hippocampal regions, with the antiepileptic drug levetiracetam can partially restore memory performance in patients with MCI (Bakker et al. 2012, 2015). Studies of mice are consistent with these observations and suggest a causal link between hippocampal hyperactivity and memory impairment. Two studies reported a partial restoration in cognitive function upon treatment of rodent disease models with levetiracetam (Koh et al. 2010, Sanchez et al. 2012).
In conclusion, accumulating evidence from studies of humans and mice suggests a causal role of hippocampal hyperactivity at the early stages of AD. However, the establishment of hippocampal hyperactivity as a viable target for disease-modifying interventions requires more validation and additional translational efforts.
SLEEP, BRAIN OSCILLATIONS, AND MEMORY CONSOLIDATION
Sleep disturbances represent a prevalent symptom in many patients with AD (Zhao et al. 2016). Remarkably, sleep disturbance can be an early symptom in individuals with Aβ plaque deposition, or other pathological markers of the disease, precede pronounced memory decline (Spira et al. 2013, Sprecher et al. 2017) and become worse as the disease progresses (Liguori et al. 2014). Most intriguingly, the magnitude of the sleep deficits is correlated with memory loss, both in healthy adults and in patients with AD, suggesting a direct link between sleep quality and memory performance (Ficca et al. 2000, Westerberg et al. 2010). Importantly, not only is sleep disturbance a passive symptom of AD but increasing evidence indicates it can influence disease progression and worsen the symptoms, such as impaired memory consolidation. Thus, sleep times of less than 6.5 h per night increase the risk for cognitive decline in healthy patients (Keage et al. 2012). Also, higher sleep fragmentation is associated with a higher risk for developing AD (Lim et al. 2013a). In APOE ε4 carriers, subjects with better sleep quality are less likely to develop AD (Lim et al. 2013b). Finally, patients with sleep disturbances have abnormal levels of Aβ in cerebrospinal fluid (CSF) (Ju et al. 2013, Sprecher et al. 2017), and there is a correlation between impaired sleep quality, including sleep duration, and cortical amyloid plaque load (Spira et al. 2013).
How are sleep disturbances linked mechanistically to AD? Studies reported that the levels of soluble Aβ in CSF or interstitial fluid fluctuate during the sleep-wake cycle in mice or humans (Huang et al. 2012, Kang et al. 2009). These diurnal fluctuations are prominent before plaque formation and disappear after Aβ plaques are formed (Huang et al. 2012, Roh et al. 2012). Increasing evidence indicates that sleep disruption itself can determine the levels of Aβ in the CSF. Thus, short-time sleep deprivation can disrupt the diurnal fluctuation cycle of Aβ in the brain, and sleep deprivation for even just one night can increase the level of Aβ in CSF in healthy subjects (Ooms et al. 2014, Wei et al. 2017). Similarly, in patients with MCI, there is a correlation between impaired slow-wave sleep and increased levels of Aβ in blood plasma (Sanchez-Espinosa et al. 2014). The diurnal fluctuations in levels of Aβ may depend on many factors, including Aβ production by active neurons and the clearance of Aβ from the brain. In patients with late-onset AD, Aβ clearance was reported to be impaired, whereas the production rate, at least in the awake state, was normal (Mawuenyega et al. 2010). Potentially groundbreaking insights came from the recent identification of a glymphatic system (Xie et al. 2013). According to this model, during the sleep phase, glia cells shrink and the extracellular space widens correspondingly. This opens pathways that facilitate the transport of extracellular waste products, such as soluble Aβ, from the interstitial space.
At the circuit level, increasing evidence supports the notion that neuronal processing, particularly during non-REM (rapid eye movement) sleep, is important for memory consolidation. During this slow-wave sleep, recently acquired memories are thought to be transferred from short-term storage sites in the hippocampus to long-term memory storage sites in the neocortex (see Diekelmann & Born 2010). Experimental evidence obtained from rodents indicates that during slow-wave sleep there is repeated replay of hippocampal signaling patterns that were learned in the awake state (Wilson & McNaughton 1994). A similar form of replay takes place in humans during slow-wave sleep, as evidenced by studies of hippocampal activities in subjects tested during virtual reality maze trips and the following stages of sleep (Peigneux et al. 2004). Various studies underscored the importance of undisturbed slow-wave sleep for effective memory consolidation in healthy adults (Mander et al. 2014, Mednick et al. 2003, Takashima et al. 2006). The electrophysiological correlate of memory consolidation in non-REM sleep is cortical slow oscillations (Steriade et al. 1993), which are inherently linked to higher-frequency EEG rhythms, such as sleep spindles and hippocampal sharp-wave ripples (Staresina et al. 2015, Steriade 2006). There is a strong bidirectional link between those oscillations, their coordination, and memory consolidation (Clemens et al. 2005, Helfrich et al. 2018). On the basis of experiments using transcranial current stimulation (Marshall et al. 2006), auditory loop stimulation (Ngo et al. 2013), or pharmacological approaches (Mednick et al. 2013) in healthy adults, evidence suggests that enhancing slow-wave activity, associated sleep spindles, or both during sleep can boost memory performance. A recent study has even suggested a beneficial effect of enhancing sleep slow oscillations by transcranial direct current stimulation on memory in patients with early AD (Ladenbauer et al. 2017). Conversely, selective disruption of slow waves during sleep without changing overall sleep time can impair memory consolidation in healthy subjects (van der Werf et al. 2009).
In AD patients, disruption of slow-wave sleep can occur at early stages of the disease. Thus, APOE ε4 carriers at risk for developing AD (Hita-Yanez et al. 2012) as well as patients suffering only from mildly impaired memory performance (Westerberg et al. 2012) showed impaired slow oscillations during sleep. Particularly relevant is a recent study in which Mander et al. (2015) combined PET, fMRI, EEG, and memory tests to study the status of slow oscillations in AD. They found that the medial prefrontal cortex, a cortical region with a high susceptibility for slow wave generation (Murphy et al. 2009), is particularly prone to massive cortical Aβ plaque deposition (Figure 2). Their findings suggest that the Aβ plaque load in the medial prefrontal cortex can predict the level of impairments of slow oscillations during sleep and the worsening of memory performance.
Figure 2.
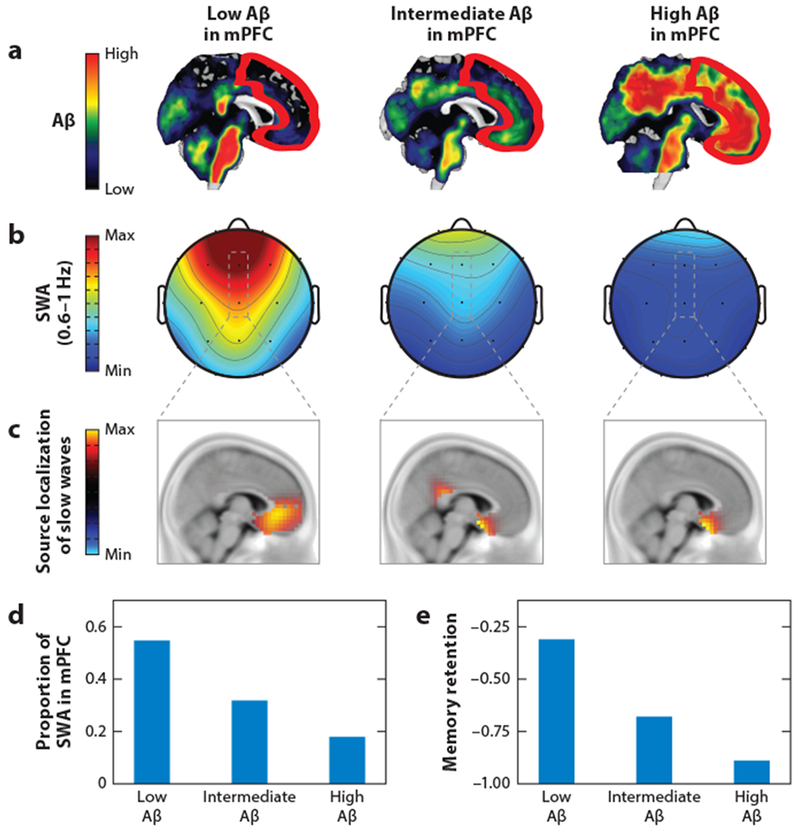
Aβ plaque load, slow oscillations during sleep, and memory performance in subjects without cognitive impairments. (a) Aβ plaque load measured with PiB-PET from subjects with low (left), intermediate (middle), and high (right) plaque load, respectively. The red outline indicates the location of the mPFC. (b) EEG-based maps of the power of sleep SWA (0.6–1 Hz) for the three subjects in panel a. (c) Corresponding source localization of mPFC slow waves for the three subjects in panel a. (d) Proportions of sleep SWA for the three subjects in panel a. (e) Memory retention for the three subjects in panel a. Panels a–e adapted from Mander et al. (2015) with permission from Macmillan Publishers Ltd. Abbreviations: Aβ, amyloid beta; mPFC, medial prefrontal cortex; PET, positron emission tomography; PiB, Pittsburgh compound B; SWA, slow-wave activity.
Independent evidence for the disruption of slow-wave activity in AD also comes from studies that were performed in mouse models of β-amyloidosis. Experiments combining electrophysiology and calcium imaging, representing a direct and accurate way to probe for neuronal activity, demonstrated that the coherence of slow waves between different cortical regions, the hippocampus, and the thalamus is completely disrupted in an APP23 × PS45 mouse model compared with wild-type controls (Figure 3a–c) (Busche et al. 2015b). Consistent with this observation, the acute application of synthetic Aβ alone could also elicit such a breakdown of slow-wave coherence in wild-type mice. Conversely, slow oscillations in both AD mouse models and wild-type mice treated with Aβ were restored by increasing synaptic inhibition with GABA receptor agonists such as benzodiazepines (Figure 3a,b). Remarkably, restoring slow oscillations also rescued normal memory function in those mice (Figure 3d). In line with these findings, the pharmacological suppression of Aβ production with a BACE (β-secretase) (Neumann et al. 2015) inhibitor effectively restored neuronal and circuit dysfunction as well as memory deficits (Keskin et al. 2017). These results provide encouraging evidence that neuronal impairments can be restored even during late-stage AD.
Figure 3.
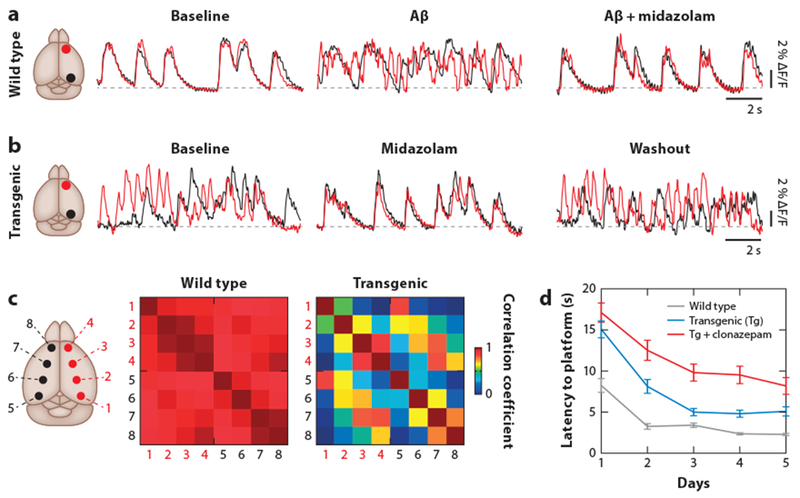
Disrupted cortical slow-wave oscillations in a transgenic mouse model of AD in vivo. (a) Overlay of SWA traces in the frontal (red) and occipital (black) cortices monitored with wide-field calcium imaging in a wild-type mouse (left). The application of soluble Aβ disrupts the synchrony of SWA (middle). Rescue of SWA by midazolam (right). (b) Disrupted SWA in the transgenic APP23 × PS45 mouse model of AD (left). The correlation can be transiently restored by application of midazolam (middle and right). The sites of recording are indicated in the corresponding schematics (leftmost images). (c) Disrupted SWA in transgenic AD mice. Cross-correlation matrices for eight cortical recording sites (as indicated) in wild-type (left) and transgenic AD mice (right). (d) Improvement of memory performance in a water maze test performed over five consecutive days in wild-type (gray), transgenic untreated (red), and clonazepam-treated transgenic (blue) mice. Panels a–d adapted from Busche et al. (2015b) with permission from Springer Nature. Abbreviations: Aβ, amyloid beta; AD, Alzheimer’s disease; APP, amyloid precursor protein; PS, presenilin-1; SWA, slow-wave activity.
ANALYSIS OF SYNAPTIC AND CELLULAR IMPAIRMENTS
Neuroanatomical and in vitro physiological studies suggest that synaptic impairment is a key pathomechanism underlying AD. In fact, synapse loss correlates more strongly with cognitive decline than do the numbers of plaques, tangles, or lost neurons (DeKosky et al. 1996, Masliah et al. 1994, Terry et al. 1991). Experimental studies even showed that synaptic dysfunction and loss (i.e., synaptic failure) (Selkoe 2002) begin to occur at the earliest stages of pathology, prior to plaque formation and independent of neurodegeneration (D’Amelio et al. 2011, Hong et al. 2016, Hsia et al. 1999, Lanz et al. 2003, Roy et al. 2016). In line with these synaptic impairments and in view of the loss of memory in demented patients, it is of major interest to know how the cellular correlates of learning and memory, long-term potentiation (LTP) and long-term depression (LTD), are affected. For such an analysis, water-soluble extracts from patients with AD were applied to rodent hippocampal slices and shown to inhibit LTP or enhance LTD (Barry et al. 2011, Klyubin et al. 2008, Li et al. 2009, Shankar et al. 2008). Removal of Aβ from the extracts prevented these effects. Moreover, the acute exposure to synthetic or naturally secreted Aβ oligomers produced similar impairments in plasticity (Freir et al. 2001, Lambert et al. 1998, Walsh et al. 2002). Accumulating evidence indicates that a small pool of low-molecular-weight (LMW) oligomers, including dimers, trimers, and Aβ*56, are more potent in producing such synaptic and cognitive impairments than are high-molecular-weight oligomers (Lesne et al. 2006, Yang et al. 2017), which are much more prevalent in the AD brain (Yang et al. 2017). Intriguingly, picomolar concentrations of Aβ increased LTP in hippocampal slices, pointing toward a potential physiological role of extremely low amounts of Aβ in learning and memory processes (Puzzo et al. 2008).
Spine loss can occur independently of the formation of Aβ plaques (Hsieh et al. 2006, Jacobsen et al. 2006, Shankar et al. 2007, Spires et al. 2005, Wei et al. 2010). In fact, in the human AD brain, the levels of soluble Aβ, rather than plaques, are strongly correlated with the extent of synaptic loss (Lue et al. 1999). Hong et al. (2016) showed, for example, that injections of LMW dimers into the ventricles of wild-type mice were sufficient to induce substantial synapse loss within 3 days. The results indicated that synapses were eliminated by phagocytic microglia cells, which were activated by the deposition of the complement factor C1q at synapses. Such a mechanism is intriguing, as recent genome-wide association studies implicate microglia and complement pathways in AD (Mhatre et al. 2015). Furthermore, in vivo ratiometric calcium imaging in wild-type mice exposed to Aβ oligomers revealed that abnormal increases in intracellular calcium levels immediately precede the loss of synapses (Arbel-Ornath et al. 2017). In later stages of AD, synapse loss appears to be maximal at locations near amyloid plaques (Spires et al. 2005). Intriguingly, Aβ oligomers are also most abundant in the vicinity of plaques and accumulate at pre- and postsynaptic sites (Koffie et al. 2009), strongly suggesting their direct role in synaptic modifications. Their actions are not restricted to neuronal cell bodies but have a pathological effect on neurites (Kuchibhotla et al. 2008), astrocytes (Kuchibhotla et al. 2009, Pekny et al. 2016), and microglia (Prinz et al. 2011).
Because synaptic failure and plasticity defects were expected to result in the silencing of neurons, it was surprising that many neurons were hyperactive under in vivo conditions in mouse models (Busche et al. 2008, 2012, 2015a,b; Grienberger et al. 2012; Keskin et al. 2017; Liebscher et al. 2016; Maier et al. 2014; Rudinskiy et al. 2012; Scala et al. 2015; Siskova et al. 2014; Xu et al. 2015) (Figure 4a,b). This hyperactivity is responsible for alterations of the sensory integration in the cortex and the corresponding behavioral defects. Thus, in the visual cortex of APP23 × PS45 mice, hyperactive neurons lose their ability to respond selectively to the orientation and direction of visual stimuli (Grienberger et al. 2012). This neuronal defect was associated with deficits in a visual pattern discrimination task. In line with these observations, electrophysiological studies of the hippocampus of aged rats revealed that hyperactivity impairs the ability of neurons to encode spatial locations (Koh et al. 2010, Wilson et al. 2005). Furthermore, hyperactivity can exacerbate and cause, in transgenic AD mouse models, epileptiform discharges as well as spontaneous, recurrent seizures (Figure 4c,d) (Born 2015, Palop et al. 2007, Verret et al. 2012). A similar susceptibility to epilepsy is often observed in patients with AD (Scarmeas et al. 2009, Vossel et al. 2016).
Figure 4.
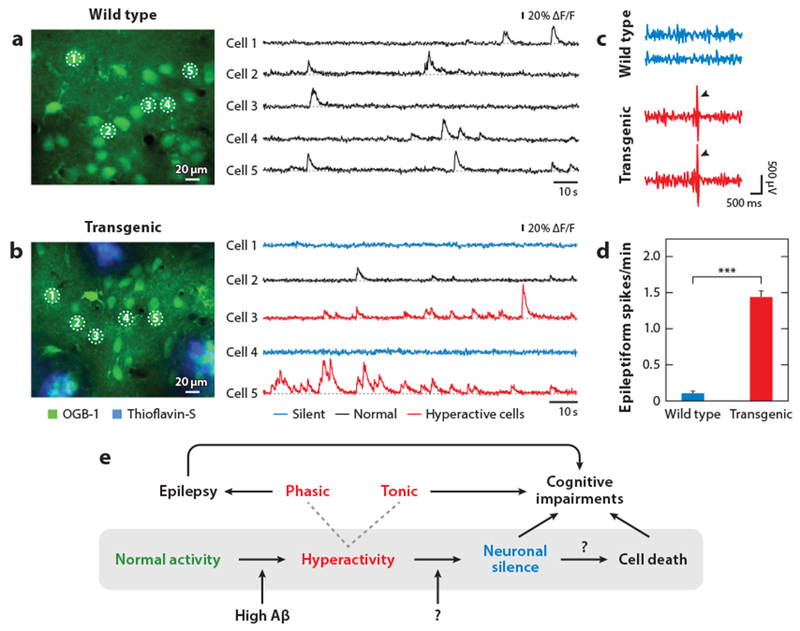
Impaired neuronal activity in mouse models of AD in vivo. (a) Two-photon calcium imaging recordings of spontaneous activity from layer-2/3 neurons (indicated as cells 1 through 5) in the frontal cortex of a wild-type mouse in vivo. The position of the neurons is indicated in the image on the left. (b) Recordings in an APP23 × PS45 transgenic mouse from a neuron with normal activity (black, cell 2), hyperactive neurons (red, cell 3 and cell 5), and silent neurons (blue, cell 1 and cell 4). In panels a and b the cortex was stained with the calcium indicator OGB-1 (green) and plaques were labeled with thioflavin-S (blue). Panels a and b adapted from Busche et al. (2008) with permission from AAAS. (c) Cortical EEG recordings from a wild-type mouse and a hAPPJ20 transgenic mouse. Arrowheads depict epileptiform spikes. (d) Epileptiform spike frequency in wild-type and transgenic mice; summary plot from panel c (***, P < 0.001). Panels c and d reprinted from Verret et al. (2012) with permission from Elsevier. (e) Schematic model indicating the sequence of events that may underlie the progression of neuronal dysfunction in AD. Reproduced from Busche & Konnerth (2015) with permission from Wiley. Abbreviations: Aβ, amyloid beta; AD, Alzheimer’s disease; APP, amyloid precursor protein; OGB-1, Oregon green-BAPTA-1; PS, presenilin-1.
There is now substantial evidence that neuronal hyperactivity, both in the hippocampus and in the cortex, is directly mediated by Aβ, most likely in the form of soluble LMW oligomers. First, brain levels of Aβ correlate strongly with the amount of hyperactive cells in the cortex (Keskin et al. 2017). Second, suppression of Aβ production by γ-secretase (acute) or β-secretase (chronic) can rescue the hyperactivity phenotype in the hippocampus and cortex (Busche et al. 2012, Keskin et al. 2017). Third, reintroduction of soluble Aβ oligomers to mice that were successfully treated with a BACE inhibitor resulted in the recurrence of cortical hyperactivity (Keskin et al. 2017). Finally, local application of A-β dimers to hippocampal neurons induced hyperactivity (Busche et al. 2012). That hyperactive neurons cluster around plaques at later stages of AD is not surprising because plaques are surrounded by a halo of Aβ oligomers (Keskin et al. 2017, Koffie et al. 2009). Calcium overload in neuronal processes also seems to be due to the action of Aβ oligomers around plaques (Arbel-Ornath et al. 2017). Experimental evidence obtained in mouse models of AD indicates that soluble Aβ produces neuron-specific alterations of the excitation/inhibition (E/I) balance (Busche & Konnerth 2016, Busche et al. 2008). The E/I balance can be restored to normal levels, as indicated by experiments in which the pharmacological enhancement of GABAergic inhibition through the administration of benzodiazepines prevented cellular hyperactivity. Moreover, this treatment restored slow-wave oscillations and ameliorated memory deficits (Figure 3b,d) (Busche et al. 2008, 2015b). A recent remarkable observation is that the interaction between oscillations and Aβ is bidirectional. There is evidence that the restoration of oscillatory activity can prevent plaque formation (Iaccarino et al. 2016, Kastanenka et al. 2017). Aβ-dependent impairments of synaptic inhibition can account for many of the known defects in the AD brain, including impairments of gamma and slow oscillations, epileptic activity and seizures, and hippocampal BOLD hyperactivation, as well as cognitive impairments (Bakker et al. 2012, Busche et al. 2015b, Palop & Mucke 2016, Verret et al. 2012). Even though Aβ can interact with multiple receptors and proteins at the synapse, other mechanisms, such as impaired glutamate homeostasis (Fogel et al. 2014, Kamenetz et al. 2003, Li et al. 2009), may shift the E/I balance toward excitation.
In addition to the presence of hyperactive neurons, a considerable fraction of neurons are hypoactive and even functionally completely silent in mouse models of AD (Figure 4a,b) (Busche et al. 2008, 2012). Actually, such hypoactivity was more in line with the original synaptic failure hypothesis (Selkoe 2002). The hypoactivity was caused partly by an increase in synaptic inhibition (Busche et al. 2008). During the progression of the disease, hypoactive neurons occurred later than hyperactive neurons and were found only after plaque formation (Busche et al. 2012). Sensory responses to external stimuli are absent in these cells, even after spontaneous activity is restored by application of a GABAA antagonist (Grienberger et al. 2012). Although the presence of amyloid plaques seems to be required for hypoactivity, Aβ does not directly promote silencing of neurons in vivo. Hypoactivity may be a consequence of excessive hyperactivity, which may result in increased but maladaptive compensatory inhibition to prevent excessive firing (Busche & Konnerth 2015). However, the molecular mechanisms underlying hypoactivity may be more complex, as recent evidence points to the presence of a previously unknown but prevalent fragment of APP, Aη-alpha, that can directly promote silencing of neurons in vivo and inhibit LTP in vitro (Willem et al. 2015). In summary, there is now ample evidence from both in vitro and in vivo studies that Aβ impairs the functions of synapses, neurons, and larger circuits. Aβ exists in multiple forms in the brain—from monomers to plaques—but the small pool of LMW oligomers appears to be responsible for most of the known neuronal deficits. Aβ oligomers attack synapses, resulting in widespread plasticity defects as well as synapse dysfunction and loss. As a consequence, there is the expected silencing of circuits but, remarkably, also neuronal hyperactivity and epileptic activity. Together these effects may lead to cognitive impairments over time (Figure 4e).
CONCLUSIONS
Neuronal circuits are functionally impaired even at early stages of AD, often before the onset of overt symptoms of dementia. The constant improvement of imaging technologies is increasingly used to develop better biomarkers for AD in PET studies and to improve fMRI analyses of those brain regions strongly affected by the disease. New variants of multimodal imaging provide better insights into the changes that occur in different cortical areas, including the DMN, and in the hippocampus. There is growing evidence that circuit disruption occurs early during the progression of the disease and that some dysfunctions are triggered by excessive levels of neuronal activity, particularly in the hippocampus. How helpful are mouse studies? Mouse models of β-amyloidosis have many limitations, related mostly to the short lifetime of mice, the lack of brain atrophy, and the uncontrolled expression of Aβ in many transgenic models. Therefore, it is surprising that some of the neuronal impairments, primarily the sustained hyperactivity and the susceptibility to epilepsy as well as the impairment of the circuits underlying brain oscillations and memory consolidation, are remarkably well conserved in mice. Thus, mouse models seem to be particularly useful for the analysis of early dysfunctions of neurons and circuits. We anticipate that concerted studies of humans and appropriately tailored mouse models will have a strong potential to clarify those pathophysiological mechanisms needed for the design of effective treatments.
ACKNOWLEDGMENTS
This work was supported by the Deutsche Forschungsgemeinschaft (SFB 870) and a European Research Council Advanced Grant (to A.K.).
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- Allen G, Barnard H, McColl R, Hester AL, Fields JA, et al. 2007. Reduced hippocampal functional connectivity in Alzheimer disease. Arch. Neurol 64:1482–87 [DOI] [PubMed] [Google Scholar]
- Alzheimer A 1907. Über eine eigenartige Erkrankung der Hirnrinde. Allg. Z. Psychiatr. Phys.-Gerichtl. Med 64:146–48 [Google Scholar]
- Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR. 1995. An English translation of Alzheimer’s 1907 paper, “Über eine eigenartige Erkankung der Hirnrinde.” Clin. Anat 8:429–31 [DOI] [PubMed] [Google Scholar]
- Arbel-Ornath M, Hudry E, Boivin JR, Hashimoto T, Takeda S, et al. 2017. Soluble oligomeric amyloid-β induces calcium dyshomeostasis that precedes synapse loss in the living mouse brain. Mol. Neurodegener 12:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai F, Watson DR, Shi Y, Wang Y, Yue C, et al. 2011. Specifically progressive deficits of brain functional marker in amnestic type mild cognitive impairment. PLOS ONE 6:e24271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker A, Albert MS, Krauss G, Speck CL, Gallagher M. 2015. Response of the medial temporal lobe network in amnestic mild cognitive impairment to therapeutic intervention assessed by fMRI and memory task performance. NeuroImage Clin 7:688–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, et al. 2012. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron 74:467–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry AE, Klyubin I, Mc Donald JM, Mably AJ, Farrell MA, et al. 2011. Alzheimer’s disease brain-derived amyloid-β-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J. Neurosci 31:7259–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, et al. 2011. Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat. Neurosci 14:750–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bookheimer SY, Strojwas MH, Cohen MS, Saunders AM, Pericak-Vance MA, et al. 2000. Patterns of brain activation in people at risk for Alzheimer’s disease. N. Engl. J. Med 343:450–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borlikova GG, Trejo M, Mably AJ, Mc Donald JM, Sala Frigerio C, et al. 2013. Alzheimer brain-derived amyloid β-protein impairs synaptic remodeling and memory consolidation. Neurobiol. Aging 34:1315–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Born HA. 2015. Seizures in Alzheimer’s disease. Neuroscience 286:251–63 [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. 1991. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–59 [DOI] [PubMed] [Google Scholar]
- Buckley RF, Schultz AP, Hedden T, Papp KV, Hanseeuw BJ, et al. 2017. Functional network integrity presages cognitive decline in preclinical Alzheimer disease. Neurology 89:29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Andrews-Hanna JR, Schacter DL. 2008. The brain’s default network: anatomy, function, and relevance to disease. Ann. N. Y. Acad. Sci 1124:1–38 [DOI] [PubMed] [Google Scholar]
- Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, et al. 2005. Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. J. Neurosci 25:7709–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche MA, Chen X, Henning HA, Reichwald J, Staufenbiel M, et al. 2012. Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. PNAS 109:8740–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, et al. 2008. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 321:1686–89 [DOI] [PubMed] [Google Scholar]
- Busche MA, Grienberger C, Keskin AD, Song B, Neumann U, et al. 2015a. Decreased amyloid-β and increased neuronal hyperactivity by immunotherapy in Alzheimer’s models. Nat. Neurosci 18:1725–27 [DOI] [PubMed] [Google Scholar]
- Busche MA, Kekuš M, Adelsberger H, Noda T, Förstl H, et al. 2015b. Rescue of long-range circuit dysfunction in Alzheimer’s disease models. Nat. Neurosci 18:1623–30 [DOI] [PubMed] [Google Scholar]
- Busche MA, Konnerth A.2015. Neuronal hyperactivity-a key defect in Alzheimer’s disease? BioEssays 37:624–32 [DOI] [PubMed] [Google Scholar]
- Busche MA, Konnerth A. 2016. Impairments of neural circuit function in Alzheimer’s disease. Philos. Trans. R. Soc. B 371:20150429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhatwal JP, Schultz AP, Johnson K, Benzinger TLS, Jack C, et al. 2013. Impaired default network functional connectivity in autosomal dominant Alzheimer disease. Neurology 81:736–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien DT, Szardenings AK, Bahri S, Walsh JC, Mu F, et al. 2014. Early clinical PET imaging results with the novel PHF-tau radioligand F18-T808. J. Alzheimers Dis 38:171–84 [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, et al. 2005. Synaptic activity regulates interstitial fluid amyloid-β levels in vivo. Neuron 48:913–22 [DOI] [PubMed] [Google Scholar]
- Clemens Z, Fabó D, Halász P. 2005. Overnight verbal memory retention correlates with the number of sleep spindles. Neuroscience 132:529–35 [DOI] [PubMed] [Google Scholar]
- D’Amelio M, Cavallucci V, Middei S, Marchetti C, Pacioni S, et al. 2011. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat. Neurosci 14:69–76 [DOI] [PubMed] [Google Scholar]
- Damoiseaux JS, Prater KE, Miller BL, Greicius MD. 2012. Functional connectivity tracks clinical deterioration in Alzheimer’s disease. Neurobiol. Aging 33:828e19–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daselaar SM, Prince SE, Cabeza R. 2004. When less means more: deactivations during encoding that predict subsequent memory. NeuroImage 23:921–27 [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Scheff SW, Styren SD. 1996. Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration 5:417–21 [DOI] [PubMed] [Google Scholar]
- Devanand DP, Pradhaban G, Liu X, Khandji A, De Santi S, et al. 2007. Hippocampal and entorhinal atrophy in mild cognitive impairment: prediction of Alzheimer’s disease. Neurology 68:828–36 [DOI] [PubMed] [Google Scholar]
- Dickerson BC, Salat DH, Greve DN, Chua EF, Rand-Giovannetti E, et al. 2005. Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology 65:404–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diekelmann S, Born J. 2010. The memory function of sleep. Nat. Rev. Neurosci 11:114–26 [DOI] [PubMed] [Google Scholar]
- Dillen KNH, Jacobs HIL, Kukolja J, Richter N, von Reutern B, et al. 2017. Functional disintegration of the default mode network in prodromal Alzheimer’s disease. J. Alzheimers Dis 59:169–87 [DOI] [PubMed] [Google Scholar]
- Dolev I, Fogel H, Milshtein H, Berdichevsky Y, Lipstein N, et al. 2013. Spike bursts increase amyloid-β 40/42 ratio by inducing a presenilin-1 conformational change. Nat. Neurosci 16:587–95 [DOI] [PubMed] [Google Scholar]
- Drzezga A, Becker JA, van Dijk KRA, Sreenivasan A, Talukdar T, et al. 2011. Neuronal dysfunction and disconnection of cortical hubs in non-demented subjects with elevated amyloid burden. Brain 134(Pt. 6):1635–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, et al. 2014. Advancing research diagnostic criteria for Alzheimer’s disease. The IWG-2 criteria. Lancet Neurol 13:614–29 [DOI] [PubMed] [Google Scholar]
- Eichenbaum H 2017. Memory: organization and control. Annu. Rev. Psychol 68:19–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris SH, de Leon MJ, Wolf AP, Farkas T, Christman DR, et al. 1980. Positron emission tomography in the study of aging and senile dementia. Neurobiol. Aging 1:127–31 [DOI] [PubMed] [Google Scholar]
- Ficca G, Lombardo P, Rossi L, Salzarulo P. 2000. Morning recall of verbal material depends on prior sleep organization. Behav. Brain Res 112:159–63 [DOI] [PubMed] [Google Scholar]
- Fletcher E, Raman M, Huebner P, Liu A, Mungas D, et al. 2013. Loss of fornix white matter volume as a predictor of cognitive impairment in cognitively normal elderly individuals. JAMA Neurol 70:1389–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel H, Frere S, Segev O, Bharill S, Shapira I, et al. 2014. APP homodimers transduce an amyloid-β-mediated increase in release probability at excitatory synapses. Cell Rep 7:1560–76 [DOI] [PubMed] [Google Scholar]
- Freir DB, Holscher C, Herron CE. 2001. Blockade of long-term potentiation by β-amyloid peptides in the CA1 region of the rat hippocampus in vivo. J. Neurophysiol 85:708–13 [DOI] [PubMed] [Google Scholar]
- Greicius MD, Srivastava G, Reiss AL, Menon V. 2004. Default-mode network activity distinguishes Alzheimer’s disease from healthy aging: evidence from functional MRI. PNAS 101:4637–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grienberger C, Rochefort NL, Adelsberger H, Henning HA, Hill DN, et al. 2012. Staged decline of neuronal function in vivo in an animal model of Alzheimer’s disease. Nat. Commun 3:774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. 2002. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297:353–56 [DOI] [PubMed] [Google Scholar]
- Hedden T, van Dijk KRA, Becker JA, Mehta A, Sperling RA, et al. 2009. Disruption of functional connectivity in clinically normal older adults harboring amyloid burden. J. Neurosci 29:12686–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfrich RF, Mander BA, Jagust WJ, Knight RT, Walker MP. 2018. Old brains come uncoupled in sleep: slow wave-spindle synchrony, brain atrophy, and forgetting. Neuron 97:221–30. e224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hita-Yañez E, Atienza M, Gil-Neciga E, Cantero JL. 2012. Disturbed sleep patterns in elders with mild cognitive impairment: the role of memory decline and ApoE ε4 genotype. Curr. Alzheimer Res 9:290–97 [DOI] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, et al. 2016. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352:712–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, et al. 1999. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. PNAS 96:3228–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, et al. 2006. AMPAR removal underlies Aβ-induced synaptic depression and dendritic spine loss. Neuron 52:831–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Potter R, Sigurdson W, Santacruz A, Shih S, et al. 2012. Effects of age and amyloid deposition on Aβ dynamics in the human central nervous system. Arch. Neurol 69:51–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huijbers W, Mormino EC, Schultz AP, Wigman S, Ward AM, et al. 2015. Amyloid-β deposition in mild cognitive impairment is associated with increased hippocampal activity, atrophy and clinical progression. Brain 138(Pt.4):1023–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaccarino HF, Singer AC, Martorell AJ, Rudenko A, Gao F, et al. 2016. Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature 540:230–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Albert MS, Knopman DS, McKhann GM, Sperling RA, et al. 2011. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7:257–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, et al. 2006. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer’s disease. PNAS 103:5161–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J. 2004. EEG dynamics in patients with Alzheimer’s disease. Clin. Neurophysiol 115:1490–505 [DOI] [PubMed] [Google Scholar]
- Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ. 2011. Soluble amyloid β-protein dimers isolated from Alzheimer cortex directly induce tau hyperphosphorylation and neuritic degeneration. PNAS 108:5819–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KA, Schultz A, Betensky RA, Becker JA, Sepulcre J, et al. 2016. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann. Neurol 79:110–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT, Knopman DS, Gunter JL, Graff-Radford J, Vemuri P, et al. 2016. Cascading network failure across the Alzheimer’s disease spectrum. Brain 139(Pt. 2):547–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju Y-ES, McLeland JS, Toedebusch CD, Xiong C, Fagan AM, et al. 2013. Sleep quality and preclinical Alzheimer disease. JAMA Neurol 70:587–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, et al. 2003APP processing and synaptic function. Neuron 37:925–37 [DOI] [PubMed] [Google Scholar]
- Kang J-E, Lim MM, Bateman RJ, Lee JJ, Smyth LP, et al. 2009. Amyloid-β dynamics are regulated by orexin and the sleep-wake cycle. Science 326:1005–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastanenka KV, Hou SS, Shakerdge N, Logan R, Feng D, et al. 2017. Optogenetic restoration of disrupted slow oscillations halts amyloid deposition and restores calcium homeostasis in an animal model of Alzheimer’s disease. PLOS ONE 12:e0170275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keage HAD, Banks S, Yang KL, Morgan K, Brayne C, Matthews FE. 2012. What sleep characteristics predict cognitive decline in the elderly? Sleep Med 13:886–92 [DOI] [PubMed] [Google Scholar]
- Keskin AD, Kekuš M, Adelsberger H, Neumann U, Shimshek DR, et al. 2017. BACE inhibition-dependent repair of Alzheimer’s pathophysiology. PNAS 114:8631–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, et al. 2004. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh compound-B. Ann. Neurol 55:306–19 [DOI] [PubMed] [Google Scholar]
- Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, et al. 2008Amyloid β protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J. Neurosci 28:4231–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, et al. 2009. Oligomeric amyloid β associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. PNAS 106:4012–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh MT, Haberman RP, Foti S, McCown TJ, Gallagher M. 2010. Treatment strategies targeting excess hippocampal activity benefit aged rats with cognitive impairment. Neuropsychopharmacology 5:1016–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. 2008. Aβ plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 59:214–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchibhotla KV, Lattarulo CR, Hyman BT, Bacskai BJ. 2009. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 323:1211–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladenbauer J, Ladenbauer J, Külzow N, de Boor R, Avramova E, et al. 2017. Promoting sleep oscillations and their functional coupling by transcranial stimulation enhances memory consolidation in mild cognitive impairment. J. Neurosci 37:7111–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed, et al. 1998. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. PNAS 95:6448–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanz TA, Carter DB, Merchant KM. 2003. Dendritic spine loss in the hippocampus of young PDAPP and Tg2576 mice and its prevention by the ApoE2 genotype. Neurobiol. Dis 13:246–53 [DOI] [PubMed] [Google Scholar]
- Leal SL, Landau SM, Bell RK, Jagust WJ. 2017. Hippocampal activation is associated with longitudinal amyloid accumulation and cognitive decline. eLife 6:e22978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, et al. 2006. A specific amyloid-β protein assembly in the brain impairs memory. Nature 440:352–57 [DOI] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. 2009. Soluble oligomers of amyloid β protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62:788–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebscher S, Keller GB, Goltstein PM, Bonhoeffer T, Hubener M. 2016. Selective persistence of sensorimotor mismatch signals in visual cortex of behaving Alzheimer’s disease mice. Curr. Biol 26:956–64 [DOI] [PubMed] [Google Scholar]
- Liguori C, Romigi A, Nuccetelli M, Zannino S, Sancesario G, et al. 2014. Orexinergic system dysregulation, sleep impairment, and cognitive decline in Alzheimer disease. JAMA Neurol 71:1498–505 [DOI] [PubMed] [Google Scholar]
- Lim ASP, Kowgier M, Yu L, Buchman AS, Bennett DA. 2013a. Sleep fragmentation and the risk of incident Alzheimer’s disease and cognitive decline in older persons. Sleep 36:1027–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ASP, Yu L, Kowgier M, Schneider JA, Buchman AS, Bennett DA. 2013b. Modification of the relationship of the apolipoprotein E ε4 allele to the risk of Alzheimer’s disease and neurofibrillary tangle density by sleep. JAMA Neurol 70:1544–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Yu C, Zhang X, Liu J, Duan Y, et al. 2014. Impaired long distance functional connectivity and weighted network architecture in Alzheimer’s disease. Cereb. Cortex 24:1422–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Zou Q, Gu H, Raichle ME, Stein EA, Yang Y. 2012. Rat brains also have a default mode network. PNAS 109:3979–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, et al. 1999. Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am. J. Pathol 155:853–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustig C, Snyder AZ, Bhakta M, O’Brien KC, McAvoy M, et al. 2003. Functional deactivations: change with age and dementia of the Alzheimer type. PNAS 100:14504–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier FC, Wehrl HF, Schmid AM, Mannheim JG, Wiehr S, et al. 2014. Longitudinal PET-MRI reveals β-amyloid deposition and rCBF dynamics and connects vascular amyloidosis to quantitative loss of perfusion. Nat. Med 20:1485–92 [DOI] [PubMed] [Google Scholar]
- Mander BA, Marks SM, Vogel JW, Rao V, Lu B, et al. 2015. β-Amyloid disrupts human NREM slow waves and related hippocampus-dependent memory consolidation. Nat. Neurosci 18:1051–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mander BA, Rao V, Lu B, Saletin JM, Ancoli-Israel S, et al. 2014. Impaired prefrontal sleep spindle regulation of hippocampal-dependent learning in older adults. Cereb. Cortex 24:3301–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall L, Helgadóttir H, Mölle M, Born J. 2006. Boosting slow oscillations during sleep potentiates memory. Nature 444:610–13 [DOI] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Hansen L, DeTeresa R, Alford M, Terry R. 1994. Synaptic and neuritic alterations during the progression of Alzheimer’s disease. Neurosci. Lett 174:67–72 [DOI] [PubMed] [Google Scholar]
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, et al. 2010. Decreased clearance of CNS β-amyloid in Alzheimer’s disease. Science 330:1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, et al. 2011. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7:263–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mednick S, Nakayama K, Stickgold R. 2003. Sleep-dependent learning: A nap is as good as a night. Nat. Neurosci 6:697–98 [DOI] [PubMed] [Google Scholar]
- Mednick SC, McDevitt EA, Walsh JK, Wamsley E, Paulus M, et al. 2013. The critical role of sleep spindles in hippocampal-dependent memory: a pharmacology study. J. Neurosci 33:4494–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mhatre SD, Tsai CA, Rubin AJ, James ML, Andreasson KI. 2015. Microglial malfunction: the third rail in the development of Alzheimer’s disease. Trends Neurosci 38:621–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SL, Celone K, DePeau K, Diamond E, Dickerson BC, et al. 2008. Age-related memory impairment associated with loss of parietal deactivation but preserved hippocampal activation. PNAS 105:2181–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mormino EC, Brandel MG, Madison CM, Marks S, Baker SL, Jagust WJ 2012. Aβ deposition in aging is associated with increases in brain activation during successful memory encoding. Cereb. Cortex 22:1813–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris E, Chalkidou A, Hammers A, Peacock J, Summers J, Keevil S. 2016. Diagnostic accuracy of18F amyloid PET tracers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Eur. J. Nucl. Med. Mol. Imaging 43:374–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller T, Meyer HE, Egensperger R, Marcus K. 2008. The amyloid precursor protein intracellular domain (AICD) as modulator of gene expression, apoptosis, and cytoskeletal dynamics-relevance for Alzheimer’s disease. Prog. Neurobiol 85:393–406 [DOI] [PubMed] [Google Scholar]
- Müller UC, Deller T, Korte M. 2017. Not just amyloid: physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci 18:281–98 [DOI] [PubMed] [Google Scholar]
- Murphy M, Riedner BA, Huber R, Massimini M, Ferrarelli F, Tononi G. 2009. Source modeling sleep slow waves. PNAS 106:1608–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers N, Pasquini L, Göttler J, Grimmer T, Koch K, et al. 2014Within-patient correspondence of amyloid-β and intrinsic network connectivity in Alzheimer’s disease. Brain 137(Pt. 7):2052–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann U, Rueeger H, Machauer R, Veenstra SJ, Lueoend RM, et al. 2015A novel BACE inhibitor NB-360 shows a superior pharmacological profile and robust reduction of amyloid-β and neuroinflammation in APP transgenic mice. Mol. Neurodegener 10:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo H-VV, Martinetz T, Born J, Mölle M.2013. Auditory closed-loop stimulation of the sleep slow oscillation enhances memory. Neuron 78:545–53 [DOI] [PubMed] [Google Scholar]
- Nir TM, Jahanshad N, Villalon-Reina JE, Toga AW, Jack CR, et al. 2013. Effectiveness of regional DTI measures in distinguishing Alzheimer’s disease, MCI, and normal aging. NeuroImage 3:180–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien JL, O’Keefe KM, LaViolette PS, DeLuca AN, Blacker D, et al. 2010. Longitudinal fMRI in elderly reveals loss of hippocampal activation with clinical decline. Neurology 74:1969–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooms S, Overeem S, Besse K, Rikkert MO, Verbeek M, et al. 2014. Effect of 1 night of total sleep deprivation on cerebrospinal fluid β-amyloid 42 in healthy middle-aged men: a randomized clinical trial. JAMA Neurol 71:971–77 [DOI] [PubMed] [Google Scholar]
- Palmqvist S, Schöll M, Strandberg O, Mattsson N, Stomrud E, et al. 2017. Earliest accumulation of β-amyloid occurs within the default-mode network and concurrently affects brain connectivity. Nat. Commun 8:1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, et al. 2007. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55:697–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. 2016. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci 17:777–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pariente J, Cole S, Henson R, Clare L, Kennedy A, et al. 2005. Alzheimer’s patients engage an alternative network during a memory task. Ann. Neurol 58:870–79 [DOI] [PubMed] [Google Scholar]
- Peigneux P, Laureys S, Fuchs S, Collette F, Perrin F, et al. 2004. Are spatial memories strengthened in the human hippocampus during slow wave sleep? Neuron 44:535–45 [DOI] [PubMed] [Google Scholar]
- Pekny M, Pekna M, Messing A, Steinhauser C, Lee JM, et al. 2016. Astrocytes: a central element in neurological diseases. Acta Neuropathol 131:323–45 [DOI] [PubMed] [Google Scholar]
- Persson J, Lind J, Larsson A, Ingvar M, Sleegers K, et al. 2008. Altered deactivation in individuals with genetic risk for Alzheimer’s disease. Neuropsychologia 46:1679–87 [DOI] [PubMed] [Google Scholar]
- Pihlajamäki M, O’Keefe K, Bertram L, Tanzi RE, Dickerson BC, et al. 2010. Evidence of altered posteromedial cortical FMRI activity in subjects at risk for Alzheimer disease. Alzheimer Dis. Assoc. Disord 24:28–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M, Priller J, Sisodia SS, Ransohoff RM. 2011. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat. Neurosci 14:1227–35 [DOI] [PubMed] [Google Scholar]
- Puzzo D, Privitera L, Leznik E, Fa M, Staniszewski A, et al. 2008. Picomolar amyloid-β positively modulates synaptic plasticity and memory in hippocampus. J. Neurosci 28:14537–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiroz YT, Budson AE, Celone K, Ruiz A, Newmark R, et al. 2010. Hippocampal hyperactivation in presymptomatic familial Alzheimer’s disease. Ann. Neurol 68:865–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raichle ME, MacLeod AM, Snyder AZ, Powers WJ, Gusnard DA, Shulman GL. 2001. A default mode of brain function. PNAS 98:676–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh JH, Huang Y, Bero AW, Kasten T, Stewart FR, Bateman RJ, Holtzman DM. 2012. Disruption of the sleep-wake cycle and diurnal fluctuation of β-amyloid in mice with Alzheimer’s disease pathology. Sci. Transl. Med 4:150ra122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy DS, Arons A, Mitchell TI, Pignatelli M, Ryan TJ, Tonegawa S. 2016. Memory retrieval by activating engram cells in mouse models of early Alzheimer’s disease. Nature 531:508–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudinskiy N, Hawkes JM, Betensky RA, Eguchi M, Yamaguchi S, et al. 2012. Orchestrated experience-driven Arc responses are disrupted in a mouse model of Alzheimer’s disease. Nat. Neurosci 15:1422–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, et al. 2012. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. PNAS 109:E2895–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Espinosa MP, Atienza M, Cantero JL. 2014. Sleep deficits in mild cognitive impairment are related to increased levels of plasma amyloid-β and cortical thinning. NeuroImage 98:395–404 [DOI] [PubMed] [Google Scholar]
- Scala F, Fusco S, Ripoli C, Piacentini R, Li Puma DD, et al. 2015. Intraneuronal Aβ accumulation induces hippocampal neuron hyperexcitability through A-type K+ current inhibition mediated by activation of caspases and GSK-3. Neurobiol. Aging 36:886–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarmeas N, Honig LS, Choi H, Cantero J, Brandt J, et al. 2009. Seizures in Alzheimer disease: who, when, and how common? Arch. Neurol 66:992–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seab JP, Jagust WJ, Wong ST, Roos MS, Reed BR, Budinger TF. 1988. Quantitative NMR measurements of hippocampal atrophy in Alzheimer’s disease. Magn. Reson. Med 8:200–8 [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. 2002. Alzheimer’s disease is a synaptic failure. Science 298:789–91 [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Hardy J. 2016. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med 8:595–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. 2011. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med 1:a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. 2007. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci 27:2866–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, et al. 2008. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med 14:837–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheline YI, Morris JC, Snyder AZ, Price JL, Yan Z, et al. 2010a. APOE4 allele disrupts resting state fMRI connectivity in the absence of amyloid plaques or decreased CSF Aβ42. J. Neurosci 30:17035–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheline YI, Raichle ME, Snyder AZ, Morris JC, Head D, et al. 2010b. Amyloid plaques disrupt resting state default mode network connectivity in cognitively normal elderly. Biol. Psychiatry 67:584–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siskova Z, Justus D, Kaneko H, Friedrichs D, Henneberg N, et al. 2014. Dendritic structural degeneration is functionally linked to cellular hyperexcitability in a mouse model of Alzheimer’s disease. Neuron 84:1023–33 [DOI] [PubMed] [Google Scholar]
- Sperling R, Chua E, Cocchiarella A, Rand-Giovannetti E, Poldrack R, et al. 2003. Putting names to faces: Successful encoding of associative memories activates the anterior hippocampal formation. NeuroImage 20:1400–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Dickerson BC, Pihlajamaki M, Vannini P, LaViolette PS, et al. 2010. Functional alterations in memory networks in early Alzheimer’s disease. Neuromol. Med 12:27–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Laviolette PS, O’Keefe K, O’Brien J, Rentz DM, et al. 2009. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron 63:178–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spira AP, Gamaldo AA, An Y, Wu MN, Simonsick EM, et al. 2013. Self-reported sleep and β-amyloid deposition in community-dwelling older adults. JAMA Neurol 70:1537–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, et al. 2005. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J. Neurosci 25:7278–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprecher KE, Koscik RL, Carlsson CM, Zetterberg H, Blennow K, et al. 2017. Poor sleep is associated with CSF biomarkers of amyloid pathology in cognitively normal adults. Neurology 89:445–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staresina BP, Bergmann TO, Bonnefond M, van der Meij R, Jensen O, et al. 2015. Hierarchical nesting of slow oscillations, spindles and ripples in the human hippocampus during sleep. Nat. Neurosci 18:1679–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M 2006. Grouping of brain rhythms in corticothalamic systems. Neuroscience 137:1087–106 [DOI] [PubMed] [Google Scholar]
- Steriade M, Nuñez A, Amzica F. 1993. A novel slow (<1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J. Neurosci 13:3252–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahmasian M, Pasquini L, Scherr M, Meng C, Förster S, et al. 2015. The lower hippocampus global connectivity, the higher its local metabolism in Alzheimer disease. Neurology 84:1956–63 [DOI] [PubMed] [Google Scholar]
- Takashima A, Petersson KM, Rutters F, Tendolkar I, Jensen O, et al. 2006. Declarative memory consolidation in humans: a prospective functional magnetic resonance imaging study. PNAS 103:756–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, et al. 1991. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol 30:572–80 [DOI] [PubMed] [Google Scholar]
- Thal DR, Rüb U, Orantes M, Braak H. 2002. Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 58:1791–800 [DOI] [PubMed] [Google Scholar]
- Uddin LQ, Kelly AM, Biswal BB, Castellanos FX, Milham MP. 2009. Functional connectivity of default mode network components: correlation, anticorrelation, and causality. Hum. Brain Mapp 30:625–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utevsky AV, Smith DV, Huettel SA. 2014. Precuneus is a functional core of the default-mode network. J. Neurosci 34:932–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Werf YD, Altena E, Schoonheim MM, Sanz-Arigita EJ, Vis JC, et al. 2009. Sleep benefits subsequent hippocampal functioning. Nat. Neurosci 12:122. [DOI] [PubMed] [Google Scholar]
- Verret L, Mann EO, Hang GB, Barth AM, Cobos I, et al. 2012. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149:708–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Ranasinghe KG, Beagle AJ, Mizuiri D, Honma SM, et al. 2016. Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease. Ann. Neurol 80:858–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, et al. 2002. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416:535–39 [DOI] [PubMed] [Google Scholar]
- Wang L, Zang Y, He Y, Liang M, Zhang X, et al. 2006. Changes in hippocampal connectivity in the early stages of Alzheimer’s disease: evidence from resting state fMRI. NeuroImage 31:496–504 [DOI] [PubMed] [Google Scholar]
- Ward AM, Schultz AP, Huijbers W, van Dijk KRA, Hedden T, Sperling RA. 2014. The parahippocampal gyrus links the default-mode cortical network with the medial temporal lobe memory system. Hum. Brain Mapp 35:1061–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei M, Zhao B, Huo K, Deng Y, Shang S, et al. 2017. Sleep deprivation induced plasma amyloid-β transport disturbance in healthy young adults. J. Alzheimers Dis 57:899–906 [DOI] [PubMed] [Google Scholar]
- Wei W, Nguyen LN, Kessels HW, Hagiwara H, Sisodia S, Malinow R. 2010. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat. Neurosci 13:190–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerberg CE, Lundgren EM, Florczak SM, Mesulam M-M, Weintraub S, et al. 2010. Sleep influences the severity of memory disruption in amnestic mild cognitive impairment: results from sleep self-assessment and continuous activity monitoring. Alzheimer Dis. Assoc. Disord 24:325–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerberg CE, Mander BA, Florczak SM, Weintraub S, Mesulam M-M, et al. 2012. Concurrent impairments in sleep and memory in amnestic mild cognitive impairment. J. Int. Neuropsychol. Soc 18:490–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willem M, Tahirovic S, Busche MA, Ovsepian SV, Chafai M, et al. 2015. η-Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature 526:443–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson IA, Ikonen S, Gallagher M, Eichenbaum H, Tanila H. 2005. Age-associated alterations of hippocampal place cells are subregion specific. J. Neurosci 25:6877–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MA, McNaughton BL. 1994. Reactivation of hippocampal ensemble memories during sleep. Science 265:676–79 [DOI] [PubMed] [Google Scholar]
- Xie L, Kang H, Xu Q, Chen MJ, Liao Y, et al. 2013. Sleep drives metabolite clearance from the adult brain. Science 342:373–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Fitzgerald S, Nixon RA, Levy E, Wilson DA. 2015. Early hyperactivity in lateral entorhinal cortex is associated with elevated levels of AβPP metabolites in the Tg2576 mouse model of Alzheimer’s disease. Exp. Neurol 264:82–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Li S, Xu H, Walsh DM, Selkoe DJ. 2017. Large soluble oligomers of amyloid β-protein from Alzheimer brain are far less neuroactive than the smaller oligomers to which they dissociate. J. Neurosci 37:152–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yassa MA, Lacy JW, Stark SM, Albert MS, Gallagher M, Stark CEL. 2011. Pattern separation deficits associated with increased hippocampal CA3 and dentate gyrus activity in nondemented older adults. Hippocampus 21:968–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan P, Grutzendler J. 2016. Attenuation of β-amyloid deposition and neurotoxicity by chemogenetic modulation of neural activity. J. Neurosci 36:632–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeineh MM, Engel SA, Thompson PM, Bookheimer SY. 2003. Dynamics of the hippocampus during encoding and retrieval of face-name pairs. Science 299:577–80 [DOI] [PubMed] [Google Scholar]
- Zhao Q-F, Tan L, Wang H-F, Jiang T, Tan M-S, et al. 2016. The prevalence of neuropsychiatric symptoms in Alzheimer’s disease: systematic review and meta-analysis. J. Aff. Disord 190:264–71 [DOI] [PubMed] [Google Scholar]
- Zhou Y, Dougherty JH Jr., Hubner KF, Bai B, Cannon RL, Hutson RK. 2008. Abnormal connectivity in the posterior cingulate and hippocampus in early Alzheimer’s disease and mild cognitive impairment. Alzheimer Dement 4:265–70 [DOI] [PubMed] [Google Scholar]