

SUMMARY
Unlike prototypical IκB proteins which are inhibitors of NF-κB RelA, cRel, and RelB dimers; the atypical IκB protein Bcl3 is primarily a transcriptional coregulator of p52 and p50 homodimers. Bcl3 exists as phospho-protein in many cancer cells. Unphosphorylated Bcl3 acts as classical IκB-like inhibitor, removes p50 and p52 from bound DNA. Neither the phosphorylation site(s) nor the kinase(s) phosphorylating Bcl3 are known. Here we show Akt, Erk2, and IKK1/2 phosphorylate Bcl3. Phosphorylation of Ser33 by Akt induces switching of K48- to K63-ubiquitination; thus promotes nuclear localization and stabilization of Bcl3. Phosphorylation by Erk2 and IKK1/2 of Ser114 and Ser446 convert Bcl3 into a transcriptional coregulator by facilitating its recruitment to DNA. Cells expressing S114A/S446A mutant have cellular proliferation and migration defects. This work links Akt and MAPK pathways to NF-κB through Bcl3 and provides mechanistic insight into how Bcl3 functions as an oncoprotein through collaboration with IKK1/2, Akt, and Erk2.
Keywords: NF-kappaB, Bcl3, phosphorylation, Akt, IKK, Erk2
eTOC Blurb
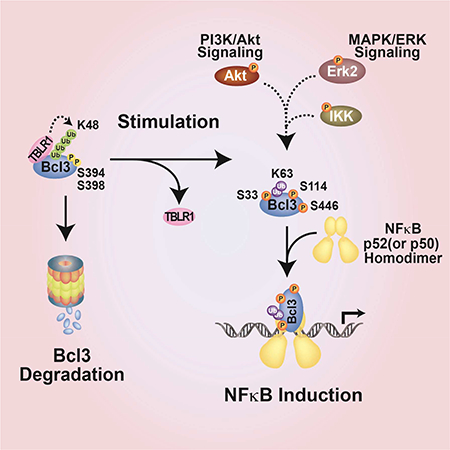
Wang et al. showed site-specific phosphorylation-induced metabolic stabilization of Bcl3 through switch of ubiquitin linkage and transcriptional coregulatory activity of Bcl3 in association with NF-κB p50 and p52. Through identification of several Bcl3 kinases targeting specific sites, this work also established convergence of three intertwined signaling pathways onto NF-κB activation.
Graphical Abstract
INTRODUCTION
B cell leukemia/lymphoma 3 (Bcl3) is an oncoprotein first found in cancers of hematological origins where Bcl3 is expressed 3–4 fold higher levels due placement of the bcl3 gene at a different location in a fused chromosome (Mathas et al., 2005; Nishikori et al., 2003; Ohno et al., 1993; Ohno et al., 1990). In many solid tumors, Bcl3 is overexpressed without undergoing chromosomal fusion suggesting other mechanisms for its activation. Oncogenic activity of Bcl3 stems from its ability to indirectly down regulate tumor suppressor p53 and to directly upregulate anti-apoptotic and proliferative genes such as cyclin D1 (Cogswell et al., 2000; Maldonado and Melendez-Zajgla, 2011; Massoumi et al., 2006; Thornburg et al., 2003; Westerheide et al., 2001). Bcl3 is shown to play a decisive role in promoting cell migration by activating genes that facilitate cell migration and suppressing genes that inhibit cell death (Massoumi et al., 2006; Massoumi et al., 2009; Wakefield et al., 2013). While excessive Bcl3 is oncogenic, the lack of Bcl3 severely compromises immunity (Zhang et al., 2007).
Bcl3 is a member of the inhibitor of NF-κB (IκB) family, which forms stable complexes with NF-κB transcription factors. IκB proteins contain three structural regions. The central region contains six to seven ankyrin repeats (AR) that fold into the ankyrin repeat domain (ARD). The ARD provides the primary contacting surface for NF-κB binding. Classical IκB proteins (IκBα, −β and −ε) prefer RelA or cRel containing NF-κB dimers. In contrast, Bcl3 prefers NF-κB p52 or p50 homodimers. Apart from the nuclear localization sequence (NLS), the N-terminus of Bcl3 is structurally featureless; the serine and proline rich C-terminus is apparently flexible.
In cancer cells as well as transiently transfected cells, Bcl3 is present predominantly in the nucleus. This has led to a mistaken assumption that Bcl3 is a constitutively nuclear protein. In many non-cancer cells such as erythroblasts, hepatocytes, and keratinocytes, Bcl3 mostly resides in the cytoplasm and requires activation prior to nuclear translocation using its NLS (Brasier et al., 2001; Massoumi et al., 2006; Zhang et al., 1998; Zhang et al., 1994). Recent studies have revealed that CYLD, a deubuquitinase enzyme that removes K63-linked ubiquitin (Ub) chain, plays an critical role in balancing nucleo-cytoplasmic distribution of Bcl3. Constitutive presence of Bcl3 in the nucleus in CYLD deficiency results in hyperproliferation and invasion of cells.
Bcl3 is identified as an extensively phosphorylated protein in different cell types (Bundy and McKeithan, 1997; Caamano et al., 1996; Nishikori et al., 2005; Viatour et al., 2004c). Bcl3 shows distinct biochemical activities depending on its phosphorylation states. Recombinant Bcl3 derived from bacteria or baculovirus with no or little phosphorylation has classical IκB-like inhibitory activity that removes p52 and p50 homodimers from κB DNAs (Fujita et al., 1993; Nolan et al., 1993). Bcl3 derived from mammalian cell associates with p52(or p50):DNA binary complexes forming ternary compplexes (Wang et al., 2012; Westerheide et al., 2001). Only two possible Bcl3 phosphorylation sites, Ser394 and Ser398, are identified by consensus sequence-based prediction method and thought to be phosphorylated by GSK3β. These two sites are apparently required for Bcl3 degradation through the proteasomal pathway (Viatour et al., 2004b; Viatour et al., 2004c).
Since little is known about the regulation of Bcl3 phosphorylation at the molecular level and its integration into gene regulatory pathways, we set out to identify Bcl3 phosphorylation sites in cells and decipher the roles of these phosphorylations in Bcl3 functions. We successfully expressed and purified the full-length human Bcl3 protein from HEK 293T cells and identified 27 phosphorylation sites using mass spectrometry (Table S1). Akt, Erk2, and IKK1/2 target at least three of these sites for phosphorylation. Bcl3 undergoes continuous degradation through K48 linked ubiquitination by E3 ligase TBLR1. Akt phosphorylation of Ser33 protects Bcl3 from degradation by altering the mode of ubiquitination from K48 to K63 linkage. Erk2 and IKK1/2 phosphorylate Ser114 and Ser446 respectively. These two phosphorylation events are critical for the transcriptional activity of Bcl3 through stabilizion of the Bcl3:p52(or p50):DNA ternary complexes. In the absence of these phosphorylation modifications, Bcl3 functions as prototypical IκB protein dissociating p52 and p50 from the p52(or p50):DNA complex.
RESULTS
Bcl3 is phosphorylated at residues Ser33 and Ser446.
During our investigation of Bcl3 activity in macrophages, we found that lipopolysaccharide (LPS) stimulation elevated Bcl3 expression and it accumulated in the nucleus (Wang et al. 2012). Since nuclear localization of many proteins is regulated at the level of protein phosphorylation, we set out to identify the phosphorylation sites that might regulate both nuclear localization and transcriptional activity of Bcl3.
Flag-tagged human Bcl3 (1–446) was expressed in and affinity purified from HEK 293T cells (Figure S1A). To determine whether the affinity purified full-length Bcl3 is phosphorylated, the protein was subjected to calf intestinal phosphatase (CIP) treatment which resulted in a faster migrating band in SDS-PAGE (Figure S1B). Our results thus confirm constitutive phosphorylation of Bcl3 in HEK 293T cells as reported previously (Bundy and Mcthien, 1996). Purified Bcl3 was then digested with trypsin and subjected to LC-MS/MS analysis. Two phosphopeptides consistent with phosphorylation at Ser33 and Ser446 were identified in this initial analysis (Figure 1A). These two evolutionary conserved serines are located within the flexible N- and C-termini of Bcl3 (Figure S1C). Most of the peptides including those corresponding to the entire ARD were not observed and therefore other possible phosphorylation sites might be missing. There are several recent publications that reported the presence of Bcl3 Ser33 and Ser446 phospho-peptides identified by whole cell phospho-proteomic analysis through mass spectrometry in different cell types (Hornbeck et al., 2015). Together these results suggest that Bcl3 phosphorylation at Ser33 and Ser446 occur in vivo and perhaps these two modifications might be essential for Bcl3 functions.
Figure 1. Akt is a Bcl3 kinase.

(A) MS/MS spectrum (left) showing phosphorylation of Bcl3 Ser33 and Ser446; fragment ion mass table (right) of the phosphopeptide. (B) In vitro kinase assay showing recombinant Akt phosphorylate Bcl3 wt and S446E, but not S33E. Commassie staining showing Bcl3 protein levels used for the kinase assay. (C) WB analysis of nuclear and cytoplasmic extracts of RAW 264.7 cells after LPS stimulation for different times. Both wt and p-Ser33 Bcl3 accumulated in the nucleus in LPS treated cells. (D) Akt Inhibitor VIII treatment decreased both the total amounts of endogenous Bcl3 as well as the p-Ser33 Bcl3 in LPS treated RAW 264.7 cells. (E) Wortmannin and Akt Inhibitor VIII inhibited Bcl3 transcriptional activity with p52 on P-Selectin reporter. (F) Luciferase assay showing p52:Bcl3 transcriptional activity was significantly decreased in Akt2 KD HeLa cells. The data was analyzed from three independent experiments performed in triplicate. RLU (Relative Luciferase Unit) *p<0.05, **p<0.01. Error bars represent standard deviation (SD).
Akt phosphorylates Bcl3 at Ser33.
To identify potential kinases targeting Bcl3, group-based prediction system (GPS 2.1) (Xue et al., 2011) was used and Akt was found to be the most probable kinase targeting Ser33. This serine is located within a near perfect Akt consensus site R-K-R-x-R-x-x-S/T (Rust and Thompson, 2011). Recombinant Akt phosphorylated E. coli-derived wt Bcl3 and S446E mutant but not S33E mutant suggesting that Akt targetted Ser33 (Figure 1B). To test if endogenous Bcl3 is phosphorylated at Ser33, affinity purified phospho (Ser33)-specific antibody (Figure S1E) was used for western blot (WB) of LPS treated RAW 264.7 cell extracts. Bcl3 expressed between 1.5 and 8 hours of induction followed by gradual reduction. Phosphorylation of Bcl3 at Ser33 was visible after 4 hours of induction, peaked at 8 hours and localized in the nucleus (Figure 1C). To further confirm whether Akt mediated phosphorylation of Ser33 is essential for Bcl3’s nuclear localization, we pre-treated RAW 264.7 cells with a specific Akt inhibitor (Inhibitor VIII) (Bain et al., 2007) followed by LPS stimulation. Only nuclear Bcl3 was phosphorylated under all conditions tested. However, the amounts of both total and phosphorylated nuclear Bcl3 were reduced in inhibitor treated cells (Figure 1D).
We next tested if phosphorylation of Bcl3 by Akt can potentially play a role in transcriptional coregulation with p52 using a reporter assay in the presence and absence of Akt inhibitor VIII. In addition, wortmannin - a potent inhibitor of PI3Ks - was also used to test if the PI3K-Akt pathway was operational in Bcl3 activation. Luciferase reporter assays were carried out in HeLa cells in which the P-Selectin reporter, a known p52-Bcl3 target gene, was co-transfected with p52 and Bcl3 expression plasmids. As described in our previous study (Wang et al. 2012) and shown in Figure S1F, Bcl3 or p52 alone did not induce the luciferase activity; however, when p52 and Bcl3 were co-expressed luciferase activity was enhanced. Addition of the inhibitors decreased luciferase activity (Figure 1E) without affecting the Akt protein levels (Figure S1D). Akt inhibitor VIII showed no effect on Bcl3 S33A mutant suggesting that the decrease of p52/Bcl3-driven luciferase activity was due to the reduction of lack of Ser33 phosphorylation in the presence of Akt inhibitor (Figure S1G). We also found that the luciferase activity was severely reduced in Akt2 knocked down (KD) HeLa cells (Figure S1H and Figure 1F ). Luciferase activity in cells over-expressing wt Akt increased but decreased in cells expressing kinase dead mutant (Figure S1I). These observations clearly suggest that Akt is a Bcl3 kinase. Collectively these results also suggest that the kinase activity of Akt is essential for transcriptional coactivator function Bcl3 through regulating the nuclear translocation of Bcl3.
Ser33 phosphorylation protects Bcl3 from degradation.
To investigate if Ser33 phosphorylation provides a signal for Bcl3 nuclear localization, phospho-mimetic (Glu) and phospho-inactive (Ala) mutants at Ser33 were generated and transiently expressed in HEK 293T cells. The S33A mutant showed very low total protein levels (Figure 2A) and was mostly localized in the cytoplasm (Figure 2B). In contrast, the phospho-mimetic S33E mutant showed enhanced protein levels, which localized both in cytoplasm and nucleus. The S446A and S446E mutants bahaved as wt in terms of protein levels and subcellular distributions (Figure S2A-B), whereas the S33A/S446A (AA) double mutant behaved similarly to the S33A mutant (Figure S2C-D) suggesting phosphorylation of Ser446 had no impact on Bcl3 sub-cellular localization. Next, we studied cellular localization of Bcl3 by immunofluorescence by using overexpressed wt, S33E, and S33A as yellow fluorescent protein (YFP) fusion proteins. Fractionation experiments revealed that these proteins expressed in HEK 293T cells behaved similarly to the corresponding Flag-tagged proteins (Figure S2E). In agreement with fractionation experiments, immunofluorescence experiments showed that wt Bcl3 and S33E mutant translocated into the nucleus whereas the S33A mutant was detected predominantly in cytoplasm (Figure 2C).
Figure 2. Phosphorylation of Bcl3 Ser33 Regulates its Protein Expression, Cellular Localization and Protein Degradation.

(A) Total protein levels of Flag-Bcl3 wt, S33E and S33A mutants. (B) fractionation of cell extract follwed by WB showing subcellular localization of various ectopically expressed Bcl3 mutants. Bcl3 S33A mutant localized in the cytoplasm, but S33E mutant localized in both cytoplasm and nucleus. (C) Immunoflorescence assay showing subcellular localization of YFP-Bcl3 wt, S33E and S33A mutants localized in cytoplasm predominantly. (D) Flag-Bcl3 wt, S33A, and S33A/S446A (AA) mutants were transiently transfected into HEK 293T cells and followed by CHX treatment for different time as indicated. The wt Bcl3 levels remained similar over the course of CHX treatment. Both S33A and AA mutants showed short half-life compare to wt Bcl3. (E) Flag-IP showing S33A mutant was covalently conjugated to higher levels of poly-Ub chains than wt Bcl3. Bcl3 wt and S33A mutant were transiently transfected into HEK 293T cells and subsequently treated with MG132. (F) Myc-IP showing S33A mutant interacts with E3 ligase TBLR1. Myc-TBLR1 was co-expressed with YFP-Bcl3 wt, S33E, and S33A mutants in HEK 293T cells.
Low protein level of S33A mutant prompted us to investigate whether the half-life of Bcl3 is modulated through phosphorylation of Ser33. Cells expressing wt Bcl3 and S33A mutant were treated with cycloheximide (CHX), an inhibitor of protein synthesis, followed by WB. Whereas the levels of wt Bcl3 remained stable over 24 hours of CHX treatment, S33A was barely detectable after 2 hours (Figure 2D). We further found that the proteasome pathway was partially responsible for the shorter half-life of S33A (Figure S2F). Lack of complete stabilization of the S33A mutant in the presence of MG132 suggested other pathways could also target the S33A mutant for degradation. Surprisingly, mutation of Ser394 and Ser398, presumed to be essential for basal degradation through phopshoryaltion by GSK3β (Viatour et al., 2004a) to Ala did not alter the stability of Bcl3. However, S394A/S398A mutant in the context of either S33A or S33E mutations showed reduced Bcl3 stability (Figure S2G) suggesting a highly complex interplay between Akt and GSK3β that control Bcl3’s stability.
Since proteasome-mediated protein degradation follows protein ubiquitination of lysine residues, we investigated the extent of poly-Ub chain conjugation on Bcl3 mutants. Flag-Bcl3 immunoprecipitation (IP) showed that the S33A mutant was significantly more poly-Ub conjugated than wt Bcl3 (Figure 2E). Since S33A protein level was extremely low, in order to achieve similar levels of Bcl3 wt and S33A mutant for IP assays we had to use 20~30 fold more cell extract expressing the S33A mutant. An earlier report demonstrated that ubiquitination of Bcl3 and subsequent degradation required TBLR1, an E3 ligase, which bound the N-terminal region of Bcl3 (Keutgens et al., 2010), leading to poly-Ub conjugation at residues Lys13 and Lys26. To test if Akt phosphorylation of Ser33 prevents TBLR1 binding, Myc-tagged TBLR1 was co-expressed with YFP-tagged Bcl3 wt, S33E, and S33A in HEK 293T cells followed by Myc-IP and immuno blot using GFP antibody to probe for YFP-tagged Bcl3. Only S33A, but not S33E or wt Bcl3, interacted with TBLR1 (Figure 2F). These results suggest that the lack of phosphorylation at Ser33 allows Bcl3 to bind TBLR1 that leads to poly ubiquitination of Bcl3 and subsequent degradation. Phosphorylation of Ser33 by Akt protects Bcl3 from TBLR1 binding, Ub-conjugation, and degradation.
Ser33 phosphorylation switches Bcl3 ubiquitination from a degradation to stabilization signal.
We examined the levels of poly-Ub conjugation of endogenous Bcl3 with or without stimulation. Akt inhibitor VIII treatment of RAW 264.7 cells, which partially blocked Ser33 phosphorylation, increased the levels of poly-Ub conjugation to Bcl3 (compare lanes 3 and 4; Figure 3A). The difference was clear only when cells were treated with MG132 along with LPS and Akt inhibitor. These results suggest that increased level of Ub-conjugation was mostly K48-linked Ub-chain. It was previously reported that cells deficient in K63-linked deubiquition enzyme, CYLD, underwent excessive nuclear accumulation of Bcl3, cyclin D1 expression, and uncontrolled proliferation (Massoumi et al., 2006). To investigate if Ser33 phosphorylation by Akt acts as a switch from a degradation to a stabilization signal leading to nuclear accumulation, wt Bcl3, S33E, and S33A mutants were expressed in HEK 293T cells and the extracts were probed with both K48- and K63- Ub linkage specific antibodies. K48-Ub antibody specifically recognized the S33A mutant but not the S33E or wt Bcl3 (Figure 3B). Conversely, K63-Ub-specific antibody recognized wt and S33E mutant more efficiently than the S33A mutant (Figure 3C). To further test the specificity of K63-ubiquitination of Bcl3, we cotransfected plasmids expressing CYLD together with wt Bcl3 in HEK 293T cells. IP experiments showed lesser amount of K63-Ub chain was conjugated to Bcl3 in the presence of CYLD (Figure 3D). These results indicate that (i) phosphorylation at Ser33 not only prevents the K48-linked ubiquitination but also facilitates K63-linked ubiquitination, and (ii) the K63-linked ubiquitination serves as a stabilization signal for Bcl3.
Figure 3. Ser33 phosphorylation switches Bcl3 Ub linkage.

(A) Bcl3-IP showing Poly-Ub conjugation of endogenous Bcl3. RAW 264.7 cells were pretreated with Akt Inhibitor VIII (5μM) for 24 hours followed by LPS stimulation for another 24 hours. MG132 (10 μM) was added for the last two hours of LPS treatment. (B) WB using K48-specific antibody showing Bcl3 S33A mutant, but not wt or S33E mutant, was linked to K48-Ub chains; (C) WB using K63-specific antibody showing that Bcl3 wt and S33E were linked with K63-Ub more efficiently than S33A. Bcl3 wt and all mutants were transiently transfected into HEK 293T cells and subsequently treated with MG132. (D) Co-expression of CYLD together with Bcl3 removes K63-Ub from Bcl3. wt Bcl3 was transfected into HEK 293T cells either alone or together with CYLD for different time as indicated. And then Bcl3 was IP out using Flag antibody followed by anti-K63-linkage specific Ub immunoblotting.
Our results in combination with prior observations provide a clear model of Bcl3 regulation. In resting cells, phosphorylation of Bcl3 by GSK3β leads to K48 ubiquitnation at N-terminal lysine residues (Lys13 and Lys26) by E3 ligase TBLR1. Akt activation stabilized Bcl3 in two ways: first, by a well established mechanism where Akt inactivated GSK3β by phosphorylation; second, by directly phosphorylating Ser33 of Bcl3 leading to non-degradative K63-ubiquitination of Bcl3 which stabilized in the nucleus (Figure S3C). We made an effort to identify the E3 ubiquitin ligase responsible for K63 ubiquitination of Bcl3. Since TRAF6 is involved in several NF-κB activation pathways, we speculated that TRAF6 might be the E3 ligase of Bcl3. TRAF6 KD HEK 293T cells with over-expressed Flag-Bcl3 did not reveal significant changes in Bcl3 protein levels suggesting that TRAF6 has no role in K63 ubiquitination of Bcl3 (Figure S3A and B).
IKK phosphorylates Bcl3 Ser446.
We turned our focus in identifing the kinase that phosphorylates Ser446. Since Bcl3 is known to function in association with NF-κB signaling, we hypothesized that pathways responsible for activating NF-κB might also be responsible for Bcl3 activation. Therefore, we tested three kinases involved in NF-κB activation pathway; NIK, IKK1, and IKK2. Both IKK1 and IKK2, but not NIK phosphorylated Bcl3 in vitro (Figure 4A). IκBδ (the C-terminal domain of NF-κB2/p100) was used as a control. To examine whether IKK1/2 are essential for Bcl3 activity in cells, we used IKK inhibitor XII, which showed equal specificity towards both IKK1 and 2 (Christopher et al., 2007). Bcl3-driven luciferase activity was reduced in the inhibitor treated HeLa cells (Figure 4B). IP using IKK1-specific antibody followed by WB with phospho-specific IKK antibody showed decreased amount of phosphorylated IKK in the inhibitor treated cells compared to the control suggesting IKK might be the Bcl3 kinase (Figure 4C). Since IKK1 and 2 remain as a complex in the cell, and the phospho-antibody recognizes both IKK1 and 2, these experiments did not clarify whether IKK1 or 2 or both target Bcl3. We generated IKK1 and IKK2 KD HeLa cells (Figure S4C) and tested the transcriptional activity of Bcl3 in these two KD cell lines by luciferase assay as described above. Luciferase activity was significantly reduced in both cell types suggesting both IKK1 and 2 were responsible for transcriptional coactivator function of Bcl3 (Figure 4D).
Figure 4. IKK is a Bcl3 kinase.

(A) In vitro kinase assay showing recombinant IKK1 and IKK2, but not NIK, phosphorylated bacterially expressed Bcl3 protein. Recombinant IκBδ was used as a control that could be phosphorylated by all three kinases. Coomassie stained SDS-PAGE showing protein amounts used for the kinase assay. (B) IKK Inhibitor XII inhibited Bcl3 transcriptional activity as indicated by luciferase reporter activity. The data was analyzed from three independent experiments performed in triplicate. *p<0.05, **p<0.01. Error bars represent SD. (C) IKK Inhibitor XII inhibited IKK1/2 kinase activity in HeLa cells. HeLa cells were left untreated (lane 1) or treated with 5–20 μM IKK Inhibitor XII (lane 2–4) for 48 hours. Cell extracts were subjected to anti-p-IKK(Ser176/180) and anti-IKK1 WB analysis. (D) Decreased levels of p52:Bcl3 transcriptional activity in both IKK1 and 2 KD HeLa cells as indicated by luciferase assay. The data was analyzed from three independent experiments performed in triplicate. *p<0.05. Error bars represent SD. (E) EMSA using Flag-Bcl3 isolated from DMSO or IKK inhibitor treated cells. Bcl3 expressed and purified from IKK inhibitor treated HEK 293T cells lost the potential to form the Bcl3:p52:DNA ternary complex (Lanes 8–11); Bcl3 wt formed the ternary complex (lanes 4–7). p52 purified from in E. coli formed the p52:DNA binary complex (lanes 2–3). Bcl3 wt purified from E. coli removed p52 from the p52:DNA binary complex (Lanes 12–13). (F) Flag-IP demonstrated the binding of recombinant p52(1–408) to different Flag-Bcl3 nuclear extracts. WB analysis showed constant amounts of Flag-Bcl3 wt, S446E, and S446A binding to increasing concentrations of p52 at various concentrations. Bound p52 was plotted against the total p52 input at various concentrations, demonstrating that S446E had greater binding affinity towards p52 homodimer than wt Bcl3 or and S446A mutant. (G) Transcriptional activity of wt and different mutants of Bcl3 monitored by luciferase assay. The data was analyzed from three independent experiments performed in triplicate. *p<0.05, **p<0.01, ***p<0.005. Error bars represent SD.
Bcl3 derived from mammalian cells is known to interact with p52 and κB DNA forming the Bcl3:p52:DNA ternary complex in Electrophoretic Mobility Shift Assay (EMSA). However, Bcl3 derived from IKK inhibitor treated cells failed to form the ternary complex further confirming IKK as an important regulator of Bcl3 (Figure 4E). To identify Bcl3 phosphorylation site(s) modified by IKK1, we carried out in vitro phosphorylation reaction using recombinant Bcl3 and IKK1 followed by phospho-peptide analysis by mass spectrometry. We found ten residues phosphorylated by IKK1 including both Ser33 and Ser446 (Table S1). While IKK1 might be a potential kinase of Bcl3, not all sites phosphorylated in vitro could be real IKK1 targets in cells. To identify sites that are phosphorylated by IKK in cells, Flag-Bcl3 was purified from transfected HEK 293T cells both in the presence or absence of IKK inhibitor XII and subjected to mass spectrometric analysis. Since these analyses were carried out in a more sensitive instrument compared to the one used for the earlier mass spectrometric analysis described above, most of the phospho Bcl3 peptides were detected. We identified as many as 27 phosphorylation sites compared to only 2 sites (Ser33 and Ser446) identified in the previous analysis (Table S1). We found that phosphorylation of only 4 high confidence sites were differentially regulated in the presence of IKK Inhibitor XII. These are, Ser91, Thr166, Ser417, and Ser446. Since Ser91 and Ser417 were not phosphorylated by IKK1 in the in vitro kinase assay, we turned our focus to Thr166 and Ser446. We mutated Thr166 to Ala or Glu either singly or in the S33E/S446E double mutant background. Surprisingly, all Thr166 mutants showed drastically reduced protein levels as well as low luciferase activity. These observations suggest that the unphosphorylated Thr166 might be important for maintaining structural integrity of Bcl3 (Figure S4A-B). In addition, IKK inhibitor showed no effect on Ser33 phosphorylation suggesting that IKK is not the cellular kinase of Ser33. Similar reporter activity between wt Bcl3 and S446A in the presence and absence of IKK inhibitor further suggests Ser446 is targeted by IKK (Figure S4D). It is unclear however if IKK1 or 2 or both target Ser446, since IKK1 and IKK2 KD independently reduced the transcriptional activity of Bcl3, they might each phosphorylate distinct residue(s).
Bcl3 phosphorylation at Ser446 activates transcription by augmenting interaction with the p52 homodimer.
Since Bcl3 regulates transcription through the formation of complexes with NF-κB p52 and p50 homodimers (Wang et al., 2012; Westerheide et al., 2001), we investigated whether Bcl3 phosphorylation affected its ability to interact with p52. Constant amounts of Flag-Bcl3 from nuclear extracts of HEK 293T cells transiently transfected with expression plasmids encoding wt and S33E, S446E, S446A and S33E/S446E (EE) mutants were mixed with increasing amounts of recombinant p52 (1–408) followed by WB using anti-p52 and anti-Bcl3 to monitor binding. Under these conditions, recombinant p52 homodimer interacted with Bcl3 wt, or S446E single or EE double mutants with higher affinity than S446A mutant (Figure 4F and S4E). Collectively, these observations indicate that phosphorylation at Ser446 enhanced protein-protein interaction between Bcl3 and p52.
Next, we examined the effect of Ser446 phosphorylation in transcription by luciferase assays using the P-Seletin reporter described above. All Bcl3 mutants - S33E, S446E, S446A, EE, and S33E/S446A (EA) - were present to a similar level in the nucleus (Figure S4H). Compared to wt Bcl3, S446E showed higher luciferase activity and S446A showed significantly reduced activity. The EE mutant showed slightly higher activity compared to that of the S446E single mutant (Figure 4G). These results suggest a Ser446 phosphorylation-mediated stabilization of the Bcl3:p52:DNA ternary complex. If Ser446 phosphorylation can stabilize the ternary complex then E.coli expressed Bcl3 EE mutant should facilitate the Bcl3:p52:DNA ternary complex formation. Conversely, phosphorylation-defective S446A mutant derived from HEK 293T cells should show deficiency in forming the ternary complex. We carried out EMSA to test the interaction between wt and mutant Bcl3 derived from both E. coli and HEK 293T cells (Figure S4F-G). Consistent with previous reports, wt Bcl3 derived from HEK 293T cells (phosphorylated form) formed the Bcl3:p52:DNA ternary complex (Figure S4F, lane 9–12) and wt Bcl3 derived from E. coli (unphosphorylated form) dissociated the p52:DNA binary complex (Figure S4F, lane 4–5). E. coli expressed EE mutant also failed to form the ternary complex (Figure S4F, lane 6–8) suggesting phosphorylation of these two residues is not sufficient to convert Bcl3 from an inhibitor into a transcriptional coregulator. In addition, Bcl3 S446A mutant derived from HEK 293T cells was still able to form the ternary complex with p52 and DNA (Figure S4G, lane 14–19). These results suggest that phosphorylation of Ser446 is necessary but not sufficient to confer complete transcriptional potential of Bcl3.
Erk2 phosphorylation at Ser114 induces transcriptional activity of Bcl3.
Several high confidence phosphorylation sites were identified by mass spectrometric analyses and phosphorylation of one or multiple of these sites could be required for the transcriptional function of Bcl3 in addition to Ser33 and Ser446 (Table S1). Our analysis of putative Bcl3 kinases also identified Erk2 as one of the possible kinases targeting some of the high confidence sites. p52/Bcl3-driven luciferase activity was reduced in the presence of Erk inhibitor II (Figure 5A left panel) as well as in Erk2 KD HeLa cells (Figure 5A right panel and Figure S5C). In vitro Erk2 kinase assay showed that recombinant Erk2 phosphorylated E. coli expressed Bcl3 but not IκBα suggesting phosphorylation specificity (Figure 5B). Mass spectrometric analysis of in vitro phosphorylated Bcl3 by Erk2 identified a few phopsho-peptides, one of which was a 35 residues long tryptic peptide spanning residues 88 to 122 that appeared several times. Although three of the four potential Erk2 sites in Bcl3 (Thr100, Ser114, and Thr129) are located within the region covered by this peptide, mass spectrometric analysis was not highly confident to determine the precise location of the phosphorylation site(s). This 35-mer peptide also contained an Erk2-specific motif (D motif) consensus in its N-terminus that was expected to stably interact with the kinase (Figure 5D) (Roskoski, 2012). Although both Thr100 and Ser114 are within the Erk2 consensus phosphorylation site, position of Thr100 is too close to the D motif to be ideally suited for phosphorylation. Moreover, a hydrophobic patch is located C-terminus to the Ser114 which might serve as the DEF site (docking site for ERK FXF, also called F motif) to bind Erk2 (Roskoski, 2012). Thus, in spite of the absence of a proline at +1 position often found in MAP kinase substrates, Ser114 appeared to be the most probable Erk2 site. Both the HEK 293T cell-derived and in vitro Erk2-derived mass spectrometric analyses of Bcl3 identified Ser282 as another potential Erk2 site for phosphorylation. We mutated each of these sites to single phospho-mimetic (Glu) mutant or as double mutants in association with S33E. Luciferase assay showed that only the S114E single and S33E/S114E double mutant were active in transcription suggesting that phosphorylation at Ser114 by Erk2 might be critical for transcriptional activity of Bcl3 (Figure S5A). These results suggest that phosphorylation of both Ser114 and Ser446 might be required for higher transcriptional activity.
Figure 5. Erk2 is a Bcl3 kinase.

(A) Erk Inhibitor II (left panel) and Erk2 KD (right panel) inhibited luciferase reporter activity as compared to the untreated or wt cells. The data was analyzed from three independent experiments performed in triplicate. *p<0.05, **p<0.01. Error bars represent SD. (B) In vitro kinase assay showing recombinant Erk2 phosphorylated bacteria expressed Bcl3 protein but not IκBα. (C) Flag-IP showing Bcl3 interacted with both wt and kinase dead Erk2 kinase; the interaction is more prominent with the kinase dead Erk2. Flag-Bcl3 co-expressed with HA-Erk2 in HEK 293T cells. (D) A 35-mer phospho-peptide identified in the mass spectrometric analysis ranging from human Bcl3(88–122) nearly matches the Erk2 consensus phosphorylation site. (E) Bacterially expressed Bcl3 protein with S33E/S114E/S446E (EEE) triple mutations formed Bcl3:p52:DNA ternary complex in EMSA (lane 8–13). The ternary complex could be further shifted up by either p52 or Bcl3 antibody (lane 15–18). Bacteria expressed Bcl3 wt protein inhibited p52 from binding to κB DNA (lane 4–7).
We prepared the recombinant phospho-mimetic S33E/S114E/S446E (EEE) triple mutant in E. coli and tested its ability to form ternary complex with p52 and κB DNA by EMSA. This triple mutant, but not the S33E/S114E double mutant, was able to form the ternary complex which could be further super-shifted by both p52 and Bcl3 antibodies (Figure 5E and 6F). Our results thus confirmed that phosphorylation by different kinases, Akt, Erk2, and IKK1/2, of at least three distinct sites, Ser33, Ser114, and Ser446, are essential to convert Bcl3 from an inhibitor to a transcriptional coregulator of p52 homodimer. Since Bcl3 could also interact with p50 homodimer, we further tested its ability to form the ternary complex with p50 and DNA. As expected, E. coli-derived wt Bcl3 functioned as an inhibitor of the p50:DNA complex (Figure S5B, lane 4–7) and the EEE triple mutant associated with the p50:DNA binary complex. Interestingly, the triple mutant was unable to fully shift the binary complex into ternary complex (Figure S5B, lane 8–13) suggesting that additional or differential modification(s) might be necessary for Bcl3 to efficiently function as a coactivator for p50 (Figure S5B).
Figure 6: Bcl3 phosphorylation is required for its cellular functions.

(A) Wound-healing scratch assays showing migration of U2OS cells. Migration was defective in CRISPR-Cas9-targeted Bcl3 KD U2OS cells. The yellow dotted line indicates the edge of the scratch showing the width of scratches. The S33E/S446E (EE) mutant rescued the migration defect, whereas the S33E/S446A (EA) and S33E/S114A/S446A (EAA) mutants only partially rescued (EA) or mostly failed to rescue (EAA). Lentivirus expressing GFP was used as a control. (B) Plots showing the average width of scratch at 24 and 48 hours after the scratch shown in (A). At the time of scratch (0 hour), the width of scratch was set to be 100%; at 24 and 48 hours, the width of scratches was measured again and divided by the initial width at 0 hour to find out the percentage. When the scratch is completely closed up, it becomes 0% that means no gap. Measurements were taken using cells growing in three individual wells at the same condition. (C) MTT assays showing U2OS cell proliferation. Proliferation was also significantly defective in cells expressing EA and EAA mutants in Bcl3 KD cells with or without TPA treatment. (D) mRNA levels of the Cyclin D1 gene in different mutant expressing U2OS Bcl3 KD cells. (E) Luciferase activity driven by Cyclin D1-κB (−39) site was impaired in Bcl3 KD U2OS cells; and only the EE mutant could rescue the activity to a level higher than the wt U2OS control cells. (F) EMSA showing the HEK 293T-derived wt and S33E/S114A/S446A (EAA) mutant could form ternary complex with p52 and κB DNA; however, for bacteria expressed Bcl3 protein, only the S33E/S114E/S446E (EEE) triple mutant but not the wt nor the S33E/S446E (EE) double mutant could form the ternary complex. Bacteria expressed wt Bcl3 inhibited p52 from binding to κB DNA (no band showed in lane 4 and 5); EE mutant failed to inhibit the p52:DNA binary complex formation, but it also failed to form stable ternary complex (lane 8–10). (G) EMSA showing only the HEK 293T-derived wt Bcl3 could form the ternary complex with p50:DNA (lane 4–5); both the HEK 293T-derived EEA and the bacteria-derived wt protein failed to form this ternary complex.
Cell proliferation and migration activities of Bcl3 require specific phosphorylation:
Two documented biological functions of Bcl3 are cell migration and cell proliferation. We monitored these functions to examine the effect of Bcl3 phosphorylation. Using CRISPR-Cas9, the bcl3 gene was KD in U2OS cells, a human bone osteosarcoma cell line (Figure S6B). Bcl3 wt and mutants (S33E/S446E, S33E/S446A, and S33E/S114A/S446A) and the GFP only control were introduced back into the KD cells by lentivirus infection. Since the guide RNA (gRNA) was generated to target Ser33, all lentivirus derived Bcl3s were generated by introducing a silent mutation at Ser33 such that exogenous Bcl3 could escape the gRNA-mediated cleavage. Our goal was to test how transcriptional regulatory activity of Bcl3 was impacted by phosphorylation. Since phosphorylation of Ser33 is required for nuclear translocation, all mutants tested contained phosphomimetic S33E mutation. Cell migration was monitored by wound-healing scratch assays. Consistent with previous reports we found that Bcl3 KD severely affected cell migration. Introduction of either wt Bcl3 or the S33E/S446E (EE) double mutant rescued the migration defect within two days after the scratch. In contrast, cells expressing the S33E/S446A (EA) and S33E/S114A/S446A (EAA) mutants showed the migration defect and the defect was greatest for EAA (Figure 6A and 6B) confirming the importance of Ser114 and Ser446 phosphorylation. The U2OS wt cells also showed migration defects upon treatment with Akt, IKK, and Erk inhibitors (Figure S6A).
We also noted that compared to other mutants, cells expressing the Bcl3 EAA mutant grew slower. To test the effect of these mutants in cell proliferation, we carried out MTT assays. U2OS cells deficient in Bcl3 showed defect in proliferation with or without TPA (12-O-Tetradecanoylphorbol-13-Acetate) treatment. More importantly, only the EE mutant, but neither the EA nor the EAA mutant, rescued this defect (Figure 6C). Since cyclin D1 is known to be essential for cell proliferation and its expression is known to be defective in Bcl3 deficient cells, we investigated whether basal expression of cyclin D1 was reduced in Bcl3 deficient U2OS cells expressing the EA or EAA mutants. Indeed, cyclin D1 expression was significantly lower in the cells expressing EA or EAA mutant compared to those expressing the EE mutant (Figure 6D). To further validate this observation we examined the reporter expression regulated by the cyclin D1-κB(−39) site, the known target of p52 and Bcl3, in U2OS cells. As shown in figure 6E, luciferase activity was about 10-fold higher in wt U2OS cells than Bcl3 KD cells. In addition, cells expressing Bcl3 EE mutant showed 16-fold higher luciferase activity than Bcl3 KD cells which confirmed the essential role of Bcl3 in cyclin D1 expression. The luciferase activity was nearly abolished in cells expressing the EAA mutant. These results further confirmed that phosphorylation at all three sites was essential for transcriptional activity of Bcl3 on cyclin D1 promoter.
Results described above suggest that the EAA mutant might be defective in ternary complex formation with p52:DNA complex. To test this, we prepared this mutant protein from HEK 293T cells and analyzed its activity by EMSA. To our surprise, we found that like the E. coli expressed EEE mutant and the HEK 293T-derived wt Bcl3, the HEK 293T-derived EAA mutant was also able to form ternary complex with p52 and DNA (Figure 6F). It appeared that the lack of phosphorylation both at Ser114 and Ser446 did not abolish Bcl3’s ability to interact with the p52:DNA complex. Bcl3 might have multiple phosphorylation states that all were able to interact with the p52:DNA binary complex and Ser114/Ser446 was one of such possible combinations. Accordingly, the EAA mutant was unable to target only selective promoters such as cyclin D1. We further tested how the EAA mutant interact with the p50:DNA complex by EMSA. We found that unlike its effect on the p52:DNA complex, the EAA mutant failed to interact with the p50:DNA binary complex (Figure 6G). It is likely that most promoters targeted by the p50:Bcl3 complex might be unable to recruit the EAA mutant. More severe effect of the EAA mutant failed to associate with the p50:DNA complex might explain its dramatic defects on cell proliferation and migration in spite of its ability to form a ternary complex with the p52:DNA complex. Overall, these results suggest that though phosphorylation at Ser114 and Ser446 is essential for transcriptional regulation of Bcl3, phosphorylation at other unknown or uncharacterized sites observed here also can play additional regulatory roles.
DISCUSSION
The present study set out to resolve a long-waited question, which is how Bcl3 functions as a transcriptional regulator through phosphorylation. We show that at least three residues of Bcl3 must be phosphorylated for its transcriptional coregulatory function with NF-κB p52 and p50 homodimers. Akt, Erk2, and IKK1/2 are the kinases catalyzing these phospho-modifications. Since both IKK1 and 2 are required and Ser446 is the only site targeted by IKK1 or 2 identified so far, there must be at least one more critical phosphorylation site on Bcl3 unless however these two kinases regulate each other with one of them targeting Ser446. Multiple mass spectrometric analyses identified as many as 27 phosphorylation sites on Bcl3 isolated from HEK 293T cells of which 17 sites are high confidence sites. 4 of these are tyrosines and 5 of them are not fully conserved between species (Table S1). We studied a total of 9 of these sites (Ser33, Thr100, Ser114, Thr129, Thr166, Ser282, Ser394, Ser398, and Ser446) which includes one low confidence site (Ser398) that was reported previously as a GSK3β site. Barring residues Ser33, Ser114, and Ser446, mutations at other sites did not reveal any clear functions on nuclear localization and/or transcription activity. Therefore, one or more of the remaining uncharacterized sites could be targeted by IKK1/2. Future experiments are required to specify the kinase and the target sites.
Our discovery adds a new axis to NF-κB activation signaling. Canonical signaling that activates RelA and cRel dimers and non-canonical signaling that activates p52 dimers have received prime attention as key NF-κB activating pathways. Numerous reports showed that Akt and MAPK signaling cascades are associated with NF-κB activation. For instance, growth factor PDGF or TNFα can also activate NF-κB through an Akt-IKK1 pathway where IKK1 is activated by Thr23 phosphorylation by Akt (Ozes et al., 1999; Romashkova and Makarov, 1999). However, how Akt-IKK1 axis activates NF-κB is not known. That is, the downstream target of IKK1 leading to NF-κB activation in this pathway remained obscure. Erk2, a MAPK, supports both cell proliferation and survival through growth factor-Ras pathway. Erk2 is also activated by LPS, through an alternative p105-Tpl2-Erk2 signaling pathway (Waterfield et al., 2003). Although many downstream targets of Erk2 are known, none of them belongs to the NF-κB-IκB family. Thus, cross talk between these pathways was often viewed as indirect, post-transcriptionally. Our work reveals a direct link between PI3K-Akt and MAPK signaling to NF-κB activation by targeting a single factor, Bcl3 (Figure 7).
Figure 7. The model of Bcl3 activation.

In resting cells, Bcl3 undergoes continuous degradation mediated by the GSK3β phosphorylation possibly at Ser394 and Ser398. Upon LPS stimulation, Akt undergoes activation through in the PI3K signaling pathway and stabilizes Bcl3 through two mechanisms; inactivating GSK3β through direct phosphorylation and inducing nuclear translocation by phosphorylation of Bcl3 at Ser33. Akt might be directly involved in IKK1 phosphorylation. IKK2 is phosphorylated by canonical NF-kB activation pathway. IKK in turn leads to Bcl3 phosphorylation at Ser446 and other residues. In addition, Erk2 also gets activated in the MAPK/ERK pathways and phosphorylates Bcl3 at Ser114. All three phosphorylation events increase Bcl3’s transcriptional potential with NF-κB p52.
Phosphorylation at three Bcl3 residues alters three of its biochemical properties: metabolic stability, nuclear localization, and transcriptional potential. Phosphorylation at Ser33 is required for both metabolic stability and nuclear localization of Bcl3. This phosphorylation switches the ubiquitination program of Bcl3 from K48 to K63 conjugation. K48 ubiquitination is required for constitutive degradation of Bcl3 similar to many other proteins that are constitutively degraded such as β-catenin. Like Bcl3, β-catenin is a transcriptional coregulator of Lef/TCF transcription factor (Cadigan and Waterman, 2012).
The third altered biochemical property of Bcl3 that is mediated through phosphorylation is directly linked to the transcriptional function where Bcl3 is converted from an NF-κB inhibitor into a transcription coactivator. Erk2 and IKK1/2 mediate the phosphorylation at Ser114 and Ser446, respectively, and these two phosphorylation are required for Bcl3:p52(or p50):DNA ternary complex formation. Both serine residues are present within the flexible segments of Bcl3. Ser114 located within the N-terminal segment and Ser446 is the last residue in the C-terminus. Bcl3 exhibits two alternative binding modes to p52 and p50 homodimers: in one mode only Bcl3:p52(or p50) binary complex is allowed and in the other mode the binary complex can accommodate specific κB DNA forming ternary complexes. How the negatively charged phosphate groups on the flexible regions of Bcl3 accommodate the DNA into the ternary complex cannot be predicted. One of the intriguing observation is the altered transcriptional regulation by differentially phosphorylated Bcl3. Identification of other phosphoryaltion sites affecting transcription is required for deeper understanding of how this is accomplished by Bcl3. While unphosphorylated Bcl3 is able to dissociate p52(or p50):DNA complexes in vitro, it is not known if Bcl3 ever exists in an unphosphorylated form in cells. However, the existence of such unphosphorylated Bcl3 cannot be ruled out perhaps by the action of signal-specific upregulation of specific phosphatases. The dephosphorylated Bcl3 would be able to down regulate transcription by removing p50 and p52 homodimers from active promoters just like the removal of p50:RelA heterodimer by IκBα. Bcl3 also associates with several other proteins such as Jab, Tip60, HDAC, and Pirin (Dechend et al., 1999). Interactions between Bcl3 and its partners might be regulated, at least in part, by phosphorylation at different sites. Future experiments are required to show how many of the phosphorylation sites identified in this study or unknown sites are functionally relevant including transcriptional regulation.
In summary, work presented here explained site-specific phosphorylation-induced metabolic stabilization of Bcl3 through switch of ubiquitin linkage, and transcriptional coregulatory activity of Bcl3 in association with NF-κB p50 and p52 homodimers. Through identification of several Bcl3 kinases, this work also established convergence of three intertwined signaling pathways onto NF-κB activation.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Gourisankar Ghosh (gghosh@ucsd.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
RAW 264.7, HEK 293T, HeLa and U2OS wt and specific gene knock down cells as indicated in the As Key Resources Table were grown in Dulbecco’s modified Eagle’s medium (CellGro) supplemented with 10% fetal bovine serum, 2 mM glutamine and antibiotics.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Flag (M2) | Sigma | Cat#F3165 |
| Rabbit polyclonal anti-Akt1/2/3 (H-136) | Santa Cruz Biotechnology | Cat#sc-8312 |
| Rabbit polyclonal anti-IKK1 (M-204) | Santa Cruz Biotechnology | Cat#sc-7184 |
| Rabbit polyclonal anti-Phospho-Akt (Ser473) | Cell Signaling Technology | Cat#9271 |
| Rabbit monoclonal anti-Phospho-IKK1/2 (Ser176/180) (16A6) | Cell Signaling Technology | Cat#2697 |
| Rabbit monoclonal anti-K63-linkage Specific Polyubiquitin (D7A11) | Cell Signaling Technology | Cat#5621 |
| Rabbit monoclonal anti-Ubiquitin, Lys48-Specific, clone Apu2 | EMD Millipore | Cat#05–1307 |
| Mouse monoclonal anti-IKK2 | IMGNEX | Cat#IMG-159 |
| Mouse monoclonal anti-Nuclear Matrix Protein p84 (5E10) | GeneTex | Cat#GTX70220 |
| Mouse monoclonal anti-GAPDH (GT239) | GeneTex | Cat#627408 |
| Rabbit anti-p100/p52 serum 1495 | Dr, Nancy Rice, NCI-Frederick Cancer Research and Development | NCI 1495 |
| Rabbit anti-Bcl3 serum 1348 | Dr, Nancy Rice, NCI-Frederick Cancer Research and Development | NCI 1348 |
| Rabbit anti-phospho-Ser33 Bcl3 serum | BioBharati LifeScience (BBL), Kolkata, India | Cat#BB-AB0090 |
| Rabbit anti-Erk2 serum | BioBharati LifeScience (BBL), Kolkata, India | Cat#BB-AB0135 |
| Rabbit anti-Ub serum | BioBharati LifeScience (BBL), Kolkata, India | Cat#BB-AB0035 |
| Rabbit anti-p50 serum | BioBharati LifeScience (BBL), Kolkata, India | Cat#BB-AB0080 |
| Ni-affinity resin | BioBharati LifeScience (BBL), Kolkata, India | Cat#BB-NA002E |
| Protein A/G plus beads | BioBharati LifeScience (BBL), Kolkata, India | Cat#BB-PAG001PD |
| Bacterial and Virus Strains | ||
| DH5a competent cells | Invitrogen | Cat#18265017 |
| BL21(DE3) competent cells - Novagen | EMD Millipore | Cat#69450 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lipopolysaccharides from Escherichia coli O55:B5 | Sigma | Cat#L6529 |
| Cycloheximide | Sigma | Cat#C1988 |
| Wortmannin | Sigma | Cat#W1628 |
| InSolution™ MG-132 | Calbiochem | Cat#474791 |
| Akt Inhibitor VIII | Calbiochem | Cat#124018 |
| IKK Inhibitor XII | Calbiochem | Cat#401491 |
| Erk Inhibitor II | Calbiochem | Cat# FR180204 |
| TPA (12-O-Tetradecanoylphorbol-13-Acetate) | Cell Signaling Technology | Cat#4174 |
| Puromycin, Dihydrochloride | EMD Millipore | Cat#540222 |
| Polybrene Infection / Transfection Reagent | EMD Millipore | Cat# TR-1003-G |
| Lipofectamine 2000 Transfection Reagent | Invitrogen | Cat#11668–019 |
| Cell Proliferation Reagent WST-1 | Roche | Cat#05015944001 |
| Alkaline Phosphatase, Calf Intestinal (CIP) | NEB | Cat#M0290 |
| Human p52(1–408) | Wang et al., 2012 | N/A |
| Mouse p50(10435) | This paper | N/A |
| Human Flag-Bcl3(1–446) | This paper | N/A |
| Human His-Bcl3(1–446) wt and various mutants | This paper | N/A |
| Rat full-length Erk2 | This paper | N/A |
| Human GST-IκBα(1–54) | This paper | N/A |
| Human His-IκBδ(482–899) | This paper | N/A |
| Human His-full-length IKK1 | Polley et al., 2013 | N/A |
| Human His-IKK2(11–700) | Polley et al., 2013 | N/A |
| Critical Commercial Assays | ||
| Dual-Luciferase Reporter Assay System | Promega | Cat#E1910 |
| RNeasy Mini Kit | QIAGEN | Cat#74106 |
| Super reverse transcriptase reaction kit | BioBharati LifeScience (BBL), Kolkata, India | Cat#BB-E0043 |
| Experimental Models: Cell Lines | ||
| RAW 264.7 cells | ATCC | TIB-71 |
| HeLa cells | ATCC | CCL-2 |
| HeLa cells with Akt2 knock down | This paper | N/A |
| HeLa cells with IKK1 knock down | This paper | N/A |
| HeLa cells with IKK2 knock down | This paper | N/A |
| HeLa cells with Erk2 knock down | This paper | N/A |
| HEK 293T cells | ATCC | CRL-3216 |
| HEK 293T cells with TRAF6 knock down | This paper | N/A |
| U2OS cells | ATCC | HTB-96 |
| U2OS cells with Bcl3 knock down | This paper | N/A |
| U2OS cells with Bcl3 knock down + S33E/S446E | This paper | N/A |
| U2OS cells with Bcl3 knock down + S33E/S446A | This paper | N/A |
| U2OS cells with Bcl3 knock down + S33E/S114A/S446A | This paper | N/A |
| Oligonucleotides | ||
| sgRNA against Bcl3 Ser33 sequence: 5’- TGCGGGCCTTGTCGTCCCCC-3’ | This paper | N/A |
| shRNA target sequences for human Erk2: 5’-CAAAGTTCGAGTAGCTATCAA-3’ | This paper | N/A |
| shRNA target sequences for human IKK1: 5’-GCGTGCCATTGATCTATATAA-3’ | This paper | N/A |
| shRNA target sequences for human IKK2: 5’-GGGCAGTCTTTGCACATCA -3’ | This paper | N/A |
| shRNA target sequences for human TRAF6: 5’-CGGAATTTCCAGGAAACTATT-3’ | This paper | N/A |
| shRNA target sequences for human Akt2: 5’-AGTCTACCCTGGTGGTCATAA-3’ | This paper | N/A |
| Recombinant DNA | ||
| Plasmids: pEYFPC1-YFP+NTFlag_Bcl3(1–446) wt and various mutants | This paper | N/A |
| Plasmids: pEYFPC1_Bcl3(1–446) wt and various mutants | This paper | N/A |
| Plasmids: pLV-NTFlag_Bcl3(1–446) wt and various mutants | This paper | N/A |
| Plasmids: pET29b_ Bcl3(1–446) wt and various mutants | This paper | N/A |
| Plasmid: pCMV-Myc_TBLR1 | Prof. Alain Chariot (University of Liege, Belgium) | N/A |
| Plasmids: pCMV6-HA-Akt1 wt and kinase dead (K179M) | Prof. Alexandra Newton (UCSD) | N/A |
| Plasmid: pcDNA3-CYLD wt | Prof. Guy Salvesen (SANFORD • BURNHAM • PREBYS Medical Discovery Institute) | N/A |
| Plasmid: pET11a_p52(1–408) | Wang et al., 2012 | N/A |
| Plasmid: pET11a_p50(1–435) | This paper | N/A |
| Plasmid: Cyclin D1 luciferase reporter | Wang et al., 2012 | N/A |
| Plasmid: P-Selectin luciferase reporter | Wang et al., 2012 | N/A |
METHOD DETAILS
Transient Protein Expression in Mammalian Cell Culture
Human Flag- or YFP-Bcl3, mouse HA-Akt, human HA-NIK, rat HA-Erk2, human Myc-TBLR1, Flag-CYLD were transfected into indicated cells using Lipofectamine 2000 (Invitrogen). Cell lysates were prepared 48 hours after transfection or as indicated.
Protein Expression and Purification
Mouse HA-Akt was a general gift from Dr. Alexandra Newton. Recombinant human p52(1–408), human His-Bcl3(1–446) wt and various mutants, and rat full-length Erk2, human GST-IκBα, human HisIκBδ(482–899) were expressed and purified from E.coli BL21(DE3) cells. Human His-full-length IKK1 and human His-IKK2(11–700) were expressed and purified from baculovirus sf9 cells as previously described (Polley et al., 2013). Baculovirus derived Akt was gift from Dr. Xiang Dong Fu. Human Flag-Bcl3(1–446) was transfected into HEK 293T cells using Lipofectamine 2000 for 48 hours and purified using Anti-Flag affinity resin. Ni-affinity resin (NTA) was obtained from BBL, Kolkata India, Flag-affinity resin (A2220) from Sigma and Glutathione Sepharose resin GE Healthcare.
Graded Dephosphorylation of Bcl3
Typically, 23 μg (70 μL in volume) of affinity purified Flag-tagged Bcl3(1–446) protein was incubated with 50 units of CIP (NEB) in NEB buffer 3 at room temperature. 10 μL aliquots of the reaction mixtures were removed from at each time point from 0 to 3 hours from the starting point of the reaction. The last aliquot was left at room temperature for overnight to see the complete de-phosphorylation of the Bcl3 protein. At each time point, the reaction was stopped by boiling the samples with SDS buffer at 95°C for 5 minutes. The samples were then analyzed by 12.5% SDS-PAGE.
Mass Spectrometric Analysis
Flag-Bcl3(1–446) was expressed in 10X 100mm dishes of HEK 293T cells in the presence or absence of 20 μM IKK Inhibitor XII for 48 hours and lysed in RIPA buffer. And then Flag affinity resin was used to purify Flag-Bcl3 proteins which were then subjected to mass spectrometric analysis.
In Gel Digest:
The gel slices were cut to 1mm by 1mm cubes and destained 3 times by first washing with 100 μl of 100 mM ammonium bicarbonate for 15 minutes, followed by addition of the same volume of acetonitrile (ACN) for 15 minutes. The supernatant was and samples were dried in a speedvac. Samples were then reduced by mixing with 200 μl of 100 mM ammonium bicarbonate-10 mM DTT and incubated at 56°C for 30 minutes. The liquid was re moved and 200 μl of 100 mM ammonium bicarbonate-55mM iodoacetamide was added to gel pieces and incubated at room temperature in the dark for 20 minutes. After the removal of the supernatant and one wash with 100 mM ammonium bicarbonate for 15 minutes, same volume of ACN was added to dehydrate the gel pieces. The solution was then removed and samples were dried in a speedvac. For digestion, enough solution of ice-cold trypsin (0.01 μg/μl) in 50 mM ammonium bicarbonate was added to cover the gel pieces and set on ice for 30 minutes. After complete rehydration, the excess trypsin solution was removed, replaced with fresh 50 mM ammonium bicarbonate, and left overnight at 37°C. The peptides were extracted twice by the addition of 50 μl of 0.2% formic acid and 5 % ACN and vortex mixing at room temperature for 30 minutes. The supernatant was removed and saved. A total of 50 μl of 50% ACN-0.2% formic acid was added to the sample, which was vortexed again at room temperature for 30 minutes. The supernatant was removed and combined with the supernatant from the first extraction. The combined extractions were analyzed directly by liquid chromatography (LC) in combination with tandem mass spectroscopy (MS/MS) using electrospray ionization.
LC-MS/MS:
Trypsin-digested peptides were analyzed by ultra high pressure liquid chromatography (UPLC) coupled with tandem mass spectroscopy (LC-MS/MS) using nano-spray ionization. The nanospray ionization experiments were performed using a TripleTof 5600 hybrid mass spectrometer (ABSCIEX) interfaced with nano-scale reversed-phase UPLC (Waters corporation nano ACQUITY ) using a 20cm-75 micron ID glass capillary packed with 2.5-μm C18 (130) CSH™ beads (Waters corporation). Peptides were eluted from the C18 column into the mass spectrometer using a linear gradient (5–80%) of ACN (Acetonitrile) at a flow rate of 250 μl/min for 1 hour. The buffers used to create the ACN gradient were: Buffer A (98% H2O, 2% ACN, 0.1% formic acid, and 0.005% TFA) and Buffer B (100% ACN, 0.1% formic acid, and 0.005% TFA). MS/MS data were acquired in a data-dependent manner in which the MS1 data was acquired for 250ms at m/z of 400 to 1250 Da and the MS/MS data was acquired from m/z of 50 to 2,000 Da. The Independent data acquisition (IDA) parameters were as follows; MS1-TOF acquisition time of 250 milliseconds, followed by 50 MS2 events of 48 milliseconds acquisition time for each event. The threshold to trigger MS2 event was set to 150 counts when the ion had the charge state +2, +3 and +4. The ion exclusion time was set to 4 seconds. Finally, the collected data were analyzed using Protein Pilot 5.0 (ABSCIEX) for peptide identifications.
Cytoplasmic/Nucleoplasmic Fractionation
Cytoplasmic/nuclear fractionation was performed as described (Savinova et al., 2009). Cells were lysed in two cell pellet volumns of cytoplasmic extract (CE) buffer containing 60 mM KCl, 10 mM HEPES-KOH pH 7.9, 1 mM EDTA, 0.5% NP-40, and 1 mM DTT supplemented with Protease Inhibitor Cocktail. Cytoplasmic extracts were collected by centrifuging at 500 × g. After collecting the cytoplasmic fractions, the nuclei were washed with additional five volumes of CE buffer and then lysed in three pellet volumes of nuclear extract buffer containing 60 mM KCl, 250 mM Tris-HCl pH 7.5, 1 mM EDTA, and 1 mM DTT supplemented with Proteasome Inhibitor Cocktail. The nuclear extracts were collected by freeze-thaw three times on dry ice and 37°C water bath and then centrifuging.
Flag-Immunoprecipitation
25 μg of Flag-tagged Bcl3(1–446) wt and different mutants’ nuclear extracts were incubated with recombinant non-tagged p52(1–408) protein in binding buffer (20 mM Tris pH 7.5, 150 mM NaCl, 0.5% NP-40, and 1 mM DTT) overnight at 4°C. Complexes of p52 with Flag-tagged Bcl3 were then immunoprecipited using 50 μL Anti-Flag affinity resin at 4°C for 2 hours. Bound complexes were washed three times with 1xTBS buffer supplemented with 0.1% NP-40, and dissolved in 4xSDS-dye by boiling at 95°C for 10 minutes followed by SDS-PAGE. Other immunoprecipitations were done using protein A plus, G plus or A/G plus agarose beads from BBL, Kolkata, India.
Luciferase Reporter Assays
HeLa cells were transciently transfected with Flag-p52(1–415) together with Flag-Bcl3(1–446) wt (or various mutants) expression vectors or empty Flag-vector, and the luciferase reporter DNA with specific κB DNA promoters (Wang et al., 2012). The total amount of plasmid DNA was kept constant for all assays. Transient transfections were carried out using Lipofectamine 2000 (Invitrogen). Renilla luciferase expression plasmid was co-transfected as an internal control. Cells were collected 48 hours after transfection. Luciferase activity assays were performed using Dual-Luciferase Reporter Assay System (Promega) following the manufacturer’s protocol. Data are represented as mean standard deviations (SD) of three independent experiments in triplicates.
EMSA
The oligonucleotides containing κB sites (Wang et al., 2012) used for EMSA were PAGE purified, end radiolabeled with 32P using T4-polynucleotide kinase (NEB) and [g-32P] ATP, and annealed to the complementary strands. Binding reaction mixtures contained indicated recombinant proteins or nuclear extracts at different concentrations, binding buffer (10mM Tris-HCl pH 7.5, 50mM NaCl, 10% (v/v) glycerol, 1% (v/v) NP-40, 1mM EDTA, 0.1mg/mL poly(dI-dC)), and ~10000cpm of 32P-labeled DNA. Reaction mixtures were incubated at room temperature for 20 minutes and analyzed by electrophoresis on a 5% (w/v) non-denatured polyacrylamide gel at 200V for 1 hour in 25mM Tris, 190mM glycine, and 1mM EDTA. The gels were then dried, exposed to a phosphorimager, and scanned by Typhoon scanner 9400 (Amershan Bioscience). Gels were quantified by using ImageQuant version5.2 (Molecular Dynamics).
Immunofluorescence
HeLa cells transfected with YFP-Bcl3 wt and different mutants for 48 hours were rinse with PBS. Cells were then imaged with YFP-LED for Bcl3 expressions. And then cells were stained with 5 μg/mL Hoechst 33342 for 5–10 minutes in dark for nuclei. Staining solution was then removed and the cells were rinsed 3 times in PBS following by imaging under microscope.
CRISPR-Cas9 Gene Editing
The Bcl3 KD U2OS cells were generated using CRISPR-Cas9 system according to the published protocol (Ran et al., 2013). In brief, the sgRNA against Bcl3 Ser33 fused with Cas9 plasmid was transfected into U2OS cells for 48 hours and then the cells were selected using 2μg/mL puromycin for 4 days for positive cells. The Bcl3 KD was validated by SURVEYOR assay. Different Bcl3 mutants cell lines were then generated using lentivirus. The viruses were generated by co-transfecting different pLV-Bcl3 mutant plasmids together with MDL, VSVG and REV plasmids in HEK 293T cells for 48 hours. The viruses were then harvested and filtered with 0.45μm filter; and used to infect the Bcl3 KD U2OS cells for 3 days. The expressions of all Bcl3 mutants were confirmed with western analysis followed by further experiments.
Wound-healing Scratch Assays
Different cells were seeded in 6-well plates at similar density and cultured until a confluent monolayer. These monolayer cells were then scored with a sterile pipette tip to leave a scratch in similar width. Wound closure was monitored by taking digitized images at various time intervals after the scratch. The images were taking using a using a Nikon Plan Apo λ 10X/0.45 objective with a 0.7x demagnifier and Nikon Eclipse Ti microscope with a sCMOS Zyla camera. The images were then analysed using Image-J software to measure the width of the scratches. All scratch assays were performed in triplicate.
Cell Proliferation Assays
Cell proliferation was analyzed using a WST-1 assay, with different cells seeded in 96-well plates at 103mL−1 in 100 μL/well. After 4 days culture, 10 μL WST-1 (Roche 05015944001) was added to each well and the incubation was continued for an additional 1 hour. The absorbance, representing the cell count in each well, was measured using a microculture plate reader (Tecan XFluor 4 Safire2) at an optical density of 450nm.
In Vitro Kinase Assays
50–100 nM of each different kinase was incubated with ~1 μg His-Bcl3 (wt or various mutants), 100 mM ATP, and 10 μCi−1 [γ−32P]-ATP in 50mM Tris-HCl (pH 7.5), 10mM MgCl, 1 mg/mL BSA, and 1mM DTT at 30°C for 30 minutes. Reactions were quenched by add ition of 4x SDS buffer and subsequent boiling; and then resolved on SDS-PAGE. The gels were dried and exposed to autoradiography film.
Generation of Stable Knock Down Cells
To generate lenti-virus, PLKO shRNA plasmids or control shRNA plasmid were transfected into HEK 293T cells together with packaging and envelope plasmids MDL, VSVG and Rev. Two days after transfection, viral supernatants were harvested and filtered through 0.45μm pore filters. Cells were infected with viral supernatants in the presence of 10μg/mL plolybrene for 24 hours twice. Cells were then selected with 1μg/mL puromycin 24 hours after infection. The knock down efficiency of target protein was confirmed by WB.
RNA isolation and RT-PCR
Total RNA was isolated using RNeasy Mini kit from QIAGEN (74106) and the RT-PCR was perfomed using a kit from BBL, Kolkata, India.
QUANTIFICATION AND STATISTICAL ANALYSIS
Where applicable, statistical parameters including sample size, precision measures (standard deviation, SD) and statistical significance are reported in the Figures and corresponding Figure Legends.
Supplementary Material
Highlights.
Identification of 27 phosphorylation sites on Bcl3
Akt phosphorylates Bcl3 promoting nuclear localization and stabilization of Bcl3
Phosphorylation by Erk2 and IKK1/2 convert Bcl3 into a transcriptional coregulator
Unphosphorylated Bcl3 have cellular proliferation and migration defects
ACKNOWLEDGEMENTS
We thank Prof. Alexandra Newton for providing the Akt expression plasmids, Prof. Guy Salvesen for the CYLD expression plasmid, and Dr. Yifeng Xia, Dr. Tushar Menon and Prof. Inder Verma for their help with the CRISPR-Cas9 system. We thank all members of the G. Ghosh laboratory for their valuable comments and discussions and Dr. Malini Sen and Mike Fernandez for reading the manuscript. V.Y.-F.W. was supported by the Science and Technology Development Fund, Macao S.A.R. (FDCT) project 023/2016/A1. This work is supported by National Institutes of Health grants GM (085490 and 071862) and AI064326.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES:
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, and Cohen P (2007). The selectivity of protein kinase inhibitors: a further update. The Biochemical journal 408, 297–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasier AR, Lu M, Hai T, Lu Y, and Boldogh I (2001). NF-kappa B-inducible BCL-3 expression is an autoregulatory loop controlling nuclear p50/NF-kappa B1 residence. The Journal of biological chemistry 276, 32080–32093. [DOI] [PubMed] [Google Scholar]
- Bundy DL, and McKeithan TW (1997). Diverse effects of BCL3 phosphorylation on its modulation of NF-kappaB p52 homodimer binding to DNA. The Journal of biological chemistry 272, 33132–33139. [DOI] [PubMed] [Google Scholar]
- Caamano JH, Perez P, Lira SA, and Bravo R (1996). Constitutive expression of Bc1–3 in thymocytes increases the DNA binding of NF-kappaB1 (p50) homodimers in vivo. Molecular and cellular biology 16, 1342–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadigan KM, and Waterman ML (2012). TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harbor perspectives in biology 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopher JA, Avitabile BG, Bamborough P, Champigny AC, Cutler GJ, Dyos SL, Grace KG, Kerns JK, Kitson JD, Mellor GW, et al. (2007). The discovery of 2-amino-3,5-diarylbenzamide inhibitors of IKK-alpha and IKK-beta kinases. Bioorganic & medicinal chemistry letters 17, 3972–3977. [DOI] [PubMed] [Google Scholar]
- Cogswell PC, Guttridge DC, Funkhouser WK, and Baldwin AS Jr. (2000). Selective activation of NF-kappa B subunits in human breast cancer: potential roles for NF-kappa B2/p52 and for Bcl-3. Oncogene 19, 1123–1131. [DOI] [PubMed] [Google Scholar]
- Dechend R, Hirano F, Lehmann K, Heissmeyer V, Ansieau S, Wulczyn FG, Scheidereit C, and Leutz A (1999). The Bcl-3 oncoprotein acts as a bridging factor between NF-kappaB/Rel and nuclear coregulators. Oncogene 18, 3316–3323. [DOI] [PubMed] [Google Scholar]
- Fujita T, Nolan GP, Liou HC, Scott ML, and Baltimore D (1993). The candidate proto-oncogene bcl-3 encodes a transcriptional coactivator that activates through NF-kappa B p50 homodimers. Genes & development 7, 1354–1363. [DOI] [PubMed] [Google Scholar]
- Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, and Skrzypek E (2015). PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic acids research 43, D512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keutgens A, Shostak K, Close P, Zhang X, Hennuy B, Aussems M, Chapelle JP, Viatour P, Gothot A, Fillet M, et al. (2010). The repressing function of the oncoprotein BCL-3 requires CtBP, while its polyubiquitination and degradation involve the E3 ligase TBLR1. Molecular and cellular biology 30, 4006–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado V, and Melendez-Zajgla J (2011). Role of Bcl-3 in solid tumors. Molecular cancer 10, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, and Fassler R (2006). Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-kappaB signaling. Cell 125, 665–677. [DOI] [PubMed] [Google Scholar]
- Massoumi R, Kuphal S, Hellerbrand C, Haas B, Wild P, Spruss T, Pfeifer A, Fassler R, and Bosserhoff AK (2009). Down-regulation of CYLD expression by Snail promotes tumor progression in malignant melanoma. The Journal of experimental medicine 206, 221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathas S, Johrens K, Joos S, Lietz A, Hummel F, Janz M, Jundt F, Anagnostopoulos I, Bommert K, Lichter P, et al. (2005). Elevated NF-kappaB p50 complex formation and Bcl-3 expression in classical Hodgkin, anaplastic large-cell, and other peripheral T-cell lymphomas. Blood 106, 4287–4293. [DOI] [PubMed] [Google Scholar]
- Nishikori M, Maesako Y, Ueda C, Kurata M, Uchiyama T, and Ohno H (2003). High-level expression of BCL3 differentiates t(2;5)(p23;q35)-positive anaplastic large cell lymphoma from Hodgkin disease. Blood 101, 2789–2796. [DOI] [PubMed] [Google Scholar]
- Nishikori M, Ohno H, Haga H, and Uchiyama T (2005). Stimulation of CD30 in anaplastic large cell lymphoma leads to production of nuclear factor-kappaB p52, which is associated with hyperphosphorylated Bcl-3. Cancer science 96, 487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan GP, Fujita T, Bhatia K, Huppi C, Liou HC, Scott ML, and Baltimore D (1993). The bcl-3 proto-oncogene encodes a nuclear I kappa B-like molecule that preferentially interacts with NF-kappa B p50 and p52 in a phosphorylation-dependent manner. Molecular and cellular biology 13, 3557–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno H, Doi S, Yabumoto K, Fukuhara S, and McKeithan TW (1993). Molecular characterization of the t(14;19)(q32;q13) translocation in chronic lymphocytic leukemia. Leukemia 7, 2057–2063. [PubMed] [Google Scholar]
- Ohno H, Takimoto G, and McKeithan TW (1990). The candidate proto-oncogene bcl-3 is related to genes implicated in cell lineage determination and cell cycle control. Cell 60, 991–997. [DOI] [PubMed] [Google Scholar]
- Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, and Donner DB (1999). NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 401, 82–85. [DOI] [PubMed] [Google Scholar]
- Polley S, Huang DB, Hauenstein AV, Fusco AJ, Zhong X, Vu D, Schrofelbauer B, Kim Y, Hoffmann A, Verma IM, et al. (2013). A structural basis for IkappaB kinase 2 activation via oligomerization-dependent trans auto-phosphorylation. PLoS biology 11, e1001581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nature protocols 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romashkova JA, and Makarov SS (1999). NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature 401, 86–90. [DOI] [PubMed] [Google Scholar]
- Roskoski R Jr. (2012). ERK1/2 MAP kinases: structure, function, and regulation. Pharmacological research 66, 105–143. [DOI] [PubMed] [Google Scholar]
- Rust HL, and Thompson PR (2011). Kinase consensus sequences: a breeding ground for crosstalk. ACS chemical biology 6, 881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savinova OV, Hoffmann A, and Ghosh G (2009). The Nfkb1 and Nfkb2 proteins p105 and p100 function as the core of high-molecular-weight heterogeneous complexes. Mol Cell 34, 591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornburg NJ, Pathmanathan R, and Raab-Traub N (2003). Activation of nuclear factor-kappaB p50 homodimer/Bcl-3 complexes in nasopharyngeal carcinoma. Cancer research 63, 8293–8301. [PubMed] [Google Scholar]
- Viatour P, Dejardin E, Warnier M, Lair F, Claudio E, Bureau F, Marine JC, Merville MP, Maurer U, Green D, et al. (2004a). GSK3-mediated BCL-3 phosphorylation modulates its degradation and its oncogenicity. Mol Cell 16, 35–45. [DOI] [PubMed] [Google Scholar]
- Viatour P, Dejardin E, Warnier M, Lair F, Claudio E, Bureau F, Marine JC, Merville MP, Maurer U, Green D, et al. (2004b). GSK3-mediated BCL-3 phosphorylation modulates its degradation and its oncogenicity. Molecular cell 16, 35–45. [DOI] [PubMed] [Google Scholar]
- Viatour P, Merville MP, Bours V, and Chariot A (2004c). Protein phosphorylation as a key mechanism for the regulation of BCL-3 activity. Cell Cycle 3, 1498–1501. [DOI] [PubMed] [Google Scholar]
- Wakefield A, Soukupova J, Montagne A, Ranger J, French R, Muller WJ, and Clarkson RW (2013). Bcl3 selectively promotes metastasis of ERBB2-driven mammary tumors. Cancer research 73, 745–755. [DOI] [PubMed] [Google Scholar]
- Wang VY, Huang W, Asagiri M, Spann N, Hoffmann A, Glass C, and Ghosh G (2012). The transcriptional specificity of NF-kappaB dimers is coded within the kappaB DNA response elements. Cell reports 2, 824–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterfield MR, Zhang M, Norman LP, and Sun SC (2003). NF-kappaB1/p105 regulates lipopolysaccharide-stimulated MAP kinase signaling by governing the stability and function of the Tpl2 kinase. Molecular cell 11, 685–694. [DOI] [PubMed] [Google Scholar]
- Westerheide SD, Mayo MW, Anest V, Hanson JL, and Baldwin AS Jr. (2001). The putative oncoprotein Bcl-3 induces cyclin D1 to stimulate G(1) transition. Molecular and cellular biology 21, 8428–8436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Liu Z, Cao J, Ma Q, Gao X, Wang Q, Jin C, Zhou Y, Wen L, and Ren J (2011). GPS 2.1: enhanced prediction of kinase-specific phosphorylation sites with an algorithm of motif length selection. Protein engineering, design & selection : PEDS 24, 255–260. [DOI] [PubMed] [Google Scholar]
- Zhang MY, Harhaj EW, Bell L, Sun SC, and Miller BA (1998). Bcl-3 expression and nuclear translocation are induced by granulocyte-macrophage colony-stimulating factor and erythropoietin in proliferating human erythroid precursors. Blood 92, 1225–1234. [PubMed] [Google Scholar]
- Zhang Q, Didonato JA, Karin M, and McKeithan TW (1994). BCL3 encodes a nuclear protein which can alter the subcellular location of NF-kappa B proteins. Molecular and cellular biology 14, 3915–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wang H, Claudio E, Brown K, and Siebenlist U (2007). A role for the IkappaB family member Bcl-3 in the control of central immunologic tolerance. Immunity 27, 438–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.