

SUMMARY
During development, hematopoietic stem and progenitor cells (HSPCs) arise from specialized endothelial cells by a process termed endothelial-to-hematopoietic transition (EHT). The genetic program driving human HSPC emergence remains largely unknown. We previously reported that the generation of hemogenic precursor cells from mouse fibroblasts recapitulates developmental hematopoiesis. Here, we demonstrate that human fibroblasts can be reprogrammed into hemogenic cells by the same transcription factors. Induced cells display dynamic EHT transcriptional programs, generate hematopoietic progeny, possess HSPC cell surface phenotype, and repopulate immunodeficient mice for 3 months. Mechanistically, GATA2 and GFI1B interact and co-occupy a cohort of targets. This cooperative binding is reflected by engagement of open enhancers and promoters, initiating silencing of fibroblast genes and activating the hemogenic program. However, GATA2 displays dominant and independent targeting activity during the early phases of reprogramming. These findings shed light on the processes controlling human HSC specification and support generation of reprogrammed HSCs for clinical applications.
Graphical Abstract:
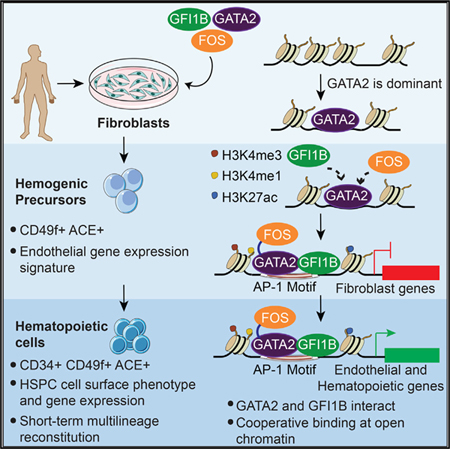
In Brief
Gomes et al. show that specification of hemogenesis in human fibroblasts is mediated by cooperative transcription factor binding. GATA2 displays dominance, interacts with GFI1B, and recruits FOS to open chromatin, simultaneously silencing the fibroblast program and initiating an endothelial-to-hematopoietic transition to definitive hematopoiesis.
INTRODUCTION
Early human blood development occurs through sequential stages in which transient hematopoietic cells support the embryo, followed by the emergence of the first hematopoietic stem cells (HSCs). HSCs are generated in the dorsal aorta of the aorta-gonad-mesonephros (AGM) region, subsequently migrate to the fetal liver, and lodge in the adult bone marrow (Ivanovs et al., 2011, 2017; Tavian et al., 2010). In addition, the placenta was identified as a site for human HSC development (Muench et al., 2017; Robin et al., 2009). Human HSCs develop from an intermediate hemogenic precursor cell with endothelial properties between days 27 and 40 (Ivanovs et al., 2017; Oberlin et al., 2002). Evidence from several non-human experimental models suggests that endothelial-to-hematopoietic transition (EHT) is a conserved developmental process (Medvinsky et al., 2011). Human HSCs bud predominantly from the endothelial floor of the dorsal aorta, co-express endothelial and hematopoietic markers, and together with non-self-renewing hematopoietic progenitors form the intra-aortic hematopoietic clusters (Tavian et al., 2010). Although there is no established phenotype that discriminates emergent human HSCs from their precursors or progenitors, some molecules have been identified that are present in developing HSCs. Angiotensin-converting enzyme (ACE) marks fetal liver HSCs (Jokubaitis et al., 2008) and ACE+CD34− cells beneath the human dorsal aorta (Sinka et al., 2012). ACE+CD34− cells may represent HSC precursors that give rise to ACE+CD34+ cells contained in aortic clusters. Human long-term repopulating HSCs reside in the CD34+CD38lowCD90+ population of umbilical cord blood (UCB) (Majeti et al., 2007). Further studies have shown that integrin alpha 6 (CD49f) (Notta et al., 2011) in UCB and GPI-80 (Prashad et al., 2015) in fetal liver further purifies self-renewing HSCs.
Directed differentiation of human pluripotent stem cells (PSCs) has provided valuable information about the transcriptional program of hematopoiesis. Human PSC-derived hemogenic cells are distinguished by expression of the transcription factors (TFs) RUNX1, GFI1, and GFI1B, which are essential for EHT (Ng et al., 2016). Recent studies have shown that SOX17-positive endothelial cells are generated during PSC differentiation and subsequently activate RUNX1 during EHT (Ng et al., 2016). Thus far, current protocols for hematopoietic differentiation of human PSCs remain skewed toward extra-embryonic hematopoiesis rather than intra-embryonic definitive HSC formation (Ditadi et al., 2017; Ng et al., 2016). TFs crucially important for hematopoietic development including GATA2 and RUNX1 are up-regulated in human intra-aortic clusters (Labastie et al., 1998). How these regulators promote definitive human hematopoiesis is unknown. Putative mechanisms include pioneer hematopoietic TFs that bind and prime closed chromatin (Soufi et al., 2012; Wapinski et al., 2013) or TFs that interact and cooperatively engage open chromatin (Chronis et al., 2017). Studies in mouse HSPCs have shown combinatorial interaction between a heptad of TFs (SCL, LYL1, LMO2, GATA2, RUNX1, ERG, and FLI-1) in hematopoietic progenitors (Wilson et al., 2010). During mouse PSC differentiation the cooperative binding of AP-1 with TEAD4 was shown to promote a hemogenic cell fate at the expense of alternative cell fates (Obier et al., 2016). As hematopoietic progenitors differentiate, GATA2 binding persists in erythroid cells, acting as a ‘‘pioneer’’ or nucleation factor for the recruitment of GATA1, indicating that GATA2 and GATA1 cooperate extensively to regulate erythroid differentiation (May et al., 2013). Difficult availability of material hinders a detailed understanding of the transcriptional control of human HSC specification.
We have previously shown the direct reprogramming of mouse fibroblasts into hemogenic precursors cells using GATA2, FOS and GFI1B with increased efficiency with ETV6 (Pereira et al., 2013). Induction leads to a dynamic process that progresses through an endothelial-like intermediate with a defined phenotype (Prom1+Sca-1+CD34+CD45−). Using this phenotype, we identified a population in vivo that expresses endothelial and early hematopoietic markers, localizes in the vascular labyrinth of mouse placenta, and upon co-culture with stromal cells will engraft primary and secondary mice (Pereira et al., 2016). Therefore, mouse hemogenic reprogramming recapitulates developmental hematopoiesis. Recently it was demonstrated that the expression of GATA2, FOS, and GFI1B within PSC-derived teratomas leads to the generation of long-term repopulating HSCs (Tsukada et al., 2017).
Here, we show that GATA2, GFI1B, and FOS reprogram human fibroblasts into hematopoietic progenitors that transit through an intermediate with endothelial properties. These cells acquire emergent HSPC-like gene expression profiles and cell surface phenotypes and will repopulate NOD-scid IL2Rg-null (NSG) mice. We have established that collaborative interactions among the three TFs engage open enhancers and promoters that mediate both the silencing of fibroblast-specific genes and activation of endothelial and hematopoietic genes. This molecular analysis of TF targeting provides insight into the regulatory machinery required for hematopoietic reprogramming and reveals the importance of TF cooperation during HSC specification.
RESULTS
Transferring Hemogenic Reprogramming to the Human System
To assess the feasibility of using the same TFs used in the mouse, we determined their combinatorial expression in human cells with in-house-developed software, GPSforGenes. We found that both GGF (GATA2, GFI1B, and FOS) and GGFE (GATA2, GFI1B, FOS, and ETV6) combinations were highly expressed in human CD34+ hematopoietic stem and progenitor cells (HSPCs) (Figures S1A and S1B). We next expressed the human TFs in adult human dermal fibroblasts (HDFs) and neonatal foreskin (BJ) fibroblasts using a doxycycline (Dox)-inducible vector system (Pereira et al., 2013). Two days after transduction with GGFE and GGF viruses, Dox was added and cultures were analyzed for more than 25 days (Figure 1A). Interestingly, in human cells the GGF minimal cocktail appears more effective than GGFE (Figure 1B). The colony formation efficiency ranged from 2.1 ± 0.2% to 2.3 ± 0.1% with GGF and from 0.4 ± 0.1% to 0.6 ± 0.1% with GGFE. An analysis of both colony number and expression of CD34 revealed that 8.4%–13.6% of the colonies contained CD34+ cells (Figure 1B). Similar morphological changes were observed in both HDF and BJ transduced cultures. The cells appeared to round up in cobblestone-like areas during the midpoint of the culture with semi-adherent and non-adherent round cells appearing later (Figure 1C). Round hematopoietic-like cells that express CD34 and CD49f by immunofluorescence were observed (Figure 1D) and further quantified by flow cytometry (Figure S1C). These changes defined the induction period; a large population of cells expressed CD49f+ (20%–25%), while CD34 expression was more restricted (0.2%–2.0%). The CD49f+ population emerges early (day 12) and is maintained, while CD34+ cells peak at day 25 and subsequently decline but do persist. No colonies, morphologic changes, or CD49f+ and CD34+ cells were observed after transduction with control viruses (Figures S1D and S1E).
Figure 1. GATA2, GFI1B, and FOS Induce CD34+ and CD49f+ Colonies in Human fibroblasts.
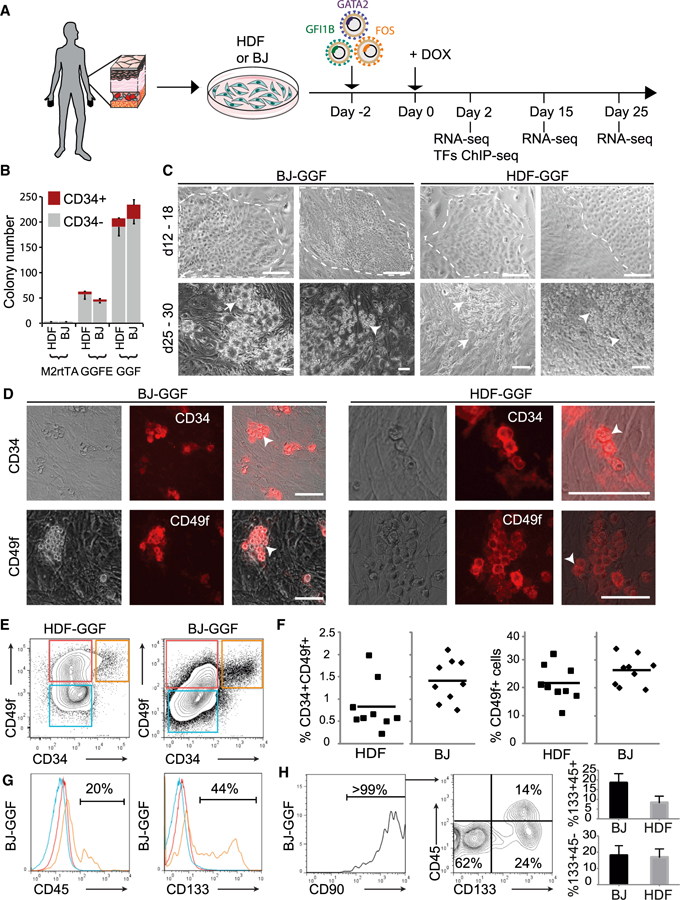
(A) Strategy for inducing hemogenesis in human dermal fibroblasts (HDFs) and neonatal foreskin fibroblasts (BJ).
(B) Cells were transduced with GATA2, GFI1B, FOS, and ETV6 (GGFE); GATA2, GFI1B, and FOS (GGF); or control M2rtTA viruses and cultured with Dox for 30 days. Colonies were stained for CD34 and counted. Colony numbers are per 10,000 transduced fibroblasts (mean ± SD, n = 3).
(C) Colony morphology 12–18 days (top, dashed line) and 25–30 days (bottom) after induction. Arrows highlight endothelial and arrowheads highlight induced hematopoietic cellular morphologies.
(D) Colonies were assayed by immunofluorescence for CD34 (top) or CD49f (bottom) 30 days after transduction. Arrowheads highlight induced hematopoietic morphologies. Scale bars, 100 μm.
(E) Analysis of CD34 and CD49f expression 26 days after transduction with GGF.
(F) Quantification of CD34+CD49f+ and CD49f+ cell populations. Each symbol represents an experiment and the horizontal bar indicates the mean.
(G) Expression of CD45 and CD133 within CD34+CD49f+ (orange lines), CD49f+ (red lines), and double-negative population (blue lines).
(H) Expression of CD90, CD45, and CD133 within the CD34+CD49f+ population. Quantification of the CD133+CD45− (bottom) and CD133+CD45+ (top) populations from three biological replicates between days 23 and 26 (mean ± SD, n = 3). See also Figures S1 and S2.
Induced Colonies Contain Cells with Human HSPC Surface Phenotypes
We examined the induced colonies for co-expression of markers that define a human HSC phenotype. Both HDF and BJ derived cells co-express CD34 and CD49f (Figure 1E). Transduced BJ cells generated greater numbers of CD49f+ (positive for CD49f and CD34-negative) and CD34+CD49f+ cells (Figure 1F), possibly because of their neonatal origin. A subpopulation of CD34+CD49f+ cells expressed CD133 (Prominin1) and low levels of CD45, which is expressed at low levels in UCB HSCs (Figures 1G, 1H, and S2A) (Jay et al., 2004). CD34+CD49f+ cells also express CD90 (Thy1) (Figures 1H and S2B). The negative markers CD38 and CD45RA were not detected (Figure S2C). These data correlate with extensive phenotypic definitions of human HSCs in UCB (Notta et al., 2011). Interestingly, GATA2 or FOS removal from the three-factor combination abolished the generation of CD34+CD49f+ cells, while removal of GFI1B increased the number of double-positive cells (Figure S2D) but diminished the percentage of CD45+ and CD133+ cells (Figure S2E), consistent with the role of GFI1B during mouse EHT (Pereira et al., 2016; Thambyrajah et al., 2016). These results demonstrate that GGF induce a human HSPC phenotype in two different types of human fibroblasts.
Comprehensive Gene Expression Analyses during Reprogramming
We interrogated the gene expression changes occurring during reprogramming by mRNA sequencing (RNA-seq) analysis of cell populations. We sorted three biological replicates of non-transduced fibroblasts, day 15 CD49f+, day 25 CD49f+, and CD34+CD49f+ cells from both types of fibroblasts (Figure 1A; Table S1). Biological replicates correlated well, and metagene analyses that represent most of the variability associated with BJ and HDF reprogramming showed (1) sets of genes expressed in fibroblasts and silenced in all other samples (Figure 2A, green lines), (2) genes transiently expressed in CD49f+ cells (Figure 2A, black lines), and (3) genes that start to be expressed in CD34+CD49f+ cells (Figure 2A, red lines). Examples of sequential changes include MMP1, a fibroblast-associated gene silenced in sorted populations; ANGPTL4, implicated in the positive regulation of angiogenesis, transiently activated in CD49f+ cells; and ACE, upregulated in CD49f+ cells with continued expression in CD34+CD49f+ cells (Figures S3A). We confirmed ACE and CD49f expression in transduced cells and found that CD34+ cells are contained within this subset (Figure S3B). Integration of the RNA-seq datasets revealed silencing of the fibroblast-associated gene signature (Yu et al., 2007), which includes GREM1, PSG5, FGF5, LUM, HAPLN1, and MAB21L (Figures 2B and S3C). Among upregulated genes in CD34+CD49f+ cells we identified proposed markers of AGM HSC precursors such as ACE, F11R, and EPCAM, the TFs RUNX1, SCL, and FOXO1, and the CD9 homing molecule. Several proangiogenic genes (JAG1, SEMA4D, VWF, FOXC2, and ETS2) are expressed in CD49f+ cells at both days 15 and 25 (Figures 2C and S3C). We asked if an angiogenic program is activated during reprogramming from a list of positive angiogenic regulators. The vast majority of these genes are activated in CD49f+ cells, and some continue to be expressed in CD34+CD49f+ cells (Figures S4A and S4B). Gene set enrichment analysis (GSEA) revealed enrichment for and regulation of angiogenesis (Figure S4C). Further analyses of genes upregulated in CD34+CD49f+ cells using the Mouse Genome Informatics (MGI) mouse mutant phenotype database showed that their genetic perturbations result in largely hematopoietic phenotypes (Figure 2D). We used GSEA to compare the transition from fibroblasts to CD34+CD49f+ cells. A significant number of HSPC gene sets were enriched (31 and 26 enriched versus 1 and 6 non-enriched gene sets) in CD34+CD49f+ cells generated from either type of fibroblasts (Figure S4D, left). Indeed, top enriched gene sets include CD34 TFs as well as the Wnt, Hedgehog, and TGFβ signaling pathways, consistent with their hemogenic roles (Figure S4D, right) (Medvinsky et al., 2011; Pereira et al., 2016). We next integrated our data with published HSC data from UCB (Notta et al., 2011). Principal-component analysis (PCA) showed that induced CD49f+ cells are the closest to CD49f+ HSCs, while both HDF and BJ fibroblasts are very distinct from all other datasets. The CD34+CD49f+ cell populations are positioned between HSCs and multipotent progenitors (MPPs) (Figure 2E). A comparison of our mouse reprogramming data (Pereira et al., 2013) with the human showed that CD34+CD49f+ cells cluster closely with mouse induced HSPCs (CD45+cKit+) (Figure 2F). To characterize the reprogramming process in more detail, we performed single-cell RNA-seq throughout induction. UCB Lin-CD34+ cells and non-transduced HDFs were profiled as controls. Genome-wide unsupervised hierarchical clustering shows that most single cells cluster according to sample group, with a clear separation of HDF from reprogrammed cells. Just 2 days after transgene activation, fibroblasts show a dramatic transcriptional change. Importantly, endogenous expression of GFI1B and GATA2 is absent at day 2 but activated in reprogrammed cells (Figure S4F), suggesting that the reprogramming process is stable and becomes independent of exogenous expression of GFI1B and GATA2. FOS is expressed in fibroblasts and continues to be expressed in reprogrammed cells. As in the PCA (Figure 2E), single CD49f+ cells are the closest to human UCB cells (Figure 2G). These data suggest that, with time in culture, these cells acquire the expression of differentiation-related genes, along with CD34. We performed GSEA with gene lists for HSCs and MPPs (Notta et al., 2011) on single-cell mRNA-seq data (Figure S4G). MPP genes are significantly enriched in the CD34+CD49f+ subset when compared to CD49f+. For the HSC gene set there is no clear enrichment in this transition, indicating that those genes are already expressed in the CD49f+ population. To provide additional information on genes that may be missing from reprogrammed CD49f+ cells, we analyzed the expression of the HSC signature genes (Notta et al., 2011) individually. We have detected 29 of 39 genes. From the 10 genes that were not detected in reprogrammed cells, only 1 (SOX18) is a TF (Table S2). In terms of signaling, pathways such as Notch and integrin alpha 6 are enriched in CD49f+ cells, while TGFβ, WNT, and T cell receptor signaling are enriched in CD34+CD49f+ (Figure S4H). To generate information on the genes and pathways changing at each transition, we performed gene list enrichment analysis (Enrichr) (Chen et al., 2013) for all possible pairwise comparisons in the single-cell data. An interactive comparative map was created using the 500 most differentially expressed genes (Figure S4E; see Table S2 for gene lists). The hyperlinks allow exploration of the data and the identification of ontologies, pathways, diseases and drugs, and so on, implicated in each cellular transition. For example, glycolysis and INFα signaling pathways (p = 2.7 × 10−5 and p = 1.2 × 10−3, respectively) were the top enriched terms for the transcriptional activation occurring in the first 2 days of reprogramming, suggesting that these genes may be direct targets of hemogenic TFs. Taken together, GGF direct hemogenic transcriptional changes in multiple types of fibroblasts of both mouse and human origins.
Figure 2. Dynamic Activation of Endothelial and HSPC-like Gene Expression Signatures in Reprogrammed Cells.
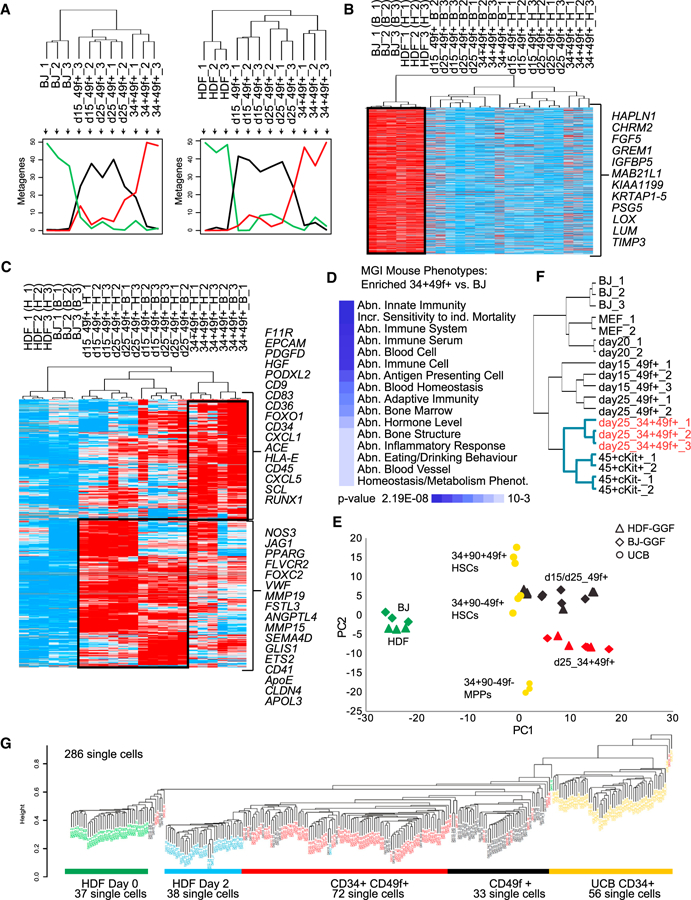
(A) Populations of non-transduced fibroblasts and GGF transduced day 15 CD49f+, day 25 CD49f+, and day 25 CD34+CD49f+ cells were profiled using RNA-seq (three biological replicates; samples that did not pass quality control were discarded). Ordered tree linkage displays clustering of the profiled samples and the metagenes that represent most of the variability associated with each cellular transition.
(B) Heatmap of genes expressed in fibroblasts and silenced in CD49f+ and CD34+CD49f+ cells.
(C) Heatmap of genes activated in CD49f+ and CD34+CD49f+ cells. Black boxes highlight the stage-specific expression of gene sets. Red indicates increased expression and blue decreased expression over the mean. Data were analyzed using Cluster 3.0 and displayed using Treeview.
(D) Gene list enrichment analysis with libraries from MGI mutant mouse phenotype ontology for genes upregulated from BJ to CD34+CD49f+. Heatmap shows enrichment p values.
(E) RNA-seq datasets were integrated with expression data from human UCB hematopoietic stem and progenitor cells (HSPCs) (from Notta et al., 2011). PCA shows the relative distances between samples.
(F) Hierarchical clustering integrating data from reprogrammed mouse and human cells (mouse data from Pereira et al., 2013); human CD34+CD49f+ cells highlighted in red. Blue lines highlight separate cluster for hematopoietic phenotypes.
(G) Non-supervised hierarchical clustering showing genome-wide gene expression data from HDF-derived single cells. UCB CD34+ single cells are marked in yellow. The number of single cells analyzed and phenotype are detailed.
See also Figures S3 and S4 and Tables S1, S2, and S4.
Induced Human Cells Engraft In Vivo
To determine if reprogrammed HSPCs were functional, we undertook xenogeneic transplants. Fibroblasts were transduced and cultured for 25 days in the presence of Dox. CD34+CD49f+ cells as well as single-positive populations were sorted and transplanted, and NSG mice were maintained on Dox for 2 weeks (Figure 3A). We detected human chimerism in peripheral blood at both 3 and 12 weeks after transplantation (Figures 3B and 3C). Although all mice had low levels of huCD45+ (0.1%–2%) at 3 weeks, the number of positive mice dropped dramatically by 3 months. We confirmed this result at week 4 by PCR using primers to human chromosome 17-alpha-satellite sequences (Figures 3D and 3E). A clear population of human CD45+ mouse CD45- was detected (Figure 3F) by flow cytometry. The huCD45+ cells contained both lymphoid and myeloid cells (Figure 3G). These results demonstrate that inducible expression of GATA2, GFI1B, and FOS converts human fibroblasts into HSPCs that repopulate for at least 3 months. Because chimerism from reprogrammed HSPCs was not sustained, we believe that it is crucial to define the TF mechanisms underlying human hemogenic induction.
Figure 3. Reprogrammed Cells Engraft In Vivo after Transplantation.
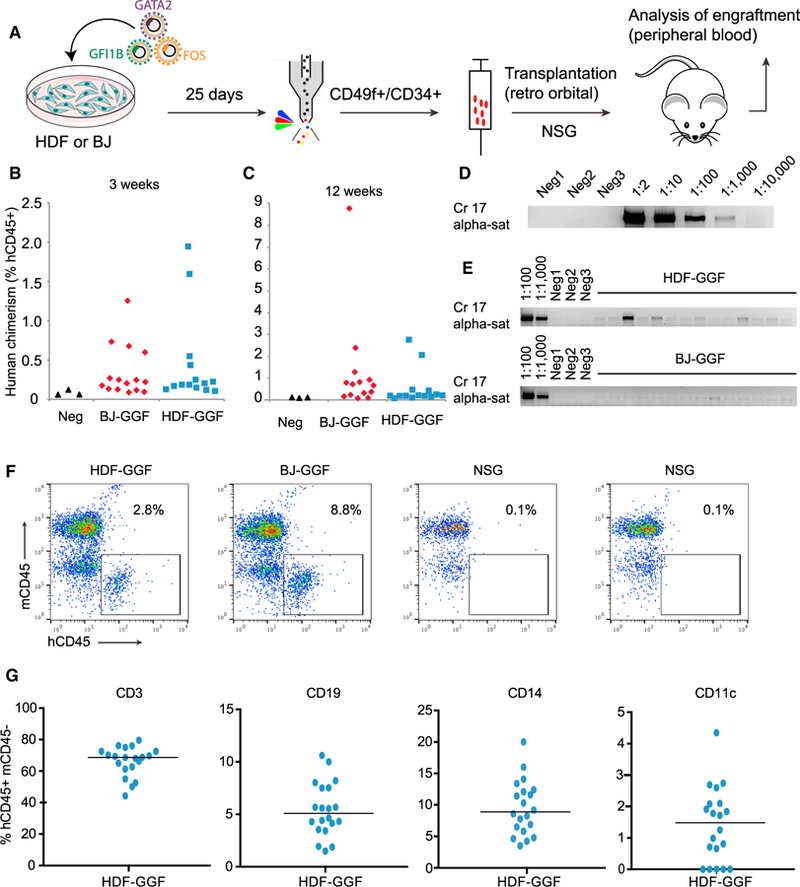
(A) Experimental design used to sort and transplant induced cells into NOD-scid IL2Rγ-null (NSG) mice. HDF or BJ fibroblasts were transduced with GGF and cultured with Dox for 25 days. Cells were dissociated, and CD49f+ (including CD34+CD49f+ double-positive cells) were sorted and then injected into 4-week-old NSG mice.
(B) Human chimerism in peripheral blood 3 weeks after transplantation with HDF-GGF (n = 15) or BJ-GGF (n = 14).
(C) Percentage of human CD45 chimerism 12 weeks after transplantation.
(D) Limit of detection of human engraftment was tested by PCR using human-specific primer pairs (chromosome 17 alpha satellite) from serial dilutions of human HDFs mixed with mouse cells. Mouse cells only were used as negative controls.
(E) Human chimerism in peripheral blood was tested using PCR 4 weeks after transplantation. Each lane represents one individual mouse 4 weeks after transplantation with HDF-GGF (top) or BJ-GGF (bottom). Blood from non-transplanted mice was used as negative controls.
(F) FACS plots showing individual mice analyzed with human CD45 (hCD45) and mouse CD45 (mCD45) antibodies.
(G) Lineage marker analysis in gated hCD45+mCD45− cells. Scatterplots show expression of human lymphoid and myeloid markers. Data from 20 mice transplanted with similar number of reprogrammed cells; the horizontal bar indicates the mean.
GATA2 Displays Dominant and Independent Targeting Capacity to Initiate Hemogenic Reprogramming
To define the molecular mechanism underlying hemogenic reprogramming, we determined where the TFs initially bind to the genome. We created tagged versions of the TFs to carry out these experiments (Figure S5A). Western blots and immunofluorescence confirmed expression and nuclear and subnuclear localization, respectively (Figures S5B–S5D) (Vassen et al., 2006). Fibroblasts were transduced with the three factors individually or in combination, after which we performed chromatin immunoprecipitation sequencing (ChIP-seq) (Figure 4A). When the three TFs are expressed in combination, GATA2 showed the most extensive binding to the fibroblast genome (6,750 peaks), followed by GFI1B (2,372 peaks) and FOS (689 peaks) (Figure 4B). Interestingly, GATA2-bound sites were similar when GATA2 was expressed alone or together with GFI1B and FOS, suggesting that GATA2 displays independent targeting capacity. In contrast, GFI1B depends on GATA2 and FOS expression to bind to the majority of its targets. FOS showed a small number of targets, suggesting that it has limited access to chromatin during the initial stages of reprogramming (Figure 4B). GATA2 displayed enrichment at promoters both when is expressed individually (Figure 4C, right) or in combination with GFI1B and FOS (Figure 4C, left) and a greater read density at transcription start sites (TSS) compared with GFI1B and FOS (Figures 4D and S5E), suggesting an important regulatory role. GATA2 targets include the CD34 gene, the HSC homing molecule CD9 (Figure 4E) (Karlsson et al., 2013), and the EHT mediator GPR56 (Figure S5F) (Solaimani Kartalaei et al., 2015). RUNX1 (Figure 4F) and BMPER (Figure S5G) (McGarvey et al., 2017), two important regulators of hematopoiesis, are targeted by both GATA2 and GFI1B in GGF-induced fibroblasts. However, GFI1B binding was lost when GATA2 was not included, suggesting cooperative binding between GATA2 and GFI1B for the induction of RUNX1 and definitive hematopoiesis. Motif prediction for GATA2 in GGF-induced fibroblasts showed that the GATA motif was strongly enriched. This analysis also identified HIF1B, BORIS, and ARID3A as regulators of hematopoietic reprogramming (Figure 4G, top). For GFI1B targets, AP-1, HIF1B, and GATA motifs were identified, but the GFI motif was strongly enriched only when GFI1B was expressed individually (Figure 4G, bottom). This suggests that GATA2 recruits GFI1B to its ‘‘natural’’ target sites. Overall these results provide insights as to how these factors engage the fibroblast genome and initiate hemogenic programming by targeting key hematopoietic regulators.
Figure 4. Analysis of TF Occupancy Reveals that GATA2 Has Both Dominant and Independent Targeting Capacity.
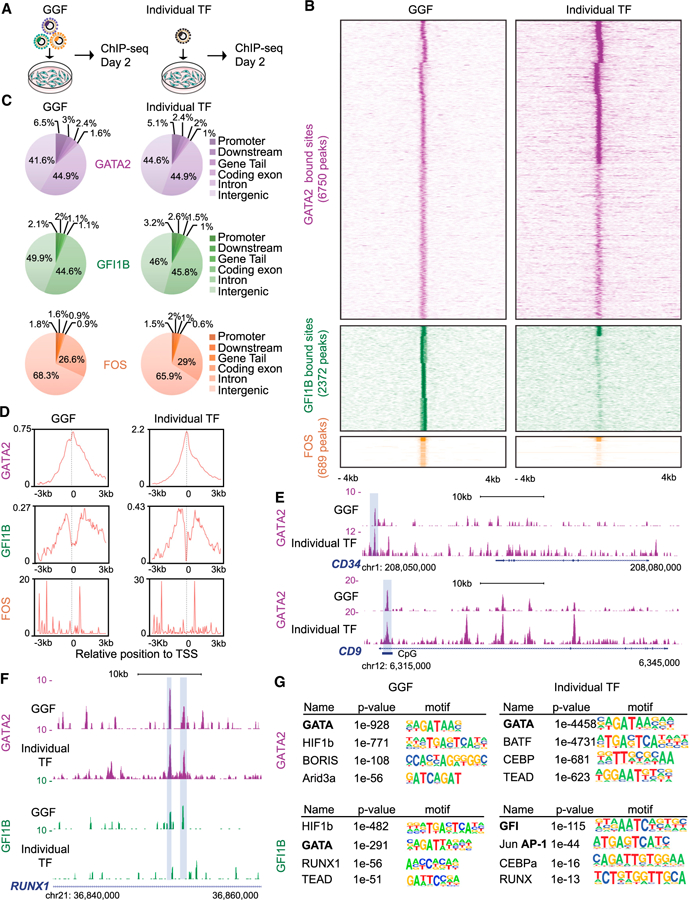
(A) Strategy for identifying GGF genomic binding sites. GGF factors were transduced in combination (left) or individually (right) and analyzed using ChIP-seq 2 days after adding Dox.
(B) Heatmaps representing genome-wide occupancy profile for GGF factors when expressed in combination or individually in HDFs. For each site, the signal is displayed within an 8 kb window centered on individual peaks.
(C) Genomic distribution of GGF and individual transcription factor (TF) peaks in transduced HDFs.
(D) ChIP-seq read density relative to transcription start site (TSS). The graph shows a plot of the average read coverage per million mapped reads centered to the TSS.
(E) Genome browser profiles illustrating GATA2-binding sites at CD34 and CD9 loci. The y axis represents the total number of mapped reads. The boxes highlight shared peaks when GATA2 is expressed individually or in combination with GFI1B and FOS.
(F) GATA2 and GFI1B occupancy profile at the RUNX1 locus. The boxes highlight shared peaks between GATA2 and GFI1B only when expressed in combination. The genomic scale is in kilobases.
(G) De novo motif prediction for GATA2 and GFI1B target sites when expressed in combination or individually. The motifs for GATA, GFI, and AP-1 factors are bolded.
GATA2 and GFI1B Interact and Share a Cohort of Target Sites
We next investigated the extent of overlap between GATA2 and GFI1B genomic targets and whether they physically interact. By displaying GATA2 and GFI1B target sites, we observed that 750 genomic positions were shared, representing 31.6% of total GFI1B targets (Figure 5A). These include HSC and EHT regulators such as PRDM1 and PODXL (Figure 5B). Motif comparison analysis showed significant similarity between GATA2 and GFI1B motifs (Jaccard similarity index = 0.1) (Vorontsov et al., 2013), supporting the interaction between the two TFs (Figure 5C). We then performed de novo motif prediction for the overlapping peaks. Interestingly, the AP-1 motif was the most enriched, followed by the GATA and GFI1 motifs, highlighting the cooperative action among the three factors during reprogramming (Figure 5D). Co-bound genes are part of pathways such as interferon-gamma signaling, inflammation, and cytoskeletal regulation by Rho GTPases (Figures 5E and S6A), processes with demonstrated relevance for HSC emergence (Pereira et al., 2013, 2016). Gene Ontology analysis of co-bound genes showed that cell motion and vasculature development were enriched terms (Figures 5F and S6B). We further interrogated our ChIP-seq data for the regulatory interactions between the three hemogenic TFs. Both GATA2 and GFI1B bind their own loci at the initial stages of reprogramming, suggesting auto-regulation as previously shown in hematopoietic progenitors (Anguita et al., 2010; May et al., 2013). In addition, GATA2 binds to a CpG island in the FOS locus and GFI1B binds to the GATA2 locus only in the presence of the other two TFs (Figure S6C). We did not detect binding of GATA2 to the GFI1B locus, suggesting that this interaction may be established later in hematopoietic progenitors (Moignard et al., 2013). To confirm physical interaction, we have performed co-immunoprecipitation (coIP) 48 hr after expression in fibroblasts. This analysis demonstrated an interaction between GATA2 and FOS and between GATA2 and GFI1B (Figure 5G). This suggests that the interplay among GGF is central for hemogenic reprogramming (Figure S6C).
Figure 5. GATA2 and GFI1B Interact and Share a Cohort of Target Sites.
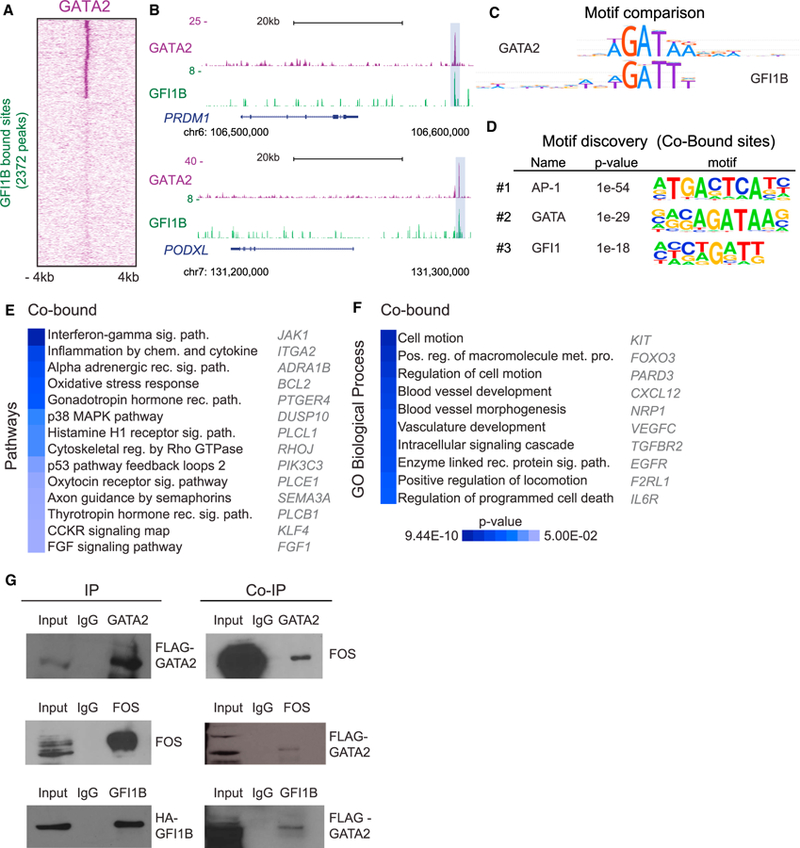
(A) Heatmap representing genome-wide occupancy profiles for GFI1B showing shared targets with GATA2 48 hr after induction of GGF with Dox. The signal at the corresponding genomic regions of TF binding is displayed across the other dataset. For each site, the signal is displayed within an 8 kb window centered on individual peaks.
(B) Genome browser profiles illustrating GATA2- and GFI1B-binding sites at PRDM1 and PODXL loci. The y axis represents the total number of mapped reads. The boxes highlight genomic positions co-occupied by GATA2 and GFI1B.
(C) Motif comparison between GATA2 and GFI1B. Jaccard similarity coefficient = 0.1.
(D) De novo motif discovery at co-bound sites. The top three most enriched motifs are shown along with p values.
(E) Panther pathway enrichment analysis of genes co-bound by GATA2 and GFI1B.
(F) Biological processes Gene Ontology (GO) terms enriched for co-bound genes. Examples of co-bound genes are shown and heatmaps display p values.
(G) Immunoblots showing immunoprecipitation (IP) of GATA2 (top), FOS (middle), and GFI1B (bottom) in HDFs 48 hr after induction of GGF (left). Right: coIP detection for FOS (top) and GATA2 (middle and bottom). Input (10%) indicates non-immunoprecipitated cell lysate, and IgG indicates control IP with isotype antibody.
GATA2 and GFI1B Engage Open Promoters and Enhancer Regions
We next asked whether GATA2 and/or GFI1B engagement correlates with gene activation or silencing during human reprogramming. We identified 1,425 significantly changing genes (across the population mRNA-seq dataset from HDF-derived cells), which were bound by either GATA2 and/or GFI1B. Specifically, 1,186 genes were bound by GATA2, and 182 were bound only by GFI1B. Fifty-seven differentially expressed genes were co-bound, targeting the cluster of genes highly expressed in fibroblasts and a second cluster of genes enriched only in CD34+CD49f+ cells (Figure 6A; Table S3) (p < 10−10, Fisher’s t test). These data suggest that GATA2 and GFI1B co-binding is involved both in the repression of fibroblast-associated genes and activation of hematopoietic-associated genes. To characterize the chromatin features associated with GATA2 and GFI1B engagement, we used previously published ChIP-seq datasets for H3K4me1, H3K4me3, H3K27ac, H3K27me3, H3K9me3, and H3K36me3 in HDFs (Table S4). GATA2- and GFI1B-bound sites in fibroblasts are enriched for marks associated with active promoters and enhancers such as H3K4me3, H3K27ac and H3K4me1 (Figure 6B). This result is consistent with the DNase I accessibility in HDFs. GATA2 and GFI1B bind mostly to DNase I-sensitive sites (Figure 6C; Table S4). These results demonstrate that GATA2 and GFI1B preferentially bind to accessible chromatin primarily in promoter and enhancer regions. We summarized the association between GATA2 and GFI1B binding and chromatin in fibroblasts using ChromHMM, a segmentation of the genome into 18 chromatin states on the basis of the combinatorial patterns of chromatin marks. We confirmed the preference of GATA2 and GFI1B in active TSS, flanking upstream TSS and active enhancers (Figure 6D, blue). In addition, we analyzed published datasets for histone marks in K562 cells (Figure S6E) and GGF TF occupancy in hematopoietic progenitor cells (HPCs) (Table S4). In contrast to GATA2 and FOS, we observed a distinct pattern for GFI1B that is strongly enriched in bivalent or poised TSS (Figure 6D, orange). This dramatic shift in GFI1B targeting suggests that the cooperative interaction between GATA2 and GFI1B may be specific for the earlier stages of hematopoietic reprogramming and EHT that is lost in downstream hematopoietic progenitors.
Figure 6. GATA2 and GFI1B Engage Open Promoters and Enhancer Regions.
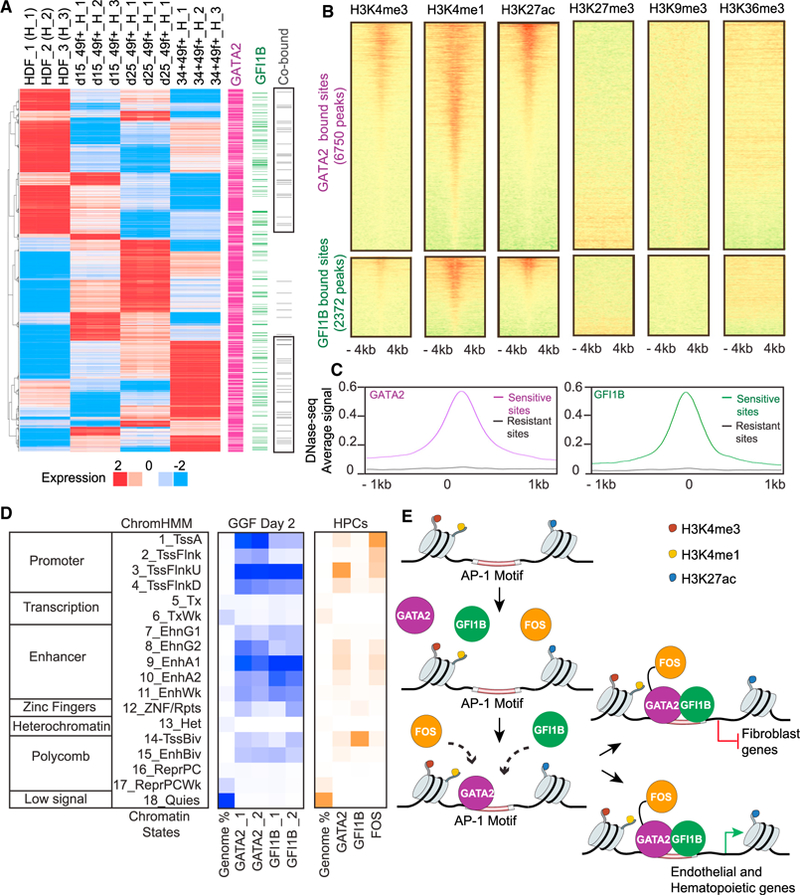
(A) Integration of RNA-seq and ChIP-seq datasets. The heatmap (left) shows 1,425 genes with at least 1.5-fold expression changes across the dataset. Genes were identified as targets of GATA2 (purple) or GFI1B (green), with co-bound targets in gray (right), on the basis of binding peaks within the range of a 10 kb window centered on the transcriptional start site.
(B) Heatmaps of normalized tag densities representing HDF chromatin marks at GATA2 and GFI1B target sites. The signal is displayed within an 8 kb window centered on the binding sites.
(C) Average DNase-seq signal of HDFs at GATA2 and GFI1B target sites. The signal is displayed within a 2 kb window.
(D) Heatmaps for chromatin-state functional enrichment. Rows represent chromatin states according to ChromHMM annotation: TssA, active promoters; TssFlnk, flanking promoters; TssFlnkU, flanking upstream promoters; TssFlnkD, flanking downstream promoters; Tx, strong transcription; TxWk, weak transcription; EnhG1/2, genic enhancers; EnhA1/2, active enhances; EnhWk, weak enhancers; ZNF/Rpts, ZNF genes and repeats; Het, heterochromatin; TssBiv, bivalent/poised TSS; EnhBiv, bivalent enhancer; ReprPC, repressed PolyComb; ReprPCWk, weak repressed PolyComb; Quies, quiescent. Blue panel shows the percentage of genome occupancy for GATA2 and GFI1B in GGF-transduced HDFs. Orange panel shows the percentage of genome occupancy for GGF in human hematopoietic progenitor cells (HPCs).
(E) Model of the mechanism of action of GGF during hemogenic induction in human fibroblasts.
DISCUSSION
We show that ectopic expression of the TFs GGF induce a hemogenic program in human fibroblasts. Induced cells exhibit endothelial and hematopoietic gene expression and HSPC surface phenotypes and engraft NSG mice to produce multilineage progeny. Mirroring mouse hemogenic induction (Pereira et al., 2013), this transition is dynamic, with initial activation of angiogenic followed by hematopoietic gene signatures. We further show that GATA2 is the dominant TF and cooperates with GFI1B to engage open chromatin regions during the initial phases of reprogramming.
We show that upon induction with GGF, we readily detect many endothelial genes, including PPARG, VWF, and FOXC2, which have defined angiogenic functions. The induction results in the generation of a large population of CD49f+ACE+ cells, while only a more restricted population activates CD34. This is consistent with the observation that ACE+CD34− cells emerge early during human hematopoietic development and localize beneath the dorsal aorta (Sinka et al., 2012). Our data support the hypothesis that these precursor cells later give rise to ACE+CD34+ cells contained in aortic clusters. Indeed, ITGA6 and CD34 are GATA2 direct targets during the initial stages of hemogenic reprogramming, providing a direct mechanistic link between human hemogenic precursor phenotype and GATA2.
To faithfully recapitulate HSC specification major interest lies in defining direct human HSC precursors during ontogeny. Our data provide useful markers: CD49f is present on human LT-HSCs (Notta et al., 2011), and we show co-expression with ACE. The VNN2 gene that encodes GPI-80 and marks fetal liver HSCs was not detected during reprogramming, nor was it a target for GATA2 or GFI1B. This is consistent with the lack of expression of GPI-80 in embryonic stem cell (ESC)-derived hemogenic endothelium (Prashad et al., 2015). Whether this gene is activated at later stages of HSC maturation remains to be investigated. It will be interesting in future studies to determine whether ACE+ cells present in the AGM (and perhaps in the first trimester human placenta) co-express CD49f during human embryonic development before they acquire CD34 expression. In addition, our datasets provide other markers that are informative and complement this phenotype such as F11R, shown to be present in HSC precursors in zebrafish (Kobayashi et al., 2014). We also found ANGPTL4 and JAG1 enriched in CD49f+ cells and then downregulated in CD34+ cells. These genes may be useful to segregate precursors from emergent human HSCs in vivo.
In addition to providing information for the identification of direct precursors of HSCs during development, our direct reprogramming strategy has identified pathways essential for EHT and HSC specification. We have shown that, in both mouse and human, GGF represent a conserved minimal TF network for hemogenic induction. In the mouse, we identified interferon signaling and inflammation as enriched pathways in hemogenic precursors (Pereira et al., 2013). Recently, multiple studies explored inflammation during EHT and HSC formation in mouse and zebrafish models (Espin-Palazon et al., 2018). Enrichment of this pathway during human hemogenic induction supports its importance for human EHT. Another example is the role of FOS and AP-1 in HSC specification. This was not revealed by classical genetic ablation, possibly because of compensatory effects. AP-1 is essential for human and mouse hemogenic induction (Lis et al., 2017; Pereira et al., 2013; Sandler et al., 2014) and has recently been implicated during EHT from mouse ESCs (Obier et al., 2016). Our studies corroborate that direct cellular reprogramming informs the process of HSC specification and offers tractable means to systematically interrogate pathways of human HSC formation. It will be interesting to address the role of other enriched pathways such as cytoskeletal regulation by Rho GTPase and the histamine H1 receptor pathway. Genes that are co-targeted during hematopoietic reprogramming by GATA2 and GFI1B are additional candidates for further study (e.g., ITGA2, PLCL1, ADRA1B, RHOJ, SEMA3A, and PLCB1; see Table S3 for comprehensive lists).
In the mouse, lineage divergence from endothelium may occur before extensive formation of intra-aortic clusters (Rybtsov et al., 2014; Swiers et al., 2013). The enrichment of angiogenesis and cell motility during human hemogenic reprogramming suggests that human hemogenic precursors may not represent a cohort of mature endothelium but more likely a different lineage with endothelial features that is committed to the hematopoietic route. This is also supported by studies of human ESC-derived hematopoiesis (Ditadi et al., 2015). Glycolysis-associated genes were rapidly activated 2 days after induction, and motif enrichment analysis identified HIF1 as a regulator of human hematopoietic reprogramming. This suggests that a metabolic shift toward glycolysis underlies hemogenic reprogramming and human stem cell formation that may be mediated by HIF and AP-1, as recently suggested in zebrafish (Harris et al., 2013; Zhang et al., 2018).
In contrast to induced PSC (iPSC) reprogramming, the mechanisms underlying direct hematopoietic reprogramming remain poorly understood. We have explored the regulatory mechanisms underlying hemogenic induction with ChIP-seq at the initial stages of reprogramming. Two different models of action have been proposed for how TFs access chromatin to initiate reprogramming: (1) pioneer TFs, as a unique class of transcriptional regulators with the capacity to bind nucleosomal DNA (closed chromatin) and activate gene regulatory networks in target cells (Soufi et al., 2012), and (2) cooperative interaction among multiple TFs (Chronis et al., 2017). In this recent study, iPSC reprogramming factors bind cooperatively to enhancer regions to direct somatic inactivation and pluripotent gene expression initiation (Chronis et al., 2017). Our data support a model (Figure 6E) whereby GGF cooperate to silence fibroblast-specific program and gradually impose the hemogenic program. GATA2 and GFI1B binding occurs at open chromatin, promoters, and enhancer regions supporting the cooperative binding model. However, GATA2 showed independent targeting capacity and was crucial for the recruitment of GFI1B to target sites. Our finding mirrors the role of GATA2 during hematopoietic progenitor differentiation by recruiting GATA1 to erythroid genes (May et al., 2013), suggesting that GATA2 nucleates the binding of the complex to initiate hematopoietic reprogramming at open chromatin regions. It is then likely that the reprogramming cascade initiated by the TFs will then rely on a stepwise process of chromatin remodeling to shut down fibroblast enhancers and open endothelial and hematopoietic enhancers, as recently demonstrated during iPSC reprogramming (Knaupp et al., 2017; Li et al., 2017). During mouse ESC differentiation, it was shown that AP-1 motifs were enriched in open chromatin regions and co-localized with TF-binding sites that were specific to hemogenic endothelial cells (Goode et al., 2016). In human endothelial cells, AP-1 cooperates with GATA2 to induce key endothelial and inflammatory genes (Kawana et al., 1995; Linnemann et al., 2011). In contrast, Gfi1b is not a part of the heptad of TFs in mouse hematopoietic progenitors (Wilson et al., 2010). Indeed, we have confirmed that in human hematopoietic progenitors, GFI1B has a very different binding pattern from GATA2 and FOS. We propose that GATA2 and GFI1B interaction is specific during hemogenic reprogramming and HSC specification. Recent ChIP-seq data from mouse ESC-derived hemogenic endothelial cells supports a similarity between GATA2 and GFI1B target sites (Goode et al., 2016). Taken together, these data highlight the cooperative action between GGF during human hematopoietic reprogramming, initially binding at fibroblast-specific genes and then activating endothelial and hematopoietic gene signatures.
Several recent publications have induced mouse and human cells into HPCs (Batta et al., 2014; Lis et al., 2017; Pereira et al., 2013; Riddell et al., 2014; Sandler et al., 2014). Two recent studies showed the generation of long-term reconstituting HSCs derived from mouse endothelial cells or human PSCs (Lis et al., 2017; Sugimura et al., 2017). In both cases, reprogrammed cells were taken into either the bone marrow niche or co-cultured on endothelial cells to promote maturation. It will be interesting to address the impact of a supportive niche on our reprogrammed cells to mature these populations. In addition, combining GATA2-, FOS-, and GFI1B-specifying factors with factors that may promote HSC maturation should be investigated. A recent study demonstrated the generation of serially engraftable murine HSCs by GGF overexpression within teratoma (Tsukada et al., 2017). This suggests that GGF are the instructive factors of HSC identity not only from fibroblasts but also from other cell types and highlights the importance of the in vivo environment as well as the need for suitable mouse models that support the maintenance of human HSCs (Cosgun et al., 2014). It will be of critical importance to develop more defined culture methods for the controlled maturation of in vitro-programmed as well as human embryo-derived nascent HSCs into definitive, fully functional HSCs.
Collectively, our results show that GGF are sufficient for the generation of hemogenic cells from human fibroblasts. Our results suggest that HSC specification is controlled by the cooperative action of TFs and underscore the importance of GATA2-GFI1B interaction and initial engagement at open chromatin regions. In summary, we demonstrate that direct cellular reprogramming provides insights into the molecular mechanisms of human HSC specification. These studies provide a plat-form for the development of patient-specific HSPCs from easily accessible HDFs.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Carlos-Filipe Pereira (filipe.pereira@med.lu.se ).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Human adult dermal fibroblasts (HDF, ScienCell), neonatal foreskin fibroblasts (BJ) and HEK293T cells (ATCC) were grown in fibroblast media (FM media; Dulbecco’s Modified Eagle Medium (Invitrogen) containing 10% fetal bovine serum (Benchmark), 1mM L-Glutamine and penicillin/streptomycin (10 μgml−1, Invitrogen) in 5% (v/v) CO2 at 37°C. Cells were grown for 2–3 days until confluence, dissociated with TrypLE Express and frozen in Fetal Bovine Serum (FBS) 10% dimethyl sulfoxide (DMSO, Sigma). HDF and BJ were used at between passages 1–4 and 10–15, respectively. All cells were maintained at 37°C and 5% (v/v) CO2. All tissue culture reagents were from Thermo Fisher Scientific unless stated otherwise.
Mice and in vivo animal studies
NSG (NOD.Cg-PrkdcScidIl2rgtm1Wjl/Sz) mice (Mus musculus) were ordered from Jackson laboratories and housed in the centralized animal care facility of the Center for Comparative Medicine and Surgery (Mount Sinai, New York). Animal experiments and procedures were approved by the Institutional Animal Care and Use Committee and conducted in accordance with the Animal Welfare Act. Mice were housed in groups of 3–5 at 22 C°–24 C° using a 12-h light/12-h dark cycle. Animals had ad libitum access to water and the regular chow diet at all times. Water was replaced by Dox supplemented water (1mg/ml) after transplantation for 2 weeks. For all experiments only female mice were used, and transplantation was performed in mice with the age of 4 weeks.
METHOD DETAILS
Molecular Cloning and Lentivirus Production
Coding regions of human GFI1B, FOS and ETV6 were individually cloned into the pFUW-tetO lentiviral vector where expression is under the control of the tetracycline operator and a minimal CMV promoter. Lentiviral vectors containing the reverse tetracycline transactivator M2rtTA under the control of a constitutively active human ubiquitin C promoter (FUW-M2rtTA) and pFUW-tetO-GATA2 have been previously described (Pereira et al., 2013). The coding region of human GFI1B was inserted into the lentiviral plasmid pLV-TRE-HA. The coding region of GATA2 was inserted into pJW321–3xFLAG and sub-cloned into the pFUW-tetO vector. The primers used for cloning are listed in the Key Resource Table. 293T cells were transfected with a mixture of viral plasmid and packaging constructs expressing the viral packaging functions and the VSV-G protein. Viral supernatants were harvested after 36, 48 and 72 hours, filtered (0.45 μm) and concentrated 40-fold with Amicon ultra centrifugal filters (Millipore).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse Anti-Human CD34 clone 581 | BD Biosciences | Cat#560710; RRID: AB_2687922 |
| Mouse Anti-Human CD34 clone 8G12 | BD Biosciences | Cat#348057; RRID: AB_2687922 |
| Rat Anti-Human CD49f clone GoH3 | BD Biosciences | Cat#555736; RRID: AB_396079 |
| Rat anti-mouse CD45 clone 30-F11 | Bio Legend | Cat#103126; RRID: AB_493536 |
| Mouse Anti-Human CD45 clone HI30 | BD Biosciences | Cat#560777; RRID: AB_1937324 |
| Mouse Anti-Human CD143 (ACE) clone BB9 | BD Biosciences | Cat#557813; RRID: AB_396883 |
| Mouse Anti-Human CD90 clone 5E10 | eBioscience | Cat#17–0909-42; RRID: AB_953611 |
| Mouse Anti-Human CD38 clone HIT2 | BD Biosciences | Cat#560676; RRID: AB_1727472 |
| Biotin Anti-Human CD133 clone AC133 | Miltenyi Biotec | Cat#130–090-664; RRID: AB_244341 |
| Mouse Anti-Human CD3 clone UCHT1 | eBioscience | Cat#47–0038-42; RRID: AB_906221 |
| Mouse Anti-Human CD19 clone HIB19 | Biologend | Cat#302254; RRID: AB_2564142 |
| Mouse Anti-Human CD11c clone 3.9 | Biologend | Cat#301618; RRID: AB_439791 |
| Mouse Anti-Human CD14 clone HDC14 | Biologend | Cat#325618; RRID: AB_830691 |
| Diamond human CD34 isolation kit | Miltenyi Biotec | Cat#130–094-531; RRID: AB_2721154 |
| Mouse Monoclonal Anti-FLAG clone M2 | Sigma Aldrich | Cat#F1804; RRID: AB_259529 |
| Rabbit polyclonal Anti-HA clone 4C12 | Abcam | Cat#ab9110; RRID: AB_10637297 |
| Rabbit polyclonal Anti-FOS clone 4 | Santa Cruz | Cat#sc-52; RRID: AB_2106783 |
| Normal rabbit IgG | Santa Cruz | Cat#sc-2025; RRID: AB_737196 |
| Mouse monoclonal β-actin clone AC-74 | Sigma Aldrich | Cat#A2228; RRID: AB_476697 |
| Bacterial and Virus Strains | ||
| Competent E. coli DH5α | NEB | Cat#C2987I |
| Biological Samples | ||
| Cord Blood | New York Blood Center | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Polybrene | Sigma | Cat#H9268 |
| Doxycycline hyclate | Sigma | Cat#D9891 |
| Dulbecco’s Modified Eagle Medium (DMEM) | GIBCO | Cat#11965–092 |
| L-Glutamine | GIBCO | Cat#25030–081 |
| Penicillin-Streptomycin Solution | GIBCO | Cat#15140–122 |
| FBS | BenchMark | Cat#100–106 |
| TryPLE Express | GIBCO | Cat#12605–010 |
| Accutase Cell detachment solution | Innovative Cell Technologies | Cat#AT104 |
| Dulbecco’s Phosphate Buffer Solution (DPBS) | GIBCO | Cat#14190–144 |
| Myelocult Media | Stem Cell Technologies | Cat#5150 |
| Hydrocortisone | Stem Cell Technologies | Cat#07904 |
| Trizol Reagent | Ambion RNA | Cat#15596026 |
| ECL | Thermo Scientific | Cat#32209 |
| Femto | Thermo Scientific | Cat#34095 |
| Protein G Agarose beads | Roche | Cat# 11719416001 |
| Protein G Dynabeads | Life Technologies | Cat#10004D |
| Proteinase K | Invitrogen | Cat#100005393 |
| RNase A | 5Prime | Cat#2900403 |
| Phenol/Chloroform/Isoamyl Alcohol | Fisher | Cat#BP1752 |
| Sodium Acetate | Sigma | Cat#S7899 |
| GlycoBlue | Life Technologies | Cat#AM9515 |
| Glycine | Fisher | Cat#G48–212 |
| Formaldehyde solution | Sigma | Cat#F8775 |
| Gelatin from Porcine Type A | Sigma | Cat#G1890–100 |
| Molecular grade water | Corning | Cat#46–000-1 |
| BSA | Fisher | Cat#BP1600 |
| BES buffered saline solution | Sigma | Cat#14280–100 |
| Protease Inhibitor cocktail tablets | Roche | Cat#1187358001 |
| Critical Commercial Assays | ||
| Nextera XT library preparation kit | Illumina | Cat#FC-121–1031 |
| TruSeq RNA Sample Prep kit | Illumina | Cat#RS-122–2001 |
| KAPA Hyper Prep Kit | KAPA Biosystems | Cat#KK8502 |
| NEBNext ChIP-seq Library Prep Master Mix Set | New England Biolabs | Cat#E6240L |
| Agilent High Sensitivity DNA kit | Agilent Technologies | Cat#5067–4626 |
| Agilent RNA 6000 Nano kit | Agilent Technologies | Cat#5067–1511 |
| Diamond CD34 Isolation kit | Miltenyi Biotec | Cat#130–094-531 |
| C1 Single-Cell Reagent kit | Fluidigm | Cat#100–6201 |
| SMARTer Ultra Low RNA kit for the Fluidigm C1 System | Clontech | Cat#634833 |
| Quant-iT PicoGreen double-stranded DNA Assay kit | Thermo Fisher Scientific | Cat#P7589 |
| AMPure XP beads | Beckman Coulter | Cat#A63881 |
| Deposited Data | ||
| Bulk RNA-seq, Single Cell RNA-seq, ChIP-seq data | This paper | Gene Expression Omnibus (GEO): GSE51025 |
| Experimental Models: Cell Lines | ||
| Human adult dermal fibroblasts (HDF) | ScienCell | Cat#3220 |
| Neonatal foreskin fibroblasts (BJ) | ATCC | Cat#CRL-2522 |
| HEK293T | ATCC | Cat#CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| NSG (NOD.Cg-PrkdcScidIl2rgtm1Wjl/Sz) | Jackson laboratories | Cat#005557; RRID: IMSR_JAX:005557 |
| Oligonucleotides | ||
| GATA2 NheI F (pJW321–3xFLAG) – AATATCGCTAGCatg gag gtg gcg ccc gag cag ccg | This paper | N/A |
| GATA2 PacI R (pJW321–3xFLAG) – GGTATCTTAATTAA tcaCTAGCCCATGGCGGTCAC | This paper | N/A |
| GFI1B HpaI F (pLV-HA) – AATATCGTTAACATGCCACG CTCCTTCCTG | This paper | N/A |
| GFI1B ClaI R (pLV-HA) – GGTATCATCGATTCACTTGA GATTGTGCTGGCT | This paper | N/A |
| GFI1B EcoRI F (pFUW) – AATATCGAATTCATGCCAC GCTCCTTCCTG | This paper | N/A |
| GFI1B EcoRI R (pFUW) – GGTATCGAATTCTCACTTG AGATTGTGCTGGCT | This paper | N/A |
| FOS EcoRI F (pFUW) – AATATCGAATTCATGATGTTC TCGGGCTTCAACGCAG | This paper | N/A |
| FOS EcoRI R (pFUW) – GGTATCGAATTCTCACAGG GCCAGCAGCGTGGG | This paper | N/A |
| CR17AS 5’ – GGGATAATTTCAGCTGACTAAACAG | This paper | N/A |
| CR17AS 3’ – TTCCGTTTAGTTAGGTGCAGTTATC | This paper | N/A |
| Recombinant DNA | ||
| pFUW-M2rtTA | Pereira et al., 2013 | N/A |
| pFUW-tetO-GATA2 | Pereira et al., 2013 | N/A |
| pFUW-tetO-FOS | This paper | N/A |
| pFUW-tetO-GFI1B | This paper | N/A |
| pFUW-tetO-ETV6 | This paper | N/A |
| pLV-tetO-HA-GFI1B | This paper | N/A |
| pFUW-tetO-3xFLAG-GATA2 | This paper | N/A |
| pJW321–3xFlag-NANOG | Given by Wang lab | N/A |
| pLV-TRE-HA-GFP | Given by Lemischka lab | N/A |
| Software and Algorithms | ||
| GPSforGenes | This paper | N/A |
| FASTX tool kit | N/A | http://hannonlab.cshl.edu/fastx_toolkit/ |
| Homer | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| Bedtools | Quinlan and Hall, 2010 | http://bedtools.readthedocs.io/en/latest/ |
| Samtools | Li et al., 2009 | http://www.htslib.org/ |
| Cufflinks | Trapnell et al., 2012 | http://cole-trapnell-lab.github.io/cufflinks/ |
| Enrichr | Chen et al., 2013 | http://amp.pharm.mssm.edu/Enrichr/ |
| MACS1.4 | Zhang et al., 2008 | http://liulab.dfci.harvard.edu/MACS/00README.html |
| TopHat | Trapnell et al., 2009 | https://ccb.jhu.edu/software/tophat/index.shtml |
| FASTQC | N/A | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| ChromHMM | Ernst and Kellis, 2012 | http://compbio.mit.edu/ChromHMM/ |
| Limma Voom | R project for Statistical Computing | http://bioconductor.org/packages/release/bioc/html/limma.html |
| Galaxy | Galaxy server | https://usegalaxy.org |
| UCSC Genome Browser | N/A | https://genome.ucsc.edu |
| Gene set enrichment analysis (GSEA) | Subramanian et al., 2005 | http://software.broadinstitute.org/gsea/index.jsp |
| DESeq2 | R project for Statistical Computing | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Ilustrator | Adobe | http://www.adobe.com/cn/products/cs6/illustrator.html |
| Photoshop | Adobe | http://www.adobe.com/cn/products/cs6/photoshop.html |
| LSRII Diva Software | BD Biosciences | http://www.bdbiosciences.com/us/home |
| FlowJo Software | FlowJo, LLC | https://www.flowjo.com/ |
| GraphPad Prism 6.0 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
Viral Transduction and Cell Culture
Fibroblasts were seeded at a density of 25,000 cells per well on 0.1% gelatin coated 6-well plates and incubated overnight with pools of pFUW lentiviruses in FM media supplemented with 8 μgml−1 polybrene. Equal MOIs of individual viral particles were applied. Transductions with mOrange in pFUW-tetO resulted in > 95% efficiency. After 16–20 hours media was replaced with fresh FM media supplemented with Doxycycline (1 μgml−1). At day 4 post-transduction cells were dissociated with TrypLE Express and 10,000 cells per well were plated on 0.1% gelatin coated 6-well plates. Reprogramming cultures were maintained in Myelocult Media (H5100; Stem Cell Technologies) supplemented with Hydrocortisone (10−6 M; Stem Cell Technologies). Media was changed every 4 days for the duration of the cultures.
Immunofluorescence
Live immunofluorescence was performed with Phycoerythrin (PE)-conjugated sterile rat monoclonal antibodies against CD49f and CD34 (Key Resource Table) at a 1:20 dilution. Emergent colonies were washed once with PBS 5% FBS and incubated with conju-gated antibodies for 30 min at room temperature in the presence of mouse serum. Cultures were then washed twice with PBS 5% FBS to remove unbound antibody. For detection of tagged TFs, fibroblasts were fixed with 2% PFA for 20 min, permeabilized with 0.4% Triton X-100, blocked for 30 min and incubated with anti-FLAG, anti-HA, anti-FOS antibodies at 1:200 dilution for 2 hours. Cells were washed, incubated with secondary antibodies conjugated with Alexa Flour 488 (Invitrogen, A12379) and nuclear counterstained with 4,6-diamidino-2-phenylindole (DAPI, 1 μgml−1, Sigma). Cells were visualized on a Leica DMI4000 microscope and processed with Leica software and Adobe Photoshop.
Flow Cytometry Analysis and Fluorescence-Activated Cell Sorting
Cell cultures were dissociated with TrypLE Express or Accutase Cell detachment solution (Innovative Cell Technologies, Inc) and stained with fluorochrome-coupled antibodies (Key Resource Table). Cell populations were isolated on an InFlux cell sorter (BD Biosciences) and immediately lysed in Trizol (Ambion) for RNA extraction, cultured on 0.1% gelatin coated 6-well plates in Myelocult media or transplanted. Flow cytometric analysis was performed on a 5-laser LSRII with Diva software (BD Biosciences) and further analyzed using FlowJo software. DAPI (1 μgml−1) was added before analysis to exclude dead cells.
Long-term Repopulation Assays
NSG (NOD.Cg-PrkdcScidIl2rgtm1Wjl/Sz, Jackson laboratories) mice were used as recipients for induced cells. For isolating a population that include both CD49f+ and CD34+ cells, cultures were dissociated with Accutase 25 days after transduction and stained with PE-conjugated anti-CD49f and PE-conjugated anti-CD34. The PE-positive cell population was isolated by FACS sorting. Animals were transplanted with cells intravenously by retro-orbital injection and administered with Dox (1mg/ml) in drinking water for 2 weeks. Up to 6 hours before transplantation with human cells 4-week old female NSG mice received a sublethal total body irradiation dose of 200 cGy. The numbers of PE+ cells transplanted were in the range of 100,000 cells per animal. Starting 3–4 weeks after transplantation, NSG mice were bled from the orbital venous plexus and human contribution was assessed by flow cytometry with mouse and human anti-CD45 antibodies and by PCR with human specific primers to the Cr17 alpha-satellite sequence. Hematopoietic lineage contribution was assessed by flow cytometry with anti-CD3, anti-CD19, anti-CD14 and anti-CD11c human antibodies.
Genomic PCR
Genomic DNA was isolated using Easy DNA extraction kit (Invitrogen). Presence of human sequences was checked by PCR using Phusion Flash (Thermo Scientific) high-fidelity PCR Master Mix (30 cycles of 98°C for 1 s; 60°C for 5 s and 72°C for 15 s) with primers for the chromosome 17 alpha satellite (Key Resource Table).
GPSforGenes
Gene expression data was downloaded from BioGPS database (GeneAtlas U133A), transformed to log-space and normalized to bring the expression values to 0–1 range for each gene across different samples. The resulting data was then searched for samples with the highest averaged expression for (GATA2 + FOS + GFI1B) and (GATA2 + FOS + GFI1B + ETV6).
mRNA-seq Library Preparation and Sequencing
FACS isolated cells were lysed in Trizol (Ambion). RNA integrity was evaluated using a Eukaryotic RNA 6000 Nano chip on an Agilent 2100 Bioanalyzer (Agilent Technologies). Up to 1 μg of total RNA from each sample was used for library preparation with the TruSeq RNA Sample Preparation Kit (Illumina). A common adaptor was used for all samples and barcode sequences present in the reverse primer were introduced by 12–20 cycles of amplification as described (Pereira et al., 2013). Each library was assessed for quality and size distribution using an Agilent High Sensitivity Assay bioanalyzer chip and quantified by real-time PCR. Equimolar amounts of each barcoded library were mixed and single-end sequenced on an Illumina HiSeq Sequencing System.
Single cell mRNA-seq Library Preparation
HDF, HDF+GGF day 2, day 15 CD49f+, day 25 CD34+CD49f+ populations were FACS sorted and collected. Umbilical cord blood was obtained from the New York Blood Center and Lin-CD34+ cells were isolated using Diamond CD34 Isolation Kit (Miltenyi Biotec) according to manufacturer’s instructions. After isolation, Lin-CD34+ cells were stored in liquid nitrogen until use. cDNA synthesis was performed following the manufacturers instruction using the C1 Single-Cell Auto Prep System (Fluidigm) (Pereira et al., 2016). The cDNA reaction products were quantified using the Quant-iT PicoGreen double-stranded DNA Assay Kit (Thermo Fisher Scientific) and then diluted to a final concentration of 0.15–0.30 μgml−1 using C1 Harvest Reagent. Sequencing libraries were prepared using the Nextera XT DNA Library Prep Kit (Illumina). 96 single-cell libraries were mixed together. The concentration of the mixed libraries was determined using Agilent Bioanalyzer. The libraries were sequenced yielding ~0.18–5.7 Million 75-nt reads on a HiSeq 2000 platform at Girihlet, Inc.
Coimmunoprecipitation (Co-IP)
Nuclear extracts were prepared from HDFs with ectopic expression of 3xFLAG-tagged GATA2, HA-tagged GFI1B and FOS and incubated with 5 μg of each antibody (Key Resource Table) The immune complexes were then washed four times with the lysis buffer by centrifugation. IP/co-IP were performed using 5% of input samples. For the control IP, we used 5 μg of rabbit IgG (Key Resource Table). Samples were heated in SDS sample buffer and processed by western blotting.
Western Blot Analysis
Cells were lysed in RIPA-B buffer (20 mM Na2HPO4 [pH 7.4], 150 mM NaCl, 1% Triton X-100) in the presence of protease inhibitors (3 μg/ml aprotinin, 750 μg/ml benzamidine, 1 mM phenylmethylsulfonyl fluoride, 5 mM NaF and 2 mM sodium orthovanadate) and incubated on ice for 30 min with occasional vortexing. Samples were centrifuged to remove cell debris and heated in SDS sample buffer. For immunoblotting, membranes were blocked with TBST buffer (10 mM Tris-HCl (pH 7.9), 150 mM NaCl, and 0.05% Tween 20) containing 3% milk, incubated with primary antibodies, washed three times with TBST, incubated with HRP-conjugated secondary antibodies, washed three times with TBST and subsequently detected by ECL or Femto (Thermo Scientific).
Chromatin Immunoprecipitation (ChIP)-seq
ChIP assays were performed in HDFs transduced with a pool of 3xFLAG-tagged-GATA2, HA-tagged-GFI1B and FOS and the transgenes were induced with Doxycycline. After 48hr, 20–50×10^6 cells were used for each experiment and crosslinking conditions were optimized for each factor. For GATA2 and GFI1B ChIP cells were fixed with 11% formaldehyde (Sigma) at room temperature on a rotating platform for 10 min. Formaldehyde was quenched by adding of 125 mM of glycine on a rotating platform for 5 min at room temperature and cross-linked cells were washed twice in ice-cold PBS. Chromatin shearing was done using the E210 Covaris to a 150–350bp range, insoluble debris was centrifuged, then sheared chromatin fragments were incubated overnight at 4°C with antibodies coupled to 50 μl Protein G dynabeads (Invitrogen). For FOS ChIP 3 μg of antibody was used per 5–10×10^6 cells and for FLAG and HA 10μg of antibody per 20–50×10^6 cells. Beads were washed five times with RIPA buffer and once with TE containing 50 mM NaCl, and complexes eluted from beads in elution buffer by heating at 65°C and shaking in a Thermomixer. Reverse cross-linking was performed overnight at 65°C. Whole cell extract DNA was treated for cross-link reversal. Immunoprecipitated and whole cell extract DNA were treated with RNaseA, proteinase K and purified using Phenol:Chloroform:Isoamyl Alcohol extraction followed by ethanol precipitation. For FOS ChIP, 5–10×10^6 cells were double crosslinked. First, cells were crosslinked in PBS supplemented with Di(N-succinimidyl) glutarate (DSG, ThermoFisher Scientific 20593) at a final concentration of 2 mM for 45 min at room temperature on a rotating platform. After 3 washes in PBS, formaldehyde crosslinking of proteins and DNA was done for 10 min at room temperature at a concentration of 11% formaldehyde (Sigma) in PBS. Formaldehyde was quenched by adding of 125 mM of glycine on a rotating platform for 5 min at room temperature and crosslinked cells were washed twice in ice-cold PBS. Libraries were prepared using either KAPA Hyper Prep Kit or NEBNext ChIP-seq Library Prep Master Mix Set for Illumina according to the manufacturer’s guidelines. Libraries were size-selected on a 2% agarose gel for a 200–400bp fragments and were sequenced on Illumina HiSeq 2000.
ChIP-seq Data Visualization
To produce the heatmaps, each feature (such as peaks of a TF, histone marks) was aligned at GATA2 or GFI1B summits and tiled the flanking up- and downstream regions within ± 4kb in 100bp bins. To control for input in our data, we computed at each bin a input-normalized value as log2(RPKMTreat) - log2(RPKMInput), where RPKMTreat is RPKM of the corresponding TF or histone and RPKMInput is RPKM of the corresponding whole genome ‘Input’. We plotted the density of DNase-seq signal within ± 1kb around the center of GATA2 or GFI1B summits and compared it to the resistant sites, which were resized to be in the same range as GATA2 or GFI1B summits.
QUANTIFICATION AND STATISTICAL ANALYSIS
mRNA-seq Analysis
For each sample 4.5–26.5 M 100-nt reads were obtained, pre-processed with the FASTX-toolkit suite and aligned to the human genome (Homo sapiens hg19 assembly) using TopHat mapper. Post alignment with TopHat release 1.4.1 against the Homo sapiens hg19 assembly using the known transcripts option. All resultant .bam files were processed using Samtools version 0.2.5 and Bed-tools version 2.16.2 and visualized on the Integrated Genome Browser version 2.1 or the UCSC Genome Browser. Transcript assembly and expression estimation was conducted with Cufflinks release 1.3.0 using a Homo sapiens hg19 reference annotation and upper quartile normalization. Cufflinks assemblies were merged and processed through Cuffdiff for gene FPKM reporting and differential expression analysis. Each library was treated as a separate non-replicate sample. Gene transcript count data from the mRNA-seq analysis was obtained by reprocessing the data through TopHat release 2.0.0 and Cufflinks and Cuffdiff release 2.0.0. Gene set enrichment analysis (GSEA) between HDF or BJ and CD34+CD49f+ was performed using the genes.fpkm.tracking file (Table S1) output from Cufflinks release 1.3.0 run against the Molecular Signatures Database version 2.0 curated gene sets (Gene set sizes 0–5000) ranked by Ratio_of_Classes. Non-negative Matrix Factorization (NMF) of the FPKM values obtained from RNA sequencing was performed on the GenePattern Platform using the NMF consensus analysis module at k. initial = 2 and k. final = 5, here we show metagene results for k = 4. Visualization of FPKM expression density and inter-sample FPKM correlation was conducted in R version 2.15.0 with the CummeRbund package. Gene list enrichment analysis with gene set libraries created from level 4 of the MGI mouse phenotype ontology was performed with Enrichr.
Single cell mRNA-seq Analysis
For single cell mRNA-Seq analysis the raw fastq files were aligned against the Ensemble GRCh38 genome using the Gencode v25 gene annotation. Gene level read counts were then calculated using featureCounts from the Subread package. Raw counts were log transformed and quantile normalized. For hierarchical clustering Pearson correlation was used as distance metric. Hierarchical clustering was plotted using the dendextend library in R.
mRNA-seq Quality Control
Scater library (McCarthy et al., 2017) was used to include samples and genes that pass quality control. For single cell analysis, we first discarded genes that are expressed in less than 1% of the cells. Then we applied Scater library function ‘isOutlier’ with 3 median absolute deviations in order to define the thresholds for following parameters: total number of counts, number of genes detected and number of counts belonging to mitochondrial genes. Values beyond this threshold are considered outliers and were not included in the analysis. As a result, we defined the following criteria: 1) total number of counts detected per sample ≥ 891,010; 2) number of genes detected in each single cell ≥ 2,619; 3) number of counts belonging to mitochondrial genes ≤ 500,224. From single cell mRNA-seq 39 cells did not pass the quality control filters and were not included in the analysis. As for genes, from 56,269 we used 24,536 for analysis. For population mRNA-seq from the initial 24 samples from BJ and HDF-derived cells 1 sample did not pass the quality control and was discarded (BJ Day 15 CD49f+).
ChIP-seq analysis
ChIP-seq analysis was performed on the raw FASTQ files. FASTQ files were mapped to the human hg19 genome using Bowtie 2 program allowing for 2 base pair mismatches. Mapped output files were processed through MACS1.4 analysis software to determine peaks. Homer software package was used for peak annotation and further analysis was performed using the Galaxy server and R statistical software.
Chromatin State Fold-Enrichment
Enrichment scores for genomic features, such as GATA2 and GFI1B Chip-seq peaks and histone marks were calculated using ChromHMM Overlap Enrichment (Chronis et al., 2017), based on public segmentation. ChromHMM segmentation, that contains 18 different chromatin states, was downloaded from Roadmap website and used for analysis. Enrichment scores were calculated as the ratio between the observed and the expected overlap for each feature and chromatin state based on their sizes and the size of the human genome.
Gene List Enrichment Analysis with Single Cell Data
Differential gene expression was calculated for all pairwise sample groups (HDF, DAY2, CD34+ CD49f+, CD49f+, UCB CD34+) using variance-stabilizing transformation combined with limma. To calculate gene set enrichment the top 500 and bottom 500 genes for each differential expression gene signature were uploaded to Enrichr. The significance of gene set overlap was measured using the Fisher Exact test.
Motif Analyses
For de novo motif discovery, findMotifsGenome.pl procedure from Homer was used on GATA2 and GFI1B separately. Co-bound regions by GFI1B and GATA2 were found using bedtools. Co-bound regions were used for de novo motif discovery using Homer and CCAT. In order to evaluate similarity of the two sets based on the intersections Jaccard statistic were used.
Integration of Independently Obtained Gene Expression and Genome Location Datasets
Data from microarray and RNA-seq experiments were adjusted using quantile normalization; genes insignificantly changing their expression across samples were removed using ANOVA with the consequent adjustment for multiple testing (Benjamini-Hochberg correction, p < 0.05). Data for the remaining genes were converted to Z-scores. Hierarchical clustering and Principal Component Analysis (PCA) of the integrated dataset was performed using Cluster 3.0 and visualized with JAVA treeview. For differential gene expression analysis, DESeq2 was used in HDF, day 15 CD49f+, day 25 CD49f+ and day 25 CD34+CD49f+ and genes were selected using the following criteria: 1) DESeq2 differential calls with an adjusted p value < 0.05 (FDR used as adjusted p value) between either of four groups of sample; 2) Absolute changes in expression between minimal and maximal expression > 1.5 fold. Intersection between the genes identified by Chip-Seq for GATA2 and GFI1B (binding peaks within ± 5kb around the transcriptional start) and mRNA-seq identified 1,425 genes, which were clustered and visualized. Those 1,425 genes we divided into 3 groups: 1) Genes bound only by GATA2; 2) Genes bound only by GFI1B; 3) Genes co-bound by GATA2 and GFI1B.
DATA AND SOFTWARE AVAILABILITY
The accession number for population, single cell mRNA-seq, and ChIP-seq data reported in this paper is Gene Expression Omnibus (GEO): GSE51025. The public datasets used in this study can be found in Table S4.
Supplementary Material
Highlights.
GATA2, GFI1B, and FOS induce a hemogenic program in human fibroblasts
Induced cells display dynamic endothelial-to-hematopoietic transition programs
GATA2 is the dominant transcription factor that initiates hemogenic reprogramming
GATA2 and GFI1B interact and bind cooperatively at open chromatin
ACKNOWLEDGMENTS
This manuscript is dedicated to Ihor Lemischka in loving memory of his contributions to our science and our lives. We also mourn the loss of Dmitri Papatsenko. His computational skills and great sense of humor will be sorely missed. We thank the members of the Lemischka and Moore laboratories for useful discussions; Y. Liu and X. Niu for laboratory management; A. Waghray, D.-F. Lee, M. Fidalgo, V.J. Valdes, and I. Kulakovskiy for assistance; and Christoph Schaniel for critical reading of the manuscript. We also thank the flow cytometry, irradiation, and mouse facilities at Mount Sinai. We specially thank the Pluripotent Stem Cell Core and S.L. D’Souza, Mount Sinai Genomics, and A. Jayaprakash for help with materials and protocols. This study was supported by funds from NIH (1R01HL119404 to K.A.M. and I.R.L.) and NIH/IAD (R33A1116191 to B.K.C), New York Dept. of Health NYSTEM (C32597GG to K.A.M.), the Charles H. Revson Foundation, the European Commission (PIIF-GA-2013–628761 to C.-F.P.), and Fundação para a Ciência e Tecnologia (SFRH/BD/51968/2012 and PTDC/BIM-MED/0075/2014 to A.M.G. and C.-F.P.). The Knut and Alice Wallenberg Foundation is acknowledged for generous support.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and four tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.11.032.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Anguita E, Villegas A, Iborra F, and Hernández A (2010). GFI1B controls its own expression binding to multiple sites. Haematologica 95, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batta K, Florkowska M, Kouskoff V, and Lacaud G (2014). Direct reprogramming of murine fibroblasts to hematopoietic progenitor cells. Cell Rep 9, 1871–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chronis C, Fiziev P, Papp B, Butz S, Bonora G, Sabri S, Ernst J, and Plath K (2017). Cooperative binding of transcription factors orchestrates reprogramming. Cell 168, 442–459.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgun KN, Rahmig S, Mende N, Reinke S, Hauber I, Schäfer C, Petzold A, Weisbach H, Heidkamp G, Purbojo A, et al. (2014). Kit regulates HSC engraftment across the human-mouse species barrier. Cell Stem Cell 15, 227–238. [DOI] [PubMed] [Google Scholar]
- Ditadi A, Sturgeon CM, Tober J, Awong G, Kennedy M, Yzaguirre AD, Azzola L, Ng ES, Stanley EG, French DL, et al. (2015). Human definitive haemogenic endothelium and arterial vascular endothelium represent distinct lineages. Nat. Cell Biol 17, 580–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditadi A, Sturgeon CM, and Keller G (2017). A view of human haematopoietic development from the Petri dish. Nat. Rev. Mol. Cell Biol 18, 56–67. [DOI] [PubMed] [Google Scholar]
- Ernst J, and Kellis M (2012). ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espin-Palazon R, Weijts B, Mulero V, and Traver D (2018). Proinflammatory signals as fuel for the fire of hematopoietic stem cell emergence. Trends Cell Biol 28, 58–66. [DOI] [PubMed] [Google Scholar]
- Goode DK, Obier N, Vijayabaskar MS, Lie-A-Ling M, Lilly AJ, Hannah R, Lichtinger M, Batta K, Florkowska M, Patel R, et al. (2016). Dynamic gene regulatory networks drive hematopoietic specification and differentiation. Dev. Cell 36, 572–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JM, Esain V, Frechette GM, Harris LJ, Cox AG, Cortes M, Garnaas MK, Carroll KJ, Cutting CC, Khan T, et al. (2013). Glucose metabolism impacts the spatiotemporal onset and magnitude of HSC induction in vivo. Blood 121, 2483–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanovs A, Rybtsov S, Welch L, Anderson RA, Turner ML, and Medvinsky A (2011). Highly potent human hematopoietic stem cells first emerge in the intraembryonic aorta-gonad-mesonephros region. J. Exp. Med 208, 2417–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanovs A, Rybtsov S, Ng ES, Stanley EG, Elefanty AG, and Medvinsky A (2017). Human haematopoietic stem cell development: from the embryo to the dish. Development 144, 2323–2337. [DOI] [PubMed] [Google Scholar]
- Jay KE, Rouleau A, Underhill TM, and Bhatia M (2004). Identification of a novel population of human cord blood cells with hema-topoietic and chondrocytic potential. Cell Res 14, 268–282. [DOI] [PubMed] [Google Scholar]
- Jokubaitis VJ, Sinka L, Driessen R, Whitty G, Haylock DN, Bertoncello I, Smith I, Péault B, Tavian M, and Simmons PJ (2008). Angiotensin-converting enzyme (CD143) marks hematopoietic stem cells in human embryonic, fetal, and adult hematopoietic tissues. Blood 111, 4055–4063. [DOI] [PubMed] [Google Scholar]
- Karlsson G, Rörby E, Pina C, Soneji S, Reckzeh K, Miharada K, Karlsson C, Guo Y, Fugazza C, Gupta R, et al. (2013). The tetraspanin CD9 affords high-purity capture of all murine hematopoietic stem cells. Cell Rep 4, 642–648. [DOI] [PubMed] [Google Scholar]
- Kawana M, Lee ME, Quertermous EE, and Quertermous T (1995). Cooperative interaction of GATA-2 and AP1 regulates transcription of the endothelin-1 gene. Mol. Cell. Biol 15, 4225–4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaupp AS, Buckberry S, Pflueger J, Lim SM, Ford E, Larcombe MR, Rossello FJ, de Mendoza A, Alaei S, Firas J, et al. (2017). Transient and permanent reconfiguration of chromatin and transcription factor occupancy drive reprogramming. Cell Stem Cell 21, 834–845.e6. [DOI] [PubMed] [Google Scholar]
- Kobayashi I, Kobayashi-Sun J, Kim AD, Pouget C, Fujita N, Suda T, and Traver D (2014). Jam1a-Jam2a interactions regulate haematopoietic stem cell fate through Notch signalling. Nature 512, 319–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labastie MC, Cortés F, Roméo PH, Dulac C, and Péault B (1998). Molecular identity of hematopoietic precursor cells emerging in the human embryo. Blood 92, 3624–3635. [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Liu J, Yang X, Zhou C, Guo J, Wu C, Qin Y, Guo L, He J, Yu S, et al. (2017). Chromatin accessibility dynamics during iPSC reprogramming. Cell Stem Cell 21, 819–833.e6. [DOI] [PubMed] [Google Scholar]
- Linnemann AK, O’Geen H, Keles S, Farnham PJ, and Bresnick EH (2011). Genetic framework for GATA factor function in vascular biology. Proc. Natl. Acad. Sci. U S A 108, 13641–13646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lis R, Karrasch CC, Poulos MG, Kunar B, Redmond D, Duran JGB, Badwe CR, Schachterle W, Ginsberg M, Xiang J, et al. (2017). Conversion of adult endothelium to immunocompetent haematopoietic stem cells. Nature 545, 439–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeti R, Park CY, and Weissman IL (2007). Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. Cell Stem Cell 1, 635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May G, Soneji S, Tipping AJ, Teles J, McGowan SJ, Wu M, Guo Y, Fugazza C, Brown J, Karlsson G, et al. (2013). Dynamic analysis of gene expression and genome-wide transcription factor binding during lineage specification of multipotent progenitors. Cell Stem Cell 13, 754–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Campbell KR, Lun AT, and Wills QF (2017). Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 33, 1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarvey AC, Rybtsov S, Souilhol C, Tamagno S, Rice R, Hills D, Godwin D, Rice D, Tomlinson SR, and Medvinsky A (2017). A molecular roadmap of the AGM region reveals BMPER as a novel regulator of HSC maturation. J. Exp. Med 214, 3731–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvinsky A, Rybtsov S, and Taoudi S (2011). Embryonic origin of the adult hematopoietic system: advances and questions. Development 138, 1017–1031. [DOI] [PubMed] [Google Scholar]
- Moignard V, Macaulay IC, Swiers G, Buettner F, Schütte J, Calero-Nieto FJ, Kinston S, Joshi A, Hannah R, Theis FJ, et al. (2013). Characterization of transcriptional networks in blood stem and progenitor cells using high-throughput single-cell gene expression analysis. Nat. Cell Biol 15, 363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muench MO, Kapidzic M, Gormley M, Gutierrez AG, Ponder KL, Fomin ME, Beyer AI, Stolp H, Qi Z, Fisher SJ, and Bárcena A (2017). The human chorion contains definitive hematopoietic stem cells from the fifteenth week of gestation. Development 144, 1399–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng ES, Azzola L, Bruveris FF, Calvanese V, Phipson B, Vlahos K, Hirst C, Jokubaitis VJ, Yu QC, Maksimovic J, et al. (2016). Differentiation of human embryonic stem cells to HOXA+ hemogenic vasculature that resembles the aorta-gonad-mesonephros. Nat. Biotechnol 34, 1168–1179. [DOI] [PubMed] [Google Scholar]
- Notta F, Doulatov S, Laurenti E, Poeppl A, Jurisica I, and Dick JE (2011). Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science 333, 218–221. [DOI] [PubMed] [Google Scholar]
- Oberlin E, Tavian M, Blazsek I, and Péult B (2002). Blood-forming potential of vascular endothelium in the human embryo. Development 129, 4147–4157. [DOI] [PubMed] [Google Scholar]
- Obier N, Cauchy P, Assi SA, Gilmour J, Lie-A-Ling M, Lichtinger M, Hoogenkamp M, Noailles L, Cockerill PN, Lacaud G, et al. (2016). Cooperative binding of AP-1 and TEAD4 modulates the balance between vascular smooth muscle and hemogenic cell fate. Development 143, 4324–4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira CF, Chang B, Qiu J, Niu X, Papatsenko D, Hendry CE, Clark NR, Nomura-Kitabayashi A, Kovacic JC, Ma’ayan A, et al. (2013). Induction of a hemogenic program in mouse fibroblasts. Cell Stem Cell 13, 205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira CF, Chang B, Gomes A, Bernitz J, Papatsenko D, Niu X, Swiers G, Azzoni E, de Bruijn MF, Schaniel C, et al. (2016). Hematopoietic reprogramming in vitro informs in vivo identification of hemogenic precursors to definitive hematopoietic stem cells. Dev. Cell 36, 525–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prashad SL, Calvanese V, Yao CY, Kaiser J, Wang Y, Sasidharan R, Crooks G, Magnusson M, and Mikkola HK (2015). GPI-80 defines self-renewal ability in hematopoietic stem cells during human development. Cell Stem Cell 16, 80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell J, Gazit R, Garrison BS, Guo G, Saadatpour A, Mandal PK, Ebina W, Volchkov P, Yuan GC, Orkin SH, and Rossi DJ (2014). Reprogramming committed murine blood cells to induced hematopoietic stem cells with defined factors. Cell 157, 549–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin C, Bollerot K, Mendes S, Haak E, Crisan M, Cerisoli F, Lauw I, Kaimakis P, Jorna R, Vermeulen M, et al. (2009). Human placenta is a potent hematopoietic niche containing hematopoietic stem and progenitor cells throughout development. Cell Stem Cell 5, 385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybtsov S, Batsivari A, Bilotkach K, Paruzina D, Senserrich J, Nerushev O, and Medvinsky A (2014). Tracing the origin of the HSC hierarchy reveals an SCF-dependent, IL-3-independent CD43(−) embryonic precursor. Stem Cell Reports 3, 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler VM, Lis R, Liu Y, Kedem A, James D, Elemento O, Butler JM, Scandura JM, and Rafii S (2014). Reprogramming human endothelial cells to haematopoietic cells requires vascular induction. Nature 511, 312–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinka L, Biasch K, Khazaal I, Páault B and Tavian M (2012). Angiotensin-converting enzyme (CD143) specifies emerging lympho-hematopoietic progenitors in the human embryo. Blood 119, 3712–3723. [DOI] [PubMed] [Google Scholar]
- Solaimani Kartalaei P, Yamada-Inagawa T, Vink CS, de Pater E, van der Linden R, Marks-Bluth J, van der Sloot A, van den Hout M, Yokomizo T, van Schaick-Solernó ML, et al. (2015). Whole-transcriptome analysis of endothelial to hematopoietic stem cell transition reveals a requirement for Gpr56 in HSC generation. J. Exp. Med 212, 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi A, Donahue G, and Zaret KS (2012). Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 151, 994–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimura R, Jha DK, Han A, Soria-Valles C, da Rocha EL, Lu YF, Goettel JA, Serrao E, Rowe RG, Malleshaiah M, et al. (2017). Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature 545, 432–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiers G, Baumann C, O’Rourke J, Giannoulatou E, Taylor S, Joshi A, Moignard V, Pina C, Bee T, Kokkaliaris KD, et al. (2013). Early dynamic fate changes in haemogenic endothelium characterized at the single-cell level. Nat. Commun 4, 2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavian M, Biasch K, Sinka L, Vallet J, and Péault B (2010). Embryonic origin of human hematopoiesis. Int. J. Dev. Biol 54, 1061–1065. [DOI] [PubMed] [Google Scholar]
- Thambyrajah R, Mazan M, Patel R, Moignard V, Stefanska M, Marinopoulou E, Li Y, Lancrin C, Clapes T, Möröy T, et al. (2016). GFI1 proteins orchestrate the emergence of haematopoietic stem cells through recruitment of LSD1. Nat. Cell Biol 18, 21–32. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada M, Ota Y, Wilkinson AC, Becker HJ, Osato M, Nakauchi H, and Yamazaki S (2017). In vivo generation of engraftable murine hematopoietic stem cells by Gfi1b, c-Fos, and Gata2 overexpression within teratoma. Stem Cell Rep 9, 1024–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassen L, Fiolka K, and Möröy T (2006). Gfi1b alters histone methylation at target gene promoters and sites of gamma-satellite containing heterochromatin. EMBO J 25, 2409–2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorontsov IE, Kulakovskiy IV, and Makeev VJ (2013). Jaccard index based similarity measure to compare transcription factor binding site models. Algorithms Mol. Biol 8, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski OL, Vierbuchen T, Qu K, Lee QY, Chanda S, Fuentes DR, Giresi PG, Ng YH, Marro S, Neff NF, et al. (2013). Hierarchical mechanisms for direct reprogramming of fibroblasts to neurons. Cell 155, 621–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NK, Foster SD, Wang X, Knezevic K, Schütte J, Kaimakis P, Chilarska PM, Kinston S, Ouwehand WH, Dzierzak E, et al. (2010). Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell 7, 532–544. [DOI] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, et al. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CY, Yin HM, Wang H, Su D, Xia Y, Yan LF, Fang B, Liu W, Wang YM, Gu AH, et al. (2018). Transforming growth factor-beta1 regulates the nascent hematopoietic stem cell niche by promoting gluconeogenesis. Leukemia 32, 479–491. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for population, single cell mRNA-seq, and ChIP-seq data reported in this paper is Gene Expression Omnibus (GEO): GSE51025. The public datasets used in this study can be found in Table S4.