

Abstract
Since the early 2000s, numerous computational tools have been created and used to predict intrinsic disorder in proteins. At the present, the output from these algorithms is difficult to interpret in the absence of standards or references for comparison. There are many reasons to establish a set of standard-based guidelines to evaluate computational protein disorder predictions. This viewpoint explores a handful of these reasons, including standardizing nomenclature to improve communication, rigor and reproducibility, and making it easier for newcomers to enter the field. We also discuss an approach for reporting predicted disorder in single proteins with respect to whole proteomes. Our suggestions are not intended to be formulaic; they should be viewed as a starting point to establish guidelines for interpreting and reporting computational protein disorder predictions.
Keywords: bioinformatics, computational biology, intrinsically disordered protein, protein sequence, protein structure, proteomics
Intrinsically disordered proteins and protein regions fail to form a stable three-dimensional structure under physiological conditions, but instead remain unstructured, existing as highly dynamic conformational ensembles that vary over time and populations.[1–5] A protein may have one, many, or no intrinsically disordered regions, and some proteins are entirely disordered. Protein disorder is a difficult property to characterize experimentally,[6–9] “since [disordered domains] do not typically ‘freeze’ while their ‘pictures are taken’”.[10] These challenges have led to the development of numerous computational tools that predict disorder from primary structure alone.[11] For decades, these tools have played a critical role in protein disorder characterization efforts.[12]
Protein disorder is connected to many aspects of biology,[13–27] human health and disease,[25,28–30] and has brought in a large number of researchers from a wide range of disciplines. These researchers are experts in their own areas, but are often unfamiliar with the methods and tools used to study protein disorder. Experimentalists often use disorder prediction algorithms and related computational tools to assess the disorder content of a protein of interest as part of either: (i) a first-stage assessment to motivate and/or justify experimental characterization of a putatively intrinsically disordered protein, or (ii) a late-stage analysis to complement or explain molecular-level results obtained experimentally.[31–33] Both cases are important, however, the former is particularly relevant to our analysis because it implies that guidelines and/or tools are available to firmly interpret disorder predictions to decide whether experimental investigation is warranted or not. While useful guidelines are available for making intrinsic disorder predictions,[34] resources for interpreting these predictions are scarce; an example is QUARTER (QUality Assessment for pRotein inTrinsic disordEr pRedictions)[35], which is a toolbox of methods designed for ten different disorder predictors. The lack of generally accepted guidelines for interpreting the results of intrinsic disorder predictions leads to inconsistencies in the reported data and often causes confusion. Although disorder predictions are based on some objective criteria used to develop the corresponding computational tools, interpreting these predictions is rather subjective. The situation is not as simple as it seems, and, in essence, it resembles a case of a glass that is halfway filled with water, which for some is half-full, being half-empty for others. Furthermore, one should keep in mind that the results generated for a given protein by different predictors can be rather dissimilar. As a precis, let us consider the classification of the global disorder status of a given protein based on its content of predicted disordered residues. The protein is considered as highly ordered, if 0 to 10% of its sequence is disordered, or moderately disordered, if 11% to 30% of the sequence is disordered, or highly disordered, if 31% to 100% of the sequence is disordered.[36] The use of such criteria can be problematic, if it is implemented universally for a broad range of different disorder predictors. To justify the universal application of classification criteria across different algorithms one would expect, at minimum, agreement for the average disorder/order classification made. However, agreement is not always found. For example, a quick assessment of predicted disorder content in the H. sapiens proteome using the Disorder Atlas[37] proteome browser tool reveals expected values for percent disorder of ~24% (IUPred) and ~39% (DisEMBL-H). Applying the aforementioned criteria leads to different interpretations of the average disorder state predicted by these algorithms. A protein of average disorder content would be predicted as moderately disordered by IUPred and highly disordered by DisEMBL-H. To recap, the protein science community must employ criteria to meaningfully interpret disorder predictions. Unfortunately, adopting universal criteria is made difficult, if not impossible, by the large number of disorder prediction algorithms, the diverse definitions of intrinsic disorder employed to predict disorder, and the differences in the prevalence of features that correlate with intrinsic disorder in nature (i.e. the properties underlying different intrinsic disorder definitions). The goal of this Viewpoint is to set the stage to address these challenges. Prior to discussing this issue in detail, we will provide a brief overview of the resources available for computationally assessing intrinsic disorder, and the output of disorder prediction algorithms.
With numerous databases and over 60 disorder prediction algorithms available,[11] deciding on an appropriate approach to computationally analyze disorder can be a daunting task. There are three core computational resources for characterizing intrinsic disorder: (i) disorder prediction algorithms (including DisEMBL,[38] ESpritz,[39] IUPred,[40–42] PONDR,[43] and many others), (ii) databases of pre-computed intrinsic disorder predictions (D2P2,[44] MobiDB,[45–48] and datasets hosted on various digital data repositories[27]), and (iii) databases of experimentally characterized intrinsically disordered proteins or proteins with intrinsically disordered regions (e.g., DisProt,[49–51] IDEAL,[52,53] etc.). Let us focus primarily on disorder prediction algorithms and the accurate evaluation of their output. Following metrology principles, we define accuracy in the context of disorder prediction algorithms as the protein disorder measurement that delivers the true value of the intended measurand (object to be measured).[54] Importantly, we are not referring to the actual computational accuracies and confidence of the disorder prediction algorithms. Instead, we are focusing our attention on finding an appropriate means that can be used to accurately interpret the disorder predictions made by existing algorithms and seek to provide standards for comparison of such results.
Disorder prediction algorithms use many different types of input variables to identify intrinsic disorder, including but not limited to the assessment of relevant physicochemical properties[55] and patterns,[56–58] scoring matrices[59,60] and scales,[61] and various applications of Shannon’s entropy[62] to assess disorder-relevant properties.[63,64] While the definitions of disorder employed are diverse, the general operation of these algorithms is straightforward. After submitting a primary sequence for disorder prediction, the residue-by-residue disorder propensity values are typically returned. Next, end users employ a binary classification system to label each residue as either ordered or disordered by comparing residue-specific scores to an algorithm-specific threshold. Figure 1 displays a summary of this classification process. After classifying the residues, users then compute the percentage of disordered residues, as well as the length and location of continuous stretches of disordered residues. Another approach used to analyze the disorder status of a given protein involves inspecting distribution peculiarities of actual per-residue disorder scores within the protein sequence, which typically range from 0 to 1 (Figure 1 displays one such plot). Here, users commonly rely on the visual analysis of the resulting disorder profiles to gain some useful information. We reiterate that both of these approaches involves subjective judgement.
Figure 1. Residue level binary classification of order/disorder.
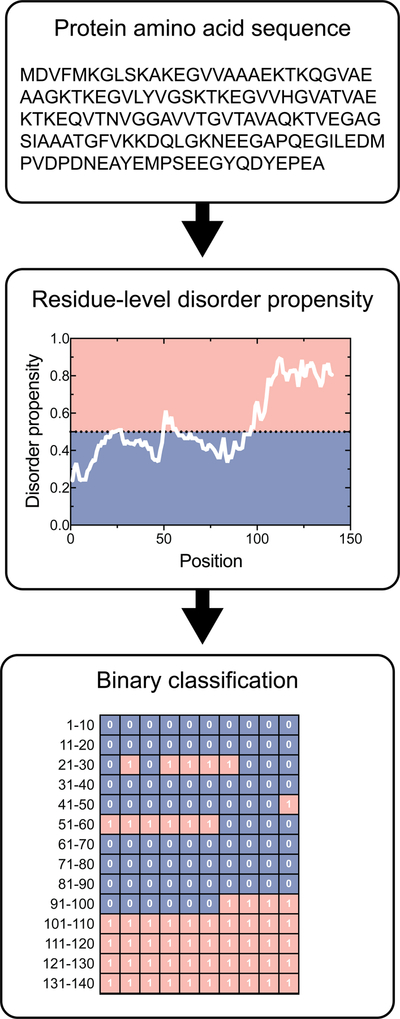
The amino acid sequence of a protein of interest is input into a disorder prediction algorithm, which returns residue-by-residue disorder propensity values. These disorder propensity values are compared against a defined threshold to classify a residue as either ordered or disordered. IUPred-disorder predictions are displayed for the H. sapiens alpha-synuclein protein (P37840). IUPred uses a threshold value of 0.5 to assign ordered and disordered residues. Residues that fall within the red region are disordered, whereas residues that fall in the blue region are ordered.
The output(s) of the aforementioned procedures lead to the central problem of this Viewpoint: How does one interpret these predictions to decide whether intrinsic disorder is a significant feature of the protein of interest? Needless to say, only experimental investigation will truly determine the role or influence of intrinsic disorder on the function of a particular protein––and these investigations require substantial time and financial investments. Nevertheless, tools and guidelines that enable the rigorous interpretation of disorder predictions are needed to help researchers decide whether experimental investigation is warranted.
To further illustrate the need for disorder interpretation tools, it is important to distinguish standards for accurate measurement from standards for comparison. Disorder prediction algorithms use standards for accurate measurement at two different stages: (i) internally-defined standards used during the initial stages of algorithm development, which is a pool of protein sequences and corresponding structural information defined by the algorithm developers themselves, and (ii) community-wide standards used at later stages of development that serves to calibrate and benchmark performance of published algorithms against a centralized pool of characterized proteins. The latter is the focus of the Critical Assessment of Structure Predictions (CASP) experiment.[65] For a given disorder prediction tool, CASP determines prediction accuracy against characterized targets to evaluate the effectiveness of different methods in predicting intrinsic disorder. Disorder prediction algorithms serve to predict some objective measure of disorder, and the CASP competition ultimately compares this predicted value against a set of known measurands (characterized structured and unstructured proteins). The balanced accuracy, specificity, sensitivity, and Matthews Correlation Coefficient are commonly used as performance metrics. CASP further provides a platform to compare the accuracy of predictions made by competing and/or complementary algorithms. The balanced accuracy (Acc) is calculated using Eq. 1. True positives, true negatives, false positives, and false negatives are denoted by pt, nt, pf, nf, respectively. The specificity is computed as the fraction of correctly predicted negatives, whereas the sensitivity is computed as the fraction of correctly predicted positives. The performance of the binary classification system is further assessed using the Matthews Correlation Coefficient (MCC), Eq. 2, which returns a value in the interval [−1, 1] describing the agreement between a predicted classification and observed classification (−1 indicates complete disagreement; +1 indicates complete agreement; 0 indicates that the prediction is no better than random). Equations (1) and (2) are:
| Eq. 1 |
| Eq. 2 |
CASP further provides a platform to compare the accuracy of predictions made by competing and/or complementary algorithms. In addition to CASP, a number of other studies have rigorously compared and ranked disorder prediction algorithms[66,67].
While the protein disorder community has established a clear standard for measurement and calibration through its inclusion in CASP, it is important to recognize that these standards are not standards for comparison because they do not facilitate the interpretation of the predictions. We also note that consensus predictions made by meta-predictors[43,68–70] do not provide this information either. While meta-predictors improve accuracy by homing in on the regions of the sequence having the greatest likelihood of disorder, they do not indicate whether the results are conspicuous and/or anomalous. Abstractly, algorithm developers use measurement standards to calibrate their tools to measure disorder, just as the developer of a mass balance instrument would use objects with a defined mass in kilograms to calibrate their instrument to measure mass. In the case of a mass balance, an individual using the balance cannot use the reported value in kilograms to determine whether they are heavy or light. Instead, this heavy versus light classification is determined by comparing the reported mass against threshold values derived from a population of measured masses. In the case of disorder prediction algorithms, where a researcher uses a high-performing algorithm to predict disorder content in a protein of interest, what do these returned values/predictions actually mean? Similar to the mass balance analogy, these predicted values are virtually meaningless in isolation, without standards for comparison that put the measurand into context. To address this problem, we proposed proteome-based quantitative guidelines for disorder prediction algorithms.[26,27] These guidelines enable users to evaluate whether a protein is disordered or not with respect to a reference standard – a central measurement (median, mean) for a specified proteome – for comparative purposes.
Resources for interpreting intrinsic disorder predictions must follow rigor and reproducibility guidelines. Scientific rigor and reproducibility is a topic that has recently attracted considerable attention. The US National Institutes of Health’s “Principles and Guidelines for Reporting Preclinical Research” to encourage the use of standards for rigorous reporting of research results.[71] In any scientific field, reproducibility can be achieved if three conditions are met:
Scientists agree on scientific methodologies based on consensus of a majority of people carrying out studies in the field.
Scientists agree on standards for measuring, reporting and comparing results based on consensus of a majority of people carrying out studies in the field.
Scientists follow the best practices agreed upon for conditions (1) and (2).
In our opinion, population-based reporting guidelines are needed to meaningfully interpret intrinsic disorder predictions. Population statistics directly provide the standing of predicted disorder features in a protein of interest (or intrinsically disordered region) relative to the rest of the proteome. A population-based solution is provided by our quantitative proteome-based guidelines for reporting and characterizing intrinsic disorder.[26] These guidelines are implemented in a web-based toolkit, known as Disorder Atlas,[71] that enables users to objectively interpret disorder predictions with respect to the whole proteome. Disorder Atlas employs a rich user interface that implements pre-computed disorder predictions together with our previously published disorder interpretation guidelines.[26] As a result, this software further enables users to conduct population-level analyses of intrinsic disorder regardless of their computational background. Figure 2 displays a simple example of using population-statistics to interpret intrinsic disorder predictions of total disorder content and long disordered regions in two disordered proteins and two ordered proteins. α-Casein[72–74] and Spinophilin[75] are characterized as intrinsically disordered proteins, whereas serum albumin[76] and myoglobin[77] are considered as ordered proteins. Figure 2 demonstrates these disorder/order classifications are captured by the relative standing with respect to the Homo sapiens proteome.
Figure 2. Population-based interpretation of protein disorder predictions.
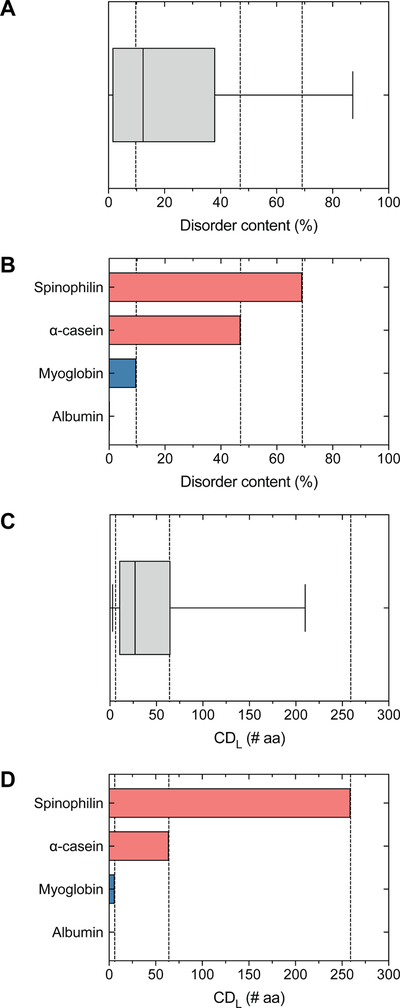
The standing of IUPred-predicted (A, B) disorder content and (C, D) the longest continuous disordered region (CDL) with respect to the Homo sapiens proteome for two ordered proteins (Albumin, P02768; Myoglobin, P02144) and two disordered proteins (alpha-casein, P47710; Spinophilin, Q96SB3). IUPred predicts Albumin contains 0.2% disorder (1st percentile) and lacks continuous disordered regions, Myoglobin contains 9.7% disorder (46th percentile) and a 6 AA CDL (15th percentile), alpha-casein contains 47.0% disorder (80th percentile) with a 64 AA CDL (74th percentile), and Spinophilin contains 69.1% disorder (90th percentile) and a 259 aa CDL (97th percentile).
In addition to improving the interpretation of disorder predictions, standards for comparison will permit the scientific community to define what intrinsic disorder is in a more quantitative and rigorous fashion. One of the hallmarks of intrinsically disordered proteins and intrinsically disordered protein regions is the amino acid composition. Disordered sequences exhibit a relatively low proportion of hydrophobic and aromatic residues, and a relatively high proportion of charged and polar residues.[4,56,78–81] Yet, for a given stretch of amino acids, it remains unclear what proportion of the total amino-acid sequence needs to be composed of hydrophobic/aromatic versus charged/polar to drive the region into a disordered regime. Is there a particular number of consecutive disordered amino acids that makes the intrinsically disordered region stand out with respect to other proteins containing intrinsically disordered regions?
More broadly, the increased interest in intrinsic disorder warrants the establishment of a central web service to facilitate the computational characterization of intrinsic disorder in a protein of interest. Experimentalists are the target user base of this web service, and as such, it must serve as a comprehensive resource that predicts intrinsic disorder from a sequence of interest and allows these predictions to be objectively interpreted with confidence. The envisioned web service would be a collaborative project built by an expert panel in the protein disorder community. A tentative list of milestones toward realizing this web service includes:
Establish a governing board comprised of experts in the disorder community.
Establish a curated list of the top metrics for predicting intrinsic disorder from protein sequences, and definitions of intrinsic disorder based on these metrics.
- Establish a community-sponsored list of recommended disorder prediction algorithms.
- Based on the agreed upon definitions, experts should publish a categorized list of merit-based recommendations for the highest performing prediction algorithms for each of primary definition(s) of intrinsic disorder. This list should be based on CASP evaluations, as well as other quality assessments within the community, and should be updated every few years as new algorithms are continuously developed and evaluated.
Agree upon and publish algorithm-specific and proteome-specific standards for comparison to objectively interpret the output of the recommended intrinsic disorder prediction algorithms.
Establish a centralized web service with recommended disorder prediction algorithms and tools for objectively interpreting the predictions made by these algorithms.
The central web service could be set-up as an addition to an existing service or by combining several existing architectures, but must provide standards for comparison for each community-recommended prediction algorithm. Without standards for comparison, we reiterate the point that the results cannot be meaningfully interpreted and are of little use to experimentalists. This service should also include features for comparing results from user-specified combinations of the supported disorder prediction algorithms, and should further include structural information where possible. While services exist that support some of these features, a comprehensive service that incorporates all of these features does not exist to our knowledge.
Given the uncertainties in the analysis and interpretation of protein disorder predictions, we present a standardization problem in this Viewpoint to open an active dialogue between researchers to develop standards for comparison. Here we discussed general procedures and available methods in one article, and encourage the use of quantitative guidelines to report and interpret results from those methods. The views expressed here are not intended as a set mandate, but instead offer suggested approaches along with descriptions of how predicted disorder can be assessed quantitatively, as well as certain caveats or cautions that users should be mindful of. For example, a general statistical standard for comparison might not correlate with biochemical function. An intrinsically disordered region might contain amino-acid residues that interact and fold upon binding a molecule in the surrounding environment, giving a critical functional importance to the intrinsically disordered region. How to effectively capture functional influence using a combination of pattern recognition, structural bioinformatics, and standards for comparison remains an open question for the intrinsic disorder community. Of importance, we note our guidelines are not meant to deter or hinder data interpretation; they are only meant to guide, not to restrict. This work is a starting point to begin establishing reference points for interpreting disorder, and through modification with more advanced metrics it is envisioned that the presented guidelines will evolve to become even more useful.
It is worth noting that guidelines are meant to build the protein disorder community and maintain a level of collegiality. Straightforward explanations of methods and interpretations opens the field to new researchers, fosters interactions among researchers to improve the guidelines, and increases rigor and reproducibility in the field. More importantly, guidelines themselves represent a community effort; establishing useful guidelines for interpreting protein disorder data cannot be achieved without surveying the expertise of many members of the protein disorder, biochemical, and biophysical communities. At this early stage, it is important that these guidelines are both simple and flexible. Flexibility is particularly important for allowing the incorporation of changes from an active field of research that is frequently cultivating new findings, methodologies, and thoughts on data interpretation.
We hope that our initial guidelines provide a starting point for standardizing protein disorder nomenclature and interpretation to enable better communication and improved reproducibility within the field. We further envision that these guidelines will have the added benefit of lowering the barrier to entry for researchers interested in beginning protein disorder investigations.
Acknowledgements
This work was partially supported by the University of Michigan Protein Folding Diseases Initiative, the University of Michigan Medical School Research Discovery Fund, and the National Institutes of Health grant R01 DK108921.
List of abbreviations:
- CASP
Critical Assessment of Structure Predictions
References
- [1].Schweers O, Schönbrunn-Hanebeck E, Marx A, Mandelkow E, J. Biol. Chem 1994, 269, 24290. [PubMed] [Google Scholar]
- [2].Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT, Biochem 1996, 35, 13709. [DOI] [PubMed] [Google Scholar]
- [3].Wright PE, Dyson HJ, J Mol Biol 1999, 293, 321. [DOI] [PubMed] [Google Scholar]
- [4].Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z, J Mol Graph Model 2001, 19, 26. [DOI] [PubMed] [Google Scholar]
- [5].Dyson HJ, Wright PE, Nat Rev Mol Cell Biol 2005, 6, 197. [DOI] [PubMed] [Google Scholar]
- [6].Uversky VN, Protein Sci 2002, 11, 739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Johnson DE, Xue B, Sickmeier MD, Meng J, Cortese MS, Oldfield CJ, Le Gall T, Dunker AK, Uversky VN, J Struct Biol 2012, 180, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Receveur-Brechot V, Durand D, Curr Protein Pept Sci 2012, 13, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Uversky VN, Biochim Biophys Acta Proteom 2013, 1834, 932. [DOI] [PubMed] [Google Scholar]
- [10].Uversky VN, Protein Sci 2013, 22, 693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Meng F, Uversky V, Kurgan L, Curr Protoc Protein Sci 2017, 88, 2.16.1. [DOI] [PubMed] [Google Scholar]
- [12].Obradovic Z, Peng K, Vucetic S, Radivojac P, Brown CJ, Dunker AK, Proteins: Structure, Function, and Bioinformatics 2003, 53, 566. [DOI] [PubMed] [Google Scholar]
- [13].Karush F, J Am Chem Soc 1950, 72, 2705. [Google Scholar]
- [14].Parker D, Ferreri K, Nakajima T, LaMorte VJ, Evans R, Koerber SC, Hoeger C, Montminy MR, Mol Cell Biol 1996, 16, 694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Richards JP, Bächinger HP, Goodman RH, Brennan RG, J. Biol. Chem 1996, 271, 13716. [DOI] [PubMed] [Google Scholar]
- [16].Iakoucheva LM, Brown CJ, Lawson JD, Obradović Z, Dunker AK, J Mol Biol 2002, 323, 573. [DOI] [PubMed] [Google Scholar]
- [17].Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT, J Mol Biol 2004, 337, 635. [DOI] [PubMed] [Google Scholar]
- [18].Gunasekaran K, Tsai C-J, Nussinov R, J Mol Biol 2004, 341, 1327. [DOI] [PubMed] [Google Scholar]
- [19].Uversky VN, Oldfield CJ, Dunker AK, J Mol Recognit 2005, 18, 343. [DOI] [PubMed] [Google Scholar]
- [20].Vucetic S, Xie H, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN, J Proteome Res 2007, 6, 1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Uversky VN, Obradovic Z, J Proteome Res 2007, 6, 1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Potoyan DA, Papoian GA, J Am Chem Soc 2011, 133, 7405. [DOI] [PubMed] [Google Scholar]
- [23].Xue B, Dunker K, Uversky V, J Biomol Struct Dyn 2012, 30, 137. [DOI] [PubMed] [Google Scholar]
- [24].Varadi M, Zsolyomi F, Guharoy M, Tompa P, PLoS ONE 2015, 10, e0139731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wang B, Merillat SA, Vincent M, Huber AK, Basrur V, Mangelberger D, Zeng L, Elenitoba-Johnson K, Miller RA, Irani DN, Dlugosz AA, Schnell S, Scaglione KM, Paulson HL, J Biol Chem 2016, 291, 3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Vincent M, Whidden M, Schnell S, Biophys Chem 2016, 213, 6. [DOI] [PubMed] [Google Scholar]
- [27].Vincent M, Schnell S, Sci Data 2016, 3, 160045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cheng Y, LeGall T, Oldfield CJ, Dunker AK, Uversky VN, Biochem 2006, 45, 10448. [DOI] [PubMed] [Google Scholar]
- [29].Uversky VN, Oldfield CJ, Dunker AK, Annu Rev Biophys 2008, 37, 215. [DOI] [PubMed] [Google Scholar]
- [30].Uversky VN, Oldfield CJ, Midic U, Xie H, Xue B, Vucetic S, Iakoucheva LM, Obradovic Z, Dunker AK, BMC Genom 2009, 10(Suppl 1), S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bracken C, Iakoucheva LM, Romero PR, Dunker AK, Curr Opin Struct Biol 2004, 14, 570. [DOI] [PubMed] [Google Scholar]
- [32].Uversky VN, Radivojac P, Iakoucheva LM, Obradovic Z, Dunker AK, Methods Mol. Biol 2007, 408, 69. [DOI] [PubMed] [Google Scholar]
- [33].Oldfield CJ, Xue B, Van Y-Y, Ulrich EL, Markley JL, Dunker AK, Uversky VN, Biochim Biophys Acta Proteom 2013, 1834, 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lieutaud P, Ferron F, Uversky AV, Kurgan L, Uversky VN, Longhi S, Intrinsically Disord Proteins 2016, 4, e1259708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hu G, Wu Z, Oldfield C, Wang C, Kurgan L, Bioinformatics 2018, DOI 10.1093/bioinformatics/bty881. [DOI] [PubMed]
- [36].Rajagopalan K, Mooney SM, Parekh N, Getzenberg RH, Kulkarni P, J Cell Biochem 2011, 112, 3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Vincent M, Schnell S, bioRxiv 2017, DOI 10.1101/060699. [DOI]
- [38].Linding R, Jensen LJ, Diella F, Bork P, Gibson TJ, Russell RB, Structure 2003, 11, 1453. [DOI] [PubMed] [Google Scholar]
- [39].Walsh I, Martin AJM, Di Domenico T, Tosatto SCE, Bioinformatics 2012, 28, 503. [DOI] [PubMed] [Google Scholar]
- [40].Dosztanyi Z, Csizmok V, Tompa P, Simon I, Bioinformatics 2005, 21, 3433. [DOI] [PubMed] [Google Scholar]
- [41].Dosztányi Z, Csizmók V, Tompa P, Simon I, J Mol Biol 2005, 347, 827. [DOI] [PubMed] [Google Scholar]
- [42].Dosztányi Z, Protein Sci 2018, 27, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Xue B, Dunbrack RL, Williams RW, Dunker AK, Uversky VN, Biochim Biophys Acta 2010, 1804, 996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Oates ME, Romero P, Ishida T, Ghalwash M, Mizianty MJ, Xue B, Dosztányi Z, Uversky VN, Obradovic Z, Kurgan L, Dunker AK, Gough J, Nucleic Acids Res 2012, 41, D508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Martin AJM, Walsh I, Tosatto SCE, Bioinformatics 2010, 26, 2916. [DOI] [PubMed] [Google Scholar]
- [46].Di Domenico T, Walsh I, Martin AJM, Tosatto SCE, Bioinformatics 2012, 28, 2080. [DOI] [PubMed] [Google Scholar]
- [47].Potenza E, Domenico TD, Walsh I, Tosatto SCE, Nucleic Acids Res 2015, 43, D315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Piovesan D, Tosatto SCE, Bioinformatics 2018, 34, 122. [DOI] [PubMed] [Google Scholar]
- [49].Vucetic S, Obradovic Z, Vacic V, Radivojac P, Peng K, Iakoucheva LM, Cortese MS, Lawson JD, Brown CJ, Sikes JG, Newton CD, Dunker AK, Bioinformatics 2005, 21, 137. [DOI] [PubMed] [Google Scholar]
- [50].Sickmeier M, Hamilton JA, LeGall T, Vacic V, Cortese MS, Tantos A, Szabo B, Tompa P, Chen J, Uversky VN, Obradovic Z, Dunker AK, Nucleic Acids Res 2007, 35, D786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Piovesan D, Tabaro F, Mičetić I, Necci M, Quaglia F, Oldfield CJ, Aspromonte MC, Davey NE, Davidović R, Dosztányi Z, Elofsson A, Gasparini A, Hatos A, Kajava AV, Kalmar L, Leonardi E, Lazar T, Macedo-Ribeiro S, Macossay-Castillo M, Meszaros A, Minervini G, Murvai N, Pujols J, Roche DB, Salladini E, Schad E, Schramm A, Szabo B, Tantos A, Tonello F, Tsirigos KD, Veljković N, Ventura S, Vranken W, Warholm P, Uversky VN, Dunker AK, Longhi S, Tompa P, Tosatto SCE, Nucleic Acids Res 2017, 45, D219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Fukuchi S, Sakamoto S, Nobe Y, Murakami SD, Amemiya T, Hosoda K, Koike R, Hiroaki H, Ota M, Nucleic Acids Res 2012, 40, D507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Fukuchi S, Amemiya T, Sakamoto S, Nobe Y, Hosoda K, Kado Y, Murakami SD, Koike R, Hiroaki H, Ota M, Nucleic Acids Res 2014, 42, D320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Joint Committee for Guides in Metrology (JCGM), International Vocabulary of Metrology - Basic and General Concepts and Associated Terms (VIM), BIPM, Sevres, France, 2012. [Google Scholar]
- [55].Kyte J, Doolittle RF, Journal of Molecular Biology 1982, 157, 105. [DOI] [PubMed] [Google Scholar]
- [56].Uversky VN, Gillespie JR, Fink AL, Proteins 2000, 41, 415. [DOI] [PubMed] [Google Scholar]
- [57].Martin EW, Holehouse AS, Grace CR, Hughes A, Pappu RV, Mittag T, J Am Chem Soc 2016, 138, 15323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Holehouse AS, Das RK, Ahad JN, Richardson MOG, Pappu RV, Biophys J 2017, 112, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Radivojac P, Obradovic Z, Brown CJ, Dunker AK, Pac Symp Biocomput 2002, 7, 589. [PubMed] [Google Scholar]
- [60].Midic U, Dunker AK, Obradovic Z, in Proceedings of the KDD-09 Workshop on Statistical and Relational Learning in Bioinformatics, New York, NY, 2009, 10.1145/1562090.1562096. [DOI] [Google Scholar]
- [61].Campen A, Williams RM, Brown CJ, Meng J, Uversky VN, Dunker AK, Protein Pept Lett 2008, 15, 956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Shannon CE, Bell System Technical Journal 1948, 27, 379. [Google Scholar]
- [63].Chen JW, Romero P, Uversky VN, Dunker AK, J. Proteome Res 2006, 5, 879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].DeForte S, Uversky VN, Protein Sci 2016, 25, 676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Moult J, Pedersen JT, Judson R, Fidelis K, Proteins 1995, 23, ii. [DOI] [PubMed] [Google Scholar]
- [66].Walsh I, Giollo M, Di Domenico T, Ferrari C, Zimmermann O, Tosatto SCE, Bioinformatics 2015, 31, 201. [DOI] [PubMed] [Google Scholar]
- [67].Peng Z-L, Kurgan L, Curr. Protein Pept. Sci 2012, 13, 6. [DOI] [PubMed] [Google Scholar]
- [68].Ishida T, Kinoshita K, Bioinformatics 2008, 24, 1344. [DOI] [PubMed] [Google Scholar]
- [69].Schlessinger A, Punta M, Yachdav G, Kajan L, Rost B, PLoS ONE 2009, 4, e4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kozlowski LP, Bujnicki JM, BMC Bioinform 2012, 13, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Principles and Guidelines for Reporting Preclinical Research, 2017. [https://www.nih.gov/research-training/rigor-reproducibility/principles-guidelines-reporting-preclinical-research]
- [72].Holt C, Sawyer L, Protein Eng Des Sel 1988, 2, 251. [DOI] [PubMed] [Google Scholar]
- [73].Sawyer L, Holt C, J Dairy Sci 1993, 76, 3062. [DOI] [PubMed] [Google Scholar]
- [74].Melnikova DL, Skirda VD, Nesmelova IV, J Phys Chem B 2017, 121, 2980. [DOI] [PubMed] [Google Scholar]
- [75].Ragusa MJ, Dancheck B, Critton DA, Nairn AC, Page R, Peti W, Nat Struct Mol Biol 2010, 17, 459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Sugio S, Kashima A, Mochizuki S, Noda M, Kobayashi K, Protein Eng 1999, 12, 439. [DOI] [PubMed] [Google Scholar]
- [77].Hubbard SR, Hendrickson WA, Lambright DG, Boxer SG, J Mol Biol 1990, 213, 215. [DOI] [PubMed] [Google Scholar]
- [78].Williams RM, Obradovi Z, Mathura V, Braun W, Garner EC, Young J, Takayama S, Brown CJ, Dunker AK, Pac Symp Biocomput 2001, 89. [DOI] [PubMed] [Google Scholar]
- [79].Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK, Proteins 2001, 42, 38. [DOI] [PubMed] [Google Scholar]
- [80].Radivojac P, Iakoucheva LM, Oldfield CJ, Obradovic Z, Uversky VN, Dunker AK, Biophys J 2007, 92, 1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Vacic V, Uversky VN, Dunker AK, Lonardi S, BMC Bioinformatics 2007, 8, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]